
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExploring the potential of gold(III) cyclometallated compounds as cytotoxic agents: variations on the C^N theme†
B.
Bertrand
ab,
S.
Spreckelmeyer
ac,
E.
Bodio
b,
F.
Cocco
d,
M.
Picquet
b,
P.
Richard
b,
P.
Le Gendre
b,
C.
Orvig
c,
M. A.
Cinellu
*d and
A.
Casini
*a
aDept. Pharmacokinetics, Toxicology and Targeting, Research Institute of Pharmacy, University of Groningen, A. Deusinglaan 1, 9713 AV Groningen, The Netherlands. E-mail: a.casini@rug.nl; Fax: +31 50 363 3274
bICMUB UMR6302, CNRS, Univ. Bourgogne Franche-Comté, F-21000 Dijon, France
cMedicinal Inorganic Chemistry Group, Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, BC V6/1Z1, Canada
dUniversità degli Studi di Sassari, Dipartimento di Chimica e Farmacia, Via Vienna, 2, Sassari, I-07100, Italy. E-mail: cinellu@uniss.it; Fax: +39 079 229559
First published on 26th May 2015
Abstract
A series of novel (C^N) cyclometallated Au(III) complexes of general formula [Au(pyb-H)L1L2]n+ (pyb-H = C^N cyclometallated 2-benzylpyridine, L1 and L2 being chlorido, phosphane or glucosethiolato ligands, n = 0 or 1) have been synthesized and fully characterized using different techniques, including NMR, IR and far-IR, mass spectrometry, as well as elemental analysis. The crystal structure of one compound has been solved using X-ray diffraction methods. All compounds were tested in vitro in five human cancer cell lines including the lung, breast, colon and ovarian cancer cells. For comparison purposes, all compounds were also tested in a model of healthy human cells from the embryonic kidney. Notably, all new compounds were more toxic than their cyclometallated precursor bearing two chlorido ligands, and the derivative bearing one phosphane ligand presented the most promising toxicity profile in our in vitro screening, displaying a p53 dependent activity in colorectal cancer HCT116 cells. Finally, for the first time C^N cyclometallated gold(III) complexes were shown to be potent inhibitors of the zinc finger protein PARP-1, involved in the mechanism of cisplatin resistance.
Introduction
Metallodrugs play an important role in the treatment of several diseases. Among them, cisplatin [cis-diamminedichlorido platinum(II)], discovered accidentally,1 is widely used in chemotherapy due to its strong effects against ovarian, testicular and lung cancer. Unfortunately, anticancer therapy with cisplatin has a range of limitations, including resistance, limited spectrum of action and severe side effects, including nephrotoxicity. For this reason, since cisplatin's discovery, numerous Pt-based anticancer agents have been developed to minimize such harmful side effects.2 However, to date, only two additional Pt(II) compounds have achieved international marketing approval, carboplatin and oxaliplatin, although not being deprived of toxic effects.3 In the effort to develop improved anticancer metallodrugs, several other metal complexes have been explored in vitro and in vivo, including compounds of Fe, Ru, Ti and Au.4–6On the basis of these chemical considerations, the peculiar properties of gold have been exploited for several applications. To date, many complexes containing Au in +1 and +3 oxidation states have shown therapeutic properties against cancer or inflammatory diseases. When exploiting the reactivity of gold derivatives against cells, one should bear in mind also the noble metal characteristics. Thus, the stabilization of the oxidation states +1 and +3 is of paramount importance to observe any kind of biological activity. Otherwise, the metal center can ultimately undergo reduction process, thus leading to the formation of Au(0).7
In this context, the development of anticancer gold organometallics of various families has identified promising candidates with anticancer properties, including Au(I) alkynyl complexes, Au(I/III) N-heterocyclic carbene (NHC) complexes and Au(III) cyclometallated complexes.8–12 The advantage of NHC and cyclometallated organometallic gold compounds is their relative stability with respect to other classical coordination complexes. There is evidence that Au compounds preferentially target proteins in biological environments, such as the Se-enzyme thioredoxin reductase (TrxR),13–16 Zn finger enzymes,17,18 as well as the membrane water and glycerol channel aquaglyceroporins.19–21
Notably, increasing interest is focused on the family of cyclometallated gold(III) complexes, in which the Au3+ ions are highly stabilized under physiological conditions. Recently, we reviewed a variety of cyclometallated gold(III) complexes of different scaffolds, namely (C^N)Au, (C^N^N)Au, and (C^N^C)Au complexes.8 For example, Fricker and coworkers reported the synthesis and biological activity of square-planar six-membered cycloaurated Au(III) compounds with a pyridinyl–phenyl linked backbone and two monodentate or one bidentate leaving group(s), which were able to inhibit the cysteine proteases cathepsins B and K in vitro.22 Structure/activity relationships were investigated by modifications to the pyridinyl–phenyl backbone, and leaving groups, demonstrating optimal activity with substitution at the 6 position of the pyridine ring. In this study, the importance of the leaving groups was also highlighted.22 Moreover, one of the derivatives containing thiosalicylate, as one of the ligands bound to the (C^N)Au scaffold, was tested in vivo against the HT29 human colon tumor xenograft model, where a modest decrease in tumor growth was observed compared to the untreated control tumor.22
Taking into account these promising studies, we report here the synthesis and characterization of a new series of four gold(III) cyclometallated compounds of the general formula [Au(pyb-H)L1L2]n+ (pyb-H = C^N cyclometallated 2-benzylpyridine, L1 and L2 being 1,3,5-triazaphosphaadamantane (PTA), thio-β-D-glucose tetraacetate (GluS−) or chlorido ligands; n = 0, 1). Notably, the PTA ligand was chosen for its good water solubility, while the GluS− ligand was selected with the aim of facilitating uptake into cancer cells modulating the lipophilic/hydrophilic character, as well as via possible interaction with GLUT1 transporters.23 Moreover, binding of Au(III) to thiolate should prevent ligand exchange reactions with biological nucleophiles leading to inactivation of the compound. The four new gold(III) compounds were obtained in good yields and fully characterized by 1H-NMR, 13C-NMR, 31P-NMR and far-IR, as well as X-ray crystallography for compound [Au(pyb-H)(PTA)Cl][PF6] (2-PF6). All complexes were tested vs. a panel of human cancer cell lines, as well as in non-tumorigenic human embryonic kidney cells HEK-293 T in vitro. Since Pt drugs have been known to elicit apoptosis in certain cell lines via a p53-dependent pathway,24 the human colorectal carcinoma HCT116 p53+/+ cells overexpressing p53, and HCT116 p53−/− cells knock-out for this gene were selected to compare the antiproliferative effects of the Au complexes with cisplatin. Finally, the selected compounds were tested as inhibitors of the enzyme poly(adenosine diphosphate [ADP-ribose]) polymerase 1 (PARP-1). It is worth mentioning that PARPs are Zn finger proteins playing a key role in DNA repair by detecting DNA strand breaks and catalyzing poly(ADP-ribosylation).25,26 Specifically, PARP-1 is involved in the cisplatin resistance mechanism in cancer cells.24
Results and discussion
Synthesis and structural characterization
The (C^N) cyclometallated precursor [Au(pyb-H)Cl2] (1) (pyb-H = C^N cyclometallated 2-benzylpyridine) was synthesized as described by Cinellu et al. by the reaction of sodium tetrachloridoaurate with 2-benzylpyridine (pyb) in refluxing MeCN/H2O overnight.27 As previously reported, replacement of one chlorido ligand by triphenylphosphine can be achieved by reacting 1 with one equivalent of PPh3 in the presence of excess KPF6 or NaBF4.27 Following this method, we prepared the new phosphane derivative [Au(pyb-H)(PTA)Cl][PF6] (2-PF6) containing 1,3,5-triazaphosphaadamantane (PTA), which is not toxic to cancer cells and is known in general to improve water solubility. Thus, by reacting 1 with one equivalent of PTA in the presence of KPF6 for 1.5 hours at room temperature, we obtained 2-PF6 in a very good yield (Scheme 1). Complexation of the phosphane ligand as well as the isomeric purity of 2-PF6 were assessed by 31P NMR showing the PF6− anion heptuplet signal centred at −144.4 ppm and a singlet of the coordinated phosphorous at −14.2 ppm, downfield shifted by 86 ppm with respect to the free phosphane ligand.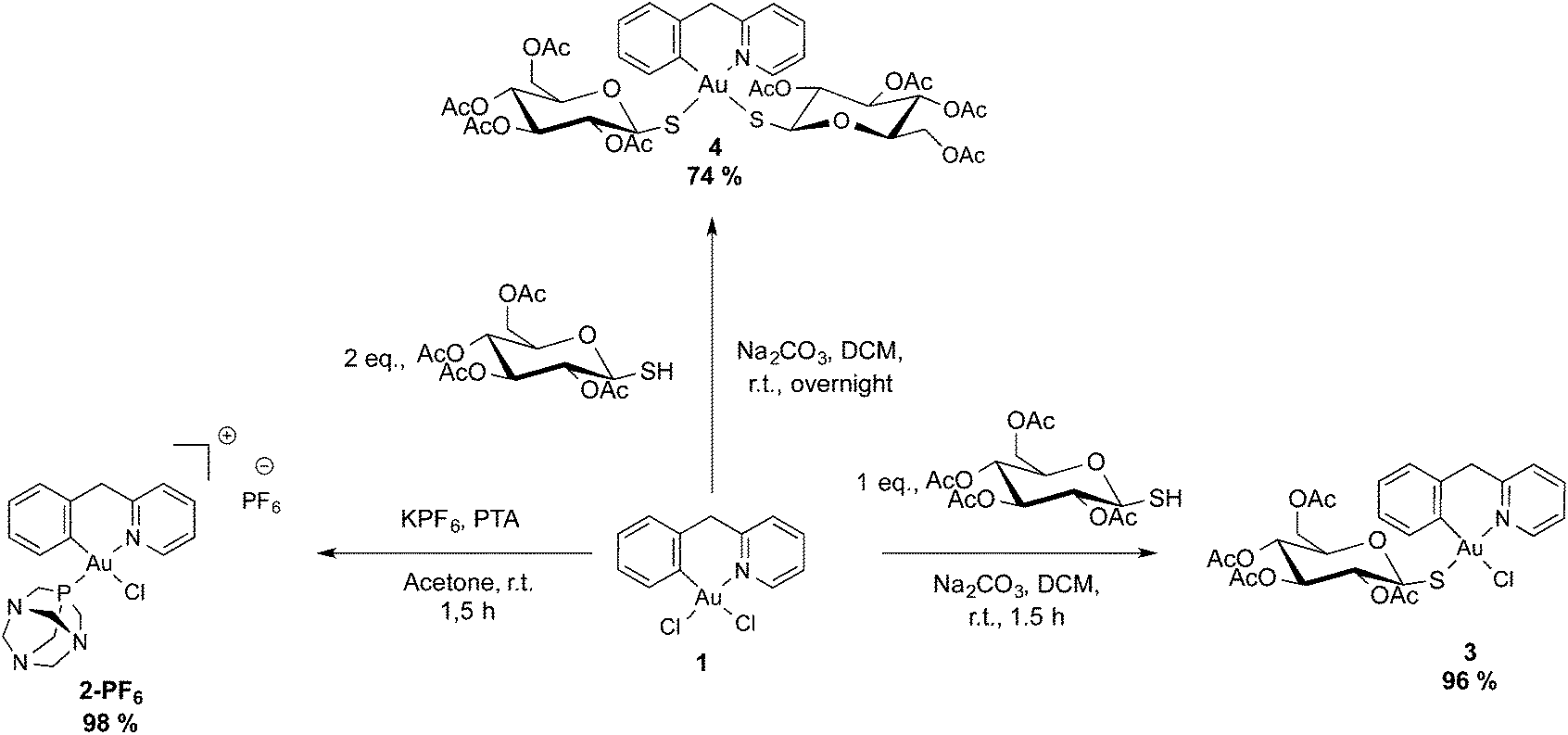 | ||
| Scheme 1 Synthesis of novel (C^N)gold(III) cyclometallated compounds. | ||
Of the two possible geometrical isomers, trans-P–Au–N or trans-P–Au–C, the IR and 1H NMR spectra, taken together, support a trans-P–Au–N arrangement. Indeed, in the IR spectrum the Au–Cl stretching vibration is observed at values consistent with a chlorine trans to a carbon atom (see the Experimental section) and in the 1H NMR spectrum the H6 proton (for numbering scheme, see Scheme 2) of the pyridine is strongly deshielded with respect to the free ligand (Δδ = 0.44 ppm), as usually observed in related complexes and reflects the through-space influence of the adjacent chloride ligand.27
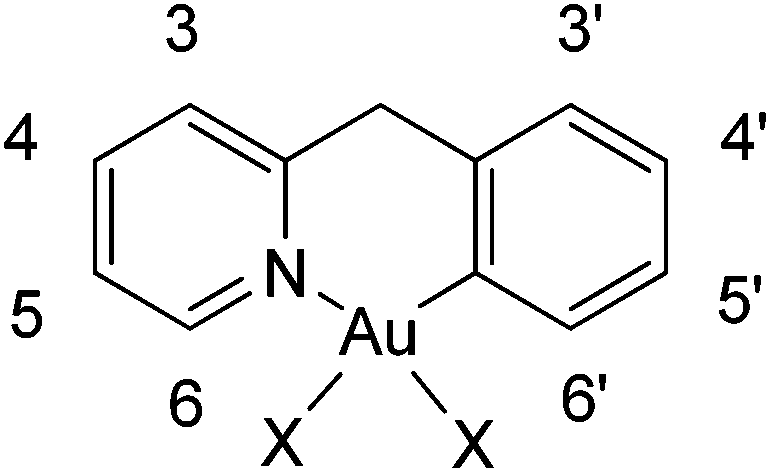 | ||
| Scheme 2 1H and 13C labelling used for the attribution of the NMR signals of the cyclometallated ligand. | ||
X-ray-quality crystals of 2-PF6 were obtained by slow diffusion of diethyl ether into an acetone solution and the structure in the solid state was solved by X-ray diffraction analysis. The solid state molecular structure of the cation is depicted in Fig. 1 with principal bond lengths and angles reported in Table 1; the corresponding bond parameters of the analogous triphenylphosphine complex [Au(pyb-H)(PPh3)Cl][BF4]28 (5-BF4) are also reported for comparison. The compound crystallizes in the P21/n monoclinic space group and the asymmetric unit contains two independent cations, two independent PF6− anions and one acetone molecule (Fig. S1 in the ESI†). The conformations of the two cations are similar and simply differ by a rotation of the PTA groups by about 19°, thus for simplicity reasons, the following description will only be with regard to one of these cations (depicted in Fig. 1). The gold atom displays an almost regular square-planar coordination, with a slight distortion in the N–Au–C angle: 87.39(7)° (Fig. 1). The bite angle of the six-membered cyclometallated ring, the bond lengths and angles involving the gold atom are comparable to those observed in 5-BF4 by Fuchita (Table 1);28 small differences – e.g. in the Au–N bond distance – may be attributed to the different electronic and steric properties of the two phosphane ligands. Moreover, the six-membered metallacycle is in a boat-like conformation (Fig. 1B) with atoms C1, C6, C8 and N1 essentially coplanar within the estimated standard deviation. This best plane forms dihedral angles with planes C6–C7–C8 and N1–Au1–C1 of 48.75(16) and 39.34(9)°, which is in line with previous observations made by Fuchita et al. for 5-BF4.28 Notably, the PTA ligand is trans to the nitrogen of the cyclometallated ligand, as previously suggested by the FIR and 1H NMR spectra.
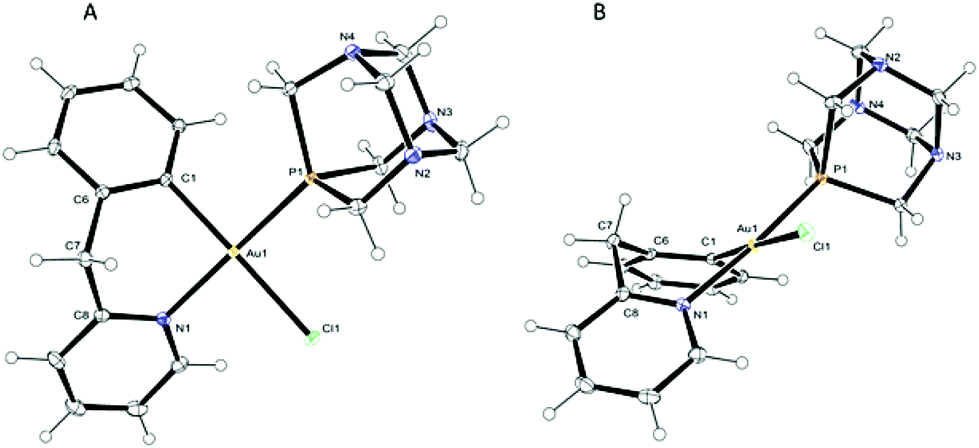 | ||
| Fig. 1 ORTEP view of the cation in complex 2-PF6; top (A) and side (B) views. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 28
28
| Distances (Å) and angles(°) | 2-PF6 (for both independent molecules) | 5-BF4 | |
|---|---|---|---|
| Au–C | 2.040(2) | 2.042(2) | 2.03(1) |
| Au–N | 2.0961(18) | 2.0998(18) | 2.079(10) |
| Au–P | 2.2709(6) | 2.2840(6) | 2.311(3) |
| Au–Cl | 2.3696(5) | 2.3612(5) | 2.362(3) |
| C–Au–N | 87.39(7) | 87.26(8) | 85.8(4) |
| C–Au–P | 93.44(6) | 91.92(6) | 94.8(3) |
| C–Au–Cl | 178.35(6) | 177.56(6) | 175.2(3) |
| N–Au–P | 175.43(5) | 177.43(5) | 176.4(3) |
| N–Au–Cl | 91.25(5) | 90.31(5) | 89.4(3) |
| P–Au–Cl | 87.985(19) | 90.512(19) | 89.9(1) |
Complex 1 was also treated with one or two equivalents of thio-β-D-glucose tetraacetate (GluSH) and sodium carbonate in dichloromethane for 1.5 hours to obtain compound 3, or overnight to obtain compound 4 in good yields. The non-planarity and the conformational stability of the cyclometallated Au(III) scaffold give rise to a planar chirality which upon coordination with optically pure tetraacetylated β-D-glucosethiolate leads to the formation of a mixture of diastereoisomers. In both the 1H and 13C NMR spectra, 3 presents doubled signals in a 50/50 ratio for protons and carbons close to the metal centre due to the equimolar presence of the two possible diastereoisomers. The largest split is observed for H6′ with a difference of 0.25 ppm between the two diastereoisomers, while the signals of H6 are separated by only 0.03 ppm, thus suggesting close proximity of H6′ to the chiral centres. The 1H NMR spectrum of 4 was quite complex, showing two signals for each proton of the cyclometallated ligand in a ratio of 1/0.3. These signals correspond neither to the precursor 1 nor to the mono-sugar 3. However, the structure of the compound was further assessed by the association of the ESI-MS spectra showing the peak of adduct [4 + Na]+ and the elemental analysis corresponding to 4·H2O.
In the absence of a crystal structure determination for complex 3, to discriminate between the two possible isomers, i.e. thio-glucose in trans position to the carbon or to the nitrogen atom (Fig. S2†), its far-IR (FIR) spectrum was compared to those of the other complexes, whose structures were known (2-PF6 and 5-BF4) or unambiguous (1 and 4). Indeed, in this range of energy we can observe the stretching bands of Au–Cl and Au–S bonds. The main FIR bands are shown in Table S2 in the ESI.†
The dichlorido complex 1 displays a medium band at 358 cm−1 and a strong one at 287 cm−1, corresponding to the stretching of the Au–Cl bonds in trans position to the nitrogen and carbon atoms, respectively. In the case of 2-PF6, the FIR spectrum shows an intense band at 310 cm−1 close to the value (305 cm−1) observed for the analogous complex with PPh3, consistent with a chlorine trans to the carbon atom of the phenyl substituent.
Compound 3 presents two bands: a broad one at 372 cm−1 corresponding to the Au–S bond trans to a nitrogen atom, and a strong one at 295 cm−1 corresponding to the Au–Cl bond in trans position to the carbon atom. As expected, the FIR spectra of 4 did not show any Au–Cl stretching band, but a broad double band with peaks at 375 and 369 cm−1, consistent, respectively, with a sulfur trans to a nitrogen and to a carbon atom.
Antiproliferative activity
Compounds 1, 2-PF6, 3 and 4 were screened for their antiproliferative properties in vitro in a panel of human cancer cell lines including ovarian adenocarcinoma (A2780), mammary carcinoma (MCF-7), lung carcinoma (A549) and colon carcinoma overexpressing p53 (HCT116 p53+/+) or p53 knock-out (HCT116 p53−/−), as well as on healthy human embryonic kidney cells (HEK-293T). The IC50 was determined after 72 hours of incubation with different concentrations of compounds using the classical MTT test. The results are summarized in Table 2.| IC50a (μM) |
||||||
|---|---|---|---|---|---|---|
| Comp. | A2780 | HCT116 p53+/+ | HCT116 p53−/− | MCF7 | A549 | HEK-293T |
| a The reported values are the mean ± SD of at least three determinations. | ||||||
| 1 | 36.1 ± 7.8 | 25.5 ± 6.6 | 21.1 ± 3.1 | 25.5 ± 4.7 | 54.4 ± 0.3 | 21.0 ± 5.1 |
| 2-PF6 | 2.7 ± 0.2 | 2.1 ± 0.7 | 14.0 ± 1.1 | 15.6 ± 4.6 | 40.5 ± 5.0 | 7.1 ± 0.8 |
| 3 | 15.7 ± 7.4 | 9.7 ± 4.8 | 18.4 ± 1.1 | 19.7 ± 3.8 | 40.0 ± 0.7 | 11.7 ± 6.1 |
| 4 | 17.4 ± 4.5 | 10.5 ± 2.0 | 18.5 ± 0.6 | 15.1 ± 3.9 | 18.2 ± 1.2 | 12.9 ± 3.1 |
| Cisplatin | 1.9 ± 0.6 | 5.3 ± 0.2 | 22.9 ± 2.3 | 20.0 ± 3.0 | 12.06 ± 0.8 | 8.6 ± 1.3 |
In general, the new cyclometallated complexes 2-PF6, 3 and 4 were more toxic than their precursor 1, containing two chlorido ligands, in all cell lines with the exception of the A549 cell line in which most of the gold complexes appeared to be poorly toxic. The phosphane-containing complex 2-PF6 presents the most interesting toxicity profile, comparable to that of cisplatin in A2780 cells (IC50 = 2.7 ± 0.2 and 1.9 ± 0.6 μM, respectively). Furthermore, complex 2-PF6 is twice as toxic as cisplatin against HCT116+/+ cells (IC50 = 2.1 ± 0.7 and 5.3 ± 0.2 μM) and poorly effective on the HCT116 p53−/− (IC50 = 14.0 ± 1.1 μM). The latter result suggests similar dependence on p53 pathways for compound 2-PF6 as for cisplatin. In terms of selectivity, 2-PF6 is also ca. 3-fold less toxic on the HEK-293T cells compared to the HCT116 p53+/+. Compounds 3 and 4 showed overall moderate antiproliferative properties, and their inactivity rules out the idea that the tetra-acetylated β-D-glucose-1-thiolato ligand may enhance the uptake of the compounds, for example through GLUT-1 transporters. Indeed, our previously reported studies on Au(I) NHC complexes with similar thio-sugar ligands also showed scarce cytotoxic effects most likely due to poor gold uptake.29
PARP-1 inhibition
In order to further investigate the mechanism of action of our organometallic compounds, and inspired by our recent results that indicate that some cytotoxic gold(III) compounds are efficient inhibitors of the zinc-finger protein PARP-1,17 we tested complexes 1 and 2-PF6 on the purified human enzyme as described in the Experimental section. Remarkably, potent PARP-1 inhibition was indeed observed with both compounds: 1 with IC50 = 1.30 ± 0.40 nM, and 2-PF6 with IC50 = 1.87 ± 0.20 nM, respectively. Notably, these values are in the same range of those previously observed for Au(III) complexes with N-donor ligands.17Conclusions
The potential of organometallic compounds for biological applications, including as anticancer agents, has been demonstrated by numerous studies.30 Here, we report the synthesis and characterization of a new series of (C^N) cyclometallated gold(III) complexes bearing different ancillary ligands selected to confer different reactivity and biological properties to the resulting compounds, such as PTA for increased water-solubility, and thio-sugar moieties to influence uptake and reduce exchange with biological nucleophiles. The X-ray structure of compound 2-PF6 was solved and revealed the typical square-planar geometry of the gold(III) cation, as well as the coordination of the phosphane ligand trans to the nitrogen. Compound 1, which is the precursor of the series, was poorly cytotoxic on all cell lines, while the phosphane-containing compound 2-PF6 shows the most promising results against the HCT116 cancer cell line overexpressing p53.Interestingly, compounds 1 and 2-PF6 inhibited the zinc-finger enzyme PARP-1 in nM concentrations, suggesting the possible design of selective inhibitors and the use of organometallic gold compounds in combination therapies with other anticancer drugs. PARP inhibitors are currently highly investigated for their selective cytotoxic properties and can be considered as DDR inhibitors,31 which can be used in combination with classical DNA damaging agents for optimizing the therapeutic outcome.
Overall, our study shows the potential for improvement of the biological properties of organometallic gold-based compounds by tuning their coordination environment. Further studies are ongoing to evaluate the mechanisms of transport and possible targets for this new series of gold compounds, including interactions with nucleic acids.
Experimental section
General remarks
All reactions were carried out under purified argon using the Schlenk techniques. Solvents were dried and distilled under argon before use. The precursor [Au(pyb-H)Cl2] was synthesized according to a literature procedure.26 All other reagents were commercially available and used as received. All the physico-chemical analyses were performed at the “Plateforme d'Analyses Chimiques et de Synthèse Moléculaire de l'Université de Bourgogne”. The identity and purity (≥95%) of the complexes were unambiguously established using high-resolution mass spectrometry and NMR. The exact mass of the synthesized complexes was obtained on a Thermo LTQ Orbitrap XL. 1H- (300.13, 500.13 or 600.23 MHz), 13C- (125.77 or 150.90 MHz) and 31P- (121.49, 202.45 or 242.94 MHz) NMR spectra were recorded on Bruker 300 Avance III, 500 Avance III or 600 Avance II spectrometers, respectively. Chemical shifts are quoted in ppm (δ) relative to TMS (1H and 13C) using the residual protonated solvent (1H) or the deuterated solvent (13C) as internal standards. 85% H3PO4 (31P) was used as an external standard. Infrared spectra were recorded on a Bruker Vector 22 FT-IR spectrophotometer (Golden Gate ATR) and far infrared spectra were recorded on a Bruker Vertex 70v FT-IR spectrophotometer (Diamant A225 ATR). X-ray diffraction data for 2 were collected on a Bruker Nonius Kappa CCD APEX II at 115 K.Synthesis of cyclometallated gold(III) complexes based on 2-benzylpyridine ligand (pyb)
:1 mixture of diastereomers (83.9 mg, 96% yield). 1H NMR (acetone-d6, 500.13 MHz, 298 K): 1.89/1.94 (2 s, 3H, CH3), 1.95 (s, 3H, CH3), 1.98/1.99 (2 s, 3H, CH3), 2.07/2.09 (2 s, 3H, CH3), 3.74 (m, 1H, CH), 4.02/4.13 (2 dd, 1H, JH–H = 12.0 Hz, 2.0 Hz, CH2-sugar), 4.22/4.26 (2 dd, 1H, JH–H = 12.0 Hz, 5.5 Hz, CH2-sugar), 4.33 (d, 1 H, JAB = 14.5 Hz, CHAHB-pyb), 4.52 (d, 1 H, JAB = 14.5 Hz, CHAHB-pyb), 4.99–5.09 (m, 2H, 2 CH), 5.17–5.25 (2 t, 1H, JH–H = 9.5 Hz, CH), 5.45/5.64 (2 d, 1H, JH–H = 9.5 Hz, CH), 7.08–7.12 (2m, 1H, JH–H = 5.5 Hz, H5′), 7.16–7.20 (m, 1H, H4′), 7.29–7.31 (2 d, 1H, JH–H = 5.5 Hz, H3′), 7.33/7.58 (2 d, 1H, JH–H = 5.5 Hz, H6′), 7.70–7.73 (m, 1H, H5), 7.98 (broad d, 1H, JH–H = 8.0 Hz, H3), 8.22 (m, 1H, JH–H = 8.0 Hz, H4), 9.17/9.20 (2 d, 1H, JH–H = 5.5 Hz, H6). 13C{1H} NMR (acetone-d6, 125.77 MHz, 300 K): 20.6–21.0 (CH3), 47.9 and 48.0 (CH2-pyb), 63.0 (CH2-sugar), 69.7/69.8, 73.0/73.3, 75.4, 76.2/76.4 and 83.0/83.1 (CH-sugar), 125.3 (C5), 127.1 (C3), 128.7 (C4′/5′), 128.8 (C4′/5′), 129.9/130.0 (C3′), 131.9–133.1 (C6′), 134.0/134.1 (C–Au), 143.1/143.2 (C4), 144.5/144.6 (Cipso), 152.0/152.1 (C6), 156.9/157.1 (Cipso), 169.8–170.9 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). ESI-MS (DMSO-MeOH), positive mode exact mass for [C26H29NO9SAuClNa]+ (786.08093): measured m/z 786.07946 [M + Na]+. IR (νmax, cm−1): 1743, 1612, 1569, 1435, 1367, 1219, 1029, 912, 753, 376, 295, 221. Anal. Calc. for C26H29NO9SAuCl: C, 40.87, H, 3.83, N, 1.83, S, 4.20%. Found: C, 40.58, H, 4.18, N, 1.85, S, 3.73%.
O); minor isomer: 20.6, 20.7, 21.1 and 21.2 (CH3), 48.4 (CH2-pyb), 63.0, 63.2, 69.6, 70.0, 73.6, 75.0, 75.3, 76.4, 76.6, 83.8 and 84.8 (CH-sugar), 123.8, 125.4, 126.9, 127.8, 131.7 and 133.4 (CH-pyb), 134.5 (C–Au), 137.2 (CH-pyb), 150.4 (Cipso-pyb), 152.9 (CH-pyb), 157.9 (Cipso-pyb), 169.8, 169.9, 170.1 and 170.8 (CO). (CH2Cl2/MeOH), positive mode exact mass for [C40H48NO18S2AuNa]+ (1114.18705): measured m/z 1114.18560 [M + Na]+. IR (νmax, cm−1): 1740, 1614, 1569, 1437, 1367, 1218, 1029, 754, 375, 369, 212. Anal. Calc. for C40H48AuNO18S2·H2O: C, 43.29, H, 4.54, N, 1.26, S, 5.78%. Found: C, 43.11, H, 4.72, N, 1.30, S, 4.78%.
O). ESI-MS (DMSO-MeOH), positive mode exact mass for [C26H29NO9SAuClNa]+ (786.08093): measured m/z 786.07946 [M + Na]+. IR (νmax, cm−1): 1743, 1612, 1569, 1435, 1367, 1219, 1029, 912, 753, 376, 295, 221. Anal. Calc. for C26H29NO9SAuCl: C, 40.87, H, 3.83, N, 1.83, S, 4.20%. Found: C, 40.58, H, 4.18, N, 1.85, S, 3.73%.
O); minor isomer: 20.6, 20.7, 21.1 and 21.2 (CH3), 48.4 (CH2-pyb), 63.0, 63.2, 69.6, 70.0, 73.6, 75.0, 75.3, 76.4, 76.6, 83.8 and 84.8 (CH-sugar), 123.8, 125.4, 126.9, 127.8, 131.7 and 133.4 (CH-pyb), 134.5 (C–Au), 137.2 (CH-pyb), 150.4 (Cipso-pyb), 152.9 (CH-pyb), 157.9 (Cipso-pyb), 169.8, 169.9, 170.1 and 170.8 (CO). (CH2Cl2/MeOH), positive mode exact mass for [C40H48NO18S2AuNa]+ (1114.18705): measured m/z 1114.18560 [M + Na]+. IR (νmax, cm−1): 1740, 1614, 1569, 1437, 1367, 1218, 1029, 754, 375, 369, 212. Anal. Calc. for C40H48AuNO18S2·H2O: C, 43.29, H, 4.54, N, 1.26, S, 5.78%. Found: C, 43.11, H, 4.72, N, 1.30, S, 4.78%.
X-ray crystallography
Crystals of 2-PF6 were obtained by slow diffusion of diethyl ether into a concentrated solution of 2-PF6 in acetone. Intensity data were collected on a Bruker Kappa CCD APEX II at 115 K. The structure was solved by direct methods (SIR92)32 and refined with full-matrix least-squares methods based on F2 (Shelx 97)33 with the aid of the Olex2 program.34 All non-hydrogen atoms were refined with anisotropic thermal parameters. Hydrogen atoms were included in their calculated positions and refined with a riding model. Crystallographic data are reported in Table S1 (ESI†).Cell viability assay
The human breast cancer cell line MCF7, the human lung cancer cell line A549 and the human ovarian cancer cell line A2780 were obtained from the European Centre of Cell Cultures ECACC, Salisbury, UK. Human colon cancer cell lines HCT116 p53+/+ and HCT116 p53−/− were a kind gift from Dr Götz Hartleben (ERIBA, Groningen, NL), while non-tumoral human embryonic kidney cells HEK-293T were kindly provided by Dr Maria Pia Rigobello (CNR, Padova, Italy). Cells were cultured in DMEM (Dulbecco's modified Eagle medium) or RPMI containing GlutaMax, supplemented with 10% FBS and 1% penicillin/streptomycin (all from Invitrogen), at 37 °C under a humidified atmosphere of 95% of air and 5% CO2 (Heraeus, Germany).For evaluation of growth inhibition, cells were seeded in 96-well plates (Costar, Integra Biosciences, Cambridge, MA) at a concentration of 10000 cells per well (A2780, MCF-7, HEK-293T) or 6000 cells per well (HCT116, A549) and grown for 24 h in complete medium. Solutions of the gold compounds were prepared by diluting a freshly prepared stock solution (10−2 M in DMSO) of the corresponding compound in aqueous media (RPMI or DMEM for the A2780 or A549, MCF-7, HCT116 p53+/+ and HEK-293T, respectively). Stability in DMSO was checked by NMR, and the compounds resulted to be stable over several hours. Cisplatin was purchased from Sigma-Aldrich and stock solutions were prepared in water. Afterwards, the intermediate dilutions of the compounds in the cell culture medium were added to the wells (200 μL) to obtain a final concentration ranging from 0 to 50 μM, and the cells were incubated for 72 h. Afterwards, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoliumbromide (MTT) was added to the cells at a final concentration of 0.5 mg ml−1 and incubated for 2 h, then the culture medium was removed and the violet formazan (artificial chromogenic precipitate of the reduction of tetrazolium salts by dehydrogenases and reductases) dissolved in DMSO. The optical density of each well (96-well plates) was quantified three times in quadruplicates at 550 nm using a multi-well plate reader, and the percentage of surviving cells was calculated from the ratio of absorbance of treated to untreated cells. The IC50 value was calculated as the concentration reducing the proliferation of the cells by 50% and it is presented as a mean (±SE) of at least three independent experiments.
PARP-1 activity determinations
PARP-1 activity was determined using Trevigen's HT Universal Colorimetric PARP assay. This assay measures the incorporation of biotinylated poly(ADP-ribose) onto histone proteins in a 96 microtiter strip well format. Recombinant human PARP-1 (high specific activity, purified from E. coli containing recombinant plasmid harboring the human PARP gene, supplied with the assay kit) was used as the enzyme source. 3-Aminobenzamide (3-AB), provided in the kit, was used as the control inhibitor. Two controls were always performed in parallel: a positive activity control for PARP-1 without inhibitors, that provided the 100% activity reference point, and a negative control, without PARP-1 to determine background absorbance. The final reaction mixture (50 μL) was treated with TACS-Sapphire, a horseradish peroxidase colorimetric substrate, and incubated in the dark for 30 min. Absorbance was read at 630 nm after 30 min. The data correspond to the mean of at least three experiments performed in triplicate ± SD.Acknowledgements
A. C. thanks the University of Groningen for funding (Rosalind Franklin Fellowship) and the University of Sassari (Visiting Professor Program). B. B. thanks the ERASMUS program for a mobility fellowship to the University of Sassari. The French Ministère de l'Enseignement Supérieur et de la Recherche is acknowledged for a Ph.D. grant to B. B. The authors acknowledge COST Action CM1105 for financial support and fruitful discussion. The authors thank the CNRS, the “Université de Bourgogne” and the “Conseil Régional de Bourgogne” through the 3MIM integrated project (“Marquage de Molécules par les Métaux pour l'Imagerie Médicale”) and PARI SSTIC no. 6 for their support. Dr Fanny Picquet, Marie José Penouilh, and Marcel Soustelle are warmly acknowledged for the physico-chemical analyses.Notes and references
- B. Rosenberg, L. Vancamp and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS PubMed.
- K. D. Mjos and C. Orvig, Chem. Rev., 2014, 114, 4540–4563 CrossRef CAS PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- G. Boscutti, L. Marchio, L. Ronconi and D. Fregona, Chem. – Eur. J., 2013, 19, 13428–13436 CrossRef CAS PubMed.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS PubMed.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- E. A. Pacheco, E. R. T. Tiekink and M. W. Whitehouse, in Gold Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2009, pp. 283–319 Search PubMed.
- B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209–4219 RSC.
- W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC.
- L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269–3284 RSC.
- E. Vergara, E. Cerrada, A. Casini, O. Zava, M. Laguna and P. J. Dyson, ChemMedChem, 2010, 29, 2596–2603 CAS.
- B. Bertrand, L. Stefan, M. Pirrotta, D. Monchaud, E. Bodio, P. Richard, P. Le Gendre, E. Warmerdam, M. H. de Jager, G. M. Groothuis, M. Picquet and A. Casini, Inorg. Chem., 2014, 53, 2296–2303 CrossRef CAS PubMed.
- R. Rubbiani, E. Schuh, A. Meyer, J. Lemke, J. Wimberg, N. Metzler-Nolte, F. Meyer, F. Mohr and I. Ott, MedChemComm, 2013, 4, 942–948 RSC.
- S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863–873 RSC.
- A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692–1707 CrossRef CAS PubMed.
- A. Meyer, L. Oehninger, Y. Geldmacher, H. Alborzinia, S. Wolfl, W. S. Sheldrick and I. Ott, ChemMedChem, 2014, 9, 1794–1800 CAS.
- F. Mendes, M. Groessl, A. A. Nazarov, Y. O. Tsybin, G. Sava, I. Santos, P. J. Dyson and A. Casini, J. Med. Chem., 2011, 54, 2196–2206 CrossRef CAS PubMed.
- M. Serratrice, F. Edafe, F. Mendes, R. Scopelliti, S. M. Zakeeruddin, M. Grätzel, I. Santos, M. A. Cinellu and A. Casini, Dalton Trans., 2012, 41, 3287–3293 RSC.
- A. De Almeida, G. Soveral and A. Casini, MedChemComm, 2014, 5, 1444–1453 RSC.
- A. P. Martins, A. Ciancetta, A. deAlmeida, A. Marrone, N. Re, G. Soveral and A. Casini, ChemMedChem, 2013, 8, 1086–1092 CrossRef CAS PubMed.
- A. P. Martins, A. Marrone, A. Ciancetta, A. G. Cobo, M. Echevarría, T. F. Moura, N. Re, A. Casini and G. Soveral, PLoS One, 2012, 7(5), e37435 CAS.
- Y. Zhu, B. R. Cameron, R. Mosi, V. Anastassov, J. Cox, L. Qin, Z. Santucci, M. Metz, R. T. Skerlj and S. P. Fricker, J. Inorg. Biochem., 2011, 105, 754–762 CrossRef CAS PubMed.
- E. Vergara, E. Cerrada, C. Clavel, A. Casini and M. Laguna, Dalton Trans., 2011, 40, 10927–10935 RSC.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- V. Schreiber, F. Dantzer, J. C. Ame and G. de Murcia, Nat. Rev. Mol. Cell Biol., 2006, 7, 517–528 CrossRef CAS PubMed.
- A. I. Anzellotti and N. P. Farrell, Chem. Soc. Rev., 2008, 37, 1629–1651 RSC.
- M. A. Cinellu, A. Zucca, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, J. Chem. Soc., Dalton Trans., 1996, 4217–4225 RSC.
- Y. Fuchita, H. Ieda, Y. Tsunemune, J. Kinoshita-Nagaoka and H. Kawano, J. Chem. Soc., Dalton Trans., 1998, 791–796 RSC.
- B. Bertrand, A. de Almeida, E. P. M. van der Burgt, M. Picquet, A. Citta, A. Folda, M. P. Rigobello, P. Le Gendre, E. Bodio and A. Casini, Eur. J. Inorg. Chem., 2014, 2014, 4410–4410 CrossRef CAS PubMed.
- M. A. Cinellu, I. Ott and A. Casini, in Bioorganometallic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2014, pp. 117–140 Search PubMed.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS PubMed.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1051849. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt01023c |
| This journal is © The Royal Society of Chemistry 2015 |