
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced bi-stability in a ruthenium alkynyl spiropyran complex†
Mark C.
Walkey
a,
Lindsay T.
Byrne
ab,
Matthew J.
Piggott
a,
Paul J.
Low
a and
George A.
Koutsantonis
*a
aSchool of Chemistry and Biochemistry, University of Western Australia, 35 Stirling Highway, Crawley, 6009, Australia. E-mail: george.koutsantonis@uwa.edu.au
bCentre for Microscopy, Characterisation and Analysis, University of Western Australia, Crawley, Western Australia 6009, Australia
First published on 15th April 2015
Abstract
The inclusion of a ligated ruthenium moiety to ethynyl spiropyran, 5′-ethynyl-1′,3′,3′-trimethyl-6-nitrospiro[chromene-2,2′-indoline], has been shown to increase the lifetime of the ring-opened merocyanine form twentyfold. Calculations suggest that the higher barrier to thermal reversion of the merocyanine form of the metal alkynyl complex arises from the capacity for greater delocalisation of charge consequent of the presence of the ruthenium moiety. The complex may provide a different switching mechanism to the 5,5′-dithienylperfluorocyclopentene electrode decoupling seen previously.
Molecular switches can be broadly defined as molecular species that undergo reversible changes between two or more distinct structures in response to some thermal, chemical, electronic or, most commonly, photochemical stimulus. In addition to the intrinsic interest in such bi- (or multi-) stable compounds, these elementary molecular machines and photochromes attract considerable attention arising from their potential applications in sensing, molecular-scale data storage and processing, and for optical or electronic signal modulation.1–4 Each of these potential application areas has different requirements in terms of the nature of the triggering stimulus and the lifetime of the ‘switched’ state. Surprisingly, despite the essentially limitless chemical space, the number of structural motifs that have been established as robust molecular switching elements is relatively sparse; prominent examples of the molecular architectures that are able to undergo structural isomerization between two or more distinct states, and have been investigated as potential molecular switches, include azobenzenes,5–7 stilbenes,8–10 spiropyrans,5,11–13 diarylethenes,14–16 and fulgides.5,17–19
Although the great majority of photochemically controlled molecular switches explored to date have been based on organic structures, the incorporation of photochromic entities into metal complexes can provide a number of advantages, allowing the combination of magnetic, electrochemical, and optical properties of the metal complexes with the photochromic reaction,20–25 providing access to a broader diversity of molecular architectures and permitting an element of control over the switching characteristics.26 For example, diarylethene moieties, specifically 5,5′-dithienylperfluorocyclopentene (DTE), which represents perhaps the archetypal molecular switch, has been incorporated into metal alkynyl complexes.23,27–34 In one example, the metal was able to mediate the switching of cubic nonlinear optical properties of the molecule utilising electro-, halo- and photochromic phenomena specific to the organometallic complex.27 In another organometallic DTE example, the metal also serves to decouple the molecule from the electrodes thereby allowing the photoreversion reaction to the closed form of the DTE to proceed efficiently.29
Whilst DTE is perhaps the best known of the photochromic molecular switches, the synthetic accessibility of the spiropyran motif, and its photochromic nature, make it an attractive basis for alternative molecular switch design and development. Like other light-actuated switches it has advantages and disadvantages; chief amongst the latter are photo-degradation, which can be ameliorated by covalent immobilisation,35 and the relatively fast rate of thermal reversion from the photo-activated open (conducting, merocyanine) state to the closed (resistive, spiropyran) form (e.g., Scheme 1).36 The factors influencing the thermal and photoinitiated opening and closing of spiropyrans have been extensively explored.37,38
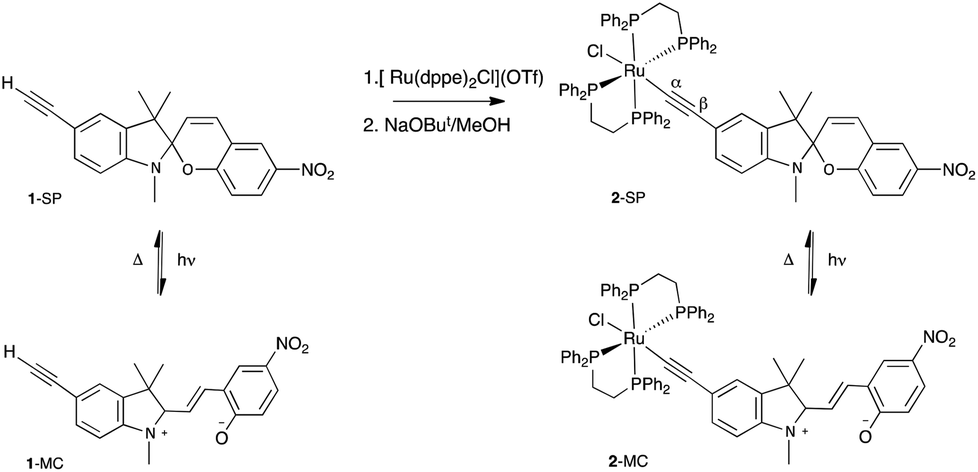 | ||
| Scheme 1 Synthesis of the ruthenium ethynylspiropyran, 2. | ||
We are exploring the incorporation of the spiropyran motif into metal complexes39via an alkynyl linkage utilising the ligand 5′-ethynyl-1′,3′,3′-trimethyl-6-nitrospiro[chromene-2,2′-indoline] (1, Scheme 1), with a view to optimising the photoswitching properties, and this communication describes our initial efforts in this area.
Ethynyl-substituted spiropyrans have been previously prepared from condensation of 5-ethynylsalicylaldehyde with Fischer's base40 or from the corresponding formyl spiropyran via a Wittig reaction with (bromomethyl)triphenylphosphonium bromide and subsequent HBr elimination.41 Ligand 1 was prepared41 for the current work by an improved procedure involving condensation of the appropriately substituted trimethylsilylethynyl indolium salt with 5-nitrosalicylaldehyde, in the presence of piperidine, in excellent yield (ESI†). Subsequent de-protection gave the required terminal alkyne.
The metal complex was prepared using established methodology;42–44 thus reaction of 1-SP with [Ru(dppe)2Cl](OTf) gave the ruthenium alkynyl complex, [Ru{C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C(1′,3′,3′-trimethyl-6-nitrospiro[chromene-2,2′-indoline])}(dppe)2] (2-SP, Scheme 1) in good yield, after deprotonation of the intermediate vinylidene species. Complex 2-SP was characterised spectroscopically, with the presence of the triple bond inferred from the absorption at 2014 cm−1 in the IR spectrum. The 1H and 13C NMR spectra of 2-SP were consistent with the amalgamation of the ligated ruthenium and ethynyl ligand. In particular, the alkynyl α-carbon was assigned to the signal observed at 127.0 ppm, showing coupling to the four equivalent phosphorus atoms, with the signal for the β-carbon at 116.3 ppm. The phosphorus nuclei gave a peak at 69.4 ppm in the 31P NMR spectrum.
C(1′,3′,3′-trimethyl-6-nitrospiro[chromene-2,2′-indoline])}(dppe)2] (2-SP, Scheme 1) in good yield, after deprotonation of the intermediate vinylidene species. Complex 2-SP was characterised spectroscopically, with the presence of the triple bond inferred from the absorption at 2014 cm−1 in the IR spectrum. The 1H and 13C NMR spectra of 2-SP were consistent with the amalgamation of the ligated ruthenium and ethynyl ligand. In particular, the alkynyl α-carbon was assigned to the signal observed at 127.0 ppm, showing coupling to the four equivalent phosphorus atoms, with the signal for the β-carbon at 116.3 ppm. The phosphorus nuclei gave a peak at 69.4 ppm in the 31P NMR spectrum.
Spiropyrans can exist in two observable isomeric states, a closed nearly, colourless spiropyran (SP) (e.g.2-SP, Scheme 1) and an open, coloured merocyanine (MC) (e.g.2-MC). In nitro-substituted spiropyrans the open, MC state is accessed photochemically with UV wavelengths via the triplet manifold, a pathway associated with the presence of the nitro group.38 The MC form reverts thermally to the closed SP state, or can be induced by irradiation with visible light. The proportions of the MC form of 1 and 2 are negligible under ambient conditions in THF solution. Polar solvents enhance the stability of the MC form of spiropyrans38 but 2 was found to be poorly soluble in EtOH and MeCN, restricting measurements here to THF solution. Whilst complex 2 was obtained as a dark green solid, a THF solution of this material rapidly becomes yellow, suggesting a measure of the MC form (2-MC) in the solid, which rapidly closes (to 2-SP) in solution. Our primary interest lies in extending the lifetime of the conducting merocyanine form, pushing towards the bi-stability required in a viable molecular electronic switch.
Irradiating THF solutions of 1-SP and 2-SP with UV light at 254 nm generated the ring-open forms 1-MC and 2-MC, characterised by absorptions in the visible region at λmax, 598 and 633 nm, respectively. The thermal reversion of complexes 1-MC and 2-MC to the closed SP forms followed first order kinetics, as determined by UV-Vis spectroscopy. The band at 633 nm (2-MC) proved to be solvatochromic, shifting to 578 nm on changing from pure THF to 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 THF:MeCN, consistent with the charge transfer nature of the transition. The rate constants and half-lives are shown in Table 1. Thus, the presence of the [Ru(dppe)2] moiety in 2-MC decreases the rate of thermal ring-closure (and thus increases the half-life) by a factor of 22.
:50 THF:MeCN, consistent with the charge transfer nature of the transition. The rate constants and half-lives are shown in Table 1. Thus, the presence of the [Ru(dppe)2] moiety in 2-MC decreases the rate of thermal ring-closure (and thus increases the half-life) by a factor of 22.
| λ max (nm) | k (s−1) | Half-life (s) | |
|---|---|---|---|
| 1-MC | 598 | 3.56 ± 0.12 × 10−2 | 19.5 ± 0.7 |
| 2-MC | 633 | 1.61 ± 0.06 × 10−3 | 430 ± 016 |
To explore the underlying electronic factors that might influence the ring-closing reactions of 1-MC and 2-MC, we turned to DFT calculations. The geometries of complexes 2-SP and 2-MC and the corresponding ligand 1-SP and 1-MC precursors were optimized at the B3LYP/3-21G* level of theory, using a CPCM dichloromethane solvent model. Whilst the basis set employed is relatively small, previous benchmarking calculations on this class of compound have demonstrated that it provides accurate electronic descriptions with pragmatic computational expense.45 Selected bond lengths and angles are summarized in Table S1, ESI,† atomic contributions to key molecular orbitals are given in Table S2 and Mulliken charges are listed in Table S3.†
The geometry of the trans-RuCl(dppe)2 fragment changes little in response to the switch of the spirocylic ligand geometry to the merocyanine form. There are, of course, greater changes evinced in the SP/MC ligand backbones, which are largely reflected in the valence bond descriptions given in Scheme 1. Whilst there are few bond lengths worthy of comment in the ring-closed/SP form, consideration of the bond lengths along the N–C9–C11–C13 chain in the ring-open/MC form, together with the planarity of the extended ligand suggests extensive delocalization, which is subtly enhanced by the presence of the organometallic fragment. Such ideas are supported by the distribution of the frontier orbitals, with HOMO and LUMO of the spirocyclic compounds 1-SP and 2-SP (Fig. 1) being rather spatially separated and localized on the phenylethynyl portion of the molecule and the LUMOs on the nitrobenzene moiety (Table S2,†Fig. 1), whilst those the MC forms are rather more delocalized over the extended molecular backbone. In addition, a more positive net Mulliken charge at Ru is predicted in the ring-open, MC form compared to the ring-closed, SP form (+0.779 vs. +0.664), consistent with a degree of extended donor–acceptor character in the MC complex. The donor effect of the metal fragment also serves to decrease the net positive charge at C(9) in the MC form of the metal complex when compared with that of the free alkyne (+0.369 vs. +0.507). There is also a small net decrease in the charge at the O-atom involved in forming the C–O spiro centre in the open (MC) form of the metal complex (−0.520) vs. the open (MC) form of the free alkyne (−0.566). The decreased rates of ring-closing in the metal complex when compared with the organic ligand is consistent with the modulation of the charge density arising from charge transfer from the metal to the alkynyl ligand and substituents, further suggesting the ring-closing reaction takes place under charge control.
 | ||
| Fig. 1 Plots of the (a) HOMO and (b) LUMO of 2-SP and (c) HOMO and (d) LUMO of 2-MC (±0.02 (e bohr−3)1/2). | ||
We have demonstrated that appending a ruthenium alkynyl unit to a spiropyran core prolongs the lifetime of the merocyanine form of the molecule more than twenty-fold. The metal enhances the extent of charge delocalisation through donation of electron density into the organic ligand. Chiefly the metal acts to decrease the charge/electron density localisation on the spirocyclic ring-forming atoms in the open form of the complex, thus slowing the electrostatically driven ring-closing reaction. Theoretical calculations suggest that the metal provides a different switching mechanism to the 5,5′-dithienylperfluorocyclopentene electrode decoupling seen previously and presages the opportunity of the electrochemical address of the bi-stability of the molecule.
This research was supported under Australian Research Council's Discovery Projects funding scheme (DP150104117) and, in part, by the Danish National Research Foundation (DNRF93). M.C.W is a recipient of an Australian Postgraduate Award. P.J.L. gratefully acknowledges the award of an ARC Future Fellowship (FT120100073).
Notes and references
- K. Szacilowski, Chem. Rev., 2008, 108, 3481–3548 CrossRef CAS PubMed.
- K. Matsuda and M. Irie, J. Photochem. Photobiol., C, 2004, 5, 169–182 CrossRef CAS.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327–336 RSC.
- B. L. Feringa, J. Org. Chem., 2007, 72, 6635–6652 CrossRef CAS PubMed.
- M. Natali and S. Giordani, Chem. Soc. Rev., 2012, 41, 4010–4029 RSC.
- D. Bléger, J. Schwarz and A. M. Brouwer, J. Am. Chem. Soc., 2012, 134, 20597–20600 CrossRef PubMed.
- N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875–1890 CrossRef CAS PubMed.
- S.-W. Choi and H. Takezoe, Phys. E., 2009, 41, 1648–1650 CrossRef CAS PubMed.
- B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874–1885 CrossRef CAS.
- A. Tissot, M.-L. Boillot, S. Pillet, E. Codjovi, K. Boukheddaden and L. M. Lawson Daku, J. Phys. Chem. C, 2010, 114, 21715–21722 CAS.
- V. I. Minkin, Russ. Chem. Rev., 2013, 82, 1–26 CrossRef PubMed.
- S. V. Paramonov, V. Lokshin and O. A. Fedorova, J. Photochem. Photobiol., C, 2011, 12, 209–236 CrossRef CAS PubMed.
- V. I. Minkin, in Molecular Switches, Wiley-VCH, 2011, vol. 1 Search PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CAS.
- V. A. Barachevsky, A. A. Khodonov, N. E. Belikov, A. V. Laptev, A. Y. Lukin, O. V. Demina, S. I. Luyksaar and M. M. Krayushkin, Dyes Pigm., 2012, 92, 831–837 CrossRef CAS PubMed.
- S. Kobatake and M. Irie, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2003, 99, 277–313 RSC.
- R. A. Khare and N. Sekar, Asian Dyer, 2006, 3, 31–34 CAS.
- P. Belser, L. De Cola, F. Hartl, V. Adamo, B. Bozic, Y. Chriqui, V. M. Iyer, R. T. F. Jukes, J. Kuhni, M. Querol, S. Roma and N. Salluce, Adv. Funct. Mater., 2006, 16, 195–208 CrossRef CAS PubMed.
- Y. Yokoyama, Chem. Rev., 2000, 100, 1717–1740 CrossRef CAS PubMed.
- V. Guerchais and H. Le Bozec, Top. Organomet. Chem., 2010, 28, 171–225 CrossRef CAS.
- V. Guerchais, L. Ordronneau and H. Le Bozec, Coord. Chem. Rev., 2010, 254, 2533–2545 CrossRef CAS PubMed.
- M. M. Paquette, R. A. Kopelman, E. Beitler and N. L. Frank, Chem. Commun., 2009, 5424–5426 RSC.
- C.-C. Ko and V. W.-W. Yam, J. Mater. Chem., 2010, 20, 2063–2070 RSC.
- J. C.-H. Chan, W. H. Lam, H.-L. Wong, N. Zhu, W.-T. Wong and V. W.-W. Yam, J. Am. Chem. Soc., 2011, 133, 12690–12705 CrossRef CAS PubMed.
- G. Duan, W.-T. Wong and V. W.-W. Yam, New J. Chem., 2011, 35, 2267–2278 RSC.
- E. C. Harvey, B. L. Feringa, J. G. Vos, W. R. Browne and M. T. Pryce, Coord. Chem. Rev., 2015, 282, 77–86 CrossRef PubMed.
- K. A. Green, M. P. Cifuentes, T. C. Corkery, M. Samoc and M. G. Humphrey, Angew. Chem., Int. Ed., 2009, 48, 7867–7870 CrossRef CAS PubMed.
- Y.-M. Hervault, C. M. Ndiaye, L. Norel, C. Lagrost and S. Rigaut, Org. Lett., 2012, 14, 4454–4457 CrossRef CAS PubMed.
- F. Meng, Y.-M. Hervault, L. Norel, K. Costuas, C. Van Dyck, V. Geskin, J. Cornil, H. H. Hng, S. Rigaut and X. Chen, Chem. Sci., 2012, 3, 3113 RSC.
- M. N. Roberts, C. J. Carling, J. K. Nagle, N. R. Branda and M. O. Wolf, J. Am. Chem. Soc., 2009, 131, 16644–16645 CrossRef CAS PubMed.
- M. N. Roberts, J. K. Nagle, J. G. Finden, N. R. Branda and M. O. Wolf, Inorg. Chem., 2009, 48, 19–21 CrossRef CAS PubMed.
- M. N. Roberts, J. K. Nagle, M. B. Majewski, J. G. Finden, N. R. Branda and M. O. Wolf, Inorg. Chem., 2011, 50, 4956–4966 CrossRef CAS PubMed.
- H.-L. Wong, C.-H. Tao, N. Zhu and V. W.-W. Yam, Inorg. Chem., 2010, 50, 471–481 CrossRef PubMed.
- H.-L. Wong, N. Zhu and V. W.-W. Yam, J. Organomet. Chem., 2014, 751, 430–437 CrossRef CAS PubMed.
- R. Klajn, Chem. Soc. Rev., 2014, 43, 148–184 RSC.
- V. I. Minkin, Russ. Chem. Bull., 2008, 57, 687–717 CrossRef CAS.
- A. K. Chibisov and H. Gorner, Phys. Chem. Chem. Phys., 2001, 3, 424–431 RSC.
- H. Gorner, Phys. Chem. Chem. Phys., 2001, 3, 416–423 RSC.
- A. Moriuchi, K. Uchida, A. Inagaki and M. Akita, Organometallics, 2005, 24, 6382–6392 CrossRef CAS.
- Y. J. Cho, K. Y. Rho, S. H. Kim, S. R. Keum and C. M. Yoon, Dyes Pigm., 1999, 44, 19–25 CrossRef CAS.
- A. V. Laptev, A. Y. Lukin, N. E. Belikov, V. A. Barachevskii, O. V. Demin, A. A. Khodonov, S. D. Varfolomeev and V. I. Shvets, Mendeleev Commun., 2013, 23, 145–146 CrossRef CAS PubMed.
- G. A. Koutsantonis, P. J. Low, C. F. R. Mackenzie, B. W. Skelton and D. S. Yufit, Organometallics, 2014, 33, 4911–4922 CrossRef CAS.
- G. A. Koutsantonis, G. I. Jenkins, P. A. Schauer, B. Szczepaniak, B. W. Skelton, C. Tan and A. H. White, Organometallics, 2009, 28, 2195–2205 CrossRef CAS.
- M. A. Fox, J. E. Harris, S. Heider, V. Perez-Gregorio, M. E. Zakrzewska, J. D. Farmer, D. S. Yufit, J. A. K. Howard and P. J. Low, J. Organomet. Chem., 2009, 694, 2350–2358 CrossRef CAS PubMed.
- M. A. Fox, R. L. Roberts, W. M. Khairul, F. Hartl and P. J. Low, J. Organomet. Chem., 2007, 692, 3277–3290 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details on synthesis, and data of compounds, UV-Vis spectra and decay plots. Detail of theoretical calculations. See DOI: 10.1039/c5dt01107h |
| This journal is © The Royal Society of Chemistry 2015 |