
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Flow-synthesis of carboxylate and phosphonate based metal–organic frameworks under non-solvothermal reaction conditions†
Steve
Waitschat
a,
Michael T.
Wharmby
*ab and
Norbert
Stock
*a
aInstitute für Anorganische Chemie, Christian-Albrechts-Universität, Max-Eyth-Straße 2, D 24118 Kiel, Germany. E-mail: michael.wharmby@diamond.ac.uk; stock@ac.uni-kiel.de
bDiamond Light Source Ltd., Diamond House, Harwell Science and Innovation Campus, Didcot, Oxfordshire, OX11 0DE, UK
First published on 15th May 2015
Abstract
A continuous flow reactor was developed for the synthesis of porous metal–organic frameworks (MOFs) under mild reaction conditions. Commodity hardware was used to assemble the device, giving it a great degree of flexibility in its configuration. The use of paraffin to encapsulate reactions and also ultrasonic treatment were employed to prevent clogging of the reactor. Reactor design was optimised through studies of the synthesis of zirconium carboxylate framework UiO-66. Synthesis of the aluminium carboxylate CAU-13 was also performed, to demonstrate the versatility of the device. Finally the reactor was used to synthesise a new cadmium phosphonate framework, bearing the STA-12 network.
Introduction
Metal–organic frameworks (MOFs) are composed of organic linkers which join metal ions or clusters, to form networks with potential porosity.1 The wide diversity of attainable topologies and the possibility of tuning properties through the addition of functional groups to the linkers make permanently porous MOFs candidate materials for applications such as gas storage,2 gas separation,3 drug-delivery4 or heat-transformation.5 Such applications would however require the preparation of large quantities of material6 and many MOF syntheses are only reported as small scale batch reactions. The upscaling of such batch reactions can be problematic, requiring much time to be expended on re-optimising the reaction conditions. One method to produce larger quantities would be to use a continuous flow reactor.7 Flow reactors are frequently utilized within the field of organic synthesis,8 especially for the synthesis of peptide & saccharide oligomers, but also for continuous production of high-value pharmaceuticals.9 In inorganic chemistry they have been extensively used for the synthesis of metal chalcogenide and oxide nanoparticles,10,11 and flow synthesis of MOFs has only been demonstrated for a select range of frameworks (Table S1.1†).7,12–17 HKUST-1, which readily forms under a wide range of parameters, has been synthesised in flow under both mild (p ∼ ambient; T = 60 °C)12 and more severe (p = 250 bar; T = 100–400 °C)13 reaction conditions. Syntheses in flow of other widely studied frameworks, including CPO-27,13 MIL-53(Al),7 MIL-88B(Fe)17 and UiO-66,15,16,18 have been reported relatively recently. It should be noted that, with the exception of MIL-88B(Fe) and HKUST-1, all other flow synthesis studies were performed under solvothermal reactions conditions, i.e. at temperatures above the boiling point of the reaction solvent and thus under increased pressure. Furthermore, to date there has only been one report of a new MOF synthesised in a flow reactor, Ce5(BDC)7.5(DMF)4.19Herein we describe the development of a flow reactor for the synthesis of MOFs under mild reaction conditions and its application to the synthesis of the known frameworks, UiO-6620 ([Zr6O4(OH)4(BDC)6], BDC = 1,4-benzenedicarboxylate) and CAU-1321 ([Al(OH)(CDC]·1.5H2O, CDC = trans-1,4-cyclohexane-dicarboxylate). In addition we were also able to prepare a new metal phosphonate, [Cd2(H2O)2L]·xH2O (H4L = N,N′-piperazinebis(methylenephosphonic acid), which is a new member of the STA-12 family of compounds.22
Experimental
X-ray crystallography
Initial characterisation was performed using a Stoe Stadi P diffractometer fitted with an xy-stage, in transmission geometry using Cu Kα1 radiation and with data collected by an image plate detector. Powder X-ray diffraction (PXRD) patterns for Pawley fitting and Rietveld refinement were measured with a Stoe Stadi P diffractometer in transmission geometry using Cu Kα1 radiation and with data collected using a Mythen detector.Data analysis for all three compounds was performed using the TOPAS-Academic V5 suite.23 The PXRD patterns of all three compounds were indexed and subsequently Pawley fits were performed to confirm the unit cell parameters as obtained from the indexing routine. For STA-12(Cd), the profile parameters, unit cell and space group were used as the starting point for a Rietveld refinement. STA-12(Mn)22 was used as the initial model for the refinement, with Mn2+ replaced by Cd2+. Restraints were applied to all coordinative bonds around the Cd2+ ion and also to all bonding distances within the organic ligand. After several cycles of refinement of the framework, all pore water molecules were removed and new positions were determined from Fourier difference maps. Occupancies of these sites were then refined. In the final cycles of refinement, framework and water positions were refined together with isotropic displacement parameters.
Physical measurements
Infrared spectra were recorded on a Bruker ALPHA-P A220/D-01 FTIR spectrometer fitted with an ATR unit, over the spectral range 4000–400 cm−1. Thermogravimetric analysis was carried out using a NETSCH STA 429 CD analyser with a heating rate of 4 K min−1 and under flowing air (flow rate 75 ml min−1) or a TA Instruments TGA Q500 with a heating rate of 5 K min−1 and under flowing air (60 ml min−1). Elemental analysis was performed using a EuroVector EuroEA elemental analyser. NMR spectroscopy was performed using a Bruker DRX 500 spectrometer. Scanning Electron Micrographs were collected using an FEI Quanta FEG 600 electron microscope, whilst N2 sorption isotherms were recorded at 77 K with a BELSORP-max apparatus (BEL Japan Inc.).Results & discussion
Design of the flow reactor
The flow reactor was designed to be easy to set up whilst having sufficient flexibility to allow a wide variety of reaction parameters to be investigated, including molar ratios and heating method (ESI S3†). The device is built up from two syringe pumps, which contain respectively the metal salt and the organic linker in solution (Fig. 1 and S3.1†). In cases where the amount of precipitate causes the reactor to block, a third pump can be added, to pump a transport medium (e.g. paraffin) to encapsulate the reaction as droplets in flow.24 Additionally an ultrasonic probe can be used to both improve mixing and also to prevent clogging of the reactor. | ||
| Fig. 1 Schematic of the flow reactor in a two pump configuration, showing the hotplate and oil bath in which the coil of PTFE tube forming the reactor is immersed. | ||
Solutions are combined by pumping along PTFE tubing to mixers, which in the simplest form of the system are either three way Y-connectors or four way X-connectors. From these mixers, the solution flows into the reactor itself, a coil of PTFE tubing immersed in an oil bath for heating. The length and internal diameter of this tube, in combination with the total pumping rate, determine the reaction time, whilst the molar ratio of the reaction is determined by varying the pumping rate of each pump individually (ESI S3†).
Zirconium terephthalate UiO-66
Zr-based UiO-66 ([Zr6O4(OH)4(BDC)6]) is one of the most intensively studied MOFs and thus was selected for the initial test of the reactor. Initial reaction conditions were chosen to mimic the reported batch syntheses (ZrCl4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2BDC = 1:1, 120 °C, 24 h).25 The first attempts utilised a two syringe pump set-up, with one pump loaded with ZrCl4 in DMF and a second containing H2BDC in DMF (Table 1). However, this design led to precipitates forming in the tube and clogging of the reactor. The design was then modified to add a third syringe containing either DMF or paraffin as a transport medium to prevent clogging.24 Amounts of solid obtained with the additional DMF were found to be too low, whilst with paraffin, although more solid was recovered, isolation of the UiO-66 product could only be achieved by repeated washing steps with dichloromethane or toluene, which resulted in significantly lower yields. BET surface area values were also found to be significantly lower than literature values (Table 2). The reactor configuration was therefore changed again, back to a two pump set-up, with an ultrasonic probe fitted which improves mixing of the reagents and most importantly prevents clogging of the reactor (Table 1). Following optimisation of the synthesis conditions, highly-crystalline, phase pure UiO-66 could be obtained using a molar ratio of 1:3 (Zr4+:H2BDC) at 120 °C in just 45 min.
:H2BDC = 1:1, 120 °C, 24 h).25 The first attempts utilised a two syringe pump set-up, with one pump loaded with ZrCl4 in DMF and a second containing H2BDC in DMF (Table 1). However, this design led to precipitates forming in the tube and clogging of the reactor. The design was then modified to add a third syringe containing either DMF or paraffin as a transport medium to prevent clogging.24 Amounts of solid obtained with the additional DMF were found to be too low, whilst with paraffin, although more solid was recovered, isolation of the UiO-66 product could only be achieved by repeated washing steps with dichloromethane or toluene, which resulted in significantly lower yields. BET surface area values were also found to be significantly lower than literature values (Table 2). The reactor configuration was therefore changed again, back to a two pump set-up, with an ultrasonic probe fitted which improves mixing of the reagents and most importantly prevents clogging of the reactor (Table 1). Following optimisation of the synthesis conditions, highly-crystalline, phase pure UiO-66 could be obtained using a molar ratio of 1:3 (Zr4+:H2BDC) at 120 °C in just 45 min.
| Reactor volume/ml | ZrCl4 flowrate/ml min−1 | H2BDC flowrate/ml min−1 | Transporta flowrate/ml min−1 | Temp./°C | Time/min | Ultra sound | |
|---|---|---|---|---|---|---|---|
| Cycle | Amplitude | ||||||
|
a A transport medium in addition to the reaction solvent, used to prevent clogging of the reactor.
b This reaction produced the most crystalline and phase pure UiO-66, which is further analysed in this work. This is equivalent to a reaction ratio of 1:3 (ZrCl4:H2BDC).
|
|||||||
| 16.87 | 0.094 | 0.281 | — | 120 | 45 | — | — |
| 16.80 | 0.070 | 0.210 | — | 120 | 60 | — | — |
| 14.45 | 0.040 | 0.120 | 0.161 (Para.) | 120 | 45 | — | — |
| 16.83 | 0.047 | 0.140 | 0.187 (DMF) | 130 | 45 | — | — |
| 16.87b | 0.094 | 0.281 | — | 120 | 45 | 0.25 | 100% |
| 16.80 | 0.070 | 0.210 | — | 120 | 60 | 0.25 | 100% |
Characterisation
The phase purity of the UiO-66 material was confirmed by Pawley fitting of the laboratory powder X-ray diffraction (PXRD) data. A good fit to the data was obtained, however in addition to the expected UiO-66 reflections, two symmetry forbidden reflections were also observed (Fig. 2). These have been attributed to lower symmetry defect rich regions incorporated within the perfect UiO-66 structure.26,27 | ||
Fig. 2 Pawley fit of UiO-66 obtained from a flow reactor in a two-syringe configuration. Cell refined to a = 20.7367(4) Å (space group: Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m; Rwp = 5.59%, χ2 = 3.644), consistent with reported lattice parameters.20 Green arrows indicate forbidden reflections. m; Rwp = 5.59%, χ2 = 3.644), consistent with reported lattice parameters.20 Green arrows indicate forbidden reflections. | ||
Given the low reaction temperature, the presence of defects in flow reactor prepared UiO-66 is unsurprising. Evidence for their presence is also found from TGA, EDX and adsorption experiments. TGA data (Fig. S4.3†) found a cluster:linker ratio of 1:3.5, significantly lower than the ideal 1:6 ratio of perfect UiO-66.27 Defects have previously been linked to lower thermal stability of UiO-66, however TGA data (Fig. S4.3†) indicate our material is stable to in excess of 400 °C. Samples of the material were activated for adsorption measurements (220 °C, p < 10−2 kPa, 12 h) and these were found by elemental analysis to still contain nitrogen, which may indicate that missing linker sites are partially occupied by DMF molecules.25 Chloride ions occupying these sites have also been suggested, and EDX measurements indicate the presence of up to two chloride ions per zirconium in our sample.27 The presence of defects in the structure influences the BET surface area (1263 m2 g−1), which is significantly higher than the surface area of the defect-free compound (1125 m2 g−1) (Fig. 3).27
 | ||
| Fig. 3 N2 adsorption and desorption isotherms measured at 77 K on samples of UiO-66 produced in a two-pump (ultrasonic probe; green & red traces) and three-pump (paraffin transport; orange & blue traces) flow reactor. | ||
We also provide a comparison of the surface areas of other flow reactor prepared UiO-66 materials (Table 2). The presence of defects is potentially beneficial for applications in catalysis.28
Aluminium cyclohexanedicarboxylate CAU-13
To demonstrate the versatility of the flow reactor set-up for the preparation of a variety of different MOFs, the synthesis of CAU-13 ([Al(OH)(CDC]·1.5H2O) was investigated.21 The reported batch synthesis of CAU-13 is performed using a 1:1 AlCl3·6H2O:H2CDC molar ratio, and a 4:1 (vol.) DMF:water mixture, which is heated at 130 °C for 12 hours. As these conditions would lead to a build-up of pressure, the synthesis was modified to use pure DMF as the solvent (Table S5.1†). A two syringe pump set-up was again chosen. One syringe was loaded with a 0.5 M AlCl3·6H2O in DMF solution and a second with a 0.5 M H2CDC and 4.5 M acetic acid in DMF solution. Acetic acid was added to act as a modulator, to favour the formation of the –{Al–O–Al–O}– chains. Using a temperature of 130 °C and a reaction time of 20 min, it was possible to prepare microcrystalline CAU-13.
Characterisation
Phase purity was confirmed by Pawley fitting of the PXRD pattern (Fig. 4), with CAU-13 crystallizing in its open form. TGA and elemental analysis results were consistent with the reported CAU-13 composition and the BET surface area was slightly higher than reported (measured: 401 m2 g−1; reported: 378 m2 g−1) (Fig. 5 and ESI S5.7†). | ||
Fig. 4 Pawley fit of CAU-13 obtained from a flow reactor in a two-syringe configuration. Cell refined to a = 6.6111(4) Å, b = 9.4498(6) Å, c = 9.4652(5) Å, α = 107.652(4)°, β = 107.690(6)° and γ = 93.185(5)° (space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ; Rwp = 4.71%, χ2 = 1.080), consistent with reported lattice parameters.21 ; Rwp = 4.71%, χ2 = 1.080), consistent with reported lattice parameters.21 | ||
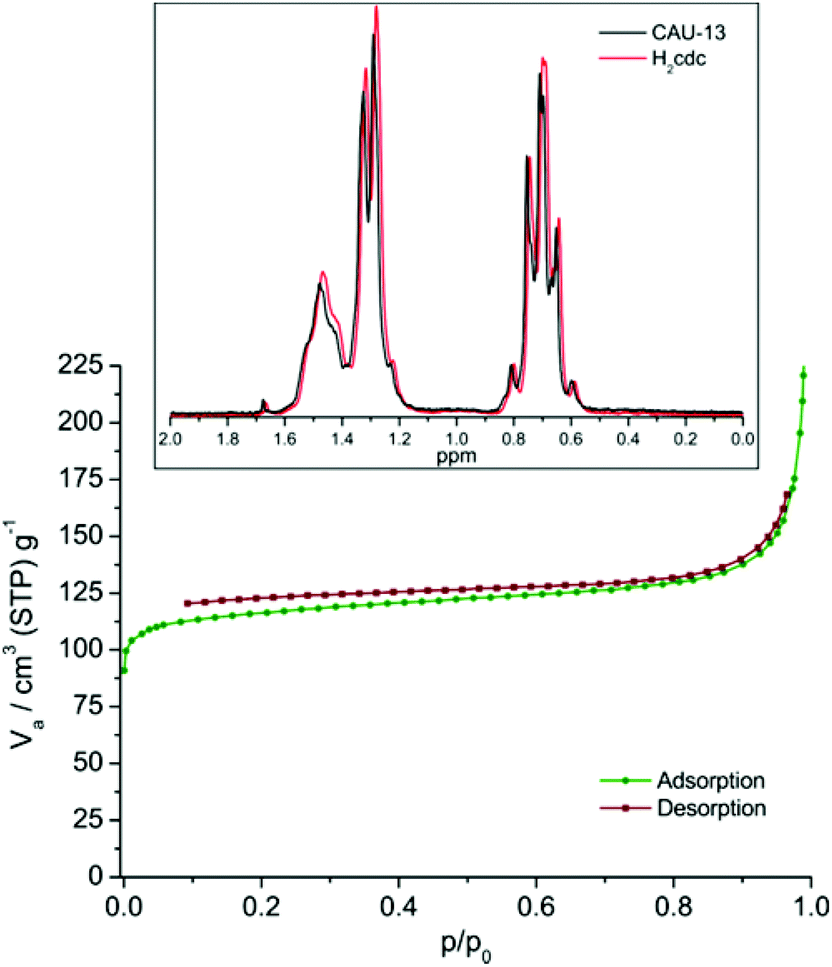 | ||
| Fig. 5 N2 adsorption isotherm measured for CAU-13 – aBET = 401 m2 g−1. Inset shows the solution state 1H NMR spectrum of CAU-13; only resonances attributable to H2cdc were identified and thus no acetic acid was incorporated. | ||
To determine whether this slight increase in BET surface area was due to the presence of defect sites, occupied by acetic acid modulator from the synthesis, a sample of CAU-13 was digested in base and analysed by solution state NMR. No additional resonances were observed in the spectra (Fig. 5) and thus no additional acetic acid molecules are thought to be incorporated, in agreement with the elemental analysis.
Synthesis scaling
Once the reaction is running, the space time yield for syntheses of UiO-66 and CAU-13 is 428 kg m−3 d−1 (yield: 194 mg, 87%) and 3049 kg m−3 d−1 (yield: 615 mg, 53%), respectively. Although in this study only product amounts of 200–600 mg per synthesis were obtained, this set-up can be used, in principle, for obtaining larger amounts by running the flow synthesis in a steady state for a longer period of time.Cadmium phosphonate STA-12(Cd)
Beyond optimization and scaled up synthesis, the flow reactor set-up is well suited for the discovery of new compounds. STA-12 is a porous metal phosphonate framework, obtained with a wide range of divalent metal cations (e.g. Mg, Mn, Fe, Co, Ni),22 however it remains unknown with Cd. STA-12 is usually synthesized with a metal to ligand ratio of 2:1 and using reaction conditions of 160–220 °C for 18–72 h in water. Alternative reaction conditions were needed for the synthesis using the flow reactor, as under these hydrothermal conditions, high pressures would be developed. At lower temperature however, the linker, N,N′-piperazinebis(methylenephosphonic acid) (H4L), is insoluble in water. To overcome this problem, H4L was neutralized with KOH to form its soluble sodium salt. A 0.1 M aqueous solution of this was loaded into one syringe of a two syringe pump set-up, whilst the other syringe was charged with a 0.1 M metal solution. Reaction conditions, determined from studies of the known compounds STA-12(Co) and STA-12(Ni), were carried out by screening the reaction temperature, time and metal to linker ratios (ESI S6, Table S6.1 and S6.2†).20,26 The best conditions are a reaction temperature of 70 °C and a M:L ratio of 1:2 for STA-12(Co) and 1:1 for STA-12(Ni). For the synthesis of STA-12(Cd) the parameters of STA-12(Ni) were chosen and an aqueous solution of Cd(AcO)2·2H2O was used. The reaction time was varied between 5–20 min by controlling the total pumping rate (Table S6.3†). Crystallinity increases with greater residence time (up to 15 min – Fig. 6).
 | ||
| Fig. 6 Samples of STA-12(Cd) prepared in the flow reactor with reaction times increasing from 5 to 20 minutes. Dashed box indicates the (140) reflection, which narrows with increasing reaction time, indicating an increase in particle size. | ||
Characterisation
The final product was analysed by PXRD and the structure was confirmed by Rietveld refinement (Fig. 7, left); the final refinement gave Rwp = 6.43%, RBragg = 1.56% and χ2 = 1.141 (Table S6.4†). STA-12(Cd) was refined in a trigonal unit cell in the space group R and unit cell parameters of a = b = 27.4100(8) Å c = 6.7279(2) Å. The structure consists of helical Cd2+ phosphonate chains, parallel to the c-axis, bridged by piperazinyl linker units, which coordinate the Cd2+ ions through the N atoms (Fig. 7, right). The structure differs slightly from previously reported STA-12 frameworks in that the piperazinyl ring plane is in the c-direction, whereas in other structures it is perpendicular.22 STA-12(Cd) is porous to N2 (Fig. S6.8†), with a pore volume of 0.08 cm3 g−1 and a BET surface area of 134 m2 g−1. N2 uptake is lower than the Co2+ and Ni2+ compounds, which is partly attributed to the greater mass of the framework forming cation.
 | ||
| Fig. 7 Rietveld plot of STA-12(Cd) (left) obtained from flow reactor and the structure determined (right). The final cycle of refinement gave Rwp = 6.43%, RBragg = 1.56% and χ2 = 1.141. | ||
Conclusions
We have developed a highly flexible and easy to set up flow reactor system for the preparation of porous MOFs under mild (i.e. sub-solvothermal) conditions, using commodity hardware. Using this device we have demonstrated the synthesis of three MOFs: two reported metal carboxylate compounds (UiO-66 and CAU-13) and a new porous metal phosphonate (STA-12(Cd)), the structure of which was confirmed by Rietveld refinement. All three compounds are porous to N2 gas, and UiO-66 shows a higher than expected BET surface area, attributed to defects, which may make it interesting for catalytic applications. The flow reactor design should allow simple and cheap scale-up of the syntheses of all three MOFs, making its development significant for industrial applications of this class of compounds.Acknowledgements
Dr G. Lampronti (University of Cambridge) is thanked for collection of SEM images; Achim Fölster (CAU, Kiel) is thanked for the preparation of Fig. 1.Notes and references
- S. R. Batten, N. R. Champness, X.-M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Öhrström, M. O'Keeffe, M. Paik Suh and J. Reedijk, Pure Appl. Chem., 2013, 85, 1715–1724 CrossRef CAS.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637–20640 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2011, 112, 1232–1268 CrossRef PubMed.
- S. K. Henninger, F. Jeremias, H. Kummer and C. Janiak, Eur. J. Inorg. Chem., 2012, 2012, 2625–2634 CrossRef CAS.
- A. U. Czaja, N. Trukhan and U. Müller, Chem. Soc. Rev., 2009, 38, 1284–1293 RSC.
- P. A. Bayliss, I. A. Ibarra, E. Perez, S. Yang, C. C. Tang, M. Poliakoff and M. Schroder, Green Chem., 2014, 16, 3796–3802 RSC.
- I. R. Baxendale, S. V. Ley, C. D. Smith and G. K. Tranmer, Chem. Commun., 2006, 4835–4837 RSC.
- F. Lévesque and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 1706–1709 CrossRef PubMed.
- J. B. Edel, R. Fortt, J. C. deMello and A. J. deMello, Chem. Commun., 2002, 1136–1137 RSC.
- A. Abou-Hassan, O. Sandre and V. Cabuil, Angew. Chem., Int. Ed., 2010, 49, 6268–6286 CrossRef CAS PubMed.
- R. Ameloot, F. Vermoortele, W. Vanhove, M. B. J. Roeffaers, B. F. Sels and D. E. De Vos, Nat. Chem., 2011, 3, 382–387 CrossRef CAS PubMed.
- M. Gimeno-Fabra, A. S. Munn, L. A. Stevens, T. C. Drage, D. M. Grant, R. J. Kashtiban, J. Sloan, E. Lester and R. I. Walton, Chem. Commun., 2012, 48, 10642–10644 RSC.
- K.-J. Kim, Y. J. Li, P. B. Kreider, C.-H. Chang, N. Wannenmacher, P. K. Thallapally and H.-G. Ahn, Chem. Commun., 2013, 49, 11518–11520 RSC.
- M. Rubio-Martinez, M. P. Batten, A. Polyzos, K.-C. Carey, J. I. Mardel, K.-S. Lim and M. R. Hill, Sci. Rep., 2014, 4 Search PubMed.
- M. Faustini, J. Kim, G.-Y. Jeong, J. Y. Kim, H. R. Moon, W.-S. Ahn and D.-P. Kim, J. Am. Chem. Soc., 2013, 135, 14619–14626 CrossRef CAS PubMed.
- L. Paseta, B. Seoane, D. Julve, V. Sebastián, C. Téllez and J. Coronas, ACS Appl. Mater. Interfaces, 2013, 5, 9405–9410 CAS.
- P. M. Schoenecker, G. A. Belancik, B. E. Grabicka and K. S. Walton, AIChE J., 2013, 59, 1255–1262 CrossRef CAS.
- L. D'Arras, C. Sassoye, L. Rozes, C. Sanchez, J. Marrot, S. Marre and C. Aymonier, New J. Chem., 2014, 38, 1477–1483 RSC.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- F. Niekiel, M. Ackermann, P. Guerrier, A. Rothkirch and N. Stock, Inorg. Chem., 2013, 52, 8699–8705 CrossRef CAS PubMed.
- M. T. Wharmby, G. M. Pearce, J. P. S. Mowat, J. M. Griffin, S. E. Ashbrook, P. A. Wright, L.-H. Schilling, A. Lieb, N. Stock, S. Chavan, S. Bordiga, E. Garcia, G. D. Pirngruber, M. Vreeke and L. Gora, Microporous Mesoporous Mater., 2012, 157, 3–17 CrossRef CAS.
- A. Coelho, TOPAS-Academic v5, Coehlo Software, Brisbane, Australia, 2012 Search PubMed.
- H. Song, J. D. Tice and R. F. Ismagilov, Angew. Chem., Int. Ed., 2003, 42, 768–772 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700–1718 CrossRef CAS.
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CAS.
- G. C. Shearer, S. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068–4071 CrossRef CAS.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Description of the flow reactor system, in depth description of the synthesis and characterisation of the title compounds, crystallographic data of STA-12(Cd), CCDC reference number 1051836. See DOI: 10.1039/c5dt01100k |
| This journal is © The Royal Society of Chemistry 2015 |