
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Strong effect of copper(II) coordination on antiproliferative activity of thiosemicarbazone–piperazine and thiosemicarbazone–morpholine hybrids†
Felix
Bacher
a,
Orsolya
Dömötör
b,
Anastasia
Chugunova
a,
Nóra V.
Nagy
c,
Lana
Filipović
d,
Siniša
Radulović
d,
Éva A.
Enyedy
*e and
Vladimir B.
Arion
*a
aUniversity of Vienna, Faculty of Chemistry, Institute of Inorganic Chemistry, Währinger Strasse 42, A-1090 Vienna, Austria. E-mail: vladimir.arion@univie.ac.at
bMTA-SZTE Bioinorganic Chemistry Research Group, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary
cInstitute of Organic Chemistry, Research Centre for Natural Sciences, Hungarian Academy of Sciences, Magyar Tudósok körútja 2, H-1117 Budapest, Hungary
dInstitute for Oncology and Radiology of Serbia, Pasterova 14, 11000 Belgrade, Serbia
eDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary. E-mail: enyedy@chem.u-szeged.hu
First published on 13th April 2015
Abstract
In this study, 2-formylpyridine thiosemicarbazones and three different heterocyclic pharmacophores were combined to prepare thiosemicarbazone–piperazine mPip-FTSC (HL1) and mPip-dm-FTSC (HL2), thiosemicarbazone–morpholine Morph-FTSC (HL3) and Morph-dm-FTSC (HL4), thiosemicarbazone–methylpyrrole-2-carboxylate hybrids mPyrr-FTSC (HL5) and mPyrr-dm-FTSC (HL6) as well as their copper(II) complexes [CuCl(mPipH-FTSC-H)]Cl (1 + H)Cl, [CuCl(mPipH-dm-FTSC-H)]Cl (2 + H)Cl, [CuCl(Morph-FTSC-H)] (3), [CuCl(Morph-dm-FTSC-H)] (4), [CuCl(mPyrr-FTSC-H)(H2O)] (5) and [CuCl(mPyrr-dm-FTSC-H)(H2O)] (6). The substances were characterized by elemental analysis, one- and two-dimensional NMR spectroscopy (HL1–HL6), ESI mass spectrometry, IR and UV–vis spectroscopy and single crystal X-ray diffraction (1–5). All compounds were prepared in an effort to generate potential antitumor agents with an improved therapeutic index. In addition, the effect of structural alterations with organic hybrids on aqueous solubility and copper(II) coordination ability was investigated. Complexation of ligands HL2 and HL4 with copper(II) was studied in aqueous solution by pH-potentiometry, UV–vis spectrophotometry and EPR spectroscopy. Proton dissociation processes of HL2 and HL4 were also characterized in detail and microscopic constants for the Z/E isomers were determined. While the hybrids HL5, HL6 and their copper(II) complexes 5 and 6 proved to be insoluble in aqueous solution, precluding antiproliferative activity studies, the thiosemicarbazone–piperazine and thiosemicarbazone–morpholine hybrids HL1–HL4, as well as copper(II) complexes 1–4 were soluble in water enabling cytotoxicity assays. Interestingly, the metal-free hybrids showed very low or even a lack of cytotoxicity (IC50 values > 300 μM) in two human cancer cell lines HeLa (cervical carcinoma) and A549 (alveolar basal adenocarcinoma), whereas their copper(II) complexes were cytotoxic showing IC50 values from 25.5 to 65.1 μM and 42.8 to 208.0 μM, respectively in the same human cancer cell lines after 48 h of incubation. However, the most sensitive for HL4 and complexes 1–4 proved to be the human cancer cell line LS174 (colon carcinoma) as indicated by the calculated IC50 values varying from 13.1 to 17.5 μM.
Introduction
Thiosemicarbazones (TSCs) are known as potent metal chelators with high affinity for first row transition metals.1,2 TSCs and their metal complexes possess a variety of biological activities, such as antifungal, antiviral, antibacterial, antimalarial and anticancer.3–8 The anticancer activity of α-N-heterocyclic TSCs (HCTs) has been known since the 1950s when 2-formylpyridine thiosemicarbazone (FTSC) showed antileukemic activity in a mice model.9 To date, the best-studied HCT is 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP or Triapine). Several clinical phase I and II trials revealed that Triapine is ineffective against a variety of solid tumors but very promising against hematologic malignancies such as leukemia.10–18 The outcome of a recent clinical phase II study including 37 patients with aggressive myeloproliferative neoplasms, with a response rate of 49% and complete remission in 24% of all patients, has recently been reported.19 Ribonucleotide reductase (RNR),20,21 an enzyme catalyzing the reduction of ribonucleotides to the corresponding 2′-deoxyribonucleotides, which is the rate determining step in DNA synthesis,22 and topoisomerase IIα (Topo IIα), an enzyme that controls the DNA topology during cell division by introducing temporary double strand breaks have been considered as possible targets for this class of compounds.23–26 New insights into the mechanism of action for RNR inhibiting HCTs and especially Triapine were recently reported.27–30 The enzymes ATP binding pocket was suggested as major target for Topo IIα inhibiting HCTs.31 The reaction of copper(II) with HCTs leading to square-planar complexes markedly enhances the Topo IIα inhibition rate.322-Acetylpyridine thiosemicarbazones possess very high cytotoxicity in human cancer cell lines with IC50 values in the nanomolar concentration range and the ability to destroy the tyrosyl radical of the mammalian RNR R2 protein under the slightly reducing conditions typical for tumors.33,34 However, high general toxicity and, consequently, the low therapeutic index along with low aqueous solubility for these and other related thiosemicarbazones prompted us to design hybrid systems, based on thiosemicarbazones and other pharmacophores. Recently, we prepared proline-TSC hybrids (3-methyl-(S)-pyrrolidine-2-carboxylate-2-formylpyridine thiosemicarbazone (L-Pro-FTSC) and 3-methyl-(R)-pyrrolidine-2-carboxylate-2-formylpyridine thiosemicarbazone (D-Pro-FTSC)) and their copper(II) complexes.35 These new compounds are highly water soluble but exhibit very low cytotoxicity, most probably because of their very low lipophilicity. We decided to extend our work and use other pharmacophoric groups for attachment at the 6-position of the TSCs pyridine ring, in order to increase the lipophilicity and modulate the antiproliferative activity. We attached the six-membered rings methylpiperazine and morpholine as well as methylpyrrole-2-carboxylate containing a five-membered planar heterocycle. It is well-known that the attachment of a piperazine moiety on a hydrophobic scaffold has a favourable effect on its water solubility,36–39 moreover the piperazine heterocycle is found in a broad variety of biologically active compounds, some of which are currently used in clinical therapy.40–49 Biologically active metal-based compounds containing a piperazine ring have also been reported.50–54 Morpholine is another well-known water-solubilizing unit incorporated in structures of biologically active compounds, showing often favorable pharmacologic effects.55–57 In particular, a morpholine moiety is also present in the approved anticancer drugs Gefitinib (against certain breast, lung and other cancers) and Carfilzomib (against multiple myeloma).58,59 A series of TSCs with different substituents at position 4 of the pyridine ring was tested on mice bearing sarcoma 180 ascites cells. Intriguingly, the 4-morpholino-2-formylpyridine thiosemicarbazone was the most effective compound, increasing the average survival time of tumor bearing mice from 13.8 to 38 days.60 The methylpyrrole-2-carboxylate ring was chosen as third possible option since it resembles proline.
Herein, we report the synthesis of six new organic compounds, namely HL1–6, representing three types of potential hybrid ligands for transition metals, as well as six copper(II) complexes all shown in Chart 1. The compounds were characterized by analytical and spectroscopic methods (1H and 13C NMR, UV–vis, IR) and X-ray diffraction (1–5). Solution equilibria of the copper(II) complexes formed with HL2 and HL4 were studied by pH-potentiometry, UV–vis and EPR spectroscopy and the thermodynamic stability data were compared to those for other related hybrid and non-hybrid systems. The antiproliferative activity of four ligands and four copper(II) complexes has been assayed. The cytotoxicity of 1–4 is markedly lower than that of the parent 2-acetylpyridine and 2-formylpyridine thiosemicarbazones, but significantly higher than that of thiosemicarbazone–proline hybrids and their copper(II) complexes making them pertinent for further development as potential anticancer drugs.
 | ||
Chart 1 Hybrid ligands and their copper(II) complexes studied in this work. Underlined numbers indicate complexes studied by X-ray diffraction. Co-crystallized solvent is not included in the formulas (see Experimental section). a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) This complex was crystallized and characterized by X-ray crystallography as [CuCl(mPip-FTSC-H)]·0.15CH3OH. This complex was crystallized and characterized by X-ray crystallography as [CuCl(mPip-FTSC-H)]·0.15CH3OH. | ||
Experimental
Chemicals
2,6-Dihydroxymethylpyridine, 4-methylpiperazine, morpholine and methylpyrrole-2-carboxylate were purchased from Acros Organics. 2-Hydroxymethyl-6-chloromethylpyridine and 6-chloromethylpyridine-2-carboxaldehyde were synthesized according to published protocols.61 2-(Chloromethyl)-6-(dimethoxymethyl)pyridine was prepared as described previously.35 Solvents were dried using standard procedures, if required.62 KOH, KCl, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 2-(N-morpholino)ethanesulfonic acid (MES), ethylenediaminetetraacetic acid (EDTA) were Sigma-Aldrich products, while HCl and CuCl2 were from Reanal. CuCl2 was dissolved in a known amount of HCl in order to get the copper(II) stock solution. Its concentration was determined by complexometric titrations with EDTA.Synthesis of ligands
:1 mixture of dry THF and dry dichloromethane (40 mL) in a 100 mL Schlenk tube. Methylpiperazine (1.50 mL, 13.53 mmol) and triethylamine (3.64 mL, 27.06 mmol) were added. The reaction mixture was stirred at 46 °C overnight. The next day a white precipitate of triethylammonium chloride was filtered off and washed with THF. The filtrate was concentrated under reduced pressure to yield a brown oily raw product. This was purified on a silica column using chloroform/methanol 4:1 as eluent. The solvent was removed under reduced pressure to yield the product as a yellow oil. Yield: 1.99 g, 79%. 1H NMR (500 MHz, DMSO-d6) δ 7.82 (t, J = 7.7 Hz, 1H, CH(Ar)), 7.41 (d, J = 7.7 Hz, 1H, CH(Ar)), 7.36 (d, J = 7.7 Hz, 1H, CH(Ar)), 5.26 (s, 1H, CH(OCH3)2), 3.60 (s, 2H, CH2), 3.30 (s, 6H, (OCH3)2), 2.50–2.35 (m, 8H, CH2(Pip), overlapped with residual DMSO signal), 2.24 (s, 3H, CH3(Pip)).
:1 mixture of dry THF and dry dichloromethane (20 mL) in a 50 mL Schlenk tube were added morpholine (0.52 mL, 5.96 mmol) and triethylamine (1.60 mL, 11.91 mmol). The reaction mixture was stirred at 46 °C overnight. The next day a white precipitate of triethylammonium chloride was filtered off and washed with THF. The filtrate was concentrated under reduced pressure to yield a brown oily raw product. This was purified on a silica column using chloroform/methanol 97.5:2.5 as eluent. The solvent was removed under reduced pressure to yield the product as a yellow oil. Yield: 0.93 g, 93%. 1H NMR (500 MHz, DMSO-d6) δ 7.82 (t, J = 7.7 Hz, 1H, CH(Ar)), 7.44 (d, J = 7.7 Hz, 1H, CH(Ar)), 7.36 (d, J = 7.2 Hz, 1H, CH(Ar)), 5.26 (s, 1H, CH(OCH3)2), 3.63–3.57 (m, 6H, CH2(Morph), CH2), 3.30 (s, 6H, (OCH3)2), 2.44–2.40 (m, 4H, CH2(Morph)).
:2 ethyl acetate/hexane as eluent. The product was obtained after removal of the solvent as a colorless oil. Yield: 0.42 g, 58%. 1H NMR (500 MHz, DMSO-d6) δ 7.76 (t, J = 7.8 Hz, 1H, CH(Ar)), 7.35 (d, J = 7.7 Hz, 1H, CH(Ar)), 7.31–7.26 (m, 1H, CH(Pyrr)), 6.97 (dd, J = 3.9, 1.8 Hz, 1H, CH(Pyrr)), 6.60 (d, J = 7.6 Hz, 1H, CH(Ar)), 6.24 (dd, J = 3.9, 2.6 Hz, 1H, CH(Pyrr)), 5.64 (s, 2H, CH2), 5.24 (s, 1H, CH(OCH3)2), 3.65 (s, 3H, COOCH3), 3.31 (s, 6H, (OCH3)2).
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) max): 3420, 3258, 3162, 2940, 2802, 1599, 1546, 1446, 1342, 1280, 1146, 983, 924, 832, 789, 732, 684 cm−1. UV–vis in water (52 μM), λ, nm (ε, M−1 cm−1): 315 (30681).
max): 3039, 2929, 2801, 1597, 1542, 1446, 1361, 1156, 821, 711, 618 cm−1. UV–vis in water (51 μM), λ, nm (ε, M−1 cm−1): 216 (20784), 271sh (14510), 314 (31569).
:1) to give a white powder which was filtered off, washed with water and dried in vacuo. Yield: 0.37 g, 91%. Anal. Calcd for C12H17N5O2S·0.3CH3OH·0.1H2O (M 306.78 g mol−1): C, 48.16; H, 6.05; N, 22.83; S, 10.45. Found: C, 48.11; H, 6.43; N, 23.13; S, 10.66. E-isomer: 1H NMR (500 MHz, DMSO-d6) δ 11.63 (s, 1H, H2), 8.33 (s, 1H, H3), 8.16 (m, 2H, H6, H3), 8.08–7.99 (m, 1H, H12), 7.80 (t, J = 7.8 Hz, 1H, H5), 7.42 (d, J = 7.6 Hz, 1H4), 3.66–3.52 (m, 6H, H7, H9, H10), 2.42 (m, 4H, H8, H11). 13C NMR (126 MHz, DMSO-d6) δ 178.81 (Cq, C13), 158.35 (Cq, C3), 153.20 (Cq, C1), 143.03 (CH, C12), 137.37 (CH, C5), 123.58 (CH, C4), 119.05 (CH, C6), 66.66 (2CH2, C9, C10), 64.37 (CH2, C7), 53.79 (2CH2, C8, C11). Z-isomer: 1H NMR (500 MHz, DMSO-d6) δ 14.26 (s, 1H, H2), 8.52 (s, 1H, H3), 8.16 (m, 1H, H3), 8.08–7.99 (m, 1H, H5), 7.65 (d, J = 7.5 Hz, 1H, H6), 7.58 (d, J = 7.8 Hz, 1H, H4), 7.39 (s, 1H, H12), 3.71 (s, 2H, H7), 3.66–3.52 (m, 4H, H9, H10), 2.48 (m, 4H, H8, H11). 13C NMR (126 MHz, DMSO-d6) δ 179.37 (Cq, C13), 157.51 (Cq, C3), 151.68 (Cq, C1), 139.19 (CH, C5), 133.91 (CH, C12), 125.31 (CH, C6), 124.30 (CH, C4), 66.66 (2CH2, C9, C10), 64.10 (CH2, C7), 53.69 (2CH2, C8, C11). For atom numbering and structures of E and Z isomers see ESI, Scheme S1.† Solubility in water (with 1% DMSO) ≥1.4 mg mL−1. ESI-MS (methanol), positive: m/z 280 ([M + H]+). IR (ATR, selected bands, max): 3462, 3268, 3167, 2816, 1611, 1522, 1452, 1261, 1109, 1067, 850, 645 cm−1. UV–vis in water (39 μM), λ, nm (ε, M−1 cm−1): 316 (26923).
max): 2923, 2821, 1593, 1532, 1313, 1145, 1107, 901, 821, 707, 622 cm−1. UV–vis in water (48 μM), λ, nm (ε, M−1 cm−1): 216 (19628), 273 (13430), 315 (21488).
:1 mixture of methanol/ethanol (2 mL) in a 10 mL Schlenk tube was stirred at 78 °C overnight. The next day the solvent was removed under reduced pressure and the crude product was recrystallized from water/methanol (5:1). The resulting white powder was filtered off, washed with a water/methanol 1:1 mixture and dried in vacuo. Yield: 0.06 g, 76%. Anal. Calcd for C14H15N5O2S (M 317.37 g mol−1): C, 52.98; H, 4.76; N, 22.07; S, 10.10. Found: C, 52.98; H, 4.65; N, 21.97; S, 10.00. E-isomer: 1H NMR (500 MHz, DMSO-d6) δ 11.66 (s, 1H, H2), 8.35 (s, 1H, H3), 8.20–8.11 (m, 2H, H3, H6), 8.04 (s, 1H, H13), 7.75 (t, J = 7.9 Hz, 1H, H13), 7.33–7.29 (m, 1H, H9), 7.00–6.92 (m, 1H, H11), 6.60 (d, J = 7.7 Hz, 1H, H4), 6.28–6.21 (m, 1H, H10), 5.64 (s, 2H, H7), 3.66 (s, 3H, H15). 13C NMR (126 MHz, DMSO-d6) δ 178.84 (Cq, C15), 161.08 (Cq, C12), 158.54 (Cq, C3), 153.30 (Cq, C1), 142.66 (CH, C13), 137.99 (CH, C5), 131.10 (CH, C9), 121.65 (Cq, C8), 120.41 (CH, C4), 119.08 (CH, C6), 118.61 (CH, C11), 109.05 (CH, C10), 53.37 (CH2, C7), 51.39 (CH3, C15). Z-isomer: 1H NMR (500 MHz, DMSO-d6) δ 14.05 (s, 1H, H2), 8.60 (s, 1H, H3), 8.24 (s, 1H, H3), 8.00 (t, J = 7.8 Hz, 1H, H5), 7.66 (d, J = 7.6 Hz, 1H, H6), 7.64–7.59 (m, 1H, H9), 7.40 (s, 1H, H13), 6.98–6.91 (m, 2H, H4, H11), 6.28–6.21 (m, 1H, H10), 5.69 (s, 2H, H7), 3.68 (s, 3H, H15). 13C NMR (126 MHz, DMSO-d6) δ 179.40 (Cq, C14), 161.08 (Cq, C12), 157.34 (Cq, C3), 151.83 (Cq, C1), 139.84 (CH, C5), 133.66 (CH, C13), 131.63 (CH, C9), 125.65 (CH, C6), 122.07 (CH, C4), 121.30 (Cq, C8), 118.95 (CH, C11), 109.37 (CH, C10), 53.37 (CH2, C7), 51.47 (CH3, C15). ESI-MS (methanol), positive: m/z 340 ([M + Na]+), 317 ([M + H]+). IR (ATR, selected bands, max): 3561, 3353, 3243, 2972, 1706, 1612, 1530, 1443, 1245, 723, 653, 608 cm−1. UV–vis in methanol (22 μM), λ, nm (ε, M−1 cm−1): 204 (13636), 237 (12182), 266 (17227), 324 (25545), 388 (1227).
:1 mixture of methanol/ethanol (4 mL) in a 25 mL Schlenk tube was stirred at room temperature for 6 h. The white precipitate was filtered off, washed with a water/methanol 1:1 mixture and dried in vacuo. Yield: 0.12 g, 47%. Anal. Calcd for C16H19N5O2S (M 345.42 g mol−1): C, 55.63; H, 5.54; N, 20.27; S, 9.28. Found: C, 55.43; H, 5.50; N, 20.06; S, 9.21. E-isomer: 1H NMR (500 MHz, DMSO-d6) δ 11.18 (s, 1H, H2), 8.17 (s, 1H, H13), 7.79–7.73 (m, 2H, H6, H5), 7.33–7.27 (m, 1H, H9), 6.96 (dd, J = 3.9, 1.8 Hz, 1H, H11), 6.69–6.62 (m, 1H, H4), 6.23 (dd, J = 3.9, 2.6 Hz, 1H, H10), 5.65 (s, 2H, H7), 3.66 (s, 3H, H17), 3.31 (s, 6H, H15, H16). 13C NMR (126 MHz, DMSO-d6) δ 180.99 (Cq, C14), 161.06 (Cq, C12), 158.57 (Cq, C3), 153.55 (Cq, C1), 144.09 (CH, C13), 138.14 (CH, C5), 131.12 (CH, C9), 121.66 (Cq, C8), 120.34 (CH, C4), 118.56 (CH, C11), 118.35 (CH, C6), 108.96 (CH, C10), 53.40 (CH2, C7), 51.38 (CH3, C17), 42.76 (2CH3, C15, C16). Z-isomer: 1H NMR (500 MHz, DMSO-d6) δ 14.71 (s, 1H, H2), 7.99 (t, J = 7.9 Hz, 1H, H5), 7.65 (d, J = 7.7 Hz, 1H, H6), 7.62 (s, 1H, H13), 7.40–7.35 (m, 1H, H9), 7.02 (dd, J = 4.0, 1.8 Hz, 1H, H11), 6.49 (d, J = 7.8 Hz, 1H, H4), 6.30 (dd, J = 3.9, 2.6 Hz, 1H, H10), 5.73 (s, 2H, H7), 3.64 (s, 3H, H17), 3.44 (s, 6H, H15, H16). 13C NMR (126 MHz, DMSO-d6) δ 180.60 (Cq, C14), 161.01 (Cq, C12), 158.01 (Cq, C3), 151.49 (Cq, C1), 140.11 (CH, C5), 136.28 (CH, C13), 131.12 (CH, C9), 124.83 (CH, C6), 121.66 (Cq, C8), 120.10 (CH, C4), 118.94 (CH, C11), 109.49 (CH, C10), 53.40 (CH2, C7), 51.51 (CH3, C17), 40.60 (C15, C16, overlapped with residual DMSO signal). For atom numbering and structures of E and Z isomers see ESI, Scheme S1.† ESI-MS (methanol), positive: m/z 368 ([M + Na]+), 346 ([M + H]+). IR (ATR, selected bands, max): 2837, 1693, 1541, 1316, 1252, 1106, 899, 757, 622 cm−1. UV–vis in methanol (28 μM), λ, nm (ε, M−1 cm−1): 235 (23929), 266 (27321), 321 (21786), 405 (2757).
max): 3420, 3258, 3162, 2940, 2802, 1599, 1546, 1446, 1342, 1280, 1146, 983, 924, 832, 789, 732, 684 cm−1. UV–vis in water (52 μM), λ, nm (ε, M−1 cm−1): 315 (30681).
max): 3039, 2929, 2801, 1597, 1542, 1446, 1361, 1156, 821, 711, 618 cm−1. UV–vis in water (51 μM), λ, nm (ε, M−1 cm−1): 216 (20784), 271sh (14510), 314 (31569).
:1) to give a white powder which was filtered off, washed with water and dried in vacuo. Yield: 0.37 g, 91%. Anal. Calcd for C12H17N5O2S·0.3CH3OH·0.1H2O (M 306.78 g mol−1): C, 48.16; H, 6.05; N, 22.83; S, 10.45. Found: C, 48.11; H, 6.43; N, 23.13; S, 10.66. E-isomer: 1H NMR (500 MHz, DMSO-d6) δ 11.63 (s, 1H, H2), 8.33 (s, 1H, H3), 8.16 (m, 2H, H6, H3), 8.08–7.99 (m, 1H, H12), 7.80 (t, J = 7.8 Hz, 1H, H5), 7.42 (d, J = 7.6 Hz, 1H4), 3.66–3.52 (m, 6H, H7, H9, H10), 2.42 (m, 4H, H8, H11). 13C NMR (126 MHz, DMSO-d6) δ 178.81 (Cq, C13), 158.35 (Cq, C3), 153.20 (Cq, C1), 143.03 (CH, C12), 137.37 (CH, C5), 123.58 (CH, C4), 119.05 (CH, C6), 66.66 (2CH2, C9, C10), 64.37 (CH2, C7), 53.79 (2CH2, C8, C11). Z-isomer: 1H NMR (500 MHz, DMSO-d6) δ 14.26 (s, 1H, H2), 8.52 (s, 1H, H3), 8.16 (m, 1H, H3), 8.08–7.99 (m, 1H, H5), 7.65 (d, J = 7.5 Hz, 1H, H6), 7.58 (d, J = 7.8 Hz, 1H, H4), 7.39 (s, 1H, H12), 3.71 (s, 2H, H7), 3.66–3.52 (m, 4H, H9, H10), 2.48 (m, 4H, H8, H11). 13C NMR (126 MHz, DMSO-d6) δ 179.37 (Cq, C13), 157.51 (Cq, C3), 151.68 (Cq, C1), 139.19 (CH, C5), 133.91 (CH, C12), 125.31 (CH, C6), 124.30 (CH, C4), 66.66 (2CH2, C9, C10), 64.10 (CH2, C7), 53.69 (2CH2, C8, C11). For atom numbering and structures of E and Z isomers see ESI, Scheme S1.† Solubility in water (with 1% DMSO) ≥1.4 mg mL−1. ESI-MS (methanol), positive: m/z 280 ([M + H]+). IR (ATR, selected bands, max): 3462, 3268, 3167, 2816, 1611, 1522, 1452, 1261, 1109, 1067, 850, 645 cm−1. UV–vis in water (39 μM), λ, nm (ε, M−1 cm−1): 316 (26923).
max): 2923, 2821, 1593, 1532, 1313, 1145, 1107, 901, 821, 707, 622 cm−1. UV–vis in water (48 μM), λ, nm (ε, M−1 cm−1): 216 (19628), 273 (13430), 315 (21488).
:1 mixture of methanol/ethanol (2 mL) in a 10 mL Schlenk tube was stirred at 78 °C overnight. The next day the solvent was removed under reduced pressure and the crude product was recrystallized from water/methanol (5:1). The resulting white powder was filtered off, washed with a water/methanol 1:1 mixture and dried in vacuo. Yield: 0.06 g, 76%. Anal. Calcd for C14H15N5O2S (M 317.37 g mol−1): C, 52.98; H, 4.76; N, 22.07; S, 10.10. Found: C, 52.98; H, 4.65; N, 21.97; S, 10.00. E-isomer: 1H NMR (500 MHz, DMSO-d6) δ 11.66 (s, 1H, H2), 8.35 (s, 1H, H3), 8.20–8.11 (m, 2H, H3, H6), 8.04 (s, 1H, H13), 7.75 (t, J = 7.9 Hz, 1H, H13), 7.33–7.29 (m, 1H, H9), 7.00–6.92 (m, 1H, H11), 6.60 (d, J = 7.7 Hz, 1H, H4), 6.28–6.21 (m, 1H, H10), 5.64 (s, 2H, H7), 3.66 (s, 3H, H15). 13C NMR (126 MHz, DMSO-d6) δ 178.84 (Cq, C15), 161.08 (Cq, C12), 158.54 (Cq, C3), 153.30 (Cq, C1), 142.66 (CH, C13), 137.99 (CH, C5), 131.10 (CH, C9), 121.65 (Cq, C8), 120.41 (CH, C4), 119.08 (CH, C6), 118.61 (CH, C11), 109.05 (CH, C10), 53.37 (CH2, C7), 51.39 (CH3, C15). Z-isomer: 1H NMR (500 MHz, DMSO-d6) δ 14.05 (s, 1H, H2), 8.60 (s, 1H, H3), 8.24 (s, 1H, H3), 8.00 (t, J = 7.8 Hz, 1H, H5), 7.66 (d, J = 7.6 Hz, 1H, H6), 7.64–7.59 (m, 1H, H9), 7.40 (s, 1H, H13), 6.98–6.91 (m, 2H, H4, H11), 6.28–6.21 (m, 1H, H10), 5.69 (s, 2H, H7), 3.68 (s, 3H, H15). 13C NMR (126 MHz, DMSO-d6) δ 179.40 (Cq, C14), 161.08 (Cq, C12), 157.34 (Cq, C3), 151.83 (Cq, C1), 139.84 (CH, C5), 133.66 (CH, C13), 131.63 (CH, C9), 125.65 (CH, C6), 122.07 (CH, C4), 121.30 (Cq, C8), 118.95 (CH, C11), 109.37 (CH, C10), 53.37 (CH2, C7), 51.47 (CH3, C15). ESI-MS (methanol), positive: m/z 340 ([M + Na]+), 317 ([M + H]+). IR (ATR, selected bands, max): 3561, 3353, 3243, 2972, 1706, 1612, 1530, 1443, 1245, 723, 653, 608 cm−1. UV–vis in methanol (22 μM), λ, nm (ε, M−1 cm−1): 204 (13636), 237 (12182), 266 (17227), 324 (25545), 388 (1227).
:1 mixture of methanol/ethanol (4 mL) in a 25 mL Schlenk tube was stirred at room temperature for 6 h. The white precipitate was filtered off, washed with a water/methanol 1:1 mixture and dried in vacuo. Yield: 0.12 g, 47%. Anal. Calcd for C16H19N5O2S (M 345.42 g mol−1): C, 55.63; H, 5.54; N, 20.27; S, 9.28. Found: C, 55.43; H, 5.50; N, 20.06; S, 9.21. E-isomer: 1H NMR (500 MHz, DMSO-d6) δ 11.18 (s, 1H, H2), 8.17 (s, 1H, H13), 7.79–7.73 (m, 2H, H6, H5), 7.33–7.27 (m, 1H, H9), 6.96 (dd, J = 3.9, 1.8 Hz, 1H, H11), 6.69–6.62 (m, 1H, H4), 6.23 (dd, J = 3.9, 2.6 Hz, 1H, H10), 5.65 (s, 2H, H7), 3.66 (s, 3H, H17), 3.31 (s, 6H, H15, H16). 13C NMR (126 MHz, DMSO-d6) δ 180.99 (Cq, C14), 161.06 (Cq, C12), 158.57 (Cq, C3), 153.55 (Cq, C1), 144.09 (CH, C13), 138.14 (CH, C5), 131.12 (CH, C9), 121.66 (Cq, C8), 120.34 (CH, C4), 118.56 (CH, C11), 118.35 (CH, C6), 108.96 (CH, C10), 53.40 (CH2, C7), 51.38 (CH3, C17), 42.76 (2CH3, C15, C16). Z-isomer: 1H NMR (500 MHz, DMSO-d6) δ 14.71 (s, 1H, H2), 7.99 (t, J = 7.9 Hz, 1H, H5), 7.65 (d, J = 7.7 Hz, 1H, H6), 7.62 (s, 1H, H13), 7.40–7.35 (m, 1H, H9), 7.02 (dd, J = 4.0, 1.8 Hz, 1H, H11), 6.49 (d, J = 7.8 Hz, 1H, H4), 6.30 (dd, J = 3.9, 2.6 Hz, 1H, H10), 5.73 (s, 2H, H7), 3.64 (s, 3H, H17), 3.44 (s, 6H, H15, H16). 13C NMR (126 MHz, DMSO-d6) δ 180.60 (Cq, C14), 161.01 (Cq, C12), 158.01 (Cq, C3), 151.49 (Cq, C1), 140.11 (CH, C5), 136.28 (CH, C13), 131.12 (CH, C9), 124.83 (CH, C6), 121.66 (Cq, C8), 120.10 (CH, C4), 118.94 (CH, C11), 109.49 (CH, C10), 53.40 (CH2, C7), 51.51 (CH3, C17), 40.60 (C15, C16, overlapped with residual DMSO signal). For atom numbering and structures of E and Z isomers see ESI, Scheme S1.† ESI-MS (methanol), positive: m/z 368 ([M + Na]+), 346 ([M + H]+). IR (ATR, selected bands, max): 2837, 1693, 1541, 1316, 1252, 1106, 899, 757, 622 cm−1. UV–vis in methanol (28 μM), λ, nm (ε, M−1 cm−1): 235 (23929), 266 (27321), 321 (21786), 405 (2757).
Synthesis of the copper(II) complexes
max): 3266, 3094, 1613, 1459, 1418, 1158, 1025, 978, 825, 787, 653 cm−1. UV–vis in water, λ, nm (ε, M−1 cm−1): 286 (19545), 392 (10063) (measured at 44 μM); 607 (265) (measured at 1.88 mM). X-ray diffraction quality crystals of the composition 1·0.15CH3OH were obtained after slow diffusion of diethyl ether into a methanolic solution of 1 (c ≈ 5 mg mL−1) in the presence of a small amount of triethylamine.
max): 3458, 3394, 3037, 2690, 1492, 1369, 1311, 1249, 1130, 908, 612 cm−1. UV–vis in water, λ, nm (ε, M−1 cm−1): 254 (11463), 299 (17561), 405 (15366) (measured at 41 μM); 574 (145) (measured at 1.79 mM).
max): 3431, 3367, 3109, 1677, 1640, 1462, 1419, 1166, 1114, 783, 630 cm−1. UV–vis in water, λ, nm (ε, M−1 cm−1): 284 (20179), 389 (10893) (measured at 56 μM); 598 (252) (measured at 2.83 mM).
max): 3499, 2859, 1593, 1359, 1242, 1123, 909, 869, 789 cm−1. UV–vis in water, λ, nm (ε, M−1 cm−1): 255 (10474), 299 (17207), 405 (15212) (measured at 40 μM); 575 (293) (measured at 1.78 mM).
max): 3346, 3105, 1704, 1622, 1463, 1406, 1332, 1253, 1111, 734, 635 cm−1. UV–vis in DMF, λ, nm (ε, M−1 cm−1): 298 (16447), 326 (13026, sh), 422 (11474) (measured at 76 μM); 514 (274, sh), 684 (225) (measured at 2.29 mM).
max): 3420, 3006, 1705, 1508, 1377, 1246, 1113, 913, 735, 587 cm−1. UV–vis in DMF, λ, nm (ε, M−1 cm−1): 306 (13851), 426 (15946) (measured at 74 μM); 519 (435), 648 (256) (measured at 2.21 mM).
pH-Potentiometric measurements
The purity and aqueous phase stability of the ligands mPip-dm-FTSC (HL2) and Morph-dm-FTSC (HL4) were verified and the exact concentrations of the stock solutions prepared were determined by the Gran method.63 The pH-metric measurements for determination of the protonation constants of the ligands and the overall stability constants of the copper(II) complexes were carried out at 298.0 ± 0.1 K in water and at an ionic strength of 0.10 M (KCl) in order to keep the activity coefficients constant. The titrations were performed with carbonate-free KOH solution of known concentration (0.10 M). The concentrations of the KOH and the HCl were determined by pH-potentiometric titrations. An Orion 710A pH-meter equipped with a Metrohm combined electrode (type 6.0234.100) and a Metrohm 665 Dosimat burette were used for the pH-metric measurements. The electrode system was calibrated to the pH = −log[H+] scale in water according to the method suggested by Irving et al.64 The average water ionization constant pKw, is 13.76 ± 0.01, which corresponds well to the literature data.65 The reproducibility of the titration points included in the calculations was within 0.005 pH. The pH-metric titrations were performed in the pH range 2.0–11.5. The initial volume of the samples was 5.0 mL. The concentration of the ligands was 2 mM and metal ion-to-ligand ratios of 1:1, 1:1.5, 1:2 and 1:3 were used. The accepted fitting of the titration curves was always less than 0.01 mL. Samples were deoxygenated by bubbling purified argon through them for ca. 10 min prior to the measurements and argon was also passed over the solutions during the titrations.
The protonation constants of the ligands were determined with the computer program HYPERQUAD.66 PSEQUAD67 was utilized to establish the stoichiometry of the complexes and to calculate the stability constants (logβ(MpLqHr)). β(MpLqHr) is defined for the general equilibrium pM + qL + rH ⇌ MpLqHr as β(MpLqHr) = [MpLqHr]/[M]p[L]q[H]r, where M denotes the metal ion (copper(II)) and L the completely deprotonated ligand. In all calculations exclusively titration data were used from experiments, in which no precipitate was visible in the reaction mixture.
UV–vis spectrophotometric, spectrofluorimetric and 1H NMR measurements
A Thermo Scientific Evolution 220 spectrophotometer was used to record the UV–vis spectra in the 200 to 1050 nm window. The path length was 1 or 0.5 cm. Stability constants and the individual spectra of the complexes were calculated by the computer program PSEQUAD.67 The spectrophotometric titrations were performed on samples of the copper(II) complexes 2 and 4 of ligands mPip-dm-FTSC (HL2) and Morph-dm-FTSC (HL4) over the pH range between 2 and 11.5 at an ionic strength of 0.10 M (KCl) in water at 298.0 ± 0.1 K. The concentration of the metal complexes was 2.5 mM. Measurements on the copper(II) complexes 2 and 4 were also carried out by preparing individual samples, in which the 0.1 M KCl was partially or completely replaced by HCl and pH values, varying in the range of approximately 1.0–3.0, were calculated from the HCl content. The conditional stability constants of [CuL] at pH 7.4 (10 mM HEPES) for 2 and at pH 5.6 (10 mM MES) for 4 were determined from competition titrations of the copper(II) complex of EDTA with the ligands HL2 and HL4. Samples contained 50 μM copper(II) ion and 50 μM EDTA, and the concentration of the ligands HL2 and HL4 was varied in the range of 0–170 μM. Absorbance data recorded after 1.5 h incubation time in the wavelength interval from 415 to 450 nm were used for the calculations.Three-dimensional fluorescence spectra of the ligands mPip-dm-FTSC (HL2) and Morph-dm-FTSC (HL4) and their copper(II) complexes (2 and 4) were recorded at 240–500 nm excitation and at 300–700 nm emission wavelengths for the 10 μM ligand containing samples in 1 cm quartz cell at pH 7.4 (10 mM HEPES) using 5 nm/5 nm slit widths at 0.1 M (KCl) ionic strength and 298.0 ± 0.1 K.
The pH-dependent 1H NMR studies were carried out on a Bruker Ultrashield 500 Plus instrument. 4,4-Dimethyl-4-silapentane-1-sulfonic acid was used as an internal NMR standard. Ligands mPip-dm-FTSC (HL2) and Morph-dm-FTSC (HL4) were dissolved in a 10% (v/v) D2O/H2O mixture in a concentration of 3.0 and 1.5 mM, respectively. The direct pH-meter readings were corrected according to the method of Irving et al.64 Spectra of the ligands were recorded using individual samples, in which the 0.1 M KCl was partially or completely replaced by HCl and pH values, varying in the range of approximately 1.0–2.0, were calculated from the HCl content.
Determination of the distribution coefficients (D)
D 7.4 values of ligands HL1–HL5 and their copper(II) complexes (1–5) were determined by the traditional shake-flask method in n-octanol/buffered aqueous solution at pH 7.4 at 298.0 ± 0.2 K as described previously.68,69 Two parallel experiments were performed for each sample. The ligands and the complexes were dissolved at 100 μM (∼30 μM in the case of HL5 and its complex 5) in the n-octanol pre-saturated aqueous solution of the buffer (10 mM HEPES) at constant ionic strength (0.10 M KCl). The aqueous solutions and n-octanol with 1:1 phase ratio were gently mixed with 360° vertical rotation for 3 h to avoid the emulsion formation, and the mixtures were centrifuged at 5000 rpm for 5 min by a temperature controlled centrifuge (Sanyo) at 298 K. After separation UV–vis spectra of the ligands or complexes in the aqueous phase were compared to those of the original aqueous solutions. Since measurable amounts of the ligand HL5 and its copper(II) complex 5 were not found in the aqueous phase after partitioning, their logD7.4 values were merely estimated.
EPR measurements and deconvolution of the spectra
All continuous wave (CW)-EPR spectra were recorded with a BRUKER EleXsys E500 spectrometer (microwave frequency 9.85 GHz, microwave power 10 mW, modulation amplitude 5 G, modulation frequency 100 kHz). The pH-dependent series of isotropic EPR spectra were recorded in a circulating system, at room temperature. A Heidolph Pumpdrive 5101 peristaltic pump was used to transport the solution from the titration pot through a capillary tube into a Bruker flat cell placed in the cavity of the instrument. The EPR titrations were performed over the pH range between 1.5 and 12.0 at an ionic strength of 0.10 M (KCl) under nitrogen atmosphere. Samples contained 1 mM mPip-dm-FTSC (HL2) or Morph-dm-FTSC (HL4) and 1 mM or 0.5 mM copper(II) ions. A 0.1 M KOH solution was added to the samples to adjust the pH, which was measured with an Orion 710A pH-meter equipped with a Metrohm 6.0234.100 glass electrode. For selected pH values (where predominantly complexes formed) 0.1 mL samples were introduced into quartz EPR tubes and measured individually at 77 K.Before the simulation, the measured spectra were corrected by subtracting the spectra of water measured in the same circulating system. A phase correction of −7 degree for the series of Morph-dm-FTSC (HL4) and −8 degree for the mPip-dm-FTSC (HL2) containing samples was used to correct the phase of the spectra which were probably shifted due to the not perfectly perpendicular position of the flat cell to the magnetic field. Both series of the pH-dependent isotropic CW-EPR spectra were simulated by the “two-dimensional” method using the 2D_EPR program.70 The parameters go, ACuo copper hyperfine (ICu = 3/2) and ANo nitrogen (IN = 1) superhyperfine couplings have been taking into account to describe each component curve. The relaxation parameters, α, β, and γ defined the linewidths through the equation σMI = α + βMI + γMI2, where MI denotes the magnetic quantum number of the paramagnetic metal ions. The equilibrium concentrations of the copper(II) complexes were varied by fitting their overall stability constants β(MpLqHr) defined in the section of pH-potentiometric measurements. For each spectrum, the noise-corrected regression parameter (Rj for the jth spectrum) is derived from the average square deviation (SQD) between the experimental and the calculated intensities. For the series of spectra, the fit is characterized by the overall regression coefficient R, calculated from the overall average SQD. The overall regression coefficient was 0.9933 for the series of Morph-dm-FTSC and 0.9928 for the series of mPip-dm-FTSC. The details of the statistical analysis were published previously.70 The anisotropic EPR spectra, recorded at 77 K, were analyzed individually with the aid of the EPR program.71 In case of copper(II) complexes, the anisotropic EPR parameters: rhombic g-tensor (gx, gy, gz), rhombic copper(II) hyperfine tensor (ACux, ACuy, ACuz) and rhombic nitrogen hyperfine tensor (aNx, aNy, aNz, for which x, y and z denotes the directions of the g-tensor) were fitted. For the description of the linewidth the orientation dependent α, β and γ parameters were used to set up each component spectra. Since a natural CuCl2 salt was used for the measurements, both the isotropic and anisotropic spectra were calculated as the sum of the spectra of 63Cu and 65Cu weighted by their natural abundances. The hyperfine and superhyperfine coupling constants and the relaxation parameters were obtained in field units (Gauss = 10−4 T).
Crystallographic structure determination
X-ray diffraction measurements were performed on Bruker X8 APEXII CCD and Bruker D8 VENTURE diffractometers. Single crystals were positioned at 35, 35, 40, 35 and 40 mm from the detector, and 1645, 1386, 890, 3033 and 2542 frames were measured, each for 48, 8, 48, 65 and 24 s over 0.4, 0.4, 0.4, 0.5 and 0.4° scan width for 1·0.15CH3OH, 2·2H2O, 3·0.5(C2H5)2O, 4·0.93CH3OH and 5, respectively. The data were processed using SAINT software.72 Crystal data, data collection parameters, and structure refinement details are given in Table 1. The structures were solved by direct methods and refined by full-matrix least-squares techniques. Non-H atoms were refined with anisotropic displacement parameters. H atoms were inserted in calculated positions and refined with a riding model. In the crystal structure of 1·0.15CH3OH a partly occupied (15%) co-crystallized methanol molecule position was found, while in the crystal structure of 3·0.5(C2H5)2O half molecule of diethyl ether per asymmetric unit was found to be disordered over two positions with site occupation factor (s.o.f.) 0.5:0.5. In the crystal of 4·0.93CH3OH one molecule of co-crystallized methanol is disordered over 3 positions with s.o.f. 0.4:0.35:0.25, while the second molecule position is populated to 85%. The disorder was solved by using SADI instructions implemented in SHELXL-97, while the atoms involved were refined with isotropic displacement parameters and the positions of H atoms were calculated. The following computer programs were used: structure solution, SHELXS-97 and refinement, SHELXL-97;73 molecular diagrams, ORTEP.74 CCDC 1052906–1052910.
| 1·0.15CH3OH | 2·2H2O | 3·0.5(C2H5)2O | 4·0.93CH3OH | 5 | |
|---|---|---|---|---|---|
| a R 1 = ∑||Fo| − |Fc||/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. c GOF = {∑[w(Fo2 − Fc2)2]/(n − p)}1/2, where n is the number of reflections and p is the total number of parameters refined. | |||||
| Empirical formula | C13.15H19.6ClCuN6O0.15S | C15H28Cl2CuN6O2S | C14H21ClCuN5O1.5S | C14.93H23.7ClCuN5O1.92S | C17H21ClCuN6O3S |
| Fw | 395.20 | 490.93 | 414.42 | 435.04 | 488.45 |
| Space group | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n |
P |
P |
| a [Å] | 7.6944(5) | 7.6629(3) | 7.7969(6) | 9.8676(7) | 8.0848(4) |
| b [Å] | 17.4966(10) | 11.7864(3) | 17.4139(14) | 12.0982(8) | 12.1277(6) |
| c [Å] | 13.1409(8) | 13.0949(4) | 13.145(1) | 16.606(1) | 12.3563(5) |
| α [°] | 72.114(1) | 86.450(3) | 63.8664(17) | ||
| β [°] | 102.615(2) | 73.613(1) | 103.700(2) | 74.015(3) | 83.4075(18) |
| γ [°] | 74.311(1) | 77.176(3) | 73.0771(17) | ||
| V [Å3] | 1726.40(18) | 1057.72(6) | 1734.0(2) | 1858.2(2) | 1040.45(8) |
| Z | 4 | 2 | 4 | 4 | 2 |
| λ [Å] | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| ρ calcd [g cm−3] | 1.520 | 1.541 | 1.587 | 1.555 | 1.559 |
| Crystal size, mm | 0.26 × 0.02 × 0.02 | 0.17 × 0.10 × 0.04 | 0.09 × 0.04 × 0.03 | 0.50 × 0.40 × 0.40 | 0.10 × 0.08 × 0.06 |
| T [K] | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| μ [mm−1] | 1.547 | 1.407 | 1.548 | 1.450 | 1.310 |
|
R
1a |
0.0334 | 0.0303 | 0.0424 | 0.0279 | 0.0326 |
| wR2b |
0.0753 | 0.0832 | 0.0950 | 0.0683 | 0.0745 |
| GOFc | 1.079 | 1.033 | 1.011 | 1.095 | 1.040 |
Cell lines and culture conditions
Human cervical carcinoma (HeLa), human alveolar basal adenocarcinoma (A549), human colon carcinoma (LS174) cell lines and normal human foetal lung fibroblast cell line (MRC-5) were maintained as a monolayer culture in the Roswell Park Memorial Institute (RPMI) 1640 nutrient medium (Sigma Chemicals Co, USA). RPMI 1640 nutrient medium was prepared in sterile deionized water, supplemented with penicillin (192 U mL−1), streptomycin (200 μg mL−1), HEPES (25 mM), L-glutamine (3 mM) and 10% of heat-inactivated foetal calf serum (FCS) (pH 7.2). The cells were grown at 310 K in a humidified 5% CO2 air atmosphere.MTT assay
Antiproliferative activity of the investigated ligands and complexes was determined using 3-(4,5-dimethyl-2-thiazolyl)-2,5-2H-diphenyltetrazolium bromide (MTT, Sigma-Aldrich) assay.75 Cells were seeded into 96-well cell culture plates (Thermo Scientific Nunc™), at a cell density of 4000 cells per well (HeLa), 6000 cells per well (A549), 5000 cells per well (MRC-5) and 7000 cells per well (LS174) in 100 μL of culture medium. After 24 h of growth, cells were exposed to the serial dilutions of the tested compounds. The investigated compounds were dissolved in sterile water at a concentration of 10 mM as stock solution (complexes 2 and 4), 5 mM (complex 1), or 2 mM (complex 3), and prior the use diluted with nutrient medium to the desired final concentrations (in range up to 300 μM). Ligands were dissolved in sterile water at a concentration of 10 mM HL1 and HL2 and 5 mM HL4, while ligand HL3 was dissolved in 1% DMSO at a concentration of 3 mM. Each concentration was tested in triplicates. After incubation periods of 48 h, 20 μL of MTT solution (5 mg mL−1 in phosphate buffer solution, pH 7.2) were added to each well. Samples were incubated for 4 h at 310 K, with 5% CO2 in a humidified atmosphere. Formazan crystals were dissolved in 100 μL of 10% sodium dodecyl sulfate (SDS). Absorbances were recorded after 24 h, on an enzyme-linked immunosorbent assay (ELISA) reader (Thermo Labsystems Multiskan EX 200–240 V), at the wavelength of 570 nm. The IC50 value, defined as the concentration of the compound causing 50% cell growth inhibition, was estimated from the dose–response curve.Results and discussion
These studies examined the effects of attachment of methylpiperazine, morpholine and methylpyrrole-2-carboxylate to the pyridine ring of the parent thiosemicarbazone on the aqueous solubility, lipophilicity, ability to form copper(II) complexes, their thermodynamic stability in aqueous solution, and antiproliferative activity in human cancer cell lines HeLa, A549 and LS174, as well as in nontumorigenic cell line MRC5.Synthesis and characterization of HL1–HL6
The organic hybrids were synthesized in six steps, as shown in Scheme S2.† The first four steps were described in detail previously.61,76 The key aldehydes were prepared in two steps. First, 2-(chloromethyl)-6-(dimethoxymethyl)pyridine (D) was allowed to react with methylpiperazine or morpholine in THF in the presence of triethylamine or with methylpyrrole-2-carboxylate in DMF in the presence of NaH, following a literature procedure,77 affording compounds E–G in 79, 93 and 58% yields, respectively. The aldehydes H–J were obtained by hydrolysis of species E–G in acidic aqueous solution or in acetone/water 1:5 mixture. Finally, condensation reactions of the aldehydes with thiosemicarbazide and/or 4,4-dimethyl-3-thiosemicarbazide afforded the hybrids HL1–HL6 in 47–91% yields. One- and two-dimensional 1H and 13C NMR spectra confirmed the expected structures for HL1–HL6 and the presence of E and Z isomers in DMSO. The E/Z ratio is 1:0.12, 1:0.66, 1:0.34, 1:0.62, 1:0.27 and 1:0.36 for HL1–HL6 respectively, (measured at a concentration of approximately 10 mM). The presence of E and Z isomers is typical for thiosemicarbazones and our data are in good agreement with those reported for other α-pyridyl-TSCs.78 The purity of HL1–HL6 was further evidenced by elemental analysis. The positive-ion ESI mass spectra of HL1–HL6 showed strong peaks at m/z 293, 321, 280, 308, 317 and 346, respectively, which were assigned to the [M + H]+ ion. The lipo-hydrophilic character of the ligands (HL1–HL5) is discussed in the section Solution Chemistry.
Synthesis and characterization of copper(II) complexes
By reaction of CuCl2·2H2O with HL1 and HL2 in methanol two complexes [CuCl(mPipH-FTSC-H)]Cl·0.1H2O ((1 + H)Cl·0.1H2O) and [CuCl(mPipH-dm-FTSC-H)]Cl·0.9H2O·0.5CH3OH ((2 + H)Cl·0.9H2O·0.5CH3OH) were obtained in 74 and 53% yields, respectively. The formulation of both complexes was in accord with X-ray diffraction measurements (vide infra) and elemental analyses. Re-crystallization of the first complex from methanol in the presence of a small amount of triethylamine led to crystallization of the complex with deprotonated piperazine moiety, namely [CuCl(mPip-FTSC-H)]·0.15CH3OH (1·0.15CH3OH), the structure of which was established by single crystal X-ray crystallography. ESI mass spectra of copper(II) complexes with HL1 and HL2 showed peaks with m/z 354 and 382, attributed to [M − Cl]+ ion. The copper(II) complexes [CuCl(Morph-FTSC-H)]·2H2O·0.2C2H5OH (3·2H2O·0.2C2H5OH) and [CuCl(Morph-dm-FTSC-H)]·0.2H2O·0.6CH3OH (4·0.2H2O·0.6CH3OH) were prepared in 88 and 98% yields, by reaction of copper(II) chloride with HL3 and HL4 in ethanol, and methanol, respectively. The composition of both complexes was confirmed by elemental analysis, X-ray diffraction data and ESI mass spectra. The latter showed the presence of peaks at m/z 341 and 369, assigned to [M − Cl]+ ion. Starting from copper(II) chloride and HL5 and HL6 in methanol the complexes [CuCl(mPyrr-FTSC-H)(H2O)]·0.2H2O (5·0.2H2O) and [CuCl(mPyrr-dm-FTSC-H)(H2O)] (6) were obtained in 71 and 55% yield, respectively. This was confirmed by elemental analysis and ESI mass spectra. The latter showed the presence of peaks with m/z 379 and 407, assigned to [M − Cl − H2O]+ ion. Re-crystallization of [CuCl(mPyrr-FTSC-H)(H2O)]·0.2H2O from DMF afforded the complex [CuCl(mPyrr-FTSC-H)(DMF)] (5) via replacement of coordinated water molecule by DMF, as was confirmed by single crystal X-ray diffraction analysis (see section X-ray Crystallography). Lipo-hydrophilicity data of the copper(II) complexes 1–5 are discussed in the section Solution Chemistry.X-ray crystallography
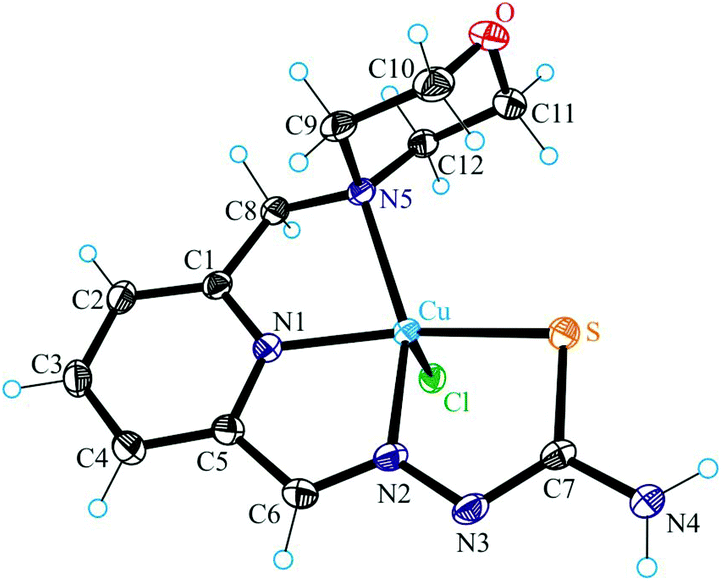
The results of single crystal X-ray diffraction studies of 1·0.15CH3OH, [2 + H]Cl·2H2O, 3·0.5(C2H5)2O, 4·0.93CH3OH and 5 are shown in Fig. 1–5. The complexes 1·0.15CH3OH and 4·0.93CH3OH crystallized in the centrosymmetric monoclinic space group P21/n, while 2·2H2O, 3·0.5(C2H5)2O and 5 crystallized in the centrosymmetric triclinic space group P. The piperazine–thiosemicarbazone and morpholine–thiosemicarbazone hybrid ligands HL1, HL3 and HL4 in 1, 3 and 4 act as tetradentate monodeprotonated ligands coordinating to copper(II) via the pyridine nitrogen atom, the azomethine nitrogen, the thiolato S atom and the piperazine or morpholine nitrogen atom, while in [2 + H]Cl·2H2O the organic ligand HL2 is overall neutral being deprotonated at N3 and protonated at N6. The coordination number of copper(II) is five in complexes 1–4 and the coordination polyhedron can be described as a square-pyramid79 (τ = 0.13, 0.11, 0.12 and 0.10 (0.07 for another crystallographically independent complex), respectively). The apical position is occupied by a chlorido ligand. Three five-membered metallocycles are formed upon binding of the monodeprotonated ligands (L1)−–(L4)− to copper(II). Two of them are essentially planar, while the N1–C1–C8–N5–Cu in 1 and 3, or N1–C1–C10–N5–Cu in 2 and 4 is markedly distorted. The dihedral angle N1–C1–C8–N5/N1–C1–C10–N5 used here as a measure of the deviation of the chelate ring from planarity is at −28.1(3) and −28.3(3)° in 1 and 3, and at −29.62(19) and −22.5(3)° in 2 and 4 (for one of the two crystallographically independent molecules), respectively. This is not surprising if one takes into account the presence of an aliphatic carbon atom (C8/C10) in this chelate ring.
 | ||
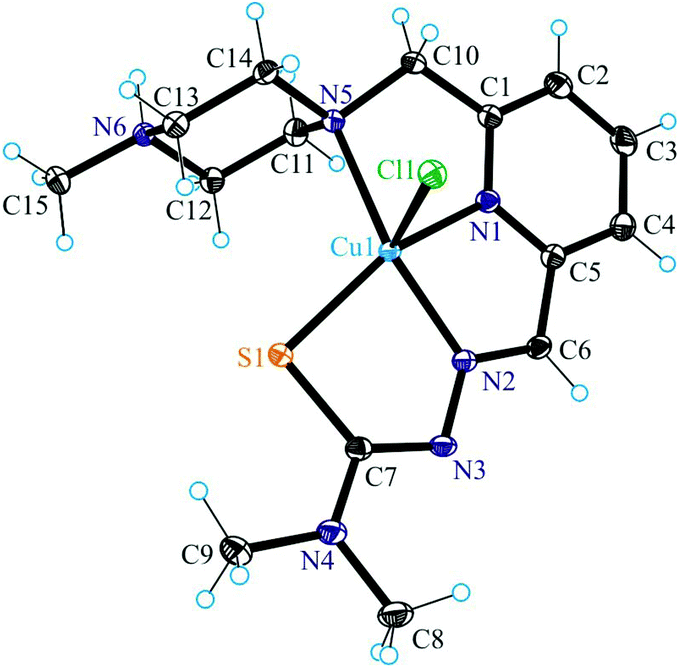
| Fig. 1 ORTEP view of 1 with thermal ellipsoids drawn at the 50% probability level. Selected bond distances (Å) and bond angles (°): Cu–N1 1.959(2), Cu–N2 2.011(2), Cu–S 2.2725(7), Cu–N5 2.172(2), Cu–Cl 2.4724(7), N2–N3 1.352(3), C7–S 1.749(3), N1–Cu–N2 79.07(9), N2–Cu–S 83.43(7), N1–Cu–N5 78.60(9), N1–Cu–S 158.26(7), N5–Cu–S 112.56(6), Cl–Cu–N1 96.58(7), Cl–Cu–N2 106.20(7), Cl–Cu–S 100.62(3), Cl–Cu–N5 95.53(6), N2–Cu–N5 150.35(9). | ||
 | ||
| Fig. 2 ORTEP view of [2 + H]+ with thermal ellipsoids drawn at the 50% probability level. Selected bond distances (Å) and bond angles (°): Cu–N1 1.9508(13), Cu–N2 1.9901(13), Cu–S 2.2719(4), Cu–N5 2.2370(13), Cu–Cl 2.4295(4), N2–N3 1.3549(18), C7–S 1.7620(16), N1–Cu–N2 79.91(6), N1–Cu–N5 77.91(2), N2–Cu–S 83.31(4), N5–Cu–S 113.45(4), Cl–Cu–N1 96.25(4), Cl–Cu–N2 109.71(4), Cl–Cu–S 101.600(15), Cl–Cu–N5 89.22(4), N1–Cu–S 158.77(4), N2–Cu–N5 152.23(5). | ||
 | ||
| Fig. 3 ORTEP view of 3 with thermal ellipsoids drawn at the 50% probability level. Selected bond distances (Å) and bond angles (°): Cu–N1 1.951(2), Cu–N2 2.004(2), Cu–S 2.2711(7), Cu–N5 2.169(2), Cu–Cl 2.4786(7), N2–N3 1.353(3), C7–S 1.744(2), N1–Cu–N2 79.09(8), N1–Cu–N5 78.88(8), N2–Cu–S 83.37(6), N5–Cu–S 112.30(6), Cl–Cu–N1 96.08(6), Cl–Cu–N2 107.42(6), Cl–Cu–S 101.66(2), Cl–Cu–N5 93.83(6), N1–Cu–S1 158.09(6), N2–Cu–N5 150.76(8). | ||
 | ||
| Fig. 4 ORTEP view of one crystallographically independent molecule of 4 with thermal ellipsoids drawn at the 50% probability level. Selected bond distances (Å) and bond angles (°): Cu1a–N1a 1.9388(17), Cu1a–N2a 1.9948(17), Cu1a–S1a 2.2523(6), Cu1a–N5a 2.1406(17), Cu1a–Cl1a 2.5088(6), N2a–N3a 1.358(2), C7a–S1a 1.754(2), N1a–Cu1a–N2a 79.68(7), N1a–Cu1a–N5a 79.90(7), N2a–Cu1a–S1a 83.88(5), N5a–Cu1a–S1a 111.88(5), Cl1a–Cu1a–N1a 91.49(5), Cl1a–Cu1a–N2a 103.19(5), Cl1a–Cu1a–S1a 103.65(2), Cl1a–Cu1a–N5a 93.40(5), N1a–Cu1a–S1a 159.83(5), N2a–Cu1a–N5a 153.91(7). (0.10 and 0.07). | ||
 | ||
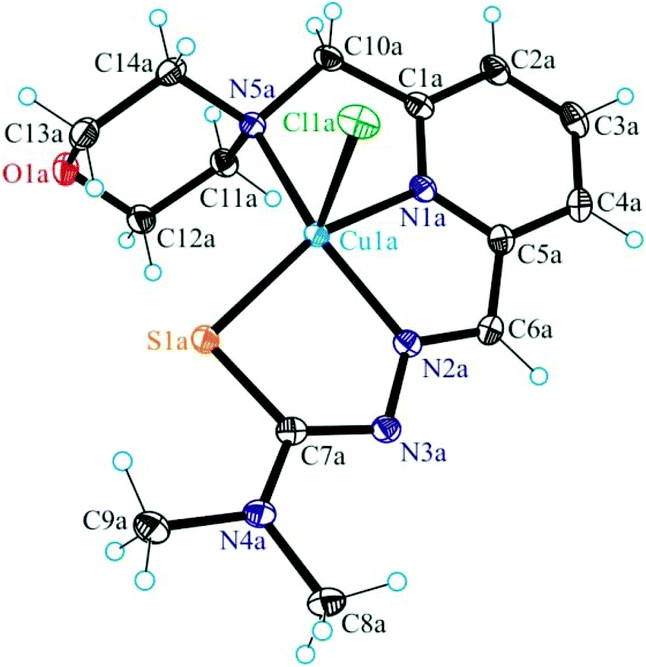
| Fig. 5 ORTEP view of 5 with thermal ellipsoids drawn at the 50% probability level. Selected bond distances (Å) and bond angles (°): Cu–N1 2.0912(14), Cu–N2 1.9820(13), Cu–S 2.2785(5), Cu–O3 2.1852(13), Cu–Cl 2.3069(4), N2–N3 1.3641(19), C7–S 1.7387(17), N1–Cu–N2 80.48(6), N2–Cu–S 83.13(4), O3–Cu–S 97.27(2), Cl–Cu–N1 98.97(4), Cl–Cu–N2 137.78(4), Cl–Cu–S 94.701(16), Cl–Cu–O3 107.32(4), N1–Cu–S 163.45(4). | ||
The terminal amine nitrogen N4 of the thiosemicarbazone moiety is involved as a proton donor in hydrogen bonding to the nitrogen atom N3i of a neighboring molecule of 1 forming pairs of molecules as displayed in Fig. S1† and in hydrogen bonding to Cl1ii, where i and ii denote the atoms generated by symmetry transformations −x + 2, −y + 1, −z + 1 and x + 1, y, z, respectively.
The protonated atom N6 acts as a proton donor to the chloride counterion with N6⋯Cl2i at 3.0837(14) Å, where i denotes atom positions generated by symmetry transformation x − 1, y + 1, z, and N6–H⋯Cl2i 159.5°. Four other hydrogen bonds are formed between the co-crystallized water molecules and the chloride counterion.
Unlike, the hybrid ligand HL5 acts as a tridentate monodeprotonated ligand binding to copper(II) via pyridine nitrogen N1, azomethine atom N2 and thiolato atom S. Like in complexes 1–4 the coordination number of the copper(II) center in 5 is five, and the coordination geometry shows a slight tendency to square-pyramidal (τ = 0.43), the remaining two places being occupied by the DMF molecule and the chlorido ligand. The pyrrol nitrogen atom N5, due to its sp2 hybridization remains unbound to copper(II). Note that sp3-hybridized proline nitrogen atom in proline–thiosemicarbazone conjugates was involved in binding to first-row transition metals and became a chiral center upon coordination.80
Solution chemistry: proton dissociation processes of ligands HL2 and HL4, lipophilicity of ligands HL1–HL5
Morph-dm-FTSC (HL4), which forms the most biologically active copper(II) complex among the studied ligands (vide infra), was chosen for the detailed solution equilibrium studies together with its methylpiperazine analogue, mPip-dm-FTSC (HL2) (Chart 1) for comparison. Deprotonation processes of these ligands were followed in aqueous solution by pH-potentiometric and 1H NMR titrations. Consecutive multiple pH-potentiometric titrations showed that no ligand decomposition occurred in the pH range studied (2.0–11.5) under an argon atmosphere. Morph-dm-FTSC (HL4) possesses three, while mPip-dm-FTSC (HL2) four functional groups which, presumably, dissociate. The proton dissociation constants determined by pH-potentiometry are listed in Table 2. The identical N-terminally dimethylated α-N-pyridyl thiosemicarbazone moiety of the ligands is expected to have a relatively low pKa value for the NpyridylH+ and a significantly higher value for the NhydrazinicH functional group based on the proton dissociation constants of structurally similar HCTs, such as 2-formylpyridine N4,N4-dimethylthiosemicarbazone (PTSC, pK1: 3.38 and pK2: 10.54) or 3-aminopyridine-2-carboxaldehyde N4,N4-dimethylthiosemicarbazone (APTSC, pK1: 4.31 and pK2: 10.29).81 Taking into account these data we attributed the pK2 of Morph-dm-FTSC to the deprotonation of the morpholinium ion. It should also be noted that the assignment of the pKa values for the methylpiperazine–thiosemicarbazone hybrid is not so straightforward. The proton dissociation steps of the ligands studied were assigned to the different functional groups by careful analysis of the results of the 1H NMR titrations and are shown in Schemes 1 and S3.† | ||
| Scheme 1 Deprotonation steps of the H3L2+ form of ligand Morph-dm-FTSC (HL4) for its E (A) and Z (B) isomers. | ||
| Method | pK1 | pK2 | pK3 | pK4 | ||
|---|---|---|---|---|---|---|
| a Estimated from the summed concentration distribution curves of the E/Z isomers in Fig. 6B and S4B. | ||||||
| mPip-dm-FTSC | pH-metry | 1.69 ± 0.02 | 3.29 ± 0.01 | 7.88 ± 0.01 | 10.23 ± 0.01 | |
| 1H NMR | Isomer E | — | 3.59 ± 0.06 | 7.94 ± 0.01 | 10.05 ± 0.03 | |
| 1H NMR | Isomer Z | — | 2.24 ± 0.05 | 7.82 ± 0.01 | >11.5 | |
| 1H NMRa | — | 3.33 | 7.90 | 10.35 | ||
| Morph-dm-FTSC | pH-metry | 2.27 ± 0.02 | 5.91 ± 0.01 | 10.18 ± 0.01 | — | |
| 1H NMR | Isomer E | 2.28 ± 0.01 | 6.08 ± 0.01 | 10.14 ± 0.01 | — | |
| 1H NMR | Isomer Z | <1 | 5.18 ± 0.01 | >11.5 | — | |
| 1H NMRa | 2.21 | 5.90 | 10.30 | — | ||
The pH-dependent 1H NMR spectra of Morph-dm-FTSC (HL4) (Fig. 7) revealed that most of the proton resonances are fairly sensitive to stepwise proton dissociation processes. In addition, the presence of Z and E isomers was observed. These were found to be involved in slow interconversion processes with regard to the NMR time scale (t1/2(obs) > ∼1 ms) in a wide pH-range. Their proton resonances were well-separated in most of the cases. However, the lines tend to broaden at pH < ∼4 due to faster isomerization around the CH12![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N1 double bond. Integrated signals of the different ligand protons belonging to the E and Z isomers were converted to molar fractions showing the predominant formation of the E isomer in the whole pH range, although the ratio of the isomers is undoubtedly changing due to the deprotonation steps (Fig. 6A). The E isomer was also found to be the major species in DMSO-d6 and its molar fraction (0.62) corresponds well to that found for aqueous solution (0.61) between pH ∼7 and ∼9, where the neutral HL form predominates. Based on the pH-dependence of the 1H NMR signals (Fig. S2†) microscopic proton dissociation constants could be computed for both Z and E isomers (Table 2). Concentration distribution curves were calculated based on these data providing the macroscopic constants as well (Table 2), which are in good agreement with the results of the pH-potentiometry. The first deprotonation process was accompanied by significant changes of the chemical shifts of the C6H pyridine ring proton and C14,15H3 terminal methyl protons. The morpholine (C8,11H2, C9,10H2) and C7H2 protons were very sensitive to the second deprotonation step, as were also the pyridine ring protons, while the chemical shifts of protons of the thiosemicarbazone moiety (C12H, C14,15H3) remain unaltered during the process. In the pH-range where the third proton dissociation occurs the signals of the last mentioned protons were shifted exclusively. These observed changes strongly support the subsequent deprotonation steps of the N4pyridylH+, N5morpholiniumH+ and N2hydrazinicH functional groups of both isomers of Morph-dm-FTSC as indicated in Scheme 1. On the other hand, marked differences are found between the pKa values of the Z and E isomers (Table 2). Most probably the hydrogen bond between the pyridyl nitrogen and the N2hydrazinicH moiety in the H2L+, HL forms of the Z isomer is responsible for these differences. Namely, it decreases pK1 of the Z isomer via stabilization of the conjugate base (H2L+) as well as pK2 due to the diminished π-electron density in the pyridine ring, which results in an easier deprotonation of the N5morpholiniumH+ group. The pK3 of the Z isomer is higher than that of the E form, since the dissociation of the N2hydrazinicH functional group participating in the hydrogen bonding is more difficult.
N1 double bond. Integrated signals of the different ligand protons belonging to the E and Z isomers were converted to molar fractions showing the predominant formation of the E isomer in the whole pH range, although the ratio of the isomers is undoubtedly changing due to the deprotonation steps (Fig. 6A). The E isomer was also found to be the major species in DMSO-d6 and its molar fraction (0.62) corresponds well to that found for aqueous solution (0.61) between pH ∼7 and ∼9, where the neutral HL form predominates. Based on the pH-dependence of the 1H NMR signals (Fig. S2†) microscopic proton dissociation constants could be computed for both Z and E isomers (Table 2). Concentration distribution curves were calculated based on these data providing the macroscopic constants as well (Table 2), which are in good agreement with the results of the pH-potentiometry. The first deprotonation process was accompanied by significant changes of the chemical shifts of the C6H pyridine ring proton and C14,15H3 terminal methyl protons. The morpholine (C8,11H2, C9,10H2) and C7H2 protons were very sensitive to the second deprotonation step, as were also the pyridine ring protons, while the chemical shifts of protons of the thiosemicarbazone moiety (C12H, C14,15H3) remain unaltered during the process. In the pH-range where the third proton dissociation occurs the signals of the last mentioned protons were shifted exclusively. These observed changes strongly support the subsequent deprotonation steps of the N4pyridylH+, N5morpholiniumH+ and N2hydrazinicH functional groups of both isomers of Morph-dm-FTSC as indicated in Scheme 1. On the other hand, marked differences are found between the pKa values of the Z and E isomers (Table 2). Most probably the hydrogen bond between the pyridyl nitrogen and the N2hydrazinicH moiety in the H2L+, HL forms of the Z isomer is responsible for these differences. Namely, it decreases pK1 of the Z isomer via stabilization of the conjugate base (H2L+) as well as pK2 due to the diminished π-electron density in the pyridine ring, which results in an easier deprotonation of the N5morpholiniumH+ group. The pK3 of the Z isomer is higher than that of the E form, since the dissociation of the N2hydrazinicH functional group participating in the hydrogen bonding is more difficult.
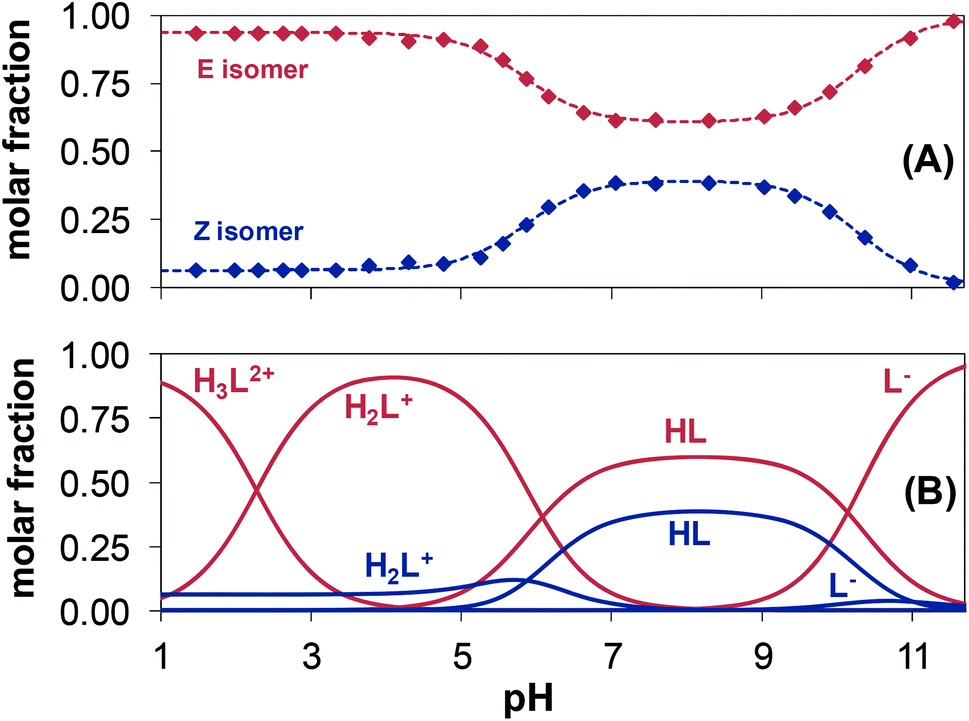 | ||
| Fig. 6 pH-Dependence of the molar fraction of the E (red symbols) and Z (blue symbols) isomers of the ligand Morph-dm-FTSC (HL4) calculated on the basis of the integrated areas of the signals of the various ligand protons (A). Concentration distribution curves for the isomeric ligand species (E: labelled in red; Z: labelled in blue) calculated with the aid of the microscopic proton dissociation constants (B). [cL = 1.5 mM; T = 298 K; I = 0.10 M (KCl); 10% D2O]. | ||
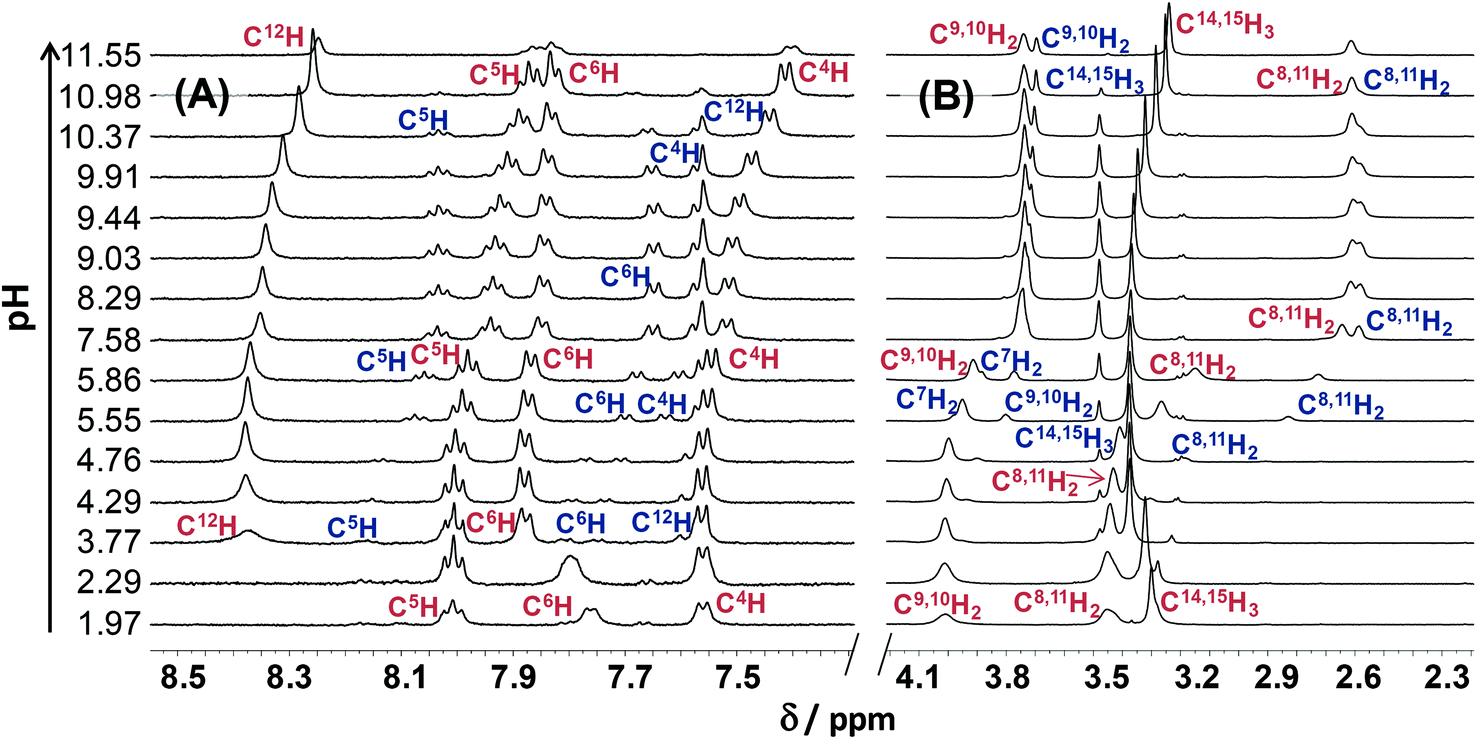 | ||
| Fig. 7 Low- (A) and high-field (B) regions of the 1H NMR spectra of Morph-dm-FTSC (HL4) at different pH values, red and blue symbols denote the peaks belonging to the protons of the major E and minor Z isomers, respectively. [cL = 1.5 mM; T = 298 K; I = 0.10 M (KCl); 10% D2O]. | ||
The pH-dependent 1H NMR spectra of mPip-dm-FTSC (Fig. S3†) and the changes of the chemical shifts of the various protons (Fig. S4†) were analyzed similarly. Data revealed that pK1 corresponds to the deprotonation of pyridinium nitrogen. However only the macroscopic constant could be determined by pH-potentiometry (Table 2) as the 1H NMR signals were fairly broadened in the pH range where this process takes place and data were not appropriate for calculation. The second deprotonation step is accompanied by significant electronic shielding effects in the case of the pyridine ring protons and a large upfield shift of the C7H2 protons. The signals belonging only to the C16H3 methyl protons are sensitive to the third proton dissociation process. These changes strongly indicate that pK2 and pK3 can be assigned to the deprotonation of the N5piperaziniumH+ and N6piperaziniumH+ groups, respectively (Scheme S3†). Protons of the thiosemicarbazone moiety were found to be sensitive to the last deprotonation step in which the N2hydrazinicH releases the proton. Comparing the microscopic constants of the E and Z isomers of the methylpiperazine–thiosemicarbazone hybrid (Table 2) it can be concluded that the lower pK2 (N5piperaziniumH+) and higher pK4 (N2hydrazinicH) values of the Z isomer are due to the presence of the hydrogen bond in the H3L2+ and HL forms (see the explanations in the case of Morph-dm-FTSC vide supra). At the same time the isomerization has no effect on the pK3 value since the N6piperaziniumH+ group is quite far from the CH12N1 double bond. The E isomer was found to be predominant in the whole pH range studied (Fig. S5†).
It is worth noting that the pKa values of the NpyridylH+ functional group of the studied thiosemicarbazone-based hybrids are significantly lower compared to those of ligands PTSC, APTSC81 due to the electron withdrawing effect of the charged morpholinium and methylpiperazinium moieties.
Both ligands mPip-dm-FTSC (HL2) and Morph-dm-FTSC (HL4) possess intrinsic fluorescence. 3-Dimensional fluorescence spectra recorded in aqueous solution at pH 7.4 (Fig. S6†) reveal their fairly similar excitation (330 nm) and emission maxima (420 nm), although the emission intensity of the morpholine–thiosemicarbazone hybrid is by a factor of 3 higher in comparison to that of HL2.
The lipo-hydrophilic character of the ligands HL1–HL5 was studied at pH 7.4 via the partitioning between n-octanol and water. The logD7.4 values determined by the analysis of the UV–vis spectra of the aqueous phases before and after separation are listed in Table 3. The results indicate a slightly higher lipophilicity of the terminally dimethylated derivatives (HL2 and HL4) compared to that of the corresponding non-methylated ligands (HL1 and HL3). Compounds containing the morpholine moiety (HL3 and HL4) possess significantly higher logD7.4 values compared to those of the methylpiperazine derivatives (HL1 and HL2) most probably due to the different protonation states of the ligands at physiological pH. According to the pKa values of the ligands studied (Table 2) mPip-dm-FTSC (HL2) is partly protonated (74% H2L+, 26% HL), while Morph-dm-FTSC (HL4) is mainly neutral (97% HL, 3% H2L+) at pH 7.4. On the other hand, the methyl ester mPyrr-FTSC (HL5) is much more lipophilic than the other ligands studied and its high logD7.4 value is manifested in a strongly reduced aqueous solubility compared to that of the corresponding proline–thiosemicarbazone conjugates (L- and D-Pro-FTSC: logD7.4 < −1.7).35 It should be also noted that all the ligands studied except mPyrr-FTSC (HL5) are more hydrophilic than Triapine (logD7.4 = +0.85)69 at physiological pH.
D7.4 values (n-octanol/water) for the ligands HL1–HL5 and for the copper(II) complexes 1–5 [T = 298 K, pH = 7.40 (10 mM HEPES) and I = 0.10 M (KCl)]
| Ligand | logD7.4 |
Complex | logD7.4 |
|
|---|---|---|---|---|
| mPip-FTSC | HL1 | −0.07 ± 0.01 | 1 | −1.53 ± 0.09 |
| mPip-dm-FTSC | HL2 | −0.03 ± 0.03 | 2 | −0.95 ± 0.10 |
| Morph-FTSC | HL3 | +0.60 ± 0.02 | 3 | −1.15 ± 0.09 |
| Morph-dm-FTSC | HL4 | +0.61 ± 0.01 | 4 | −0.90 ± 0.09 |
| mPyrr-FTSC | HL5 | >1.8 | 5 | >1.8 |
Solution chemistry: complex formation equilibria of copper(II) with ligands HL2 and HL4 and lipophilicity of the complexes
The main aim of the studies on complexation reactions of ligands mPip-dm-FTSC (HL2) and Morph-dm-FTSC (HL4) with copper(II) was monitoring the stability of the complexes 2 and 4 in aqueous solution especially at physiological pH. The complex formation processes were investigated by the combined use of pH-potentiometric, UV–vis and EPR titrations. The stoichiometries and cumulative stability constants of the complexes furnishing the best fits to the experimental data are listed in Table 4. EPR spectra were recorded at various pH values at 1:1 and 1:2 metal-to-ligand ratios at room temperature and at 77 K; the fitted experimental and simulated isotropic spectra are depicted in Fig. 8A, B and S7A, B.† The simulation of the EPR spectra resulted in the individual isotropic and anisotropic EPR spectra and parameters of the various species (Fig. 8C, S7C and S8;†Table 5). The EPR measurements at both temperatures revealed the predominant formation of mononuclear mono-ligand complexes in different protonation states. The proton displacement by the metal ion due to complex formation is almost complete already at the starting pH value of the pH-potentiometric titrations (pH ∼ 2) and a negligible amount of free copper(II) was detected by EPR at this pH, indicating the prominently high stability of the complexes formed with both ligands. Therefore, conditional stability constants for [CuL]+, which predominates in a wide pH-range, were determined by competition reactions with EDTA. The displacement of EDTA from the [Cu(EDTA)]2− complex by the ligands were followed by UV–vis spectrophotometry at pH 7.4 and 5.6, in the case of mPip-dm-FTSC (Fig. S9†) and Morph-dm-FTSC, respectively. Absorbance values recorded at λ > 415 nm were used for calculations of the conditional stability constants (logβ′) of [CuL]+, the only species contributing to the measured absorbance. The cumulative stability constants (logβ) of [CuL]+ were computed (Table 4) taking into account the protonation of the ligands at these pH values, which were kept constant during subsequent data evaluation.
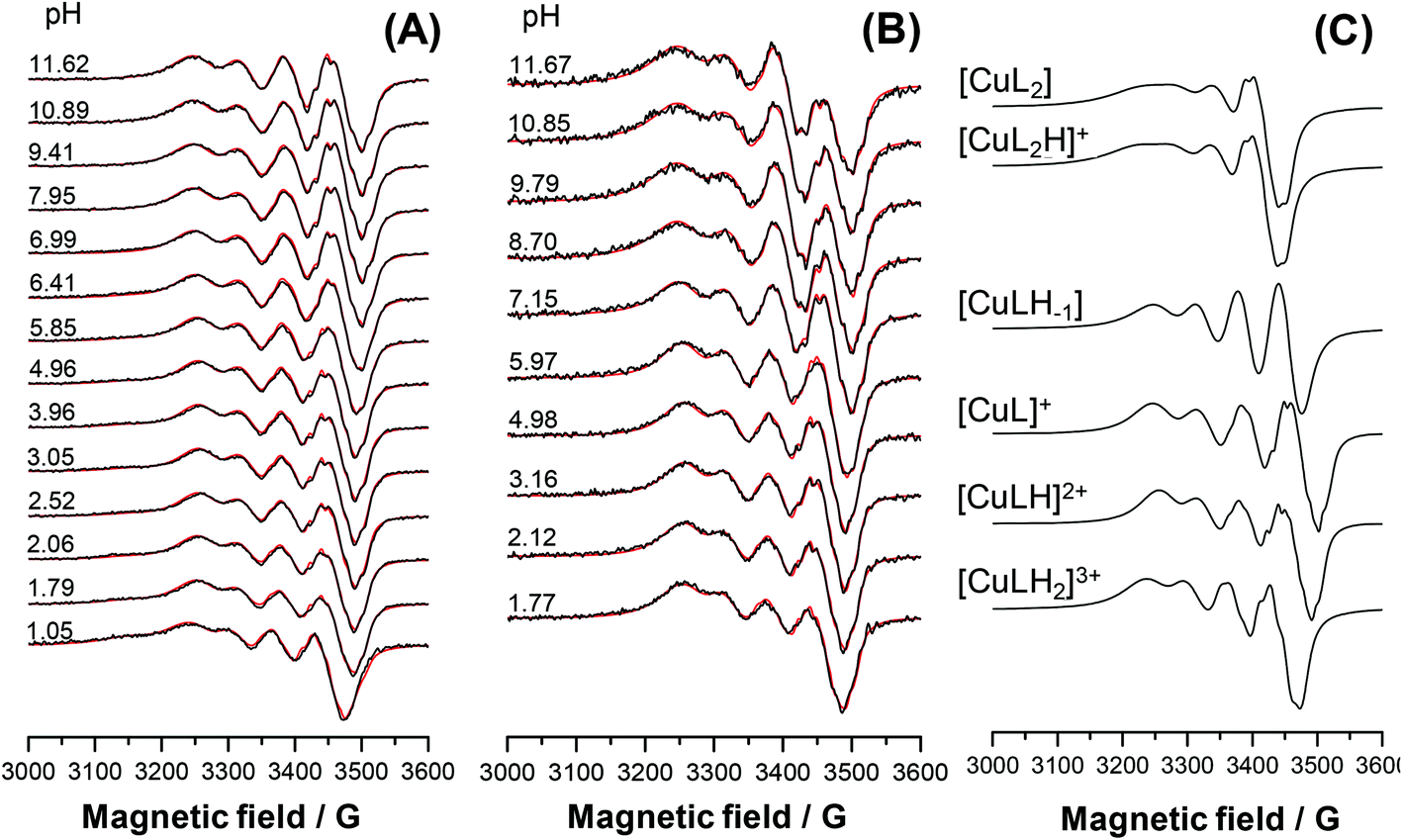 | ||
| Fig. 8 Experimental (black) and simulated (red) solution EPR spectra recorded for the copper(II)–mPip-dm-FTSC (HL2) system at 1:1 (A) and 1:2 (B) metal-to-ligand ratios. Calculated component EPR spectra obtained for the different copper(II)–mPip-dm-FTSC (HL2) species (C). [cligand = 1.0 mM; cCu = 1.0 mM (A) or cCu = 0.5 mM (B); T = 298 K; I = 0.10 M (KCl)]. | ||
β), derived and stepwise stability constants of the copper(II) complexes of ligands mPip-dm-FTSC (HL2) and Morph-dm-FTSC (HL4) determined by pH-potentiometry, UV–vis and EPR spectroscopy [T = 298 K, I = 0.10 M (KCl)]
| pH-metry | UV–vis | EPR | ||
|---|---|---|---|---|
|
a Determined via the EDTA displacement reactions by the ligand HL2 or HL4 by UV–vis spectrophotometry. Data for the pKa values of EDTA (0.9; 1.6; 2.0; 2.66; 6.16; 10.24) and logβ of the [Cu(EDTA)]2− complex (18.92) are taken from ref. 61 and conditional stability constants of [Cu(EDTA)]2− calculated for pH 7.4 and 5.6 are 16.06 and 13.61, respectively. Conditional stability constants (logβ′) of the [CuL]+ species: 16.83 ± 0.03 (HL2) at pH 7.4 (10 mM HEPES) and 13.79 ± 0.03 (HL4) at pH 5.6 (10 mM MES). β values of [CuL]+ are calculated as β = β′ × αH; where αH = 1 + ∑β (HpL) × [H+]p.
|
||||
| mPip-dm-FTSC | logβ [CuLH2]3+ |
— | 27.5 ± 0.1 | 27.9 ± 0.1 |
| logβ [CuLH]2+ |
26.53 ± 0.01 | 26.47 ± 0.01 | 26.49 ± 0.03 | |
| logβ [CuL]+ |
20.26 ± 0.03a | |||
| logβ [CuLH−1] |
8.4 ± 0.1 | 8.4 ± 0.1 | 7.8 ± 0.1 | |
| logβ [CuL2H]+ |
33.43 ± 0.02 | — | 33.84 ± 0.06 | |
| logβ [CuL2] |
— | — | 23.71 ± 0.08 | |
| pKa [CuLH2]3+ | — | 1.0 | 1.4 | |
| pKa [CuLH]2+ | 6.27 | 6.21 | 6.23 | |
| pKa [CuL]+ | 11.9 | 11.9 | 12.5 | |
| logK [CuL2] |
— | — | 3.45 | |
| Morph-dm-FTSC | logβ [CuLH]2+ |
20.9 ± 0.1 | 20.3 ± 0.1 | 20.7 ± 0.1 |
| logβ [CuL]+ |
18.86 ± 0.08a | |||
| logβ [CuLH−1] |
7.2 ± 0.1 | 7.2 ± 0.1 | 7.0 ± 0.1 | |
| logβ [CuL2H3]3+ |
40.2 ± 0.1 | — | 39.90 ± 0.08 | |
| logβ [CuL2] |
— | — | 21.71 ± 0.02 | |
| pKa [CuLH]2+ | 2.0 | 1.4 | 1.8 | |
| pKa [CuL]+ | 11.7 | 11.7 | 11.9 | |
| logK [CuL2] |
— | — | 2.85 | |
| Isotropic parametersa | Anisotropic parametersb | Calculated parametersc | |||||||
|---|---|---|---|---|---|---|---|---|---|
| g o | A o/G | a No/G | g x , gy, gz | A x , Ay, Azd/G | a N x , aNy, aNz/G | g o,calc | A o,calc/G | ||
| a Uncertainties (SD) are shown in parentheses. b The experimental errors were ±0.002 for gx and gy and ±0.001 for gz, ± 2 G for Ax and Ay and ±1 G for Az. c Isotropic values calculated via the equation go = (gx + gy + gz)/3, and Ao[MHz] = (Ax + Ay + Az)/3. d The signs of the couplings were derived from a comparison of Ao,calc with the experimental Ao values. e Higher uncertainties of anisotropic parameters were obtained for minor species. | |||||||||
| Morph-dm-FTSC | [CuLH]2+ | 2.1037(2) | 68.6(4) | 13.8(5) | 2.035, 2.058, 2.216 | 18.7, 30.4, 155.9 | 18.5, 13.9, 11.9 | 2.103 | 70.7 |
| 10.7(7) | 12.8, 15.7, 6 | ||||||||
| [CuL]+ | 2.08856(1) | 63.3(1) | 17.8(1) | 2.032, 2.053, 2.176 | 8.9, 9.8, 160.1 | 18.2, 9.6, 8.4 | 2.087 | 61.7 | |
| 11.9(1) | 10.4, 15.9, 9.8 | ||||||||
| 8.9(2) | 10.4, 15.9, 9.8 | ||||||||
| [CuLH−1]e | 2.0953(6) | 62.1(8) | 9.0(8) | 2.05, 2.07, 2.249 | 16, 19, 156 | 8, 17, 8 | 2.121 | 67.7 | |
| 9.0(8) | 15, 10, 8 | ||||||||
| [CuL2H3]3+ | 2.0745(5) | 85.5(6) | 12.3(8) | ||||||
| 12.3(8) | |||||||||
| [CuL2] | 2.106(2) | 53(2) | 17(1) | ||||||
| 14(1) | |||||||||
| mPip-dm-FTSC | [CuLH2]3+ | 2.1026(4) | 58.0(5) | 17.4(4) | 2.035, 2.059, 2.214 | −18.9, 30.4, 155.7 | 18.4, 14, 12 | 2.103 | 58.5 |
| 10.3(9) | 12, 16.3, 6 | ||||||||
| [CuLH]2+ | 2.0899(1) | 58.3(1) | 18.2(1) | 2.031, 2.055, 2.176 | 4.4, 8.3, 155.9 | 16.9, 10.9, 10.9 | 2.087 | 58.3 | |
| 11.5(2) | 11.3, 16.8, 9.7 | ||||||||
| 7.8(2) | 11.3, 16.8, 9.7 | ||||||||
| [CuL]+ | 2.0894(1) | 64.2(1) | 17.9(1) | 2.033, 2.053, 2.177 | 6.7, 10.9, 159.2 | 16.7, 9.4, 9 | 2.088 | 61.1 | |
| 12.1(2) | 11.7, 16.7, 10 | ||||||||
| 9.0(3) | 11.7, 16.7, 10 | ||||||||
| [CuLH−1]e | 2.0959(7) | 59(1) | 10(1) | 2.05, 2.07, 2.249 | 16, 19, 156 | 8, 17, 8 | 2.121 | 67.7 | |
| 10(1) | 15, 10, 8 | ||||||||
| [CuL2H]+ | 2.1144(6) | 54(1) | 15(1) | ||||||
| 12(2) | |||||||||
| [CuL2]e | 2.1118(6) | 53(1) | 16(1) | 2.03, 2.05, 2.200 | 25, −11, 150 | 17, 10, 10 | 2.093 | 57 | |
| 12(2) | 10, 17, 10 | ||||||||
In the case of Morph-dm-FTSC, [CuL]+ predominates between pH ∼4 and ∼10. This is clearly indicated by the unaltered UV–vis spectra in the wavelength range of both the d–d (Fig. 9) and CT (Fig. S10B†) bands. EPR spectra were also intact in this particular pH range (Fig. S7A†). Based on the EPR parameters of [CuL]+ (Table 5, the superhyperfine couplings to three nitrogen atoms is resolved in the spectra) the coordination of the ligand via the (S−,N1,N4,N5) donor set is the most probable in solution. The rhombic g-tensor determined from the anisotropic EPR spectra indicates a strong rhombic distortion which is probably due to the three conjugated five-membered chelate rings formed by the four donor atoms. The single-crystal X-ray crystallography revealed the same binding mode for the ligand in 4 in the solid state (Fig. 4). Upon decreasing the pH complex [CuL]+ becomes protonated and the significant UV–vis (Fig. 9 and S10B†) and EPR (Fig. S7†) spectral changes at pH < ∼3 indicate the alteration of the coordination mode. λmax values of both the d–d and CT bands are shifted to the higher wavelengths upon the formation of species [CuLH]2+ (578 nm → 690 nm, 406 nm → 410 nm, 302 nm → 323 nm). Most likely, the morpholine nitrogen is protonated and not involved in coordination in [CuLH]2+ as indicated by its higher g0 value compared to that of [CuL]+ (Table 5). On the other hand the deprotonation of [CuL]+ observed at pH > ∼10, is accompanied by only minor changes of the UV–vis spectra (see changes at ∼256 nm in Fig. S10B†). However, the decreasing ligand field (lower A0) supports the formation of a mixed hydroxido complex, [CuL(OH)], in which the ligand binds through (S−,N1,N4) donor atoms. A fairly similar deprotonation process of [CuL]+ is characteristic for mPip-dm-FTSC. The formation of [CuLH−1] ([CuL(OH)]) could be also admitted at the highly basic pH values, although additional changes could be detected in the neutral and acidic pH ranges. Namely, the inflection point of the titration curve recorded at 1:1 metal-to-ligand ratio (not shown here) at pH 6.26 strongly suggests an additional (de)protonation process which was not observed in the case of Morph-dm-FTSC. pKa of species [CuLH]2+ was also calculated on the basis of the minor changes of the d–d bands of the UV–vis spectra and the pH-dependent EPR spectra (Fig. 8A). The data obtained by the three different methods are in good agreement (Table 4). The similar g0 values of [CuLH]2+ and [CuL]+ (Table 5) indicate the same coordination mode of mPip-dm-FTSC in these complexes via a (S−,N1,N4,N5) donor atoms both in solution and in the solid state established by X-ray diffraction (Fig. 2), while the ligand field is slightly increased (somewhat higher A0) due to the deprotonation of [CuLH]2+. These results strongly indicate that the process is assigned to the deprotonation of the N6 of the methylpiperazine moiety which is not involved in the binding to copper(II). The observed UV–vis spectral changes (Fig. S10A†) and EPR parameters (Table 5) at pH < ∼3 were found to be similar to those found for the Morph-dm-FTSC system, thus the (S−,N1,N4) coordination is suggested for [CuLH2]3+ in which the methylpiperazine N5 atom is protonated.
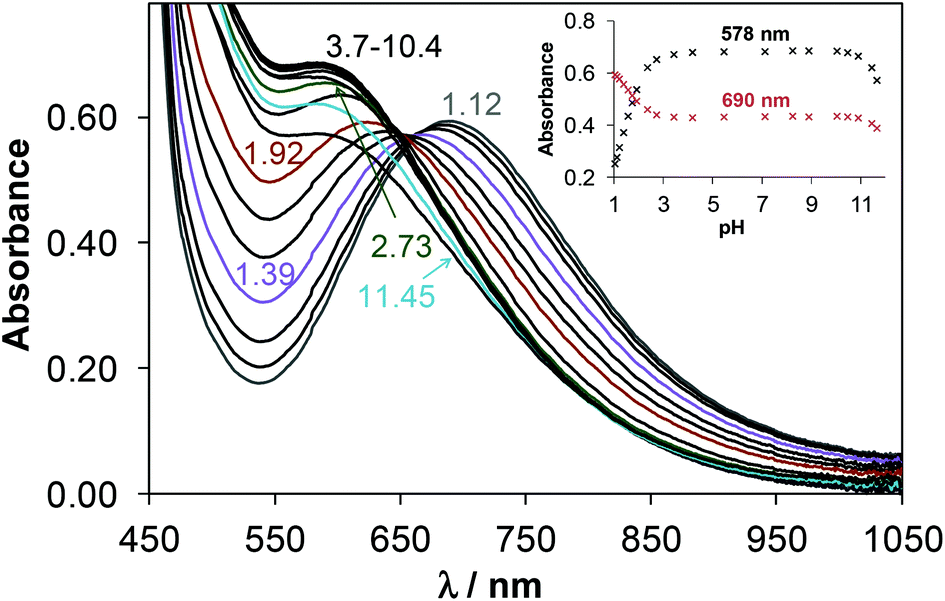 | ||
| Fig. 9 UV–vis absorbance spectra of 4 recorded in the pH range 1.1–11.7. Inset shows the absorbance values recorded at 578 and 690 nm. [ccomplex = 2.5 mM; T = 298 K; I = 0.10 M (KCl); l = 1 cm]. | ||
Formation of merely mono-ligand copper(II) complexes for HL2 and HL4 was expected. However at ligand excess (cL/cCu > 2) bis-ligand complexes were detected mainly in the basic pH range. Formation of the neutral bis-ligand complexes [CuL2] resulted in precipitation which hindered the accurate determination of their stability constants by pH-potentiometry and UV–vis spectrophotometry, although these were estimated by the EPR measurements (Table 4). The EPR data for this kind of complexes represent quite high g0 and low A0 values (Table 5) and strong rhombic distortion. The ligands in these complexes coordinate most probably via (S−,N1,N4) and (Sequatorial,N1axial) donor sets. The stepwise stability constants logK [CuL2] are lower by many orders of magnitude than logK [CuL]+ indicating the non-favored formation of the bis-ligand complexes. Constants for these minor charged bis-ligand complexes such as [CuL2H3]3+ (Morph-dm-FTSC) and [CuL2H]+ (mPip-dm-FTSC) could be calculated by pH-potentiometry as well. The former complex displays a well resolved solution EPR spectra with two coordinating N atoms, and large A0 value which indicate a symmetrical structure with (S−,N1) (S−,N1) binding mode, while the latter one has a similar coordination pattern as species [CuL2].
It is worth noting that the isotropic g and A values calculated by averaging the anisotropic values (g0,calc and A0,calc in Table 5) are in relatively good agreement with the corresponding values measured in solution, indicating that the coordination modes adopted by the ligands in solution are preserved upon freezing.
Representative concentration distribution curves were calculated by using the overall stability constants (average values obtained by the 3 methods) for the copper(II)–mPip-dm-FTSC (HL2) and copper(II)–Morph-dm-FTSC (HL4) systems at 1:1 metal-to-ligand ratio to represent the complex formation processes in the pH range studied (Fig. 10). It can be concluded that complexes [CuL]+ predominate at physiological pH even at submicromolar concentrations, although 6% of the complex is protonated in the case of mPip-dm-FTSC.
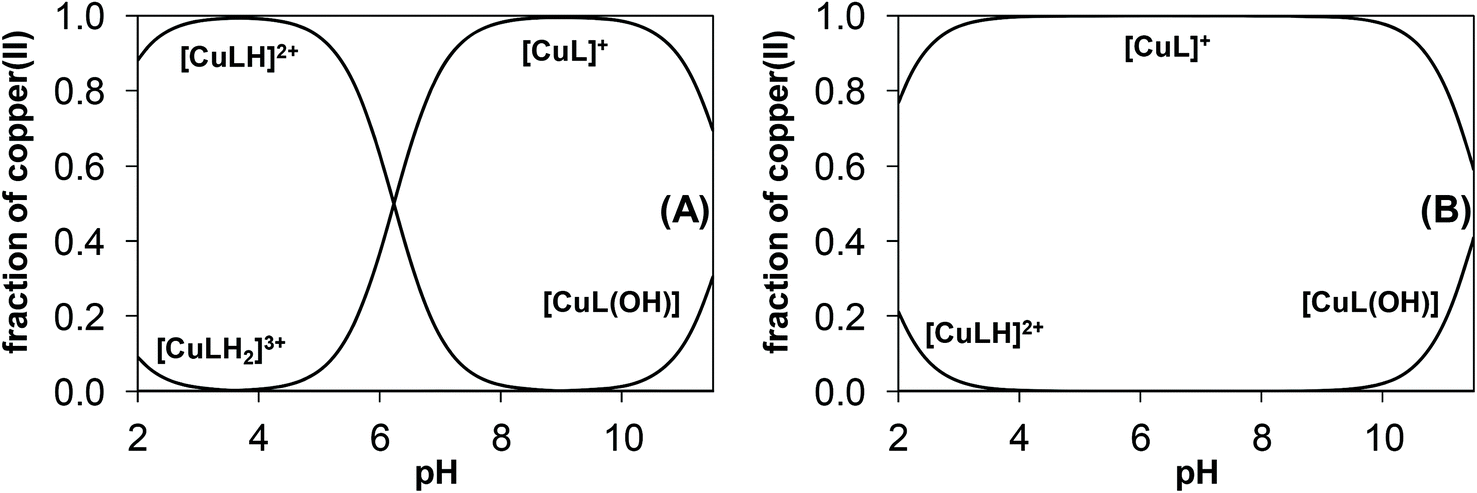 | ||
| Fig. 10 Concentration distribution curves for the copper(II)–mPip-dm-FTSC (HL2) (A) and copper(II)–Morph-dm-FTSC (HL4) (B) systems. [cL = 1.0 mM; cCu = 1.0 mM; T = 298 K; I = 0.10 M (KCl)]. | ||
In order to compare the copper(II) binding ability of mPip-dm-FTSC (HL2) and Morph-dm-FTSC (HL4) with other thiosemicarbazones pCu values (pCu = −log[Cu(II)]; cL/cCu = 10; cCu = 1 μM) have been computed at physiological pH. The higher pCu value indicates stronger chelating ability. For mPip-dm-FTSC and Morph-dm-FTSC pCu values of 17.6 and 17.0 were obtained, respectively, which are significantly higher than those reported for tridentate HCTs such as Triapine (11.6) at pH 7.4 in 30% (w/w) DMSO/H2O82 and are comparable to that of the pentadentate L-Pro-FTSC conjugate (17.5) in pure water.35
LogD7.4 values were determined for the copper(II) complexes 1–5 and are collected in Table 3 in order to characterize the hydro-lipophilic character of these species. Comparing these values to those of the metal-free ligands it can be concluded that the same lipophilicity trend is obtained. Namely, the terminal dimethylation results in somewhat increased values and complexes of the morpholine–thiosemicarbazone derivatives possess enhanced lipophilic character. Note that the copper(II) complexes are much more hydrophilic than the corresponding ligands since the positively charged [CuL]+ species predominate at physiological pH. Complex 5 is much more lipophilic than the other complexes, although its logD7.4 value cannot be determined exactly and compared to that of mPyrr-FTSC (HL5).
Cytotoxicity in cancer cell lines
The antiproliferative activity of the ligands HL1–HL4 and the copper(II) complexes 1–4 was evaluated for 48 h of continuous drug action, using colorimetric MTT assay. The study was performed in three human neoplastic cell lines, namely HeLa (cervical carcinoma), A549 (alveolar basal adenocarcinoma) and LS174 (colon carcinoma), and one human foetal lung fibroblast cell line (MRC-5), which was used as a noncancerous model for the in vitro cytotoxicity evaluation. The results for the ligands and their copper(II) complexes are summarized in Table 6 in terms of IC50 values with their standard deviations. The results revealed that compounds 1–4 exhibited significant antiproliferative activity (IC50 < 100 μM) against all cell lines used, with complex 4 showing the highest cytotoxic potential. The most sensitive to the investigated compounds was proved to be the cell line LS174, as indicated by the calculated IC50 values varying from 13.1 to 17.5 μM. In contrast, the ligands showed significantly lower activity than the parent 2-formyl- and/or 2-acetylpyridine thiosemicarbazones, except ligand HL4, which exhibited significant cytotoxic activity against both the MRC5 and LS174 cell lines (63.2 ± 4.2 and 15.9 ± 0.6 μM, respectively).| IC50a [μM] (mean ± SD) |
||||
|---|---|---|---|---|
| Compound | HeLa | A549 | LS174 | MRC5 |
| a The sign > (in front of the maximum value of the concentration) indicates that IC50 value is not reached in the examined range of concentrations. | ||||
| 1 | 38.3 ± 1.7 | 62.7 ± 4.7 | 16.4 ± 4.2 | 50.6 ± 3.5 |
| 2 | 65.1 ± 5.7 | 131.3 ± 3.9 | 17.4 ± 0.2 | 38.6 ± 5.9 |
| 3 | 63.3 ± 2.7 | 208.0 ± 0.1 | 17.5 ± 1.6 | 132.1 ± 9.2 |
| 4 | 25.5 ± 5.3 | 42.8 ± 3.7 | 13.1 ± 2.1 | 28.3 ± 3.8 |
| HL1 | >300 | >300 | >300 | >300 |
| HL2 | >300 | >300 | >300 | >300 |
| HL3 | >300 | >300 | >300 | >300 |
| HL4 | >300 | >300 | 15.9 ± 0.6 | 63.2 ± 4.2 |
The metal-free 2-formylpyridine thiosemicarbazone (FTSC) showed high cytotoxicity against human cancer cell lines 41M (ovarian carcinoma), SK-BR-3 (mammary carcinoma), SW480 (colon carcinoma) and HL60 (leukemia) after 96 or 72 h treatment with IC50 values of 2.9 ± 0.6, 3.2 ± 0.6, 10.6 ± 0.1 and 3.3 ± 0.5 μM, respectively.83,84 The effect of substitution of azomethine hydrogen atom by a methyl group is cell line dependent. While there was no change in antiproliferative activity for 2-acetylpyridine thiosemicarbazone (APTSC) in the first two cancer cell lines, a considerable increase was observed against the other two cell lines (IC50 = 0.4 ± 0.01 and 0.2 ± 0.02 μM). Terminal N4-dimethylation of FTSC resulted in a very strong enhancement of antiproliferative activity reaching IC50 values of 0.0040 ± 0.0009 and 0.0098 ± 0.0011 μM in 41M and SK-BR-3 cells after exposure for 96 h.83 The favorable effect of N4-dimethylation is also well-documented for other related α-heterocyclic thiosemicarbazones.31 The coordination of FTSC to copper(II) was reported to increase or decrease the activity depending on the cell type.85–88 In particular, [Cu(FTSC)Cl2] revealed an increase of cytotoxicity by a factor of 3 in SW480 cells when compared to that of FTSC, while against HL60 cells the activity of FTSC and the copper(II) complex was very similar.84 The proline-FTSC hybrids, we synthesized previously,80 showed a different activity compared to the compounds reported herein. Hybrids that were not methylated at N4 (L- and D-Pro-FTSC) and their nickel(II), copper(II) and zinc(II) complexes lacked activity (IC50 > 300 μM) in both the studied human cancer cell lines HeLa (cervical carcinoma) and A549 (adenocarcinoma), as well as in the non-carcinogenic cell line MRC5 (foetal human fibroblast). The terminally dimethylated hybrid dm-L-Pro-FTSC showed moderate to low anticancer activity with IC50 values of 224.6 ± 6.4, 204.3 ± 4.8 and 178.4 ± 1.5 μM in the HeLa, A549 and MRC5 cell lines respectively. Complex formation with copper(II) led to an increased cytotoxicity with IC50 values of 93.3 ± 5.5, 176 ± 1.7 and 69.4 ± 4.7 μM in the same cell lines, respectively. Complex formation with zinc(II) or nickel(II) had no favorable effect on the activity. It should be also noted that the copper(II) complex of dm-L-Pro-FTSC showed significant RNR-inhibition activity under reductive conditions at a concentration of 20 μM.80
Comparison of IC50 values for 3 and 4 indicates that terminal N4-dimethylation enhances the cytotoxicity in accord with the general trend observed in the literature.31,83 In contrast, the effect is opposite, although also cell type dependent, if the activity of compounds 1 and 2 is compared. The observed divergent effects of terminal N4-dimethylation suggest that structural modifications at the pyridine ring (coupling to piperazine and morpholine moieties which increases the denticity of the ligands) play an important role in structure–activity relationships.
Conclusions
The synthesis of new hybrid species as potential ligands for transition metals permitted the study of the effects of the methylpiperazine, morpholine and methylpyrrole-2-carboxylate attachment to the parent 2-formylpyridine thiosemicarbazone on the aqueous solubility, lipophilicity, ability to form copper(II) complexes, their thermodynamic stability and antiproliferative activity in human cancer cell lines HeLa, A549 and LS174, as well as in nontumorigenic cell line MRC5. The hybrid species HL5 and HL6 proved to be almost insoluble in water precluding any biological investigations. Attempts to hydrolyze the ester group to –COOH in these two compounds in order to improve the aqueous solubility and ascertain the biological potency of new compounds are undergoing in our laboratory. In contrast, the hybrids HL1–HL4 possess excellent water solubility. The solution speciation of copper(II) complexes of HL2 and HL4 has been characterized in pure aqueous solution via a combined approach using pH-potentiometry, EPR spectroscopy und UV–vis spectrophotometry. The two hybrid compounds were found to act as tetradentate ligands in solution coordinating to copper(II) via the (Npy,N,S−,Nhetero) donor atoms. This binding mode was confirmed by X-ray crystallography in the case of complexes 1–4. Predominant formation of highly stable [CuL]+ complexes was found at pH 7.4 in aqueous solution and based on the stability constants their decomposition cannot occur even at biologically more relevant micromolar concentrations. The morpholine derivatives HL3 and HL4 possess markedly higher logD7.4 values compared to those of the piperazine counterparts HL1 and HL2 most probably due to the different protonation states of the hybrid ligands at physiological pH. At the same time they are more hydrophilic than Triapine. Compounds prepared in this work were tested for antiproliferative activity in different human cancer cell lines. Coordination of hybrid ligands HL1–HL4 to copper(II) significantly increased the cytotoxicity in vitro. While HL1–HL4 possess low cytotoxicity with IC50 > 300 μM, their copper(II) complexes revealed high antiproliferative activity. The most active compound 4 exhibited IC50 values in the range 13.1–42.8 μM in all three human cancer cell lines. Nevertheless the toxicity of the most active complex remains considerably lower when compared to parent 2-formylpyridine and 2-acetylpyridine thiosemicarbazones and their copper(II) complexes which showed IC50 values in the nanomolar concentration range and are characterized by very high general toxicity, and, as a consequence have a low therapeutic index. Further experimental work to get insight into the mechanism of action of the prepared copper(II) complexes with hybrid ligands is required to ascertain whether they are really good candidates for further development as potential anticancer drugs.
Acknowledgements
This work was supported by the Hungarian Research Foundation OTKA project PD103905. We are thankful to the Ministry of Science and Technology of Serbia for financial support from grant no. III41026. We thank Prof. M. Galanski for measuring the two-dimensional NMR spectra and Dr Michael Malarek for reading the manuscript and his comments.Notes and references
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271–5294 CrossRef CAS PubMed.
- J. S. Casas, M. S. Garcia-Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef CAS.
- H. Beraldo and D. Gambino, Mini-Rev. Med. Chem., 2004, 4, 31–39 CrossRef CAS.
- D. C. Reis, A. A. R. Despaigne, J. G. Da Silva, N. F. Silva, C. F. Vilela, I. C. Mendes, J. A. Takahashi and H. Beraldo, Molecules, 2013, 18, 12645–12662 CrossRef CAS PubMed.
- D. C. Quenelle, K. A. Keith and E. R. Kern, Antiviral Res., 2006, 71, 24–30 CrossRef CAS PubMed.
- M. A. Salam, M. A. Affan, M. A. Arafat, R. Saha and R. Nasrin, Heteroat. Chem., 2013, 24, 43–52 CrossRef CAS PubMed.
- A. Molter, J. Rust, C. W. Lehmann, G. Deepa, P. Chiba and F. Mohr, Dalton Trans., 2011, 40, 9810–9820 RSC.
- S. Arora, S. Agarwal and S. Singhal, Int. J. Pharm. Pharm. Sci., 2014, 6, 34–41 CAS.
- R. W. Brockman, J. R. Thomson, M. J. Bell and H. E. Skipper, Cancer Res., 1956, 16, 167–170 CAS.
- S. Wadler, D. Makower, C. Clairmont, P. Lambert, K. Fehn and M. Sznol, J. Clin. Oncol., 2004, 22, 1553–1563 CrossRef CAS PubMed.
- J. Kolesar, R. C. Brundage, M. Pomplun, D. Alberti, K. Holen, A. Traynor, P. Ivy and G. Wilding, Cancer Chemother. Pharmacol., 2011, 67, 393–400 CrossRef CAS PubMed.
- C. M. Nutting, H. C. M. L. van, A. B. Miah, S. A. Bhide, J. P. Machiels, J. Buter, C. Kelly, R. D. de and K. J. Harrington, Annu. Oncol., 2009, 20, 1275–1279 CrossRef CAS PubMed.
- M. J. Mackenzie, D. Saltman, H. Hirte, J. Low, C. Johnson, G. Pond and M. J. Moore, Invest. New Drugs, 2007, 25, 553–558 CrossRef CAS PubMed.
- S. Attia, J. Kolesar, M. R. Mahoney, H. C. Pitot, D. Laheru, J. Heun, W. Huang, J. Eickhoff, C. Erlichman and K. D. Holen, Invest. New Drugs, 2008, 26, 369–379 CrossRef CAS PubMed.
- A. M. Traynor, J.-W. Lee, G. K. Bayer, J. M. Tate, S. P. Thomas, M. Mazurczak, D. L. Graham, J. M. Kolesar and J. H. Schiller, Invest. New Drugs, 2010, 28, 91–97 CrossRef CAS PubMed.
- J. J. Knox, S. J. Hotte, C. Kollmannsberger, E. Winquist, B. Fisher and E. A. Eisenhauer, Invest. New Drugs, 2007, 25, 471–477 CrossRef CAS PubMed.
- F. J. Giles, P. M. Fracasso, H. M. Kantarjian, J. E. Cortes, R. A. Brown, S. Verstovsek, Y. Alvarado, D. A. Thomas, S. Faderl, G. Garcia-Manero, L. P. Wright, T. Samson, A. Cahill, P. Lambert, W. Plunkett, M. Sznol, J. F. DiPersio and V. Gandhi, Leuk. Res., 2003, 27, 1077–1083 CrossRef CAS.
- J. E. Karp, F. J. Giles, I. Gojo, L. Morris, J. Greer, B. Johnson, M. Thein, M. Sznol and J. Low, Leuk. Res., 2008, 32, 71–77 CrossRef CAS PubMed.
- J. F. Zeidner, J. E. Karp, A. L. Blackford, B. D. Smith, I. Gojo, S. D. Gore, M. J. Levis, H. E. Carraway, J. M. Greer, S. P. Ivy, K. W. Pratz and M. A. McDevitt, Haematologica, 2014, 99, 672–678 CrossRef CAS PubMed.
- A. C. Sartorelli, E. C. Moore, M. S. Zedeck and K. C. Agrawal, Biochemistry, 1970, 9, 4492–4498 CrossRef CAS.
- F. A. French, E. J. Blanz Jr., S. C. Shaddix and R. W. Brockman, J. Med. Chem., 1974, 17, 172–181 CrossRef CAS.
- M. Kolberg, K. R. Strand, P. Graff and K. Kristoffer Andersson, Biochim. Biophys. Acta, Proteins Proteomics, 2004, 1699, 1–34 CrossRef CAS PubMed.
- J. L. Nitiss, Nat. Rev. Cancer, 2009, 9, 327–337 CrossRef CAS PubMed.
- A. J. Schoeffler and J. M. Berger, Q. Rev. Biophys., 2008, 41, 41–101 CrossRef CAS PubMed.
- J. Easmon, G. Puerstinger, G. Heinisch, T. Roth, H. H. Fiebig, W. Holzer, W. Jaeger, M. Jenny and J. Hofmann, J. Med. Chem., 2001, 44, 2164–2171 CrossRef CAS PubMed.
- L. Wei, J. Easmon, R. K. Nagi, B. D. Muegge, L. A. Meyer and J. S. Lewis, J. Nucl. Med., 2006, 47, 2034–2041 CAS.
- A. M. Merlot, D. S. Kalinowski and D. R. Richardson, Antioxid. Redox Signaling, 2013, 18, 973–1006 CrossRef CAS PubMed.
- L. Zhu, B. Zhou, X. Chen, H. Jiang, J. Shao and Y. Yun, Biochem. Pharmacol., 2009, 78, 1178–1185 CrossRef CAS PubMed.
- A. Popovic-Bijelic, C. R. Kowol, M. E. S. Lind, J. Luo, F. Himo, E. A. Enyedy, V. B. Arion and A. Graeslund, J. Inorg. Biochem., 2011, 105, 1422–1431 CrossRef CAS PubMed.
- Y. Aye, M. J. C. Long and J. Stubbe, J. Biol. Chem., 2012, 287, 35768–35778 CrossRef CAS PubMed.
- H. Huang, Q. Chen, K. Xin, L. Meng, L. Lin, X. Wang, C. Zhu, Y. Wang, Z. Chen, M. Li, H. Jiang, K. Chen, J. Ding and H. Liu, J. Med. Chem., 2010, 53, 3048–3064 CrossRef CAS PubMed.
- B. M. Zeglis, V. Divilov and J. S. Lewis, J. Med. Chem., 2011, 54, 2391–2398 CrossRef CAS PubMed.
- C. R. Kowol, R. Eichinger, M. A. Jakupec, M. Galanski, V. B. Arion and B. K. Keppler, J. Inorg. Biochem., 2007, 101, 1946–1957 CrossRef CAS PubMed.
- C. R. Kowol, R. Berger, R. Eichinger, A. Roller, M. A. Jakupec, P. P. Schmidt, V. B. Arion and B. K. Keppler, J. Med. Chem., 2007, 50, 1254–1265 CrossRef CAS PubMed.
- F. Bacher, E. A. Enyedy, N. V. Nagy, A. Rockenbauer, G. M. Bognar, R. Trondl, M. S. Novak, E. Klapproth, T. Kiss and V. B. Arion, Inorg. Chem., 2013, 52, 8895–8908 CrossRef CAS PubMed.
- O. A. Troshina, P. A. Troshin, A. S. Peregudov, V. I. Kozlovski and R. N. Lyubovskaya, Chem. – Eur. J., 2006, 12, 5569–5577 CrossRef CAS PubMed.
- C. Antonello, E. Uriarte, M. Palumbo, S. Valisena, C. Parolin and G. Palu, Eur. J. Med. Chem., 1993, 28, 291–296 CrossRef CAS.
- T. W. Moore, K. Sana, D. Yan, P. Thepchatri, J. M. Ndungu, M. T. Saindane, M. A. Lockwood, M. G. Natchus, D. C. Liotta, R. K. Plemper, J. P. Snyder and A. Sun, Beilstein J. Org. Chem., 2013, 9, 197–203 CrossRef CAS PubMed.
- L. K. Filak, D. S. Kalinowski, T. J. Bauer, D. R. Richardson and V. B. Arion, Inorg. Chem., 2014, 53, 6934–6943 CrossRef CAS PubMed.
- N. Ae and Y. Fujiwara, US Pat., US20110263847A1, 2011 Search PubMed.
- Y. Kakiya and M. Oda, WO Pat., WO2005009999A1, 2005 Search PubMed.
- T. Ochi, M. Sakamoto, A. Minamida, K. Suzuki, T. Ueda, T. Une, H. Toda, K. Matsumoto and Y. Terauchi, Bioorg. Med. Chem. Lett., 2005, 15, 1055–1059 CrossRef CAS PubMed.
- B.-C. Chen, R. Droghini, J. Lajeunesse, J. D. Dimarco, M. Galella and C. Ramakrishnan, WO Pat., WO2005077945A2, 2005 Search PubMed.
- R. Kharb, K. Bansal and A. K. Sharma, Pharm. Chem., 2012, 4, 2470–2488 CAS.
- J. Faist, W. Seebacher, R. Saf, R. Brun, M. Kaiser and R. Weis, Eur. J. Med. Chem., 2012, 47, 510–519 CrossRef CAS PubMed.
- K. Kulig, J. Sapa, D. Maciag, B. Filipek and B. Malawska, Arch. Pharm., 2007, 340, 466–475 CrossRef CAS PubMed.
- J. M. Nelson, T. M. Chiller, J. H. Powers and F. J. Angulo, Clin. Infect. Dis., 2007, 44, 977–980 CrossRef CAS PubMed.
- J. M. Burka, K. S. Bower, R. C. Vanroekel, R. D. Stutzman, C. P. Kuzmowych and R. S. Howard, Am. J. Ophthalmol., 2005, 140, 83–87 CrossRef CAS PubMed.
- D. S. Schiller and H. B. Fung, Clin. Ther., 2007, 29, 1862–1886 CrossRef CAS PubMed.
- B. Macias, M. V. Villa, I. Rubio, A. Castineiras and J. Borras, J. Inorg. Biochem., 2001, 84, 163–170 CrossRef CAS.
- P. Drevensek, J. Kosmrlj, G. Giester, T. Skauge, E. Sletten, K. Sepcic and I. Turel, J. Inorg. Biochem., 2006, 100, 1755–1763 CrossRef CAS PubMed.
- I. Turel, J. Kljun, F. Perdih, E. Morozova, V. Bakulev, N. Kasyanenko, J. A. W. Byl and N. Osheroff, Inorg. Chem., 2010, 49, 10750–10752 CrossRef CAS PubMed.
- A. Tarushi, E. Polatoglou, J. Kljun, I. Turel, G. Psomas and D. P. Kessissoglou, Dalton Trans., 2011, 40, 9461–9473 RSC.
- R. Zia ur, N. Muhammad, S. Ali, I. S. Butler and A. Meetsma, Inorg. Chim. Acta, 2011, 376, 381–388 CrossRef PubMed.
- Q.-P. Shi, Z.-H. Shi, N.-G. Li, Y.-P. Tang, T. Hao, W. Zhang, M.-Z. Shen, Z.-X. Dong, P.-X. Zhang, J.-P. Yang and J.-A. Duan, Lett. Org. Chem., 2014, 11, 590–595 CrossRef CAS.
- C. Cano, K. Saravanan, C. Bailey, J. Bardos, N. J. Curtin, M. Frigerio, B. T. Golding, I. R. Hardcastle, M. G. Hummersone, K. A. Menear, D. R. Newell, C. J. Richardson, K. Shea, G. C. M. Smith, P. Thommes, A. Ting and R. J. Griffin, J. Med. Chem., 2013, 56, 6386–6401 CrossRef CAS PubMed.
- C. Cano, O. R. Barbeau, C. Bailey, X.-L. Cockcroft, N. J. Curtin, H. Duggan, M. Frigerio, B. T. Golding, I. R. Hardcastle, M. G. Hummersone, C. Knights, K. A. Menear, D. R. Newell, C. J. Richardson, G. C. M. Smith, B. Spittle and R. J. Griffin, J. Med. Chem., 2010, 53, 8498–8507 CrossRef CAS PubMed.
- K. B. Kim and C. M. Crews, Nat. Prod. Rep., 2013, 30, 600–604 RSC.
- J. Wang, M. Sanchez-Rosello, J. L. Acena, C. del Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- K. C. Agrawal, B. A. Booth, S. M. DeNuzzo and A. C. Sartorelli, J. Med. Chem., 1976, 19, 1209–1214 CrossRef CAS.
- G. Paolucci, A. Zanella, M. Bortoluzzi, S. Sostero, P. Longo and M. Napoli, J. Mol. Catal. A: Chem., 2007, 272, 258–264 CrossRef CAS PubMed.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
- G. Gran, Acta Chem. Scand., 1950, 4, 559–577 CrossRef CAS PubMed.
- H. M. N. H. Irving, M. G. Miles and L. D. Pettit, Anal. Chim. Acta, 1967, 38, 475–488 CrossRef CAS.
- SCQuery, The IUPAC Stability Constants Database, Academic Software (Version 5.5) Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS.
- L. Zékány and I. Nagypál, in Computational Methods for the Determination of Stability Constants, ed. D. L. Leggett, Plenum Press, New York, 1985, pp. 291–353 Search PubMed.
- E. A. Enyedy, E. Zsigo, N. V. Nagy, C. R. Kowol, A. Roller, B. K. Keppler and T. Kiss, Eur. J. Inorg. Chem., 2012, 4036–4047 CrossRef CAS PubMed.
- M. N. M. Milunovic, E. A. Enyedy, N. V. Nagy, T. Kiss, R. Trondl, M. A. Jakupec, B. K. Keppler, R. Krachler, G. Novitchi and V. B. Arion, Inorg. Chem., 2012, 51, 9309–9321 CrossRef CAS PubMed.
- A. Rockenbauer, T. Szabo-Planka, Z. Arkosi and L. Korecz, J. Am. Chem. Soc., 2001, 123, 7646–7654 CrossRef CAS PubMed.
- A. Rockenbauer and L. Korecz, Appl. Magn. Reson., 1996, 10, 29–43 CrossRef CAS.
- SAINT-Plus, version 7.06a and APEX2, Bruker-Nonius AXS Inc., Madison, WI, 2004 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- R. Supino, in In Vitro Toxicity Testing Protocols, ed. S. O'Hare and C. Atterwill, Humana Press, 1995, vol. 43, ch. 16, pp. 137–149 Search PubMed.
- A. Ojida, T. Sakamoto, M.-a. Inoue, S.-h. Fujishima, G. Lippens and I. Hamachi, J. Am. Chem. Soc., 2009, 131, 6543–6548 CrossRef CAS PubMed.
- T. Watanabe, A. Kobayashi, M. Nishiura, H. Takahashi, T. Usui, I. Kamiyama, N. Mochizuki, K. Noritake, Y. Yokoyama and Y. Murakami, Chem. Pharm. Bull., 1991, 39, 1152–1156 CrossRef CAS.
- M. M. B. Pessoa, G. F. S. Andrade, V. R. Paoli Monteiro and M. L. A. Temperini, Polyhedron, 2001, 20, 3133–3141 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. Van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- F. Bacher, O. Doemoetoer, M. Kaltenbrunner, M. Mojovic, A. Popovic-Bijelic, A. Graeslund, A. Ozarowski, L. Filipovic, S. Radulovic, E. A. Enyedy and V. B. Arion, Inorg. Chem., 2014, 53, 12595–12609 CrossRef CAS PubMed.
- E. A. Enyedy, M. F. Primik, C. R. Kowol, V. B. Arion, T. Kiss and B. K. Keppler, Dalton Trans., 2011, 40, 5895–5905 RSC.
- E. A. Enyedy, N. V. Nagy, E. Zsigo, C. R. Kowol, V. B. Arion, B. K. Keppler and T. Kiss, Eur. J. Inorg. Chem., 2010, 1717–1728 CrossRef CAS PubMed.
- C. R. Kowol, R. Trondl, P. Heffeter, V. B. Arion, M. A. Jakupec, A. Roller, M. Galanski, W. Berger and B. K. Keppler, J. Med. Chem., 2009, 52, 5032–5043 CrossRef CAS PubMed.
- C. R. Kowol, P. Heffeter, W. Miklos, L. Gille, R. Trondl, L. Cappellacci, W. Berger and B. K. Keppler, JBIC, J. Biol. Inorg. Chem., 2012, 17, 409–423 CrossRef CAS PubMed.
- L. A. Saryan, E. Ankel, C. Krishnamurti, D. H. Petering and H. Elford, J. Med. Chem., 1979, 22, 1218–1221 CrossRef CAS.
- E. W. Ainscough, A. M. Brodie, W. A. Denny, G. J. Finlay and J. D. Ranford, J. Inorg. Biochem., 1998, 70, 175–185 CrossRef CAS.
- J. Garcia-Tojal, A. Garcia-Orad, A. Alvarez Diaz, J. L. Serra, M. K. Urtiaga, M. I. Arriortua and T. Rojo, J. Inorg. Biochem., 2001, 84, 271–278 CrossRef CAS.
- G. Pelosi, Open Crystallogr. J., 2010, 3, 16–28 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR numbering scheme for HL1–HL6 (Scheme S1), synthesis scheme for HL1–HL6 (Scheme S2), deprotonation steps of HL2 (Scheme S3), part of the crystal structure of 1 showing complex pairing via intermolecular hydrogen bonding interactions (Fig. S1), pH-dependence of the chemical shifts of various protons of HL4 (Fig. S2), low- (A) and high-field (B) regions of the 1H NMR spectra HL2 at different pH values (Fig. S3), pH-dependence of the chemical shifts of various protons of HL2 in the low- (A) and in the high-field (B) regions (Fig. S4), pH-dependence of the molar fraction of the E and Z isomers of HL2 (Fig. S5), 3-dimensional fluorescence spectra of HL2 and HL4 (Fig. S6), experimental and simulated solution EPR spectra recorded for the copper(II) – HL4 system at 1:1 (A) and 1:2 (B) metal-to-ligand ratio (Fig. S7), calculated component EPR spectra obtained for copper(II) complexes of HL2 and HL4 in frozen solution (Fig. S8), UV–vis spectra of [Cu(EDTA)]2− in the presence of HL2 (Fig. S9), UV–vis spectra of the copper(II) – HL2 and HL4 systems at 1:1 metal-to-ligand ratio (Fig. S10). CCDC 1052906–1052910. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01076d |
| This journal is © The Royal Society of Chemistry 2015 |