
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA mechanistic investigation of the ruthenium porphyrin catalysed aziridination of olefins by aryl azides†
P.
Zardi
a,
A.
Pozzoli‡
ab,
F.
Ferretti
a,
G.
Manca
*c,
C.
Mealli
c and
E.
Gallo
*a
aDepartment of Chemistry, University of Milan, Via Golgi 19, I-20133 Milan, Italy. E-mail: emma.gallo@unimi.it
bConsorzio Interuniversitario Reattività Chimica e Catalisi (CIRCC), Via Celso Ulpiani, 27, 70126 Bari, Italy
cIstituto di Chimica dei Composti OrganoMetallici, ICCOM-CNR, Via Madonna del Piano 10, I-50019 Sesto Fiorentino, Italy. E-mail: gabriele.manca@iccom.cnr.it
First published on 5th May 2015
Abstract
A mechanism for the aziridination of olefins by aryl azides (ArN3), promoted by ruthenium(II) porphyrin complexes, is proposed on the basis of kinetic and theoretical studies. All the recorded data support the involvement of a mono-imido ruthenium complex as the active intermediate in the transfer of the nitrene moiety “ArN” to the olefin. The selectivity of the aziridination vs. the uncatalysed triazoline formation can be enhanced by fine-tuning the electronic features of the porphyrin ligand and the olefin/azide catalytic ratio. The DFT study highlights the importance of an accessible triplet ground state of the intermediate ruthenium mono-imido complex to allow the evolution of the aziridination process.
Introduction
Aziridines are important building blocks in organic synthesis because their highly strained three-membered ring affords an epoxide-like reactivity through an easy C–N bond cleavage.1–5 The relevant biological activity of these heterocycles, which are present in several natural compounds and antitumor/antimicrobial agents,6–10 suggests their usage as precursors of important aza-compounds. Hence, efficient aziridine synthetic strategies have become important.5,11–17One of the well-established methodologies for the aziridine preparation is the transfer of transition metal-catalysed nitrene from a nitrogen source to an unsaturated hydrocarbon.18,19 In the last few decades a large number of examples of this reaction were reported using iminoiodinanes (PhI = NR) as nitrene sources in the presence of a variety of metal catalysts, e.g., porphyrin, bis-oxazoline and Schiff base complexes.20 Several efficient asymmetric versions of this reaction were also developed.11,12
More recently, organic azides (RN3) have been extensively used as nitrene precursors thanks to their atom efficiency related to the formation of N2 as the only by-product of the nitrene transfer reaction.21–28 Many reports were published concerning the aziridination of olefins by organic azides26,29–35 and excellent results in terms of yields and enantioselectivities were achieved by Katsuki's36–38 and Zhang's39,40 groups.
Since the mechanistic understanding is generally useful to optimise the efficiency of catalytic systems, there is still much debate on how the olefin aziridination works.41,42 Many different variables can be involved in the aziridine formation (Scheme 1),12,43 generally the reaction mechanism is strongly catalyst- and substrate-dependent and often available experimental data appear insufficient to determine a unique pathway.
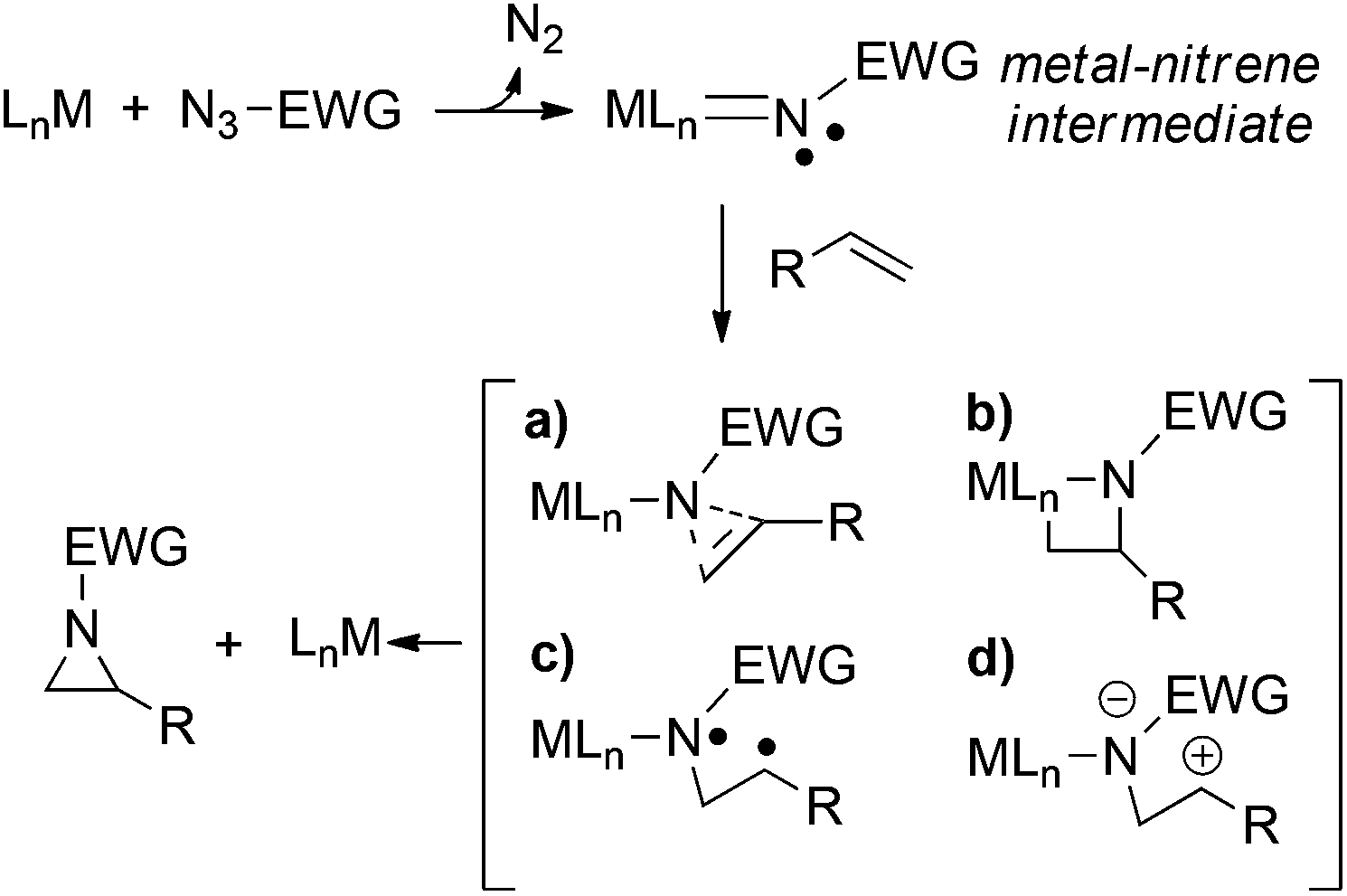 | ||
| Scheme 1 Selection of possible pathways for the olefin aziridination by organic azides: (a) concerted insertion, (b) via azametallacyclobutane, (c) stepwise radical reaction, (d) stepwise carbocationic reaction. | ||
For these reasons, theoretical analyses (e.g. DFT-based calculations) have become an essential tool to shed some light on the aziridination mechanism. For instance, theoretical approaches were fundamental in suggesting a mechanism for the cobalt(II) porphyrin-catalysed aziridination of olefins by organic azides.44,45 A radical mechanism was finally proposed in which a cobalt(III) radical nitrene complex plays the role of the active intermediate in the catalytic reaction.
Since 1999 Cenini/Gallo's group has employed aryl azides to perform the aziridination of olefins promoted by porphyrin complexes32,46–49 and a high activity of ruthenium(II) carbonyl porphyrins was reported.31,50,51 The reaction scope was investigated31 and a preliminary mechanistic proposal was formulated on the basis of experimental data.48
This work presents a more careful investigation of the aziridination mechanism of olefins by aryl azides catalysed by ruthenium(II) porphyrin complexes by combining the experimental, kinetic studies and DFT analysis.
Results and discussion
Our previous studies31 indicated that aziridination of olefins catalysed by Ru(porphyrin)CO occurred in quantitative yields and short reaction times using terminal styrenes and aryl azides bearing electron-withdrawing groups (EWGs) on the aryl moiety. It was also observed that, by running these reactions at high olefin concentrations,48 a major catalytic inhibition occurred due to the formation of 1-(4-nitrophenyl)-5-methyl-5-phenyl-1,2,3-triazoline 1a by the uncatalysed 1,3-cycloaddition between the 4-nitrophenyl azide (ArN3) and α-methylstyrene (Scheme 2). The so-formed triazoline species 1a binds the ruthenium centre of the catalyst Ru(TPP)CO (2a) (TPP = anion of tetraphenylporphyrin) and affords the coordinatively saturated complex 3a (Scheme 2). Complex 3a was fully characterised which also included a single crystal X-ray analysis.48 The poor catalytic activity of 3a has already been discussed in the previous work and, since a vacant coordinative site is required for the aryl azide coordination and activation, it was suggested that triazoline species 1a acts in a competitive inhibitor-like fashion through the catalytic cycle.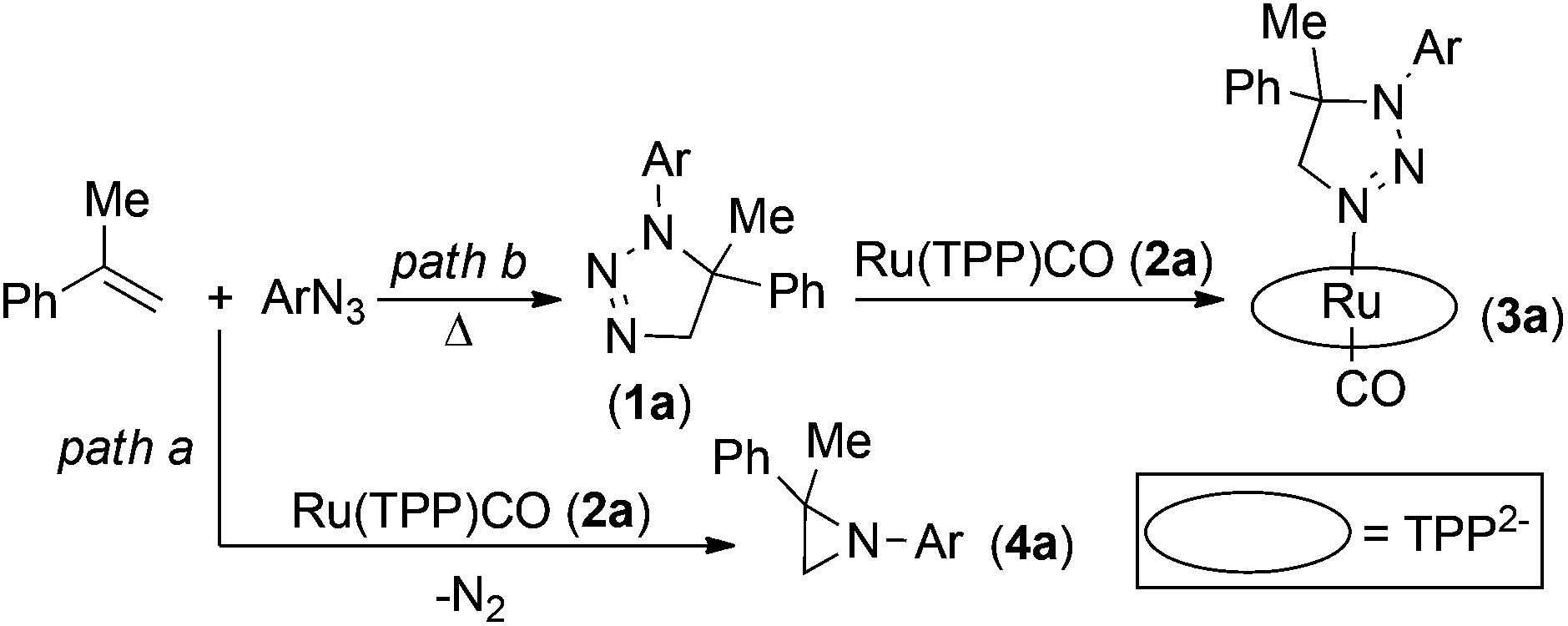 | ||
| Scheme 2 Catalysed (path a) and uncatalysed (path b) reaction between α-methylstyrene and ArN3. | ||
Preliminary kinetic studies48 indicated a first order dependence with respect to both the catalyst and 4-nitrophenyl azide (ArN3) in the 2a-catalysed reaction (Scheme 2, path a). The kinetic order with respect to the α-methylstyrene concentration was ambiguous since a first order dependence, followed by a region of rate inhibition, was observed over a given range of olefin concentrations. This non-linear behaviour can be explained by the formation of triazoline species 1a, which competes with aryl azide for coordination to the ruthenium centre.
The relationships between the two alternative processes illustrated in Scheme 2 (i.e., the catalytic aziridination vs. the stoichiometric formation of the ruthenium-triazoline complex 3a) are here clarified by a series of kinetic experiments and DFT calculations, also aiming at comparing the energetic profiles of the processes.
Kinetic study
Previous studies suggested that performance of a catalyst is critically governed by its electronic properties,31 and accordingly a series of kinetic experiments was carried out on the same substrate of the model reaction (Scheme 2) in the presence of the different ruthenium porphyrin catalysts 2a–g. Interestingly, we found that the functionalisation at the para position of the meso-aryl of the porphyrin ligand of the catalyst modifies the kinetic order with respect to the aryl azide (Table 1). In most cases, a first kinetic order with respect to the aryl azide was observed for ligands bearing electron-donating groups (EDGs) (entries 1, 2 and 4, Table 1), while the zero kinetic order emerged on functionalising the porphyrin with EWGs (entries 5–7, Table 1).|
|
|||||
|---|---|---|---|---|---|
| Entry | X | Catalyst | t (h) | Conv.b | Kinetic orderc |
a Experimental conditions: 1.2 × 10−2 mmol of the catalyst, cat./ArN3/α-methylstyrene ratio = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50:250, benzene as the reaction solvent, nitrogen atmosphere, 75 °C.
b Measured by IR spectroscopy (ν(N :50:250, benzene as the reaction solvent, nitrogen atmosphere, 75 °C.
b Measured by IR spectroscopy (ν(N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) signal of ArN3 at 2150–2100 cm−1).
c Kinetic order with respect to the aryl azide concentration. N) signal of ArN3 at 2150–2100 cm−1).
c Kinetic order with respect to the aryl azide concentration.
|
|||||
| 1 | H | 2a | 3.5 | 100% | 1 |
| 2 | MeO | 2b | 6 | 95% | 1 |
| 3 | Cl | 2c | 8 | 85% | 1 |
| 4 | t Bu | 2d | 2 | 77% | 1 |
| 5 | CF3 | 2e | 1.5 | 100% | 0 |
| 6 | COOBu | 2f | 1.5 | 100% | 0 |
| 7 | F | 2g | 2 | 100% | 0 |
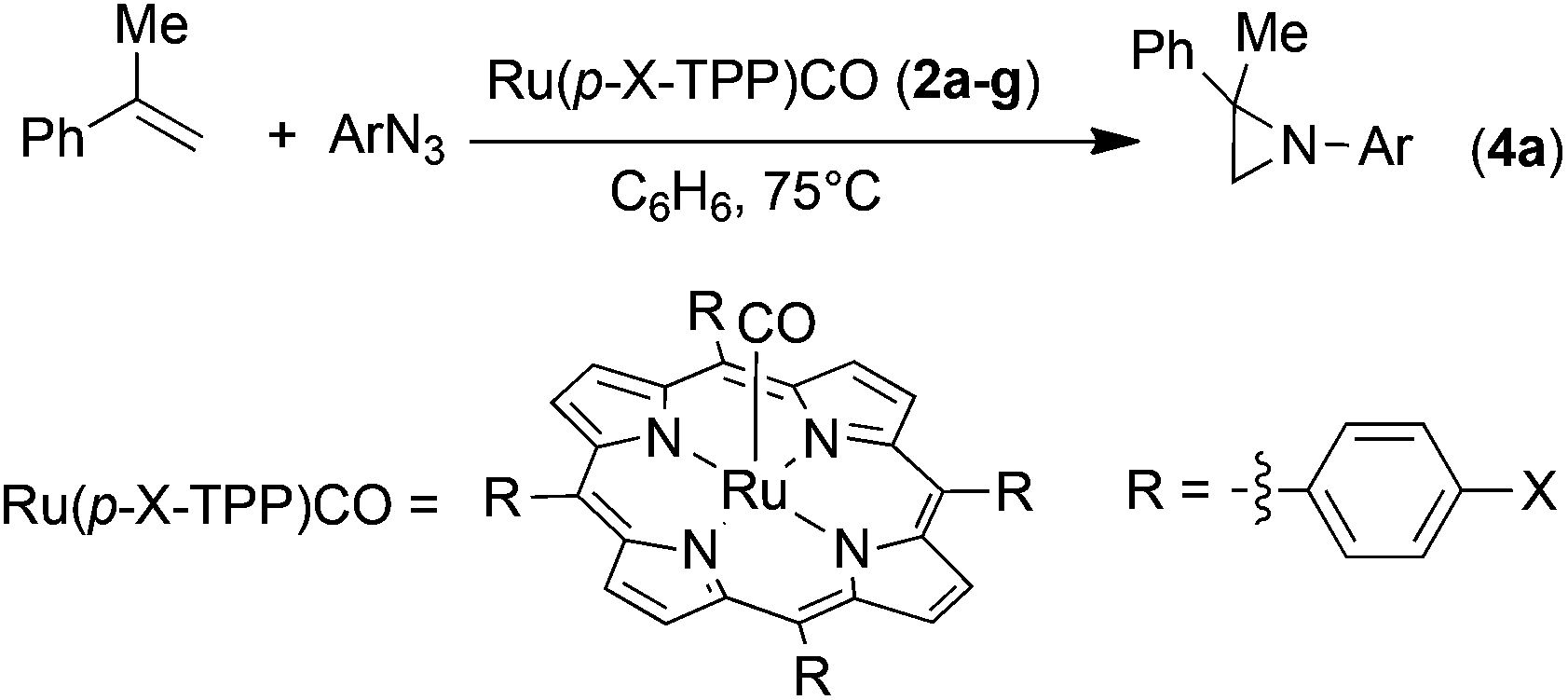
The discrepancy in the kinetic order can be explained by the dependence of the catalyst's coordination capability on the electronic properties of the porphyrin ligand. It should be noted that the dependence of the reaction kinetics on the electronic characteristics of aryl azides in the conversion of rhenium–azide adducts into corresponding imido derivatives has already been reported.52
The bond between the ruthenium(II) centre and the aryl azide is naturally weak53 and the equilibrium between the Ru(porphyrin)CO and Ru(porphyrin)(ArN3)(CO) complexes is the first step in influencing the rest of the olefin aziridination. Importantly, the equilibrium can be shifted more toward the azide coordination in the presence of a EWG on the porphyrin skeleton, which reduces the electron density on the metal centre and favours the coordination of the azide α-nitrogen atom. The last process is further facilitated by the presence of a strong aryl azide excess and as a consequence a zero kinetic order with respect to the ArN3 concentration was observed. On the other hand, the coordination capability of the metal decreases in the presence of an EDG on the ligand, disfavouring the initial anchorage of the azide. In this case, the left-shifted equilibrium can be responsible for the observed apparent first kinetic order with respect to the azide concentration.
It must be underlined that under the experimental conditions reported in Table 1 (low olefin concentration), the triazoline species does not form and hence it cannot act as an inhibiting agent.
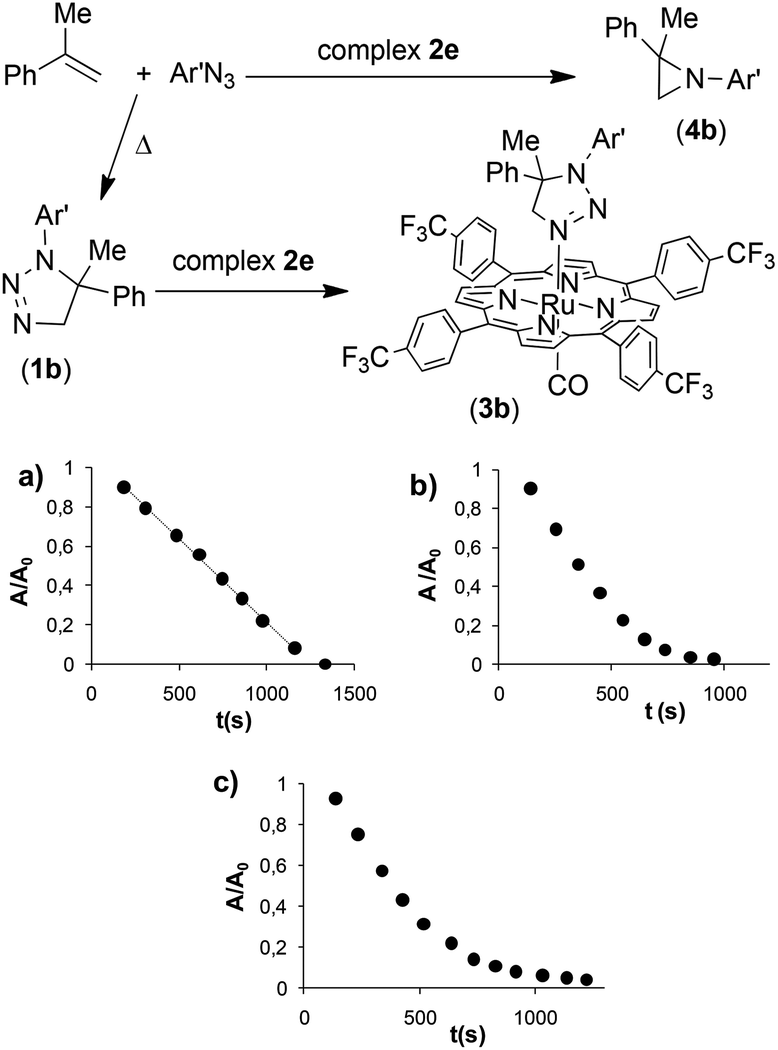
A second series of kinetic experiments was carried out by employing α-methylstyrene, as the olefin, 3,5-bis(trifluoromethyl)phenyl azide (Ar′N3) and Ru(p-CF3-TPP)CO (2e) as the catalyst, to ensure a favourable azide coordination with a consequent zero kinetic order in the azide itself. We investigated the rate constant and kinetic order in aryl azide upon increasing the α-methylstyrene concentration. Under the typical catalytic conditions (catalyst/azide/olefin = 1:50:250)31 the kinetic order with respect to aryl azide was zero (Fig. 1, graph a), while for higher α-methylstyrene concentrations (greater than or equal to 1.5 M, 20% v/v) the reaction started with a zero kinetic order and gradually converted into a first kinetic order with respect to the aryl azide concentration (Fig. 1, graph b). Finally, when the olefin amount was raised to 40% v/v (3.0 M) a clean first order dependence with respect to the aryl azide concentration was observed (Fig. 1, graph c). In addition, a first kinetic order in the catalyst 2e was observed (see the ESI†).
 | ||
| Fig. 1 Kinetic dependence on Ar′N3 using [α-methylstyrene] = 0.1 M (a), 2.1 M (b) and 3.0 M (c). | ||
A plot of the reaction rate (−Δ[Ar′N3]/Δt) vs. the olefin concentration revealed a dependence very similar to that observed in the reaction between α-methylstyrene and 4-nitrophenyl azide (ArN3).48 A first kinetic order in α-methylstyrene at low concentrations (up to 0.77 M) was followed by a drop in the reaction rate at high concentrations. The loss of linearity in the reaction rate vs. the olefin concentration dependence and the observation of a change in the aryl azide kinetic order from zero to mixed zero/first were almost coincident. Fig. 2 shows the dependence of the reaction rate (calculated at 90% conversion) with respect to the olefin concentration for the cases of 4-nitrophenyl azide (ArN3) (Fig. 2a) and 3,5-bis(trifluoromethyl)phenyl azide (Ar′N3) (Fig. 2b).
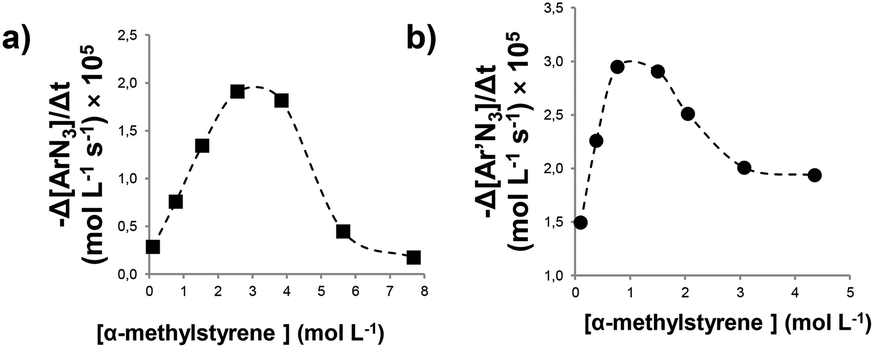 | ||
| Fig. 2 Dependence of the reaction rate with respect to α-methylstyrene concentration for the aziridination by (a) ArN3 catalysed by 2a and (b) Ar′N3 catalysed by 2e. | ||
Based on previous results, it is logical that by using 3,5-bis(trifluoromethyl)phenyl azide (Ar′N3) also at a high olefin concentration, the rate of the uncatalysed reaction becomes relevant. In fact, the triazoline species 1b is formed and its accumulation in the reaction medium leads to the formation of complex 3b, as witnessed by the presence of the typical methylene signal at −1.2 ppm of the triazoline complex in the NMR spectrum of the reaction crude product.
Complex 3b, which is structurally analogous to 3a, is responsible for the change from 0 to 1 of the kinetic order with respect to azide (Fig. 1b) when, at the end of the catalytic reaction, an appreciable amount of 1b is accumulated. Considering that 1b must release the coordination site on the metal to allow the azide anchorage, this substitution reaction seems to guide the full catalytic process in line with the registered first kinetic order with respect to the azide concentration (last part of the graphic reported in Fig. 1b). It should be noted that the coordination of triazolines to Ru(porphyrin)CO complexes is a general reaction, since other three complexes were isolated by reacting 1a with ruthenium(II) porphyrin species 2b, 2c and 2e (see the ESI†).
The comparison of the two graphics in Fig. 2 reveals that, when the catalyst Ru(p-CF3-TPP)CO (2e) is used in place of Ru(TPP)CO (2a), the inhibition process occurs at a lower α-methylstyrene concentration. This trend is in accord with the high coordination ability of ruthenium complexes with EWGs at the porphyrin ligand as stated above. For this reason, triazoline complex 3b is appreciably formed during the reaction at a lower triazoline concentration with respect to 3a. As a consequence, the inhibition of the process appears already relevant at an olefin concentration of about 1–2 M (Fig. 2).
Even if different aryl azides (ArN3 or Ar′N3) were employed in the ruthenium-catalysed reaction with α-methylstyrene, the rate of the uncatalysed reactions leading to 1a or 1b was unaffected. In fact, the rate was almost independent from the used aryl azide, as proven by the comparisons of the two kinetic constants measured in the absence of the catalyst (ratio ksynthesis of 1a/ksynthesis of 1b = 0.86). The 1,3 cycloaddition can be favoured by the presence of EWGs on both the azides employed (i.e. NO2 in ArN3 and CF3 in Ar′N3) (see DFT study for the dependence of the triazoline formation on electronic properties of reactants).
In addition, the coordination power of triazolines 1a and 1b was similar, as proved by an NMR experiment run by adding an equimolar amount of 1a to preformed triazoline complex 3b. The NMR analysis of the reaction mixture after three hours at 70 °C showed the presence of Ru(p-CF3-TPP)(1a)CO complex (3c) and the calculated 3c/3b ratio of 0.5 showed the coordination ability of both the triazolines 1a and 1b to Ru(p-CF3-TPP)CO (2e) (see the ESI†). This last experiment confirms that the coordination of an axial L ligand to the ruthenium(II) porphyrin is governed by the electronic features of the complex more than the nature of the incoming ligand L.
To confirm the inhibiting role of triazoline, complex 3b was synthesised to be used as the catalyst for the reaction between α-methylstyrene and 3,5-bis(trifluoromethyl)phenyl azide (Ar′N3). No zero kinetic order was observed with respect to the aryl azide concentration, as in the case of the reaction catalysed by 2e. The very slow reaction follows a clean first kinetic order with respect to azide at any substrate concentration (Fig. 3) confirming that the kinetics of the entire process is strongly conditioned by the presence of an axial ligand L such as triazoline.
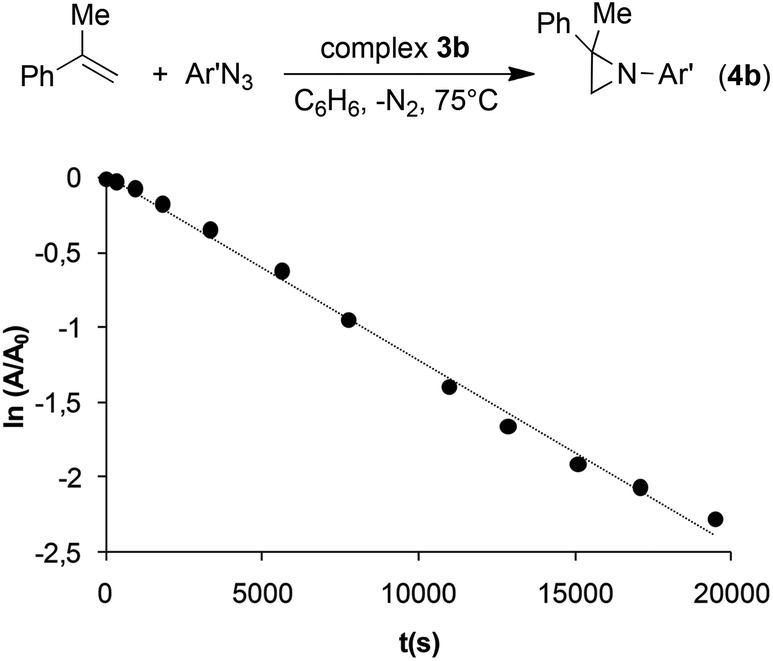 | ||
| Fig. 3 The Ar′N3 consumption by using catalyst 3b and [α-methylstyrene] = 0.1 M. | ||
It is worth noting that the same effect on the reaction rate was also observed by performing the catalytic reaction shown in Scheme 2, in the presence of “wet” Ru(TPP)CO. In fact, when catalyst 2a was immediately used after the chromatographic purification (without drying it for two hours at 120 °C under vacuum) the rate of azide conversion decreased by 70%. This effect is due to the presence of H2O on the ruthenium centre that inhibits the azide coordination.54
Based on data reported up to now, the catalytic cycle in Scheme 3 can be proposed, in which the coordination of the azide represents a crucial step.
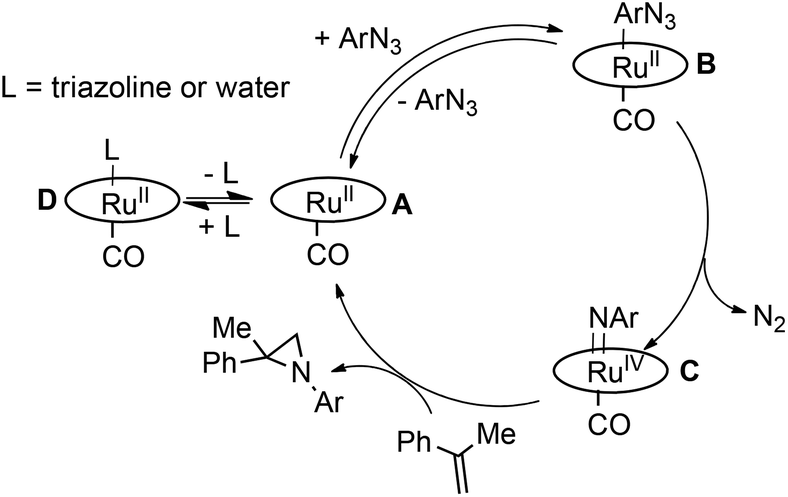 | ||
| Scheme 3 Suggested mechanism for the aziridination of α-methylstyrene by aryl azide. | ||
We propose that the reversible coordination of the azide is influenced by two factors, namely (i) the presence of competitive ligands (L) such as triazoline species or water and (ii) the electronic properties of the porphyrin skeleton. When the reaction is run in the absence of any coordinating ligand L (low α-methylstyrene concentration and anhydrous conditions) and with electron deficient catalysts, the equilibrium of the formation of species B is right-shifted and the reaction rate does not depend on the azide concentration (a zero kinetic order was registered). On the other hand, when either the triazoline is formed during the catalysis (high olefin concentration) or the coordination ability of the catalyst drops off due to the presence of EDGs on the porphyrin, the equilibrium of the formation of B is left-shifted, with this step becoming rate determining. As a consequence a first kinetic order with respect to the azide concentration was observed.
In the mechanistic proposal, the active role of the mono-imido ruthenium complex C is implied. Conversely, the presence of ruthenium bis-imido species, which are considered active catalytic intermediates in the amination of C–H bonds, can be excluded.53,55–58 This statement is supported by the analysis of the reaction crude. At the end of the catalytic reactions, Ru(porphyrin)CO complex was detected by IR spectroscopy (see the ESI†) and the presence of bis-imido ruthenium complexes was never revealed by NMR spectroscopy.
DFT study
The mechanism for the allylic amination of cyclohexene catalysed by complex [Ru](CO) (A) ([Ru] = Ru(porphine)) was proposed53 and it is generically summarised in Scheme 4.59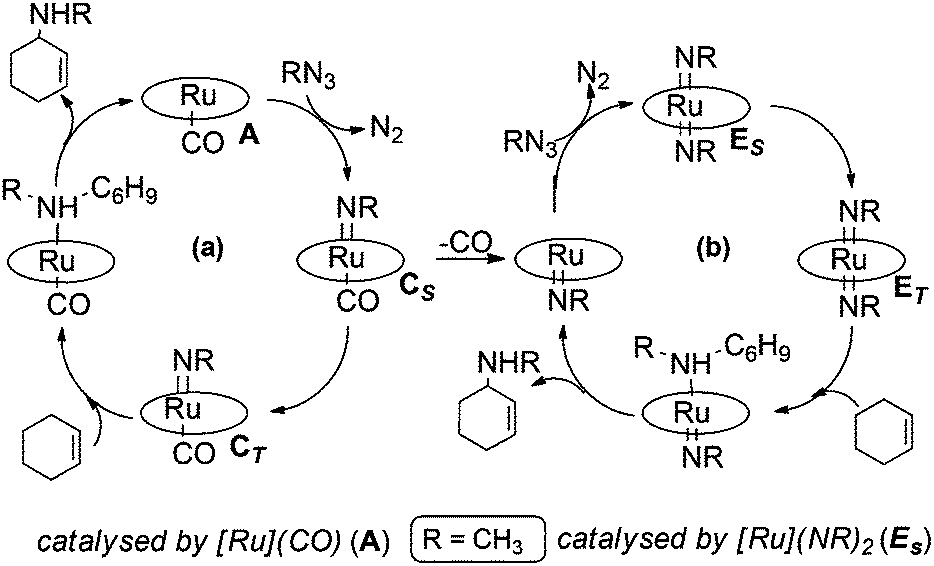 | ||
| Scheme 4 Overall mechanism of the [Ru](CO)-catalysed allylic amination of cyclohexene by RN3. | ||
After anchoring to the metal, the azide RN3 is activated with the eco-friendly N2 dismissal to form the mono-imido [Ru](NR)(CO) complex C which represents the key intermediate in any subsequent catalytic process. In particular, two alternative reactivity pathways are possible depending on whether a singlet ground state is maintained (CS) or an intersystem crossing to the triplet CT occurs. This high spin isomer is somewhat more stable (ΔG = −3.7 kcal mol−1) than the singlet one and it has a higher concentration of localised unpaired spin at the coordinated imido nitrogen atom than at the metal itself. A two step radical reactivity is promoted, as indicated in cycle (a) of Scheme 4. Formation and release of the desired allylic amine product occurs through a radical “rebound” mechanism.53,60,61 As mentioned, also the singlet isomer CS allows an alternative process, since the already weaker Ru–CO bond is consistent with a CO release, hence a second azide activation may occur at the vacated metal site (cycle (b) of Scheme 4). The resulting bis-imido complex [Ru](NR)2 (E) was experimentally and computationally proven to act as a catalyst upon another singlet (ES) → triplet (ET) spin crossing. In ET, the diradical character is initially shared by both the imido N atoms, but spin flipping can allow the rebound mechanism at only one NR ligand. After the departure of allylic amine, the regenerated five coordinated mono-imido singlet [Ru](NR) can activate another azide molecule to restart the catalytic cycle.
In conclusion, the cycles (a) and (b) of Scheme 4 are clearly interconnected and, based on experimental data, the preferred catalytic cycle depends on the azide/organic substrate concentration ratio.53
Our understanding of the allylic amination reaction is fundamental in interpreting the mechanism of the aziridination reaction, which occurs when the same [Ru](CO) (A) catalyst activates an azide molecule in the presence of an olefin. As in the cycle (a) of Scheme 4, the mono-imido species [Ru](NR)(CO) CS is formed but in this case the subsequent reactivity proceeds only from the CT spin isomer. In fact, IR spectra of the reaction crude clearly indicated that the CO ligand never detached from [Ru](CO) (A) which was always present at the end of the catalytic reaction while there was no trace of the bis-imido species (E). As before,53 our computations were carried out by using both the 3,5-bis(trifluoromethyl)phenyl azide (Ar′N3) and CH3N3 with consistent energy profiles, as indicated in Schemes 5 and S11.† In the following discussion, the results will refer to the azide Ar′N3 except for a specific case to be indicated. The initial part of Scheme 5 confirms the azide anchoring to the metal and the highest barrier to be bypassed (ΔG = +20.4 kcal mol−1) to reach the mono-imido CS and CT spin isomers (see the ESI† for the corresponding optimised structures) The latter diradical triggers the olefin's reactivity by easily approaching a triplet 5TS species with a barrier of only +4.3 kcal mol−1.
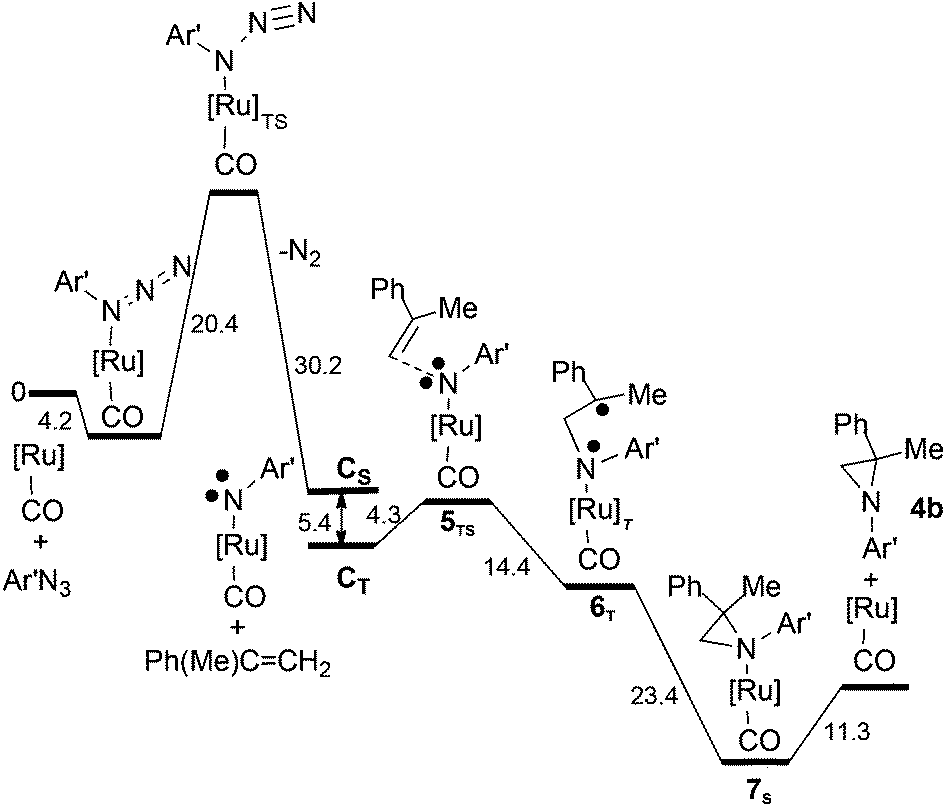 | ||
| Scheme 5 The overall energy profile for the aziridine 4b synthesis from Ar′N3 and α-methylstyrene, Ph(Me)CCH2. | ||
The 5TS structure in Fig. 4 indicates an incipient C⋯N bond (2.16 Å) between the terminal olefin C atom and the N imido one. Full bond formation (1.48 Å) is instead attained at the next triplet minimum [Ru](Ar′NCH2C(Me)Ph)(CO)T, 6T, which lies −14.4 kcal mol−1 deeper in free energy.
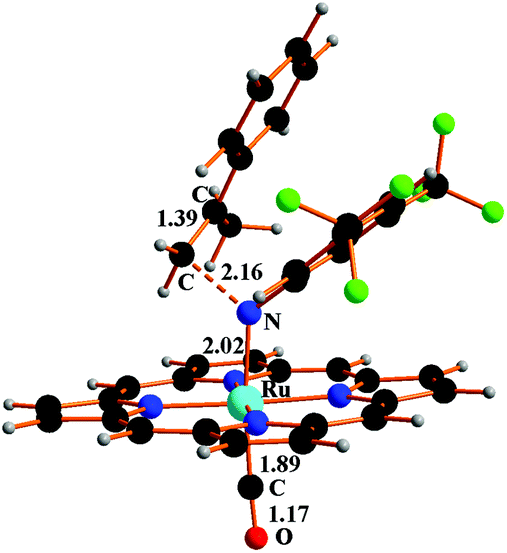 | ||
| Fig. 4 Structure of 5TS resulting from the incipient interaction between the triplet imido complex CT and α-methylstyrene Ph(Me)CCH2. | ||
In the structure of Fig. 5, the atoms with formal unpaired electrons are again the nitrogen coordinated to the metal and the second olefin carbon atom. The separation is relatively small (2.50 Å), but coupling is not featured, because the tubular spin density shapes (also shown in Fig. 5) are almost orthogonally oriented. The integrated values are +0.75 and +0.71 e2 bohr−3, respectively, while the spin density at the metal is only residual (+0.19 e2 bohr−3).
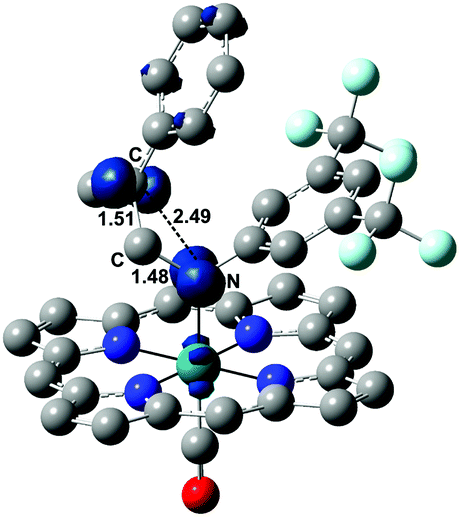 | ||
| Fig. 5 Structure of 6T with the associated spin density shapes. | ||
If the given stereochemistry of 6T momentarily prevents a prompt electron coupling, some combined torsions about the N–C and C–C single bonds of the pending Ru–N–C–C chain may easily trigger electron pairing and the ring's closure. The corresponding C–N distance is optimised to be 1.54 Å in the singlet aziridine complex [Ru](4b)(CO) (7S) (see Fig. 6), whose formation is largely exergonic (−23.4 kcal mol−1).
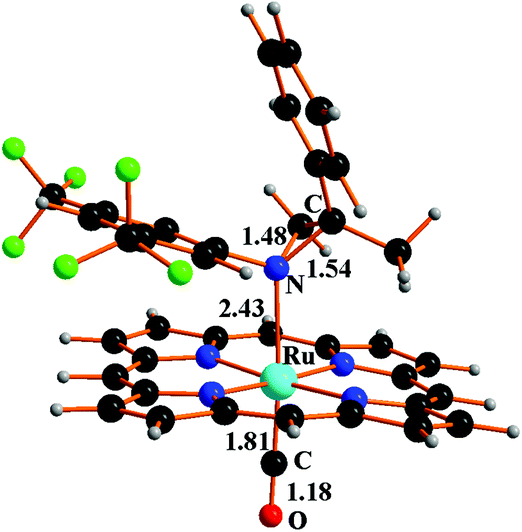 | ||
| Fig. 6 Structure of compound [Ru](4b)(CO) 7S. | ||
Similarly to the allylic amine ruthenium complex formed in cycle (b) of Scheme 4,53 the N-coordinated aziridine ligand trans to the CO one is weakly bound, the Ru–Naziridine distance being 2.43 Å in 7S. Nonetheless, there is an additional cost of +11.3 kcal mol−1 for the aziridine separation (last step in Scheme 5). It has been already remarked how the anchoring of a given N ligand to the [Ru](CO) unit is largely attributable to dispersion forces which extend to the whole TPP moiety.53 Whatever can be the nature of the attraction, the separation of the catalytic product (amine or aziridine) cannot represent a major problem under the used experimental conditions and, in particular, the ∼80 °C reaction temperature.
Since a major aspect the catalytic processes promoted by [Ru](CO) involves an intersystem crossing either at the CS → CT interconversion or the aziridine ring closure, attempts have been made to evaluate the Minimum Energy Crossing Point (MECP)62,63 by calculations. The CS → CT process, which is of importance also in the catalytic aziridination (Scheme 5) was previously analysed in detail.53 New attempts were then made to establish the MECP for spin quenching during the transformation 6T → 7S. For simplicity, calculations were performed by using the corresponding models 6T′ and 7S′ derived from the analogous methyl azide and isobutene reactants (i.e., CH3N3 and H2CC(CH3)2). Scans on both singlet and triplet Potential Energy Surfaces (PES) allowed monitoring the energy variations as a function of the C–N key distance. The results are presented in Fig. 7. The triplet has its minimum at 2.56 Å, the separation being slightly larger with respect to the optimised value of the diradical structure 6T (Fig. 5). The triplet curve crosses the singlet one near the minimum, with a pronounced trend of the species to close the aziridine molecule and transform 6T′ into 7S′. This occurs thanks to other stereochemical rearrangements, in particular the mentioned torsion about the two linkages of the N–C–C chain. It may be concluded that the MECP for the intersystem crossing is null in this case. A similar highly favoured ring-closure through an intermolecular rebound mechanism has already been reported.44,45
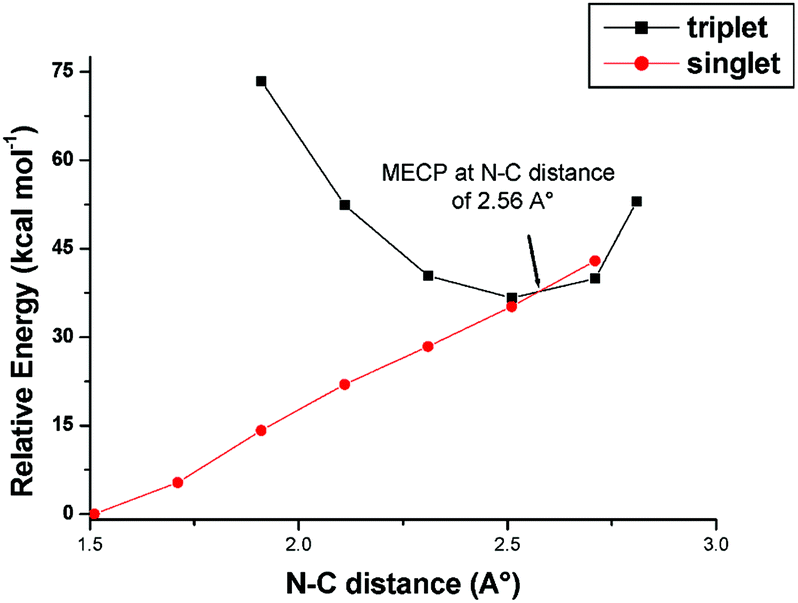 | ||
| Fig. 7 Relaxed scan plots over triplet and singlet potential energy surface (electronic energy) for the models 6T′ and 7S′, derived from methyl azide and isobutene reactants. | ||
As reported in the Experimental part, aziridine is not the only possible reaction product, since the triazoline 1b may also form from the 1,3-cycloaddition reaction of Ar′N3 with α-methylstyrene. The kinetic studies indicated that, under the catalytic conditions, the increase of the 1b yield is related to the presence of high olefin concentrations. To find out how competitive in energy the two processes can be, the energy profile for triazoline 1b generation was compared with that of the aziridination process (Scheme 6vs. Scheme 5).
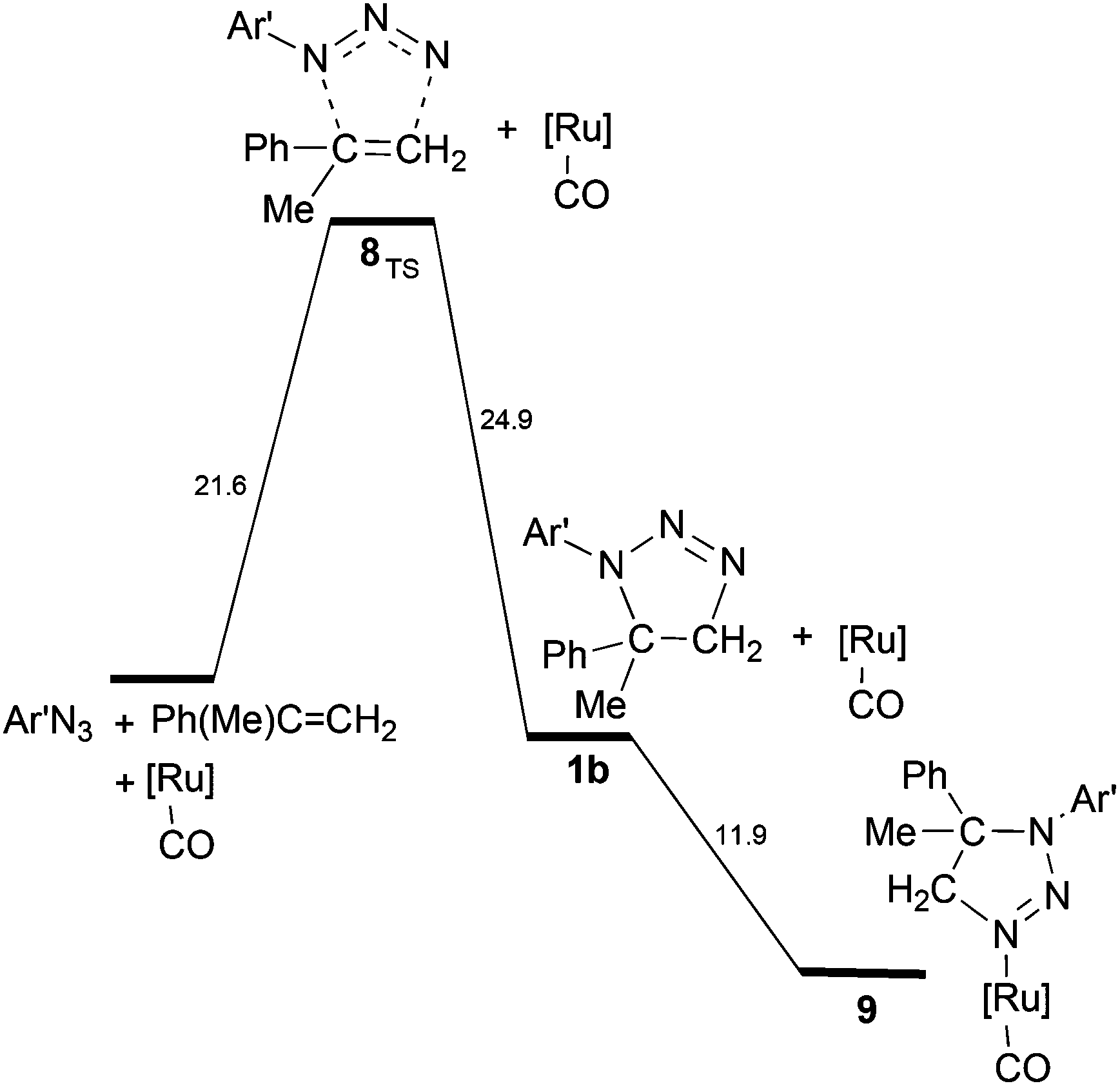 | ||
| Scheme 6 Energy profile for the formation of triazoline 1b. The eventual coordination of 1b to [Ru](CO) is exergonic. | ||
The metal unassisted triazoline 1b formation proceeds in the singlet PES through the transition state 8TS along a 1,3-cycloaddition pathway (Fig. 8).
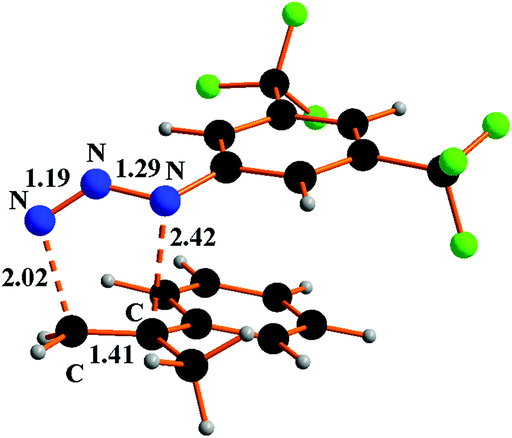 | ||
| Fig. 8 Optimised structure of 8TS species in the 1,3-cycloaddition between Ar′N3 and α-methylstyrene. | ||
The corresponding barrier of +21.6 kcal mol−1 is quantitatively similar to that of the first encountered TS in the metal promoted activation of the azide (+20.4 kcal mol−1 in Scheme 5). From the thermodynamic viewpoint, the two alternative processes are comparably difficult at least in the initial parts, none being potentially hindered under the high working temperature. The structure of 8TS indicates that the two C⋯N linkages do not form at equal speed, the one between the CH2 group and the terminal azide Nγ atom being definitely more robust (2.02 Å) than the one between the second olefin carbon atom and the Nα atom of Ar′N3 (2.42 Å). This TS feature corroborates a significant stereoselectivity of the 1,3-cycloaddition, which does not emerge from the structure of the final triazoline 1b (Fig. S17a†) which presents two identical C–N linkages of 1.49 Å. The stereoselectivity is further corroborated by the computed pathway leading to the alternative triazoline isomer 1b′ with inverted C–N linkages (Fig. S17b†). The latter isomer is insignificantly favoured (−0.8 kcal mol−1) but the barrier at 8′TS (Fig. S18†) is +5 kcal mol−1 higher. Possibly, the difference is attributable to a combination of less favourable steric interactions, which also determines the stereoselectivity.
Potentially, the triazoline formation can reduce the catalytic efficiency of the system for two reasons: (i) it subtracts starting materials from the aziridination reaction and (ii) inhibits the catalytic reaction by coordinating ruthenium of the unsaturated [Ru](CO) catalyst giving the complex [Ru](1b)(CO) (9), shown in Fig. 9, which corresponds to the last step in Scheme 6 and to the species D in Scheme 3.
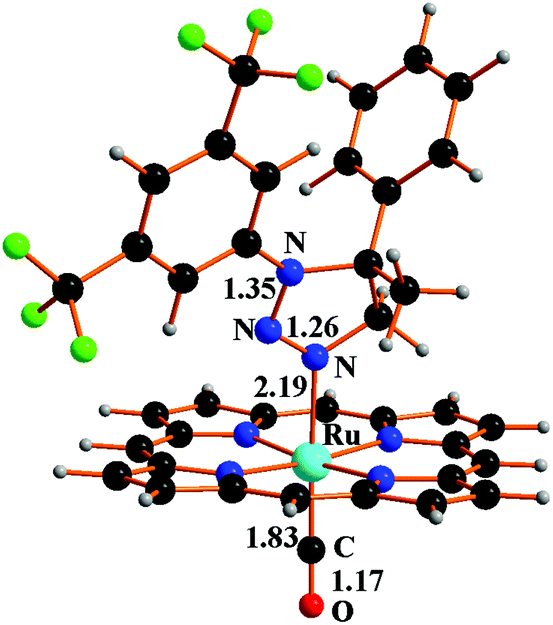 | ||
| Fig. 9 Optimised structure of complex 9. | ||
The stabilisation energy of −11.9 kcal mol−1 of complex 9 formation compares with that of the aziridine complex 7S (−11.3 kcal mol−1 in Scheme 5). Consequently, 9 does not in principle deactivate metal catalysis but it reduces its efficiency, as suggested by kinetic studies.
In the presence of [Ru](CO) the free aziridine molecules are likely favoured over that of triazoline, possibly because the overall energy balance is more exergonic by −15.2 kcal mol−1. Considering that the two processes have initial comparable barriers, the triazoline formation is thermodynamically possible, whereas the kinetic studies indicate that the alternative reactivities depend on the olefin concentration. When the latter is low (as under the catalytic conditions of choice), the olefin aziridination is essentially a unique process. Conversely, when the olefin concentration increases, the 1,3-cycloaddition yielding triazoline takes place and at least in part it disfavours the aziridination process.
Conclusion
The mechanism of the aziridination of olefins promoted by ruthenium(II) porphyrin complexes has been critically analysed from both experimental and computational viewpoints. The catalytic reaction is efficiently performed by using aryl azides as nitrogen sources with the eco-friendly N2 elimination. Experimental data, kinetic measurements and theoretical calculations all underline the active role played by a ruthenium mono-imido intermediate in the aziridination process.The mechanistic proposal of this paper is similar, at least in the early stages, to that reported for the amination of allylic C–H bonds. In fact, the aryl azide activation over the [Ru](CO) catalyst is common to these processes and yields a diradical mono-imido [Ru](NR)(CO) metal species. In the catalytic allylic amination, this species activates the allylic C–H bond allowing access to a rebound mechanism, which ultimately leads to the amine product.53 In the presence of an olefin rather than an allylic substrate, the same diradical favours the catalytic aziridination which uniquely restores the ruthenium mono-imido complex and the bis-imido derivatives, active intermediates in the allylic amination, were never involved in the catalytic reaction. As a matter of fact, the mono-imido diradical transfers to the distal olefin carbon atom one of the nitrogen unpaired electrons. A metastable and diradical N–C–C open chain is first attained, which, through minor stereochemical rearrangements, undergoes spin coupling with the consequent aziridine ring closure.
In addition to the above findings, the catalytic role of an axial ligand L was elucidated by a kinetic study (in part corroborated by the calculated energies). Ligand L (either triazoline or water) seems to interfere with the catalytic reaction mechanism and introduces an additional step into the cycle (Scheme 3, D → A transformation). In fact, the relatively stable six-coordinated ruthenium(II) complex can temporarily prevent another azide coordination and its subsequent activation. Considering that the rate of the triazoline formation depends on the olefin concentration, this last parameter must be taken into great consideration in order to enhance the reaction selectivity in aziridination.
Experimental
General conditions
Unless otherwise specified, all reactions were carried out under a nitrogen atmosphere employing standard Schlenk techniques and vacuum-line manipulations.Solvents and reagents
Benzene was dried by M. Braun SPS-800 solvent purification system whilst α-methylstyrene was purified by distillation under nitrogen over sodium. Aryl azides,64,65para-substituted-meso-tetraphenylporphyrin66 and their ruthenium carbonyl complexes31,54,67 were synthesised by methods reported in the literature. All the other starting materials are commercial products and were used as received. The synthesis and characterisation of aziridines (4a and 4b),31 triazoline 1a48 and complex 3a48 was performed as reported.
Instruments
Infrared spectra were recorded on a Varian Scimitar FTS 1000 spectrophotometer using Perkin-Elmer CaF2 cells of 0.5 mm or 0.1 mm thickness. NMR spectra were recorded at 300 K on Bruker Avance 300-DRX, operating at 300 MHz for 1H, at 75 MHz for 13C and at 282 MHz for 19F; or on a Bruker Avance 400-DRX spectrometer, operating at 400 MHz for 1H, 100 MHz for 13C and at 376 MHz for 19F. Chemical shifts (ppm) are reported relative to TMS. The 1H NMR signals of the compounds described in the following have been attributed by COSY and NOESY techniques. Assignments of resonances in 13C NMR were made by using the APT pulse sequence and HSQC and HMBC techniques.Computational details
All the calculations were carried out with the Gaussian 09 package68 at the B97D-DFT69 level of theory. All the optimised structures were validated as minima and/or transition states by computed vibrational frequencies. All the calculations were based on the CPCM70,71 model for the benzene solvent, the same was used in the experiments. The effective Stuttgart/Dresden core potential (SDD)72 was adopted for the ruthenium centre, while for all the other atomic species the basis set was 6-31G, with the addition of the polarization functions (d,p). The coordinates of all the optimised structures are reported in the ESI.†:1 mixture of α-methylstyrene and benzene (14.0 mL) and refluxed for 16 hours until the complete consumption of the aryl azide. The solution was evaporated to dryness to give a pure orange oil (220 mg, 99%). 1H NMR (300 MHz, CDCl3): δ 7.50–7.20 (8H, m, HAr), 4.71 (1H, d, J = 17.4 Hz, CHH), 4.50 (1H, d, J = 17.4 Hz, CHH), 1.77 (3H, s, CH3). 19F NMR (282 MHz, CDCl3): δ −63.66 (CF3). 13C NMR (75 MHz, CDCl3) δ 142.1 (C), 140.9 (C), 132.6 (q, J = 33.4 Hz, C-CF3), 129.7 (CHAr), 128.5 (CHAr), 125.5 (CHAr), 123.4 (q, J = 272.9 Hz, CF3), 115.5 (m, CHAr), 115.2 (q, J = 3.6 Hz, CHAr), 85.2 (CH2), 63.2 (C), 22.0 (CH3).
Kinetic measurements
N) signal at 2150–2100 cm−1. Rate constants with respect to the aryl azide concentration were determined from the specific variation of A with respect to time.
Acknowledgements
We thank Prof Fabio Ragaini for his valuable scientific suggestions.C. M. and G. M. acknowledge the ISCRA-CINECA HP grant “HP10BEG2NO” and CREA (Centro Ricerche Energia e Ambiente) of Colle Val d'Elsa (Siena, Italy) for computational resources.
Notes and references
- J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247–258 RSC.
- X. E. Hu, Tetrahedron, 2004, 60, 2701–2743 CrossRef CAS PubMed.
- J. B. Sweeney and A. Yudin, Aziridines and Epoxides in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2006 Search PubMed.
- H. Ohno, Chem. Rev., 2014, 114, 7784–7814 CrossRef CAS PubMed.
- G. S. Singh, M. D'Hooghe and N. De Kimpe, Chem. Rev., 2007, 107, 2080–2135 CrossRef CAS PubMed.
- P. A. S. Lowden, in Aziridines and Epoxides in Organic Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 399–442 Search PubMed.
- S. Ballereau, N. Andrieu-Abadie, N. Saffon and Y. Génisson, Tetrahedron, 2011, 67, 2570–2578 CrossRef CAS PubMed.
- T. Tsuchida, H. Iinuma, N. Kinoshita, T. Ikeda, T. Sawa, M. Hamada and T. Takeuchi, J. Antibiot., 1995, 48, 217–221 CrossRef CAS.
- F. M. D. Ismail, D. O. Levitsky and V. M. Dembitsky, Eur. J. Med. Chem., 2009, 44, 3373–3387 CrossRef CAS PubMed.
- T. Tsuchida, R. Sawa, Y. Takahashi, H. Iinuma, T. Sawa, H. Naganawa and T. Takeuchi, J. Antibiot., 1995, 48, 1148–1152 CrossRef CAS.
- H. Pellissier, Adv. Synth. Catal., 2014, 356, 1899–1935 CrossRef CAS PubMed.
- L. Degennaro, P. Trinchera and R. Luisi, Chem. Rev., 2014, 114, 7881–7929 CrossRef CAS PubMed.
- H.-Y. Lee, S.-B. Song, T. Kang, J. Kim Yoon and J. Geum Su, Pure Appl. Chem., 2013, 85, 741–753 CrossRef CAS.
- J. L. Jat, M. P. Paudyal, H. Gao, Q.-L. Xu, M. Yousufuddin, D. Devarajan, D. H. Ess, L. Kürti and J. R. Falck, Science, 2014, 343, 61–65 CrossRef CAS PubMed.
- P. Lu, Tetrahedron, 2010, 66, 2549–2560 CrossRef CAS PubMed.
- R. Chawla, A. K. Singh and L. D. S. Yadav, RSC Adv., 2013, 3, 11385–11403 RSC.
- S. Fantauzzi, E. Gallo, A. Caselli, C. Piangiolino, F. Ragaini, N. Re and S. Cenini, Chem. – Eur. J., 2009, 15, 1241–1251 CrossRef CAS PubMed.
- H. Jiang and X. P. Zhang, in Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, pp. 168–182 Search PubMed.
- S. Fantauzzi, A. Caselli and E. Gallo, Dalton Trans., 2009, 5434–5443 RSC.
- J. W. W. Chang, T. M. U. Ton and P. W. H. Chan, Chem. Rec., 2011, 11, 331–357 CrossRef CAS PubMed.
- D. M. Jenkins, Synlett, 2012, 1267–1270 CrossRef CAS PubMed.
- S. Braese, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- D. Intrieri, P. Zardi, A. Caselli and E. Gallo, Chem. Commun., 2014, 50, 11440–11453 CAS.
- T. Uchida and T. Katsuki, Chem. Rec., 2014, 14, 117–129 CrossRef CAS PubMed.
- T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831–3846 CAS.
- S. A. Cramer and D. M. Jenkins, J. Am. Chem. Soc., 2011, 133, 19342–19345 CrossRef CAS PubMed.
- S. Cenini, F. Ragaini, E. Gallo and A. Caselli, Curr. Org. Chem., 2011, 15, 1578–1592 CrossRef CAS.
- S. Cenini, E. Gallo, A. Caselli, F. Ragaini, S. Fantauzzi and C. Piangiolino, Coord. Chem. Rev., 2006, 250, 1234–1253 CrossRef CAS PubMed.
- J. E. Jones, J. V. Ruppel, G.-Y. Gao, T. M. Moore and X. P. Zhang, J. Org. Chem., 2008, 73, 7260–7265 CrossRef CAS PubMed.
- G.-Y. Gao, J. E. Jones, R. Vyas, J. D. Harden and X. P. Zhang, J. Org. Chem., 2006, 71, 6655–6658 CrossRef CAS PubMed.
- S. Fantauzzi, E. Gallo, A. Caselli, C. Piangiolino, F. Ragaini and S. Cenini, Eur. J. Org. Chem., 2007, 6053–6059 CrossRef CAS PubMed.
- A. Caselli, E. Gallo, S. Fantauzzi, S. Morlacchi, F. Ragaini and S. Cenini, Eur. J. Inorg. Chem., 2008, 3009–3019 CrossRef CAS PubMed.
- S. Cenini, S. Tollari, A. Penoni and C. Cereda, J. Mol. Catal. A: Chem., 1999, 137, 135–146 CrossRef CAS.
- Y. Liu and C.-M. Che, Chem. – Eur. J., 2010, 16, 10494–10501 CrossRef CAS PubMed.
- T. W.-S. Chow, G.-Q. Chen, Y. Liu, C.-Y. Zhou and C.-M. Che, Pure Appl. Chem., 2012, 84, 1685–1704 CrossRef CAS.
- Y. Fukunaga, T. Uchida, Y. Ito, K. Matsumoto and T. Katsuki, Org. Lett., 2012, 14, 4658–4661 CrossRef CAS PubMed.
- C. Kim, T. Uchida and T. Katsuki, Chem. Commun., 2012, 48, 7188–7190 RSC.
- H. Kawabata, K. Omura, T. Uchida and T. Katsuki, Chem. – Asian J., 2007, 2, 248–256 CrossRef CAS PubMed.
- L.-M. Jin, X. Xu, H. Lu, X. Cui, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2013, 52, 5309–5313 CrossRef CAS PubMed.
- J. V. Ruppel, J. E. Jones, C. A. Huff, R. M. Kamble, Y. Chen and X. P. Zhang, Org. Lett., 2008, 10, 1995–1998 CrossRef CAS PubMed.
- M. J. Zdilla and M. M. Abu-Omar, J. Am. Chem. Soc., 2006, 128, 16971–16979 CrossRef CAS PubMed.
- M. M. Montero-Campillo and M. N. D. S. Cordeiro, Int. J. Quantum Chem., 2013, 113, 2002–2011 CrossRef CAS PubMed.
- A. Sze-Man, J. S. Huang, W. Y. Yu, W. H. Fung and C. M. Che, J. Am. Chem. Soc., 1999, 121, 9120 CrossRef.
- K. H. Hopmann and A. Ghosh, ACS Catal., 2011, 1, 597–600 CrossRef CAS.
- A. I. O. Suarez, H. Jiang, X. P. Zhang and B. de Bruin, Dalton Trans., 2011, 40, 5697–5705 RSC.
- S. Cenini, S. Tollari, A. Penoni and C. Cereda, J. Mol. Catal. A: Chem., 1999, 137, 135–146 CrossRef CAS.
- C. Piangiolino, E. Gallo, A. Caselli, S. Fantauzzi, F. Ragaini and S. Cenini, Eur. J. Org. Chem., 2007, 743–750 CrossRef CAS PubMed.
- S. Fantauzzi, E. Gallo, A. Caselli, F. Ragaini, P. Macchi, N. Casati and S. Cenini, Organometallics, 2005, 24, 4710–4713 CrossRef CAS.
- A. Caselli, E. Gallo, F. Ragaini, F. Ricatto, G. Abbiati and S. Cenini, Inorg. Chim. Acta, 2006, 359, 2924–2932 CrossRef CAS PubMed.
- E. Gallo, M. G. Buonomenna, L. Vigano, F. Ragaini, A. Caselli, S. Fantauzzi, S. Cenini and E. Drioli, J. Mol. Catal. A: Chem., 2008, 282, 85–91 CrossRef CAS PubMed.
- C. Piangiolino, E. Gallo, A. Caselli, S. Fantauzzi, F. Ragaini and S. Cenini, Eur. J. Org. Chem., 2007, 743 CrossRef CAS PubMed.
- N. E. Travia, Z. Xu, J. M. Keith, E. A. Ison, P. E. Fanwick, M. B. Hall and M. M. Abu-Omar, Inorg. Chem., 2011, 50, 10505–10514 CrossRef CAS PubMed.
- G. Manca, E. Gallo, D. Intrieri and C. Mealli, ACS Catal., 2014, 4, 823–832 CrossRef CAS.
- E. Gallo, A. Caselli, F. Ragaini, S. Fantauzzi, N. Masciocchi, A. Sironi and S. Cenini, Inorg. Chem., 2005, 44, 2039–2049 CrossRef CAS PubMed.
- S. Fantauzzi, E. Gallo, A. Caselli, F. Ragaini, N. Casati, P. Macchi and S. Cenini, Chem. Commun., 2009, 3952–3954 RSC.
- D. Intrieri, A. Caselli, F. Ragaini, P. Macchi, N. Casati and E. Gallo, Eur. J. Inorg. Chem., 2012, 569–580 CrossRef CAS PubMed.
- D. Intrieri, A. Caselli, F. Ragaini, S. Cenini and E. Gallo, J. Porphyrins Phthalocyanines, 2010, 14, 732–740 CrossRef CAS.
- S. K.-Y. Leung, W.-M. Tsui, J.-S. Huang, C.-M. Che, J.-L. Liang and N. Zhu, J. Am. Chem. Soc., 2005, 127, 16629–16640 CrossRef CAS PubMed.
- For the sake of simplicity, the computed allylic amination reaction was illustrated for CH3N3, although 3,5-bis(trifluoromethyl)phenyl azide (Ar′N3) was experimentally employed. Such a reactant, also computationally evaluated, corresponded to a key barrier about 25% lower, but its energy profile was quite consistent in view of the functionality of the same azide. On the other hand, Ar′N3 has a milder behaviour over the metal centre, since it has more difficulty in being transformed into diazene Ar′NNAr′. This point emerges from comparing the energies for the reaction 2N3R → 2N2 + RNNR, which are as different as −105.2 and −71.4 kcal mol−1 for R = CH3 and R = Ar′, respectively. For all these reasons, the present study of the aziridination mechanism generally refers to Ar′N3 as the reactant of choice.
- J. T. Groves, J. Chem. Educ., 1985, 62, 928 CrossRef CAS.
- D. Balcells, C. Raynaud, R. H. Crabtree and O. Eisenstein, Chem. Commun., 2009, 1772–1774 RSC.
- J. N. Harvey, R. Poli and K. M. Smith, Coord. Chem. Rev., 2003, 238–239, 347–361 CrossRef CAS.
- J. N. Harvey and M. Aschi, Faraday Discuss., 2003, 124, 129–143 RSC.
- M. Tanno, S. Sueyoshi and S. Kamiya, Chem. Pharm. Bull., 1982, 30, 3125–3132 CrossRef CAS.
- S. Wiese, M. J. B. Aguila, E. Kogut and T. H. Warren, Organometallics, 2013, 32, 2300–2308 CrossRef CAS.
- A. D. Adler, F. R. Longo, J. D. Finarelli, J. Goldmacher, J. Assour and L. Korsakoff, J. Org. Chem., 1967, 32, 476 CrossRef CAS.
- D. P. Rillema, J. K. Nagle, L. F. Barringer and T. J. Meyer, J. Am. Chem. Soc., 1981, 103, 56–62 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision B.01), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- S. Grimme, J. Chem. Phys., 2006, 3952–3954 Search PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- M. Dolg, H. Stoll, H. Preuss and R. M. Pitzer, J. Phys. Chem., 1993, 97, 5852–5859 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt00951k |
| ‡ Present address: School of Pharmacy and Biomolecular Sciences, Liverpool John Moores University, James Parsons Building, Byrom Street, Liverpool, L3 3AF, UK. |
| This journal is © The Royal Society of Chemistry 2015 |