DOI:
10.1039/C5DT00947B
(Perspective)
Dalton Trans., 2015,
44, 16195-16211
An ethane-bridged porphyrin dimer as a model of di-heme proteins: inorganic and bioinorganic perspectives and consequences of heme–heme interactions†
Received
9th March 2015
, Accepted 2nd June 2015
First published on 4th June 2015
Abstract
Interaction between heme centers has been cleverly implemented by Nature in order to regulate different properties of multiheme cytochromes, thereby allowing them to perform a wide variety of functions. Our broad interest lies in unmasking the roles played by heme–heme interactions in modulating different properties viz., metal spin state, redox potential etc., of the individual heme centers using an ethane-bridged porphyrin dimer as a synthetic model of dihemes. The large differences in the structure and properties of the diheme complexes, as compared to the monoheme analogs, provide unequivocal evidence of the role played by heme–heme interactions in the dihemes. This Perspective provides a brief account of our recent efforts to explore these interesting aspects and the subsequent outcomes.
 Debangsu Sil | Debangsu Sil earned his bachelor's degree in chemistry from the University of Burdwan in 2007 and master's degree from the Indian Institute of Technology, Kanpur in 2009. He is currently pursuing his PhD, studying diiron and dimanganese bisporphyrins as models for diheme cytochromes, in Prof. S. P. Rath's laboratory at the Indian Institute of Technology, Kanpur. |
 Sankar Prasad Rath | Sankar Prasad Rath received his bachelor and master degrees from the Calcutta University in 1992 and 1994, respectively, and obtained his PhD with Prof. A. Chakravorty in 1999 from the Indian Association for the Cultivation of Science (IACS), Kolkata. After a postdoctoral study with Prof. A. L. Balch at the University of California, Davis, he joined the Department of Chemistry at the Indian Institute of Technology Kanpur in December 2004 where he is a full professor since 2014. He is a recipient of P. K. Kelkar Research Fellowship for young faculty (2009–12), Alexander von Humboldt Fellowship for Experienced Researcher (2012) and Chemical Research Society of India (CRSI) Bronze medal (2014). His current research interests are bioinorganic modeling of multi-heme proteins/enzymes, binding and activation of small molecules, electron and energy transfer, and probing molecular chirality using Exciton Coupled Circular Dichroism. |
1. Introduction
1.1 Multiheme cytochromes and enzymes
The family of the multiheme cytochrome c represents an extensive class of proteins with vital roles in electron transfer and enzymatic catalysis.1–3 The majority of the multiheme cytochromes and enzymes that are known today belong to the family of cytochrome c in which the heme unit is covalently connected to the polypeptide chain via formation of two thioether bonds between two cysteine residues and the vinyl side-chains of the porphyrin ring. The two cysteine residues form a typical amino acid sequence motif CXXCH, which indicates heme c ligation. Some cytochromes have three or four residues between the two cysteines.1,2 Although the first c-type cytochromes were found in aerobic or facultative anaerobic organisms, these proteins are present in anaerobes in an appreciable diversity.4a,b The first cytochromes that were identified, consisted of fewer heme co-factors but a survey of the currently available genomic sequences disclosed that the number of such co-factors per multiheme cytochrome c varies significantly. Two, three, and four are the most common numbers, followed by sequences containing five, eight or ten binding motifs.1–3 The protein structure with the maximum number of hemes per polypeptide is a hexadecaheme cytochrome,4c however, genes coding for polypeptide chains containing 43 and 45 heme binding motifs have been identified in Geobacter uraniumreducens and Anaeromyxobacter dehalogenans, respectively.2b Almost all major groups of bacteria and archaea have multiheme cytochrome c. In some of these organisms, such as representatives of Geobacter, Shewanella, Anaeromyxobacter or Desulfovibrio genera, multiheme cytochromes are present in very large numbers that correspond to a high percentage of their proteome, leading to the creation of the term ‘cytochromome’.1,2
The multiheme c cytochromes and enzymes form families that are recognizably different from the monoheme c cytochromes.1–3 They are characterized by a low amino acid residue to heme ratio and bis-histidine coordination, with few exceptions. The relative heme arrangements in these multiheme cytochromes are critical for their functional properties and each redox center can be tuned to be active at a different reduction potential thereby allowing a larger range of redox activity. In contrast, proteins with a single redox center have a range of redox activity restricted by the Nernst curve. Various factors are, however, there to influence the reduction potentials of hemes thereby allowing them to be tuned over a wide range. The relevant factors are: the heme environment (namely charges of amino acids, dipoles and exposure to solvents), the nature and relative orientations of the axial ligands, distortions of the porphyrin macrocycle, thermodynamic proton coupling (redox-Bohr effect), and heme–heme interactions.1–4
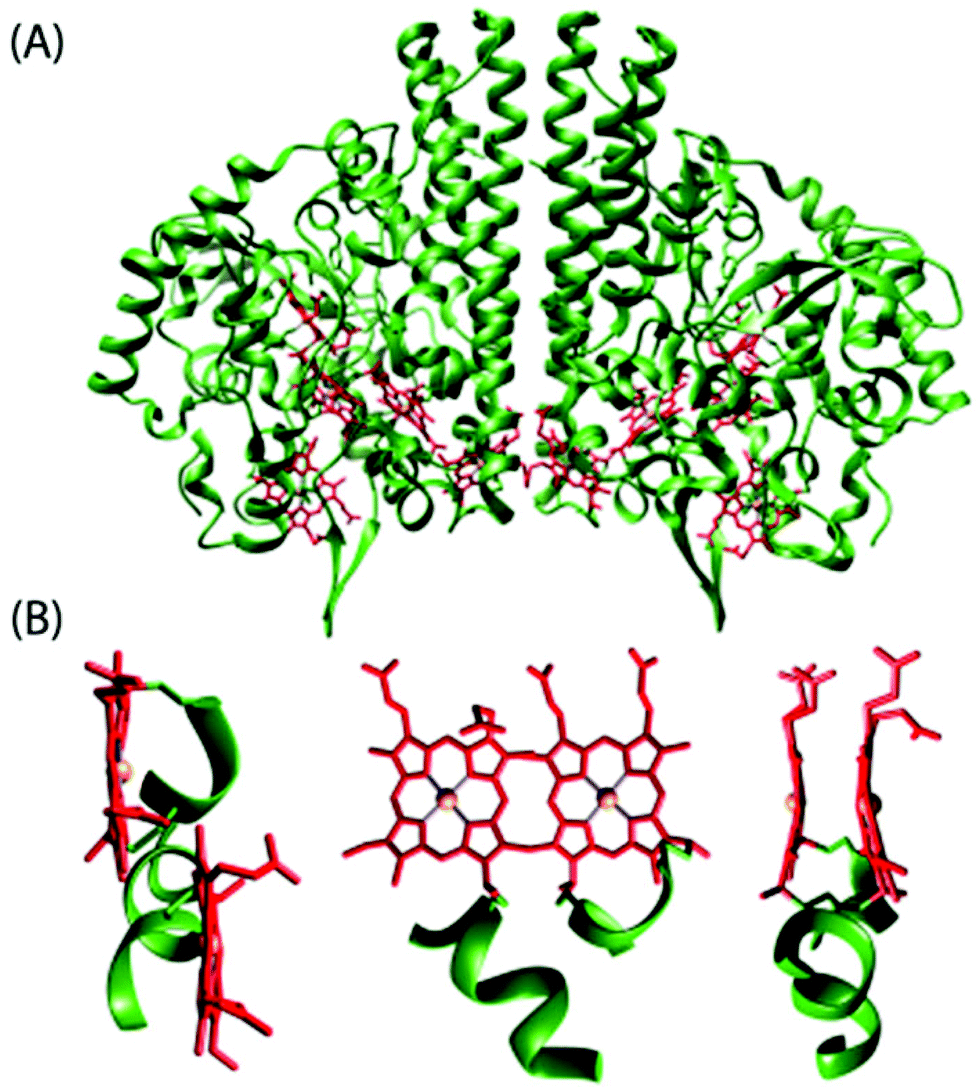
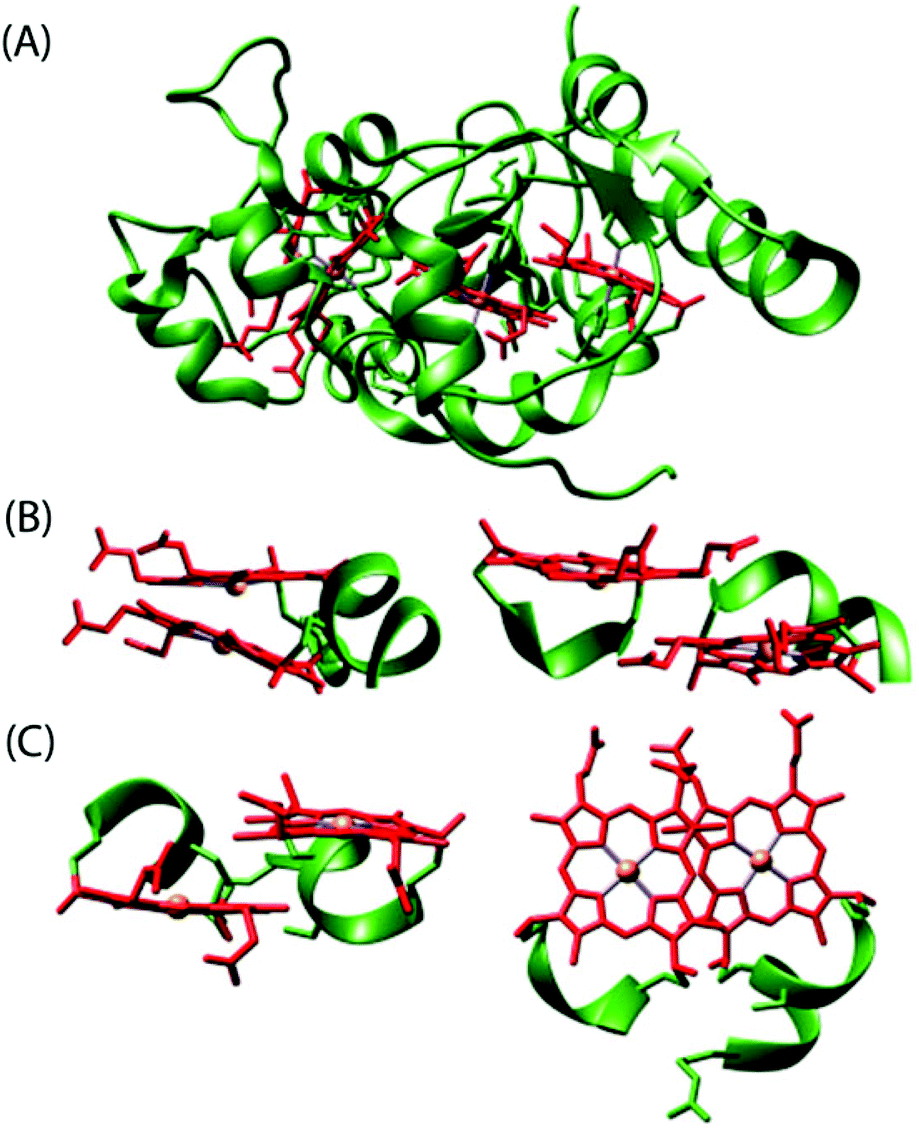
Among all the structures of multiheme proteins reported to date, a conspicuous feature is that the hemes are found clustered in structural arrangements.1–3 Certainly, these multiheme proteins have majority of their heme units closely packed forming diheme packing motifs with the two heme planes either parallel but offset (‘heme stacking motif’) or perpendicular to each other (‘diheme elbow motif’); Fig. 1–4 demonstrate some representative examples.1–3,5 The inter-heme iron distances in the parallel motif are around 10 Å and the heme planes are separated by a distance of less than 4 Å while the heme iron distances in the perpendicular motif are around 12 Å.1j Moreover, these diheme motifs form almost superimposable, larger structural heme motifs and the similarity may also extend to the folding of the polypeptide chain surrounding the hemes. Although the functional importance of the spatial disposition of these heme centers is not yet understood, they possibly reveal favorable arrangements to regulate the heme redox potential and/or permit very fast electron transfer. Understanding the importance of these structural arrangements is critical for the interpretation of the functional properties of multiheme cytochrome c which is, however, complicated by the presence of a large number and efficient coupling of the individual heme centers thereby masking their individual properties.1–3,5 The structural, thermodynamic, and kinetic study of some of the multiheme cytochromes have also been reviewed in recent times.1
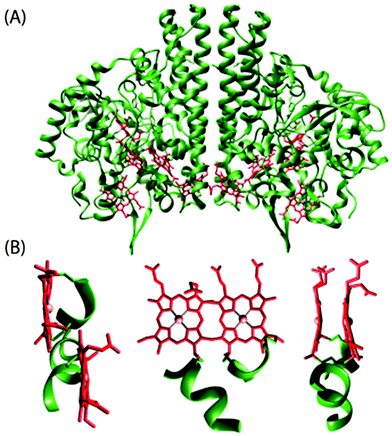 |
| | Fig. 1 (A) Structure of cytochrome c nitrite reductase (PDB code 1QDB)5a in which the protein chain and the heme groups have been colored green and red, respectively. (B) Different views of the diheme motif formed by heme 3 and heme 4 in the nitrite reductase. | |
 |
| | Fig. 2 (A) Structure of the oxidized state of tetraheme cytochrome c554 from Nitrosomonas europaea (PDB code 1FT5).5b The protein chain and the heme groups have been colored green and red, respectively. Different views of the diheme motifs formed by (B) hemes I and III and (C) hemes II and IV. | |
 |
| | Fig. 3 (A) Structure of the diheme cytochrome c, DHC2, from Geobacter sulfurreducens (PDB code 2CZS)5c containing 2 monomers per asymmetric unit. Each monomer has two heme groups covalently attached to the protein chain. The protein chain and the heme groups have been colored green and red, respectively. (B) Different views of structural arrangements of the heme groups. | |
 |
| | Fig. 4 Left, (A) structure of the diheme cytochrome c, NapB, from Haemophilus influenzae (PDB code 1JNI);3f (B) side and (C) top views of diheme motifs therein. Right, (A) structure of the aerobic form of the split-Soret diheme cytochrome c from Desulfovibrio desulfuricans ATCC 27774 (PDB code 1H21);3h (B) side and (C) top views of diheme motifs therein. The protein chain and the heme groups have been colored green and red, respectively. | |
1.2 Diheme cytochromes
A simplest member of the multi-heme family is the diheme cytochrome c (DHC2) from G. sulfurreducens, which has two spectroscopically different heme groups, having different redox properties, attached through a single polypeptide chain.5c The heme groups are covalently connected to the protein via thioether bonds to cysteine side chains with the sequence C–X1–X2–C–H. Fig. 3 shows the stereo view of the overall structure of DHC2 and arrangements of two heme groups therein.5c An Fe⋯Fe distance of 9.4 Å has been observed which, however, results in an efficient coupling between two heme centers. The imidazole planes have an interplanar angle of 36° in the axial histidine ligands of heme group I, whereas imidazoles in heme group II are nearly coplanar. Moreover, the porphyrin macrocycle of heme group II is strongly ruffle-distorted while the macrocycle of heme group I is nearly planar. The observed differences in the axial ligand orientations and porphyrin ring deformations between two heme centers in DHC2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5c have been proposed to be the consequences of heme–heme interactions. Another diheme cytochrome c with a similar heme arrangement as of DHC2 is the small subunit (NapB) of the periplasmic nitrate reductase (Fig. 4) from Haemophilus influenzae.3f Here, the two heme groups are nearly parallel and stacked at the van der Waals distances with an Fe⋯Fe distance of 9.9 Å, similar to the split-Soret diheme cytochrome c (Fig. 4) from Desulfovibrio desulfuricans ATCC 27774.3h
5c have been proposed to be the consequences of heme–heme interactions. Another diheme cytochrome c with a similar heme arrangement as of DHC2 is the small subunit (NapB) of the periplasmic nitrate reductase (Fig. 4) from Haemophilus influenzae.3f Here, the two heme groups are nearly parallel and stacked at the van der Waals distances with an Fe⋯Fe distance of 9.9 Å, similar to the split-Soret diheme cytochrome c (Fig. 4) from Desulfovibrio desulfuricans ATCC 27774.3h
The structural and functional uniqueness of the di/multi-heme cytochromes has inspired us to investigate the effect of inter-heme interactions at the molecular level. The sheer decline in intramolecular interactions with distance makes close proximity of the redox centers indispensable to significantly modify their intrinsic properties.1d However, the studies of structure–function relationships of heme proteins are complicated by the presence of huge protein chains. Synthetic chemists, therefore, have been engaged in studying metalloporphyrins in vitro, unencumbered by any biological superstructure, which eventually provide convenient systems for testing various mechanistic hypotheses regarding the fundamental processes. These attractive features have prompted us to thoroughly investigate the structure, function, and properties of the diheme and related complexes as a part of our ongoing research activities.6–13
This Perspective documents our endeavor and progress in the investigation of the dihemes and the outcome of the heme–heme interaction operational therein. In the subsequent sections, we will discuss the logical design and the conformational flexibility of the synthetic model of diheme centers followed by the illustration of the heme–heme interaction in the form of alteration of ligand field strength, spin state variation and redox potential modulation, and finally the usefulness of such diheme architectures in dioxygen activation.
2. Ethane-bridged porphyrin dimer as a model of diheme proteins
In order to mimic the heme centers in diheme cytochrome c (DHC2), a covalently linked porphyrin dimer with a highly flexible ethane spacer has been used in which two porphyrin rings can take conformations that are widely seen in the dihemes. Such a porphyrin dimer has been synthesized from the meso-formyl-octaethylporphyrinatonickel(II) upon reduction with NaBH4 and subsequent self condensation in N,N-dimethylformamide using concentrated sulfuric acid as the catalyst.14 Interestingly, the Fe⋯Fe non-bonding distances observed in such a model diheme range from 9.39–10.10 Å (vide infra),7–9 which are also very close to the Fe⋯Fe distances of 9.4 and 9.9 Å observed in the diheme cytochromes isolated from Geobacter sulfurreducens and Haemophilus influenza, respectively. The similar iron-to-iron distance and heme structural arrangement makes the ethane-bridged porphyrin dimer an ideal choice (Fig. 5) for exploring different aspects of heme–heme interactions in dihemes.
 |
| | Fig. 5 A schematic diagram overlaying the diheme cytochrome c (DHC2) with the model ethane-bridged diheme architecture. | |
2.1 Conformational flexibility
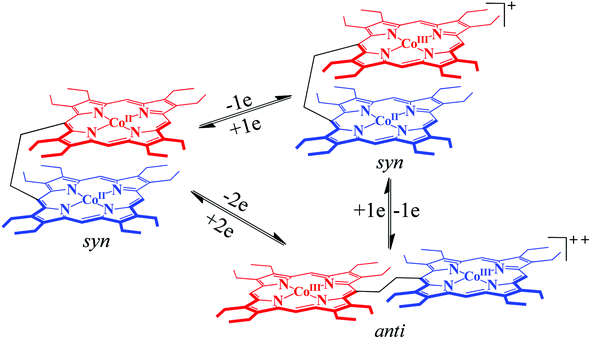
The conformational variations of porphyrinoids are known to modulate a wide variety of chemical, physicochemical and spectral properties.15 The supple ethane linker allows an easy interconversion between the syn and anti conformations (Scheme 1) of the porphyrin dimer triggered by axial ligation, π–π interaction, oxidation/reduction, and treatment with acids/bases. UV-visible spectra can be used as a powerful tool to determine the conformation of the complexes in solution.6,16 Splitting of the Soret band into two well-resolved transitions, where the low energy transition is assigned to the transition dipole moment running through the 5,15-meso carbons (B||) and the high energy transition is associated with the transition dipole moment running through the 10,20-meso carbons (B⊥), is in good agreement with Kasha's exciton coupling theory17 and is associated with the anti-form of the complex in solution.
 |
| | Scheme 1 | |
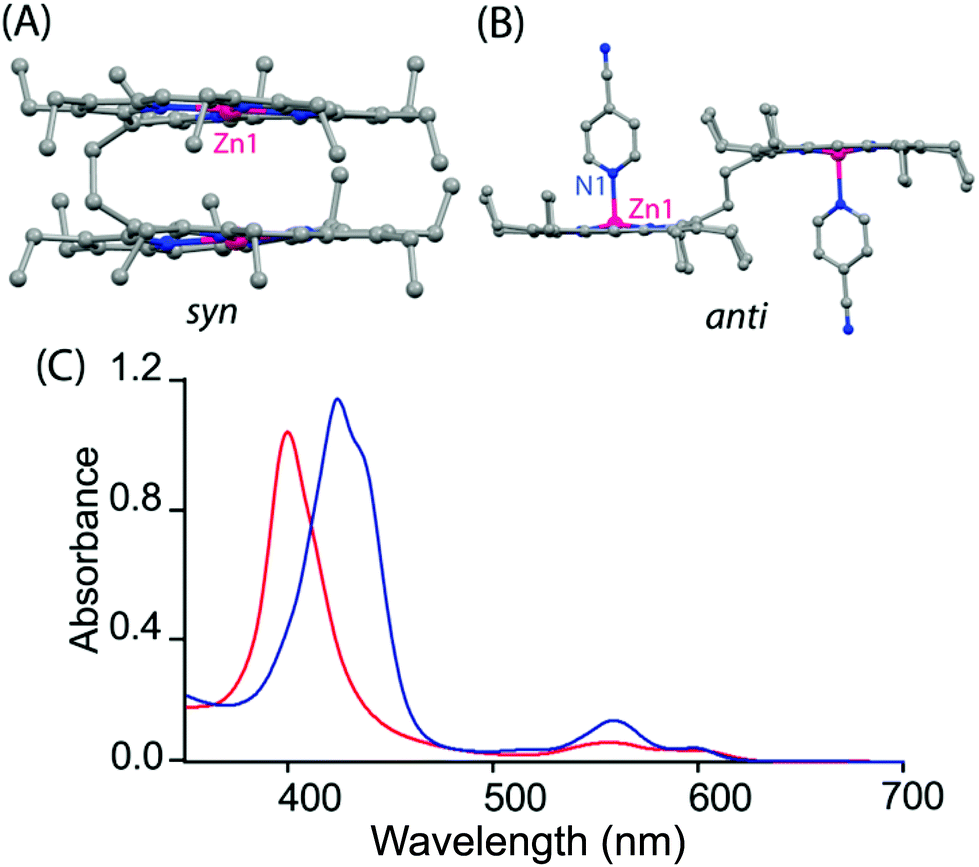
As displayed in Scheme 1, the ethane-bridged porphyrin dimer is known to switch between the syn and anti form induced by the external stimuli.6 It has been found that the free-base ligand and its Zn and cobalt complexes adopt a syn conformation in a variety of solvents due to strong π–π interactions between the two rings.6 Apart from axial ligation, strong π–π interactions between the porphyrin and π-electron rich systems (e.g. perylene) lead to the conversion of the syn to anti form. Fig. 6 shows the molecular structures of the zinc analog in syn and anti (upon axial coordination of 4-cyanopyridine) forms along with their UV-visible spectra. A split in the Soret band is the indication of anti-conformation. Moreover, syn–anti conformational switching can also be triggered by the step-wise addition/removal of electrons as depicted in Scheme 2.6b
 |
| | Fig. 6 Molecular structures of (A) the Zn(II) ethane-bridged porphyrin dimer18 in the syn form and (B) its 1:2 complex in the anti form with 4-cyanopyridine showing all non-hydrogen atoms, (C) UV-visible spectra in chloroform of the Zn(II) ethane-bridged porphyrin dimer in the syn form (red line) and the 1:2 anti form (blue line) with 4-cyanopyridine at 295 K. | |
 |
| | Scheme 2 | |
2.2 Model diheme centers
2.2.1 Anionic axial ligand.
In an effort to assess the structure–function correlation using the ethane-bridged porphyrin dimer as a model for diheme centers, a series of anti-diiron(III)bisporphyrin 1·X (X: Cl, Br, I, ClO4 and SO3CF3) have been synthesized (Scheme 3).7,19 All the complexes have been spectroscopically characterized and two of them (with X = ClO4 and SO3CF3) are also structurally characterized. While all the complexes are found to stabilize the high-spin (S = 5/2) state of iron, the complex with the SO3CF3 axial ligand is in a purely intermediate spin (S = 3/2) state. Also, Fe⋯Fe separations are found to be 10.03 and 9.77 Å, respectively, for the Fe(III) porphyrin dimers with ClO4 and SO3CF3 axial ligands.
 |
| | Scheme 3 | |
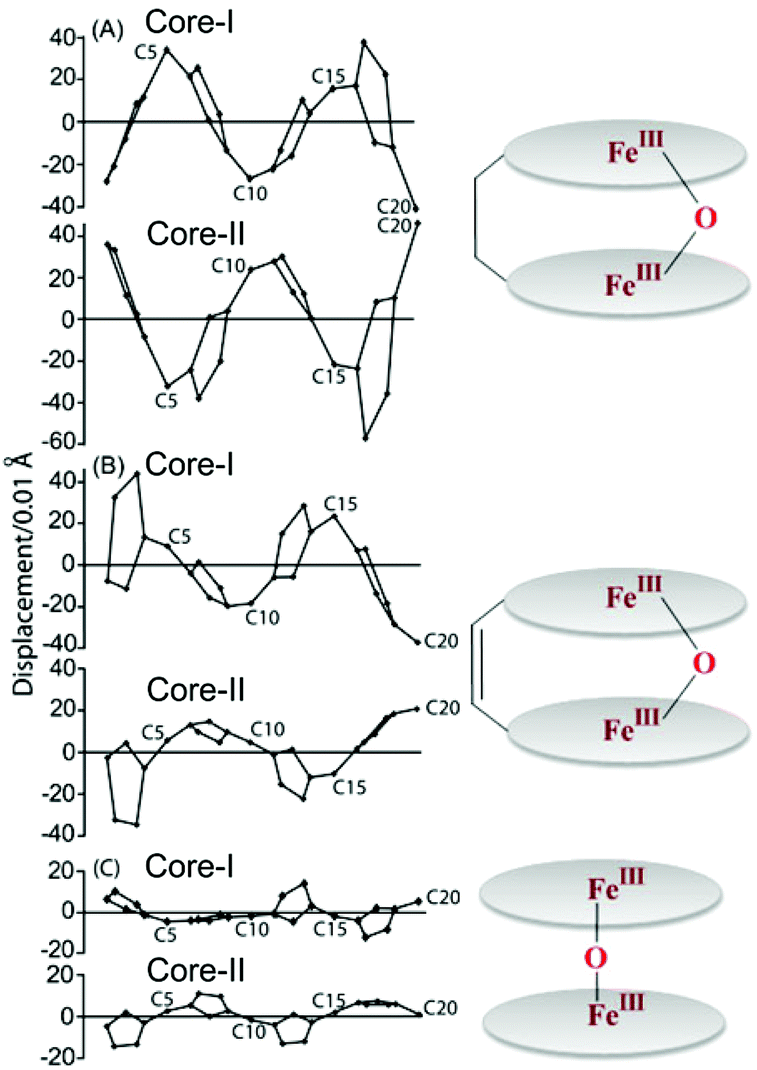
Structural parameters such as the displacement of the iron from the mean plane of the C20N4 porphyrinato core (Fe⋯Ctp) and the average Fe–Np distance are very characteristics of the iron spin states. For a typical high-spin complex, the Fe–Np and Fe⋯Ctp distances are ≥2.045 and ≥0.42 Å, respectively, whereas values reported for spin-admixed complexes are in the range of 1.961–2.038 Å for Fe–Np and 0.10–0.36 Å for Fe⋯Ctp, depending on the exact amount of S = 3/2,5/2 character present.7,20 From the X-ray structural parameters of the iron(III) porphyrin dimer with perchlorate and triflate as axial ligands, a high and intermediate-spin state of iron, respectively, has been assigned. However, porphyrin macrocycles are much more distorted in 1·SO3CF3 compared to 1·ClO4.7a
It would be interesting to compare the X-ray structure and geometrical parameters of the iron(III) porphyrin dimers with those of monoporphyrin analogs. In the dimeric complex 1·ClO4, the Fe–Np and Fe⋯Ctp distances are, respectively, 2.062(9) and 0.50 Å (for core-I) and 2.057(9) Å and 0.48 Å (for core-II) while the corresponding Fe–O distances are 1.873(9) and 1.940(7) Å.7b In contrast, the monomeric analog FeIII(OEP)ClO4 has provided the Fe–Np, Fe⋯Ctp and Fe–OClO3 distances of 1.994(10), 0.26 and 2.067(9) Å, respectively.21a Thus, while 1·ClO4 is a typical high-spin (S = 5/2) complex, its monomeric analog stabilizes the intermediate spin (S = 3/2) state of iron. The porphyrin ring has been found to be highly distorted in the dimeric complex (Fig. 7).
 |
| | Fig. 7 Out-of-plane displacements (in units of 0.01 Å) of the porphyrin core atoms of (A) 1·ClO4 and (B) FeIII(OEP)ClO421a from the mean plane of the C20N4 porphyrinato core. The horizontal axis shows the bond connectivity between atoms. Molecular structures (at 100 K) of the corresponding complexes showing all non-hydrogen atoms are given on the right. | |
Fig. 8 also displays the X-ray structures and out-of-plane displacement plots of 1·SO3CF37a and its monomeric counterpart FeIII(OEP)(SO3CF3).21b For 1·SO3CF3, the Fe–Np and Fe⋯Ctp distances are 1.978(3) Å and 0.26 Å, respectively, while the distances of 1.999(2) and 0.22 Å are observed for the monomeric complex, which suggests an intermediate spin (S = 3/2) state of iron for both the complexes. Moreover, Fe–SO3CF3 distances of 2.066(3) and 2.0392(14) have been reported for dimeric and monomeric complexes, respectively. As can be seen, porphyrin ring deformation is more in the dimeric complex compared to that in the monomeric analog.
 |
| | Fig. 8 Out-of-plane displacements (in units of 0.01 Å) of the porphyrin core atoms of (A) 1·SO3CF3 and (B) FeIII(OEP)(SO3CF3)21b from the mean plane of the C20N4 porphyrinato core. The horizontal axis shows the bond connectivity between atoms. Molecular structures (at 100 K) of the corresponding complexes showing all non-hydrogen atoms are given on the right. | |
The molecular structures of the complexes in solution are obtained from their 1H NMR spectra. The chemical shifts of the methylene, methyl and meso protons are highly sensitive to the spin states. For a typical five-coordinate high-spin Fe(III) complex with an OEP-type ligand, –CH2 proton signals are shifted downfield to ∼+40 ppm (average) while the meso protons appear at the far upfield to ∼–60 ppm (average). As iron moves towards the porphyrin mean plane in spin-admixed complexes, meso proton signals move more and more downfield while –CH2 protons move upfield. It has also been found that the high-spin complexes exhibit –CH3 signals more downfield than the complexes with intermediate spin as the unpaired spins can be delocalized up to –CH3 through σ-bonds in the former.
Fig. 9 shows the 1H NMR spectra of 1·ClO4 and 1·SO3CF3. The highly downfield shifted methylene (34.0 to 58.9 ppm) and bridging –CH2 (64.5 ppm) resonances as well as the highly upfield shifted meso proton signals (−35.2 to −52.4 ppm) clearly indicate the presence of a high-spin (S = 5/2) Fe(III) center in 1·ClO4. In contrast, the 1H NMR of its monomeric analog FeIII(OEP)ClO4 shows a downfield shifted methylene proton signal at 35.5 ppm and an upfield shifted meso proton signal at −5.5 ppm, which indicates an intermediate-spin state (S = 3/2) of Fe(III).21c In 1·SO3CF3, however, eight sharp methylene peaks have been observed in the relatively narrow region (from 16.0 to 21.6 ppm) along with two sharp meso resonances at −3.7 and −15.6 ppm indicating a pure intermediate state (S = 3/2) of Fe(III). In contrast, two downfield shifted methylene resonances (at 34.6 and 49.4 ppm) and an upfield shifted meso resonance (−24.6 ppm) suggest the presence of a nearly admixed high-spin Fe(III) center in FeIII(OEP)(SO3CF3), the monoheme analog of 1·SO3CF3.21c The spin states of iron have been further confirmed by EPR and magnetic susceptibility measurements of the polycrystalline samples also. Thus, as observed in solid, iron(III) porphyrin dimers with perchlorate and triflate as axial ligands stabilize a high and intermediate-spin of iron, respectively, which is, however, opposite to what has been expected for the monomeric complex.
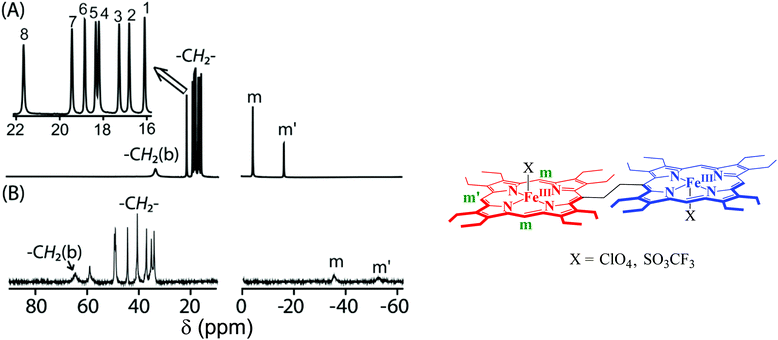 |
| | Fig. 9
1H NMR spectra (in CDCl3 at 295 K) of (A) 1·SO3CF3 and (B) 1·ClO4. | |
The factors that affect the conversion from the high-spin to an intermediate-spin state deserve comment. Studies on the model heme complexes suggest that a weak field axial ligand (e.g. ClO4−) stabilizes the intermediate-spin state of Fe(III) while CF3SO3− forms a mostly admixed high-spin complex.20,21 However, the porphyrin ring deformation is also known to play an important role in controlling the spins of iron(III) while a large deformation favours stabilization of an intermediate spin state.20 The porphyrin ring in 1·SO3CF3 has been found to be more ruffle-distorted compared to 1·ClO4. The ruffling of the porphyrin ring affects the energy level of d orbitals; the dxy orbital is destabilized due to its interaction with the a2u orbital of porphyrin, while the contraction of the equatorial Fe–Np bonds destabilizes the dx2−y2 orbitals which are, in fact, responsible for the stabilization of the pure intermediate spin of iron in 1·SO3CF3.7a In contrast, the destabilization of the dxy and dx2−y2 orbitals in 1·ClO4 is not sufficient enough for such a change in the spin state. Thus, while 1·ClO4 is a typical high-spin complex, the larger size of the triflate induces greater ruffling of the porphyrin macrocycle resulting in the stabilization of pure intermediate-spin in 1·SO3CF3 although according to the axial ligand strengths (‘magnetochemical series’22) reverse order of spin stabilization was expected.7a Thus, the complete reversal of the ligand field strength between ClO4− and CF3SO3− in the magnetochemical series has been obtained using a bisporphyrin framework.7a
2.2.2 Axial phenoxide coordination.
Instigated by the interesting results, we undertook further investigation using a variety of axial ligands in order to understand the effects of heme–heme interactions on the individual heme centers. Therefore, a series of diiron(III)bisporphyrin complexes with axial phenoxide coordination (Scheme 4) have been investigated and their structures and properties are compared with the corresponding monoporphyrin analogs.8 The complexes have been successfully characterized including single crystal X-ray structure determinations. Fig. 10 compares the structure of 2e and its monomeric counterpart FeIII(OEP){O-2,4,6-(NO2)3C6H2}.
 |
| | Scheme 4 | |
 |
| | Fig. 10 Molecular structures (at 100 K) of (A) FeIII(OEP){O-2,4,6-(NO2)3C6H2} and (B) 2e showing all non-hydrogen atoms. | |
The average Fe–Np distances are in the range of 2.057 to 2.077 Å and the Fe⋯Ctp lies within the value of 0.51 to 0.56 Å for complexes 2a, 2b, and 2c, which indicate the presence of a high-spin Fe(III) center in the complexes. However, 2e has an average Fe–Np distance of 1.972(3) Å, the shortest among all the phenolato complexes reported so far, while the Fe⋯Ctp distance is 0.23 Å suggesting a pure intermediate-spin Fe(III) center. In sharp contrast, the average Fe–Np and Fe⋯Ctp distances for FeIII(OEP){O-2,4,6-(NO2)3C6H2} (which is the monoporphyrin analog of 2e) are 2.049(2) and 0.40 Å, respectively, which confirms the stabilization of the high-spin state.8a Also, Fe⋯Fe separation within the molecules in the dimeric complexes varies within the range of 9.40 to 10.10 Å. The room temperature Mössbauer parameters of 2b [δ (ΔEq): 0.28 (0.66) mm s−1] and 2e [δ (ΔEq): 0.27 (2.7) mm s−1] also support the Fe(III) spin states as indicated by the X-ray structure of the complexes.8a
Fe–Np and Fe–O distances of 1.972(3) Å and 2.000(2) Å, observed for 2e, are the shortest and longest distances, respectively, in the series. Interestingly, 2e is the first phenoxide coordinated Fe(III)porphyrin having a pure intermediate state of iron while all other complexes stabilize in the high-spin state only.8,23 Also, the binding of 2,4,6-trinitrophenol to the iron(III) monoporphyrin, FeIII(OEP){O-2,4,6-(NO2)3C6H2}, stabilizes the high-spin state of iron.8a Among all the phenolato complexes, the porphyrin ring is most distorted in 2e while FeIII(OEP){O-2,4,6-(NO2)3C6H2}, which is the monomeric analog of 2e, shows a nearly planar porphyrin core (Fig. 11).8a The large increase in ring deformation in 2e, compared to its monomeric analog, is a result of the heme–heme interactions in dihemes, which eventually results in the stabilization of the intermediate-spin state of Fe(III).
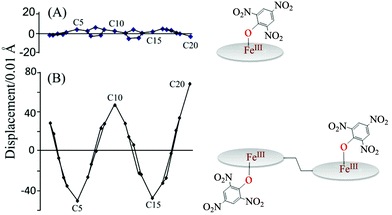 |
| | Fig. 11 Out-of-plane displacements (in units of 0.01 Å) of the porphyrin core atoms of (A) FeIII(OEP){O-2,4,6-(NO2)3C6H2} and (B) 2e from the mean plane of the C20N4 porphyrinato core. | |
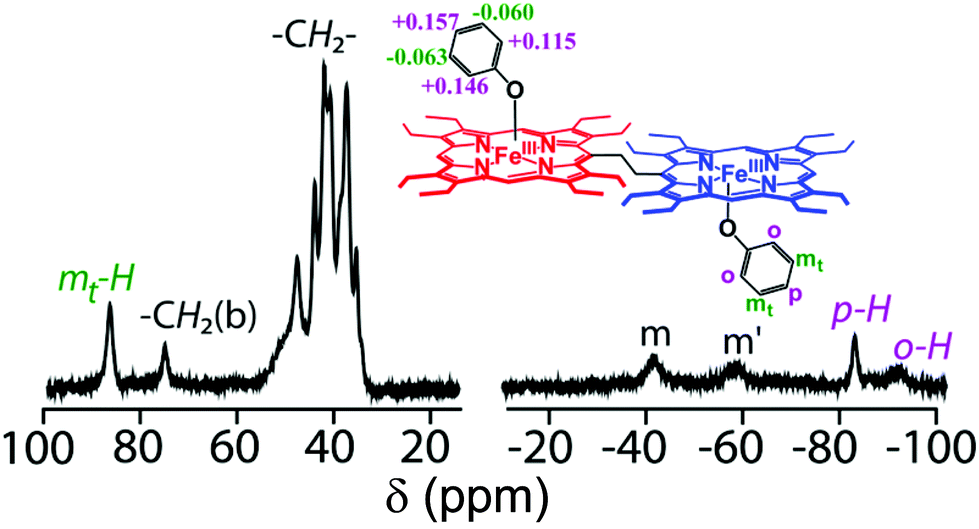
The nature and positioning of methylene and meso proton signals in the 1H NMR spectra of complexes 2a to 2d confirm the high-spin (S = 5/2) state of Fe(III) in the complexes in solution. In 2e, however, the 1H NMR spectrum suggests the presence of an intermediate-spin Fe(III) center in solution as was also observed in the solid.8a The phenolato resonances (Fig. 12) are, however, shifted to both upfield and downfield regions in the 1H NMR spectra which have been rationalized on the basis of Mulliken spin density calculation. Positive spin densities at the ortho- and para-carbons of the phenolato ring result in upfield shifting of the corresponding protons, while a negative spin density at the meta-position has an opposite effect. The alternating signs of spin densities at the ortho-, meta-, and para-carbons are also indicative of π-spin delocalization from the Fe(III) center to the phenolato ligand.8a
 |
| | Fig. 12
1H NMR spectrum (in CDCl3 at 295 K) of 2a; inset shows the Mulliken spin densities and the proton-numbering scheme on phenolato carbons. | |
Fig. 13 compares the 1H NMR spectra of 2e along with its monoporphyrin analogs: FeIII(OEP){O-2,4,6-(NO2)3C6H2} and FeIII(Me-OEP){O-2,4,6-(NO2)3C6H2}.8a Traces B and C show the 1H NMR spectra of FeIII(Me-OEP){O-2,4,6-(NO2)3C6H2} and FeIII(OEP){O-2,4,6-(NO2)3C6H2}, respectively, and, as can be seen, the nature and position of the methylene and meso resonances are characteristics of the pure high-spin state of Fe(III) in the complexes. Trace A, however, shows the spectra of 2e in which the methylene and meso signals have remarkably shifted upfield and downfield, respectively, as compared to their monomeric analogs. Therefore, axial coordination of 2,4,6-trinitrophenol to the corresponding Fe(III) complex of monoporphyrins stabilizes only the pure high-spin state (S = 5/2) while the intermediate-spin state is stabilized in the diheme analog (2e). The alteration of the spin state from high-spin to intermediate-spin on moving from FeIII(OEP){O-2,4,6-(NO2)3C6H2} to the diheme analog 2e, has been attributed to the effect of heme–heme interactions.8a
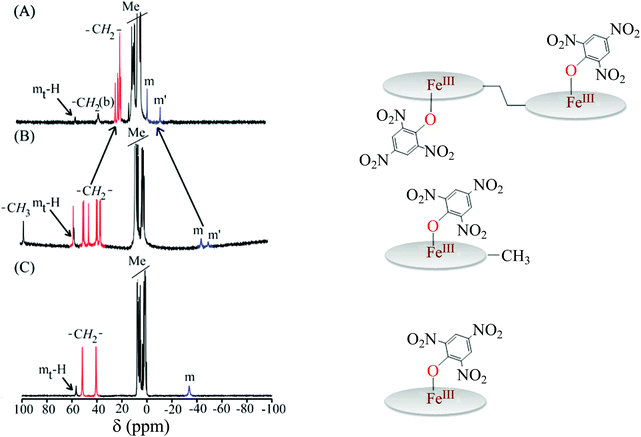 |
| | Fig. 13
1H NMR spectra (at 295 K in CDCl3) of (A) 2e, (B) FeIII(Me-OEP){O-2,4,6-(NO2)3C6H2} and (C) FeIII(OEP){O-2,4,6-(NO2)3C6H2}. | |
2.2.3 Axial thiophenolate coordination.
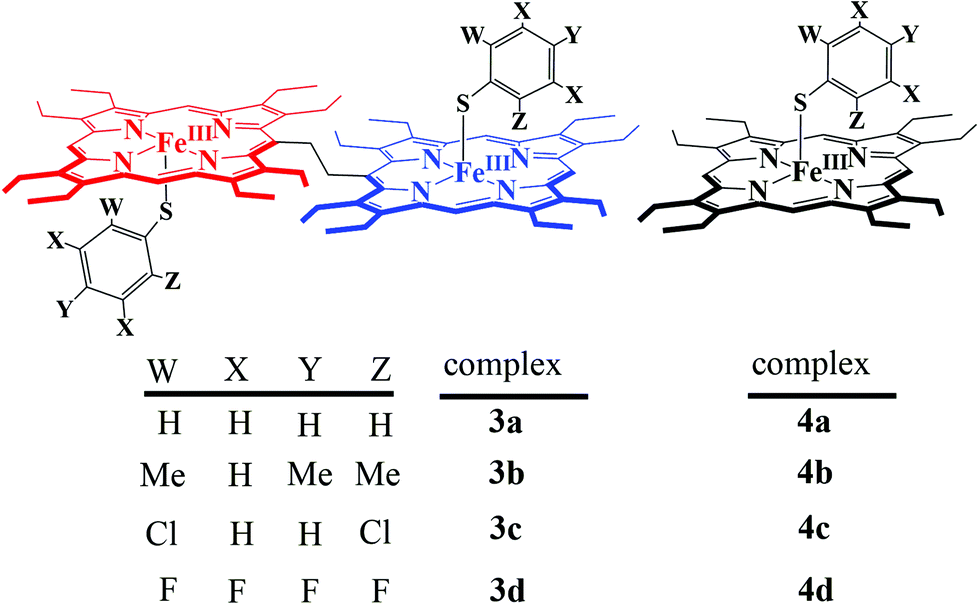
What followed immediately, is a series of thiophenolato coordinated diiron(III) bisporphyrins along with their monomeric counterparts (Scheme 5).9
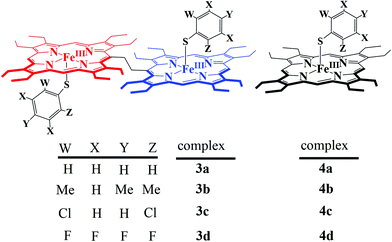 |
| | Scheme 5 | |
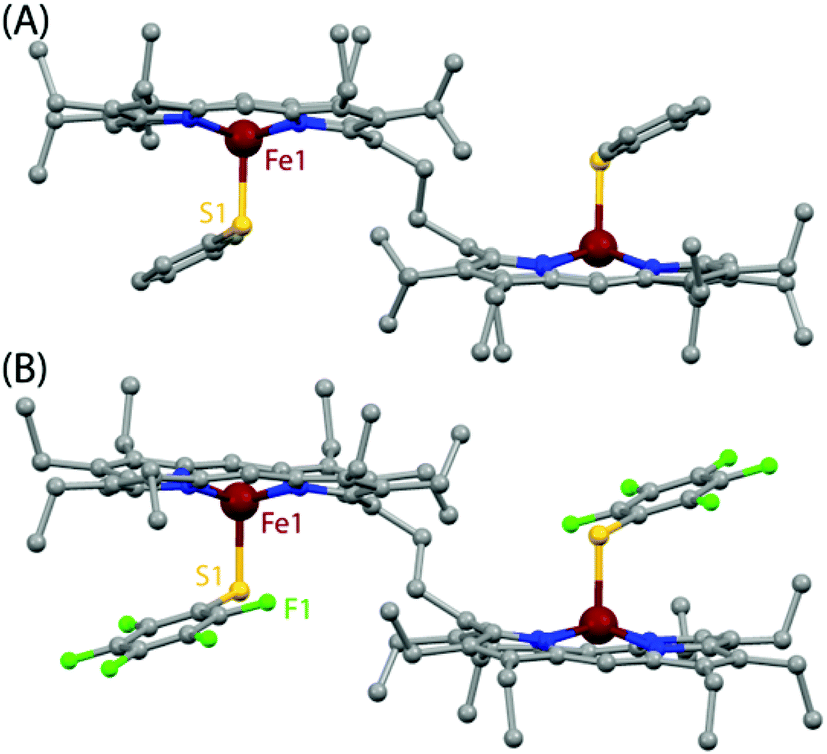
All the thiophenolato diiron(III)bisporphyrin complexes, shown in Scheme 5, have been structurally characterized. Fig. 14 displays the molecular structures of 3a and 3d as two representative cases.9 The Fe⋯Fe nonbonding distances within the molecules in the dimeric complexes have been found to vary within the range of 9.46 to 9.76 Å. The average Fe–Np (2.052 to 2.059 Å) and Fe⋯Ctp (0.41 to 0.61 Å) distances suggest a high-spin Fe(III) center for all the complexes. The Fe–S bond distances have been found to be dependent on the electron donating/withdrawing nature of the substituent on the thiophenols. The Fe–S bond distances in 3a–3d are larger compared to the Fe–O distances observed in their phenolato counterparts (vide supra). This indicates that thiophenols are weaker donors than phenols. The Fe–S–C angle in the complexes 3a–3d, though much smaller than the Fe–O–C angles in their phenolato counterparts, is similar to the Fe–S–C angles in the monoporphyrin complexes.
 |
| | Fig. 14 Molecular structures (at 100 K) of (A) 3a and (B) 3d showing all non-hydrogen atoms. | |
The porphyrin core deformations of the thiophenolato complexes are similar to their phenolato counterparts and can be best appreciated by turning to Fig. 15 where the out-of-plane displacements of the porphyrin core atoms of 3a, 2a and 4a are compared.9 While the porphyrin macrocycle in thiophenolato and phenolato complexes of the porphyrin dimer is highly distorted, the ring is nearly planar in the corresponding monomeric thiophenolato complex 4a.24 The interaction between the two rings in the model diheme results in larger ring deformation of the individual porphyrin centers. It is also interesting to note here that both the average displacement of the porphyrin core atoms and Fe⋯Ctp are greater in the thiophenolato complexes compared to the corresponding phenolato complexes of diiron(III)bisporphyrin.
 |
| | Fig. 15 Out-of-plane displacements (in units of 0.01 Å of the porphyrin core atoms) from the least-squares plane of the C20N4 porphyrinato core of (A) 4a, (B) 2a, and (C) 3a. | |
EPR spectra of all the thiophenolato diiron(III)bisporphyrin complexes are axially symmetric with g⊥ ∼ 6.0 and gII ∼ 2.0 due to the high-spin (S = 5/2) state of iron in the complexes, both in solid and solution phases.9 The 1H NMR spectra of the complexes (3a–3d) are also similar with the related phenolato analogs containing the high-spin state of iron.9 Similar to the phenolato diiron(III)bisporphyrin complexes, 2a–2e, the ortho- and para-protons show upfield shifts in the 1H NMR spectra while meta-protons move downfield, which is indicative of π-spin delocalization on the thiophenolato ligand.
Addition of excess thiophenol to the dichloromethane solution of 3 results in the formation of six-coordinate complex 5 (Scheme 6).9 The complexes, 5a–5d, produce axial EPR spectra (Fig. 16), which is typical for low-spin complexes at 120 K. It has been proposed that one axial ligand binds as thiophenolate while the other one as thiophenol. However, the formation of such six-coordinate complexes doesn't take place upon the addition of excess phenols to the related phenolato diiron(III)bisporphyrin complexes, 2a–2e.
 |
| | Scheme 6 | |
 |
| | Fig. 16 X-band EPR spectra in CH2Cl2 (at 120 K) of (A) 3a, (B) 5a and (C) 5b. | |
2.2.4 Metal redox.
Fig. 17 compares the cyclic voltammograms of both five and six-coordinate complexes. The study reveals a large positive shift of the Fe(III)/Fe(II) redox couple for the complexes going from 1·Cl (E1/2 = −1.34 V)7b to 2a (E1/2 = −0.92 V)8a to 3a (E1/2 = −0.76 V).9 A single point energy calculation clearly shows that in the HOMO for 2a, 3a and 1·Cl the electron density on the axial ligand decreases as we move from thiophenol to phenol to chloride.9 This explains the easier reduction of the Fe(III) center in the order: Cl− < PhO− < PhS−. Thiophenol is a weaker electron-donor than phenol due to back-donation of electron density from the iron center to an empty d-orbital of sulfur.
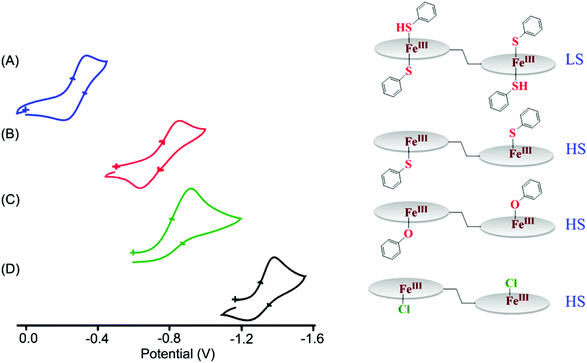 |
| | Fig. 17 Fe(III)/Fe(II) redox couple (at 298 K in CH2Cl2) of (A) 5a, (B) 3a, (C) 2a and (D) 1·Cl. The reference electrode was Ag/AgCl. | |
The Fe(III)/Fe(II) redox potential of the six-coordinate complex, 5a, was observed at −0.27 V.9 The large positive shift of 490 mV on moving from the five-coordinate high-spin complex (3a) to the six-coordinate low-spin complex (5a) suggests easier reduction of the iron center upon thiophenol coordination at the sixth position. Such a positive shift in the Fe(III)/Fe(II) redox potential has been attributed to the contributions from the sixth axial coordination as well as the spin state change from high to low.8a As can be seen, a variation of ∼1.10 V for the Fe(III)/Fe(II) redox potential can be achieved by the combination of axial coordination and metal spin. The control of heme redox potential by the spin state of iron is a key step in cytochrome P450 catalysis.25
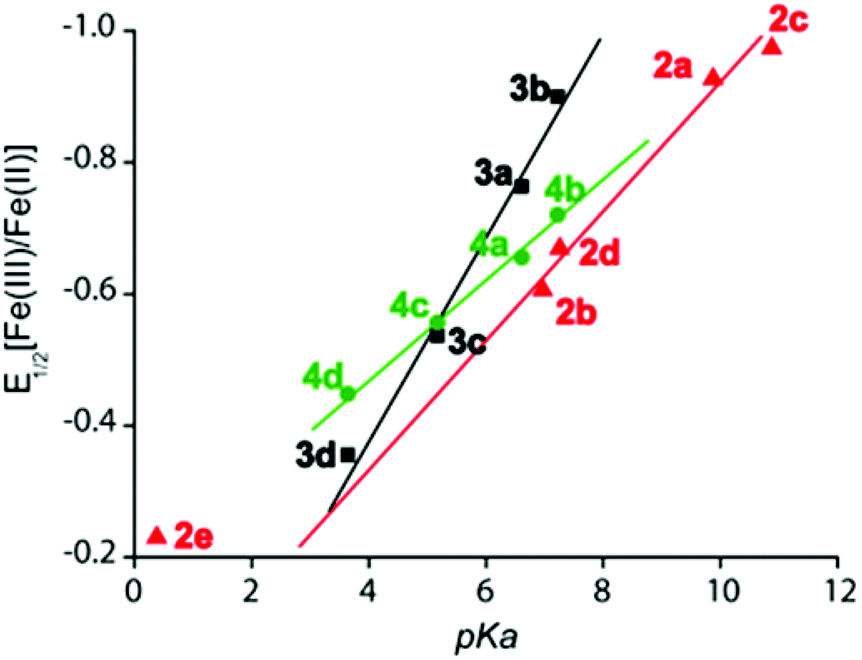
A plot of Fe(III)/Fe(II) potential of complexes 3a–3d and 4a–4d against pKa of the corresponding thiophenols (Fig. 18) has revealed a linear relationship.9 Similar pKa dependence of the Fe(III)/Fe(II) redox couple has been also observed for the phenolato analogs 2a–2e, where the deviation of 2e from linearity was demonstrated in terms of spin state change.8a,9 It is however interesting to note the steepness of the slope for the diheme complexes 3a–3d as compared to their monoheme analogs 4a–4d. A potential window of 540 mV for the Fe(III)/Fe(II) redox couple has been observed in the diheme series 3a–3d in contrast to the potential window of only 270 mV for the monomeric analogs 4a–4d. Such an increase in the potential range on going from monoheme to diheme can be attributed to the intermacrocyclic interaction in the dihemes.9
 |
| | Fig. 18 Plots of pKa of thiophenol/phenol vs. E1/2 [Fe(III)/Fe(II)] in V of thiophenolato diiron(III)bisporphyrins (■), phenolato diiron(III)bisporphyrins ( ) and thiophenolato iron(III)monoporphyrins ( ) and thiophenolato iron(III)monoporphyrins ( ). ). | |
The Fe(III)/Fe(II) redox couple has been found to be influenced by various factors including axial coordination, porphyrin ring deformation, change in the metal spin state and inter-heme interactions. However, it would be difficult to separate out the individual contributions alone since many of the factors are interrelated. A clear visualization of the outcome of inter-heme interactions can be obtained by comparing the cyclic voltammetric responses of dicobalt(II)bisporphyrin (which is found to be cofacial) and its monomeric analog, CoIIOEP (Fig. 19) under identical conditions and in the absence of any axial coordination.6b The increased number of oxidative responses, from three to four, in the cyclic voltammograms of dicobalt(II)bisporphyrin as compared to CoIIOEP indicates strong through space electronic communication in the complex. One CoIIOEP unit of the dimeric complex is oxidized at 0.25 V to generate CoIIIOEP at a potential much lower than that of monomeric CoIIOEP alone which is due to the increased electron density in the bisporphyrin scaffold. The second CoIIOEP is then oxidized at a significantly higher potential of 0.83 V due to strong inter-macrocyclic interactions in the dicobalt complex.6b
 |
| | Fig. 19 Comparison between cyclic voltammograms of the Co(II) ethane-bridged porphyrin dimer (blue-line) and monomeric CoIIOEP (red-line) in CH2Cl2 at 298 K. The reference electrode was Ag/AgCl. | |
2.3 μ-Oxo dimers
Addition of 5% aq. NaOH solution to the dichloromethane solution of 1·X changes the color from dark red to green immediately due to the formation of diiron(III)-μ-oxo bisporphyrin, 6a with a conformational change from anti to syn (Scheme 7).7b,10 Such an acid–base controlled conformational switching is also found to be reversible in nature. Similarly, cis 1,2-bis[chloroiron(III)5-(2,3,7,8,12,13,17,18-octaethylporphyrinyl]ethene, which also possesses great horizontal and vertical flexibility,26 transforms to cis diiron(III)-μ-oxo bisporphyrin, 6b (Scheme 7).10a
 |
| | Scheme 7 | |
The complex 6a crystallizes as two different solvates: one with CH3CN and the other one with toluene, however, both are very similar.7b,10b Molecular structures of the complexes are shown in Fig. 20. The Fe–O–Fe unit has been found to be remarkably bent with an angle of 147.9(1)°. There are at least six C⋯C nonbonding distances (<3.4 Å) between two porphyrin rings which are less than their van der Waals radii; the closest being the two bridging meso carbons which are separated by 3.016(5) Å. Strong π–π interactions must have contributed in the stabilization of highly bent and extremely strained geometry. Structural analysis of 1·ClO4 and 6a reflects a very high vertical flexibility of over 6.5 Å.7b,10b However, the rigid alkene spacer in 6b restricts the rotation of the individual porphyrin cores resulting in a fully eclipsed face-to-face ring orientation with a highly bent Fe–O–Fe unit with an angle of 150.9(2)°.10a The porphyrin rings of 6a and 6b are highly distorted compared to their unbridged dimer, [FeIII(OEP)]2O,27 which can be appreciated by referring to Fig. 21. As can be seen, the meso carbons (C20) that are connected by the linker are displaced most from the mean porphyrin plane in 6a/6b. Structural parameters of the complexes are compared in Table 1.
 |
| | Fig. 20 Molecular structures (at 100 K) of (A) 6a and (B) 6b showing all non-hydrogen atoms. Diagrams on the right showing the close contacts between two rings (hydrogen and ethyl groups are omitted for clarity). | |
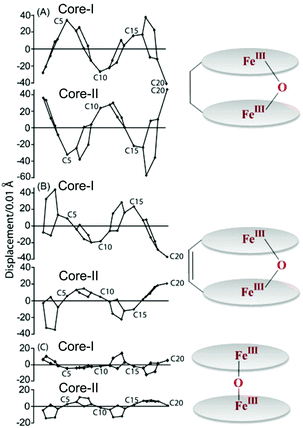 |
| | Fig. 21 Out-of-plane displacements (in units of 0.01 Å) of the porphyrin core atoms from the mean porphyrin plane of the C20N4 porphyrinato core of (A) 6a, (B) 6b and (c) [FeIII(OEP)]2O27 (monoclinic). | |
Table 1 Selected geometrical parameters
| Complex |
Fe–Npa |
Fe–Ob |
Fe–O–Fec |
Fe⋯Fed |
Δ
Fe24e |
Δ
24f |
Φ
|
Θ
|
Closest contactb |
Ref. |
|
Average value in Å.
Distance (in Å).
Angle (in °).
Nonbonding distance (in Å) between two Fe(III) centers in a molecule.
Displacement of iron (in Å) from the least-squares plane of the C20N4 porphyrinato core.
Average displacement of the 24 atoms (in Å) from the least-squares plane of the C20N4 porphyrinato core.
Average of the four N–Fe–Fe′–N′ dihedral angles (in °).
Angle (in °) between the two least-squares planes of the C20N4 porphyrinato core.
|
|
6a·CH3CN |
2.070(3) |
1.774(2) |
147.9(1) |
3.409 |
0.57 |
0.20 |
16.1 |
25.0 |
3.016(5) |
7b,10b
|
|
6a·C6H5CH3 |
2.076(3) |
1.767(2) |
151.1(1) |
3.423 |
0.54 |
0.18 |
16.4 |
21.2 |
3.042(5) |
7b,10b
|
|
6b
|
Core-I |
2.089(3) |
1.785(3) |
150.9(2) |
3.453 |
0.61 |
0.14 |
0.6 |
27.7 |
3.111(8) |
7b,10b
|
| Core-II |
2.081(4) |
1.781(3) |
0.65 |
| [FeIII(OEP)]2O (monoclinic) |
2.080(5) |
1.755(10) |
176.2(2) |
3.508 |
0.54 |
0.03 |
16.8 |
2.7 |
3.680 |
27
|
| [FeIII(OEP)]2O (triclinic) |
2.077(3) |
1.756(3) |
172.2(2) |
3.503 |
0.50 |
0.05 |
17.0 |
7.3 |
3.744 |
27
|
Both the Fe(III) centers are high-spin in nature in 6a and 6b as reflected in their X-ray structures and also in their Mössbauer spectra. Their 1H NMR spectra indicate that the syn orientation is preserved in solution.10 The cyclic voltammogram of 6a recorded at 25 °C in CH2Cl2 with 0.1 M tetrabutylammonium perchlorate (TBAP) as the supporting electrolyte and Ag/AgCl as the reference electrode shows four reversible/quasi-reversible one-electron oxidations at 0.57, 0.87, 1.27, and 1.39 V and one reductive response at −1.39 V. Under identical conditions, 6b also produces a similar voltammetric response; four one electron oxidations at 0.50, 0.84, 1.28 and 1.38 V and one-electron reduction at −1.36 V. It would be interesting to note here that under identical conditions, their unbridged counterpart [Fe(OEP)]2O shows similar oxidative responses at relatively higher potentials (0.65, 0.93, 1.30, 1.54 V) and reduction at slightly lower potentials (−1.35 V). The presence of two porphyrin macrocycles within a short distance in 6a/6b makes the porphyrin core highly deformed as well as more electron rich that is responsible for its easier oxidations.
The highly strained geometry around oxygen allows 6a and 6b to show some unusual reactivity that is not observed in the related unbridged complex [FeIII(OEP)]2O. The complex 6b upon photoirradiation (λexc > 365 nm, O–Fe LMCT transition) with excess phosphite in benzene under anaerobic conditions undergoes an immediate change in the absorption spectra (Fig. 22); the peaks corresponding to diiron(III)-μ-oxo bisporphyrin decrease while peaks corresponding to diiron(II)bisporphyrin keep increasing with time.10a Moreover, each photoreaction produces a stoichiometric amount of O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) P(OR)3 and diiron(II)bisporphyrin complex which reforms 6b in the presence of O2 to re-enter into the catalytic cycle (Scheme 8). A highly oxidized ferryl intermediate (PFeIVO) has been proposed to be responsible for the oxidation of the substrate.28 Similar results are also obtained with 6a but with less catalytic efficiency.10b
P(OR)3 and diiron(II)bisporphyrin complex which reforms 6b in the presence of O2 to re-enter into the catalytic cycle (Scheme 8). A highly oxidized ferryl intermediate (PFeIVO) has been proposed to be responsible for the oxidation of the substrate.28 Similar results are also obtained with 6a but with less catalytic efficiency.10b
 |
| | Fig. 22 Time-evolution spectral changes of 6b (red line) in benzene (∼10−6 M) in the presence of triethylphosphite (∼0.3 M) at 295 K under nitrogen. The arrows indicate the disappearance of 6b and appearance of diiron(II) bisporphyrin species (blue line). | |
 |
| | Scheme 8 | |
2.4 μ-Hydroxo dimers: unusual stabilization of two spin states of iron
Protonation of the diiron(III)-μ-oxo bisporphyrins, 6, by a strong Brønsted acid with a very weakly coordinating counterion leads to the change in color from green to red due to the formation of diiron(III)-μ-hydroxo bisporphyrins, 7 (Scheme 9). A number of μ-hydroxo complexes have been synthesized and structurally characterized. Fig. 23 shows the molecular structure of one such complex (7·I3) and its 1H NMR spectrum which clearly shows the signature of both high-spin and intermediate-spin Fe(III) centers. On going from the μ-oxo to the hydroxo complex, there is an increase in the Fe–O bond length while the Fe–O(H)–Fe unit becomes further bent which resulted in the closer approach of the two porphyrin rings. The interaction of two such rings in the μ-hydroxo complex has resulted in unequal core deformation (Fig. 24) with the eventual stabilization of two different spin states of iron in a single molecular framework.11 Also, spin states have been found to be dependent on the counter ions used and are reversibly interconvertable. The μ-hydroxo complexes exhibit weak antiferromagnetic coupling in contrast to the very strong exchange interactions in the corresponding μ-oxo dimer. However, similar μ-hydroxo complexes from the monoporphyrin counterparts have both the iron centers in the same spin state.29 These differences are believed to be due to stronger inter-heme interactions in the porphyrin dimer. Further studies are in progress.
 |
| | Scheme 9 | |
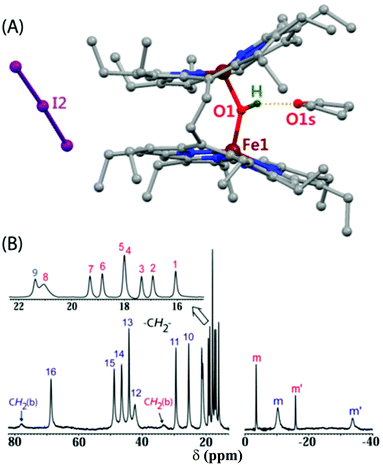 |
| | Fig. 23 (A) Molecular structure (at 100 K) of 7·I3 showing all non-hydrogen atoms (all of the hydrogens except for the μ-hydroxo proton have been omitted for clarity) and (B) its 1H NMR spectrum in CDCl3 at 295 K. | |
 |
| | Fig. 24 Out-of-plane displacements (in units of 0.01 Å) of the porphyrin core atoms from the mean porphyrin plane (24 atoms) of (A) 6a, and (B) 7·I3. | |
3. Summary and outlook
There is a high level of conservation of the heme structural arrangement throughout in various multi-heme cytochromes. The functional properties of such an important and widely distributed family are largely determined by the relative arrangement of the heme centers and their inter-macrocycle interactions which decay steeply with the Fe–Fe distance. Understanding the significance of these structural motifs is, however, hindered by the presence of a relatively large number of heme units along with the efficient coupling between them.
This Perspective presents a brief overview of our attempts to unravel the effects of heme–heme interactions using an ethane-bridged porphyrin dimer as a model of diheme centers. The porphyrin macrocycle in a diheme complex is found to be more distorted, compared to its monoheme analog. Changing the electronic nature of the axial ligand and its substituents has been found to play a crucial role in the nature of shift of the Fe(III)/Fe(II) redox couple while the inter-ring interaction decides the extent of such a shift. Such a variation of the Fe(III)/Fe(II) redox potential, as a result of heme–heme interactions, would be a very useful vehicle for tuning the redox potential of the di/multiheme proteins and their functions. From the results presented it can be concluded that inter-ring interaction is a potent tool to modulate various properties of the heme centers viz., porphyrin ring deformation, axial ligand strength, metal spin state, redox potential, and reactivity as compared to its monoheme counterparts. Thus, it can be said that the presence of more than one heme center provides Nature with a further tool to modulate various properties. However, the exact nature of such inter-heme interactions, which can be attributed to the combinations of a variety of phenomena related to steric, electrostatic, dispersion interactions etc., is still unclear and would require further investigations which are in progress.
The large differences in the structural, chemical, and electrochemical properties of the diheme, as compared to its monoheme analog, provide unequivocal evidence of the role played by heme–heme interactions. We wish to extend the work further from the diheme to triheme and so on in order to understand how the successive addition of the heme centers influences the structure and properties at the individual heme centers. Such a study would be useful in understanding various aspects of Nature's sophisticated design of multiheme centers. Also, the integration of the theoretical approach with experimental work to determine the effects of such interactions would be highly beneficial in elucidating many of the unanswered questions. We thus hope that this Perspective would attract many researchers into this important field of understanding the multiheme proteins.
Acknowledgements
The research contributions of the past and present members (Dr. Sudip Kumar Ghosh, Dr. Ranjan Patra, Dr. Arvind Chaudhary, Dr. Susovan Bhowmik, Dr. Soumyajit Dey, Dipankar Sahoo and Firoz Shah Tuglak Khan) of the group related to the project is greatfully acknowledged. The authors thank the Science and Engineering Research Board (SERB), New Delhi and the Council of Scientific and Industrial Research (CSIR), New Delhi for financial support. D.S. thanks University Grants Commission (UGC), India for his fellowship.
References
-
(a) J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian, I. Petrik, A. Bhagi and Y. Lu, Chem. Rev., 2014, 114, 4366–4469 CrossRef CAS PubMed;
(b) C. M. Paquete and R. O. Louro, Acc. Chem. Res., 2014, 47, 56–65 CrossRef CAS PubMed;
(c) C. M. Soares and A. M. Baptista, FEBS Lett., 2012, 586, 510–518 CrossRef CAS PubMed;
(d) B. M. Fonseca, C. M. Paquete, C. A. Salgueiro and R. O. Louro, FEBS Lett., 2012, 586, 504–509 CrossRef CAS PubMed;
(e) J. A. Mayfield, C. A. Dehner and J. L. DuBois, Curr. Opin. Chem. Biol., 2011, 15, 260–266 CrossRef CAS PubMed;
(f) C. M. Paquete and R. O. Louro, Dalton Trans., 2010, 39, 4259–4266 RSC;
(g) H. Akutsu and Y. Takayama, Acc. Chem. Res., 2007, 40, 171–178 CrossRef CAS PubMed;
(h) L. Shi, T. C. Squier, J. M. Zachara and J. K. Fredrickson, Mol. Microbiol., 2007, 65, 12–20 CrossRef CAS PubMed;
(i) C. G. Mowat and S. K. Chapman, Dalton Trans., 2005, 3381–3389 RSC;
(j)
I. A. Pereira and A. V. Xavier, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, John Wiley & Sons, New York, 2005, vol. 5, pp. 3360–3376 Search PubMed;
(k) J. M. Stevens, O. Daltrop, J. W. A. Allen and S. J. Ferguson, Acc. Chem. Res., 2004, 37, 999–1007 CrossRef CAS PubMed.
-
(a) M. Breuer, K. M. Rosso and J. Blumberger, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 611–616 CrossRef CAS PubMed;
(b) S. Sharma, G. Cavallaro and A. Rosato, J. Biol. Inorg. Chem., 2010, 15, 559–571 CrossRef CAS PubMed;
(c) M. Pessanha, E. L. Rothery, C. S. Miles, G. A. Reid, S. K. Chapman, R. O. Louro, D. L. Turner, C. A. Salgueiro and A. V. Xavier, Biochim. Biophys. Acta, 2009, 1787, 113–120 CrossRef CAS PubMed;
(d) C. M. Paquete, D. L. Turner, R. O. Louro, A. V. Xavier and T. Catarino, Biochim. Biophys. Acta, 2007, 1767, 1169–1179 CrossRef CAS PubMed.
-
(a) K. E. Ellis, K. E. Frato and S. J. Elliott, Biochemistry, 2012, 51, 10008–10016 CrossRef CAS PubMed;
(b) G. S. Pulcu, K. E. Frato, R. Gupta, H.-R. Hsu, G. A. Levine, M. P. Hendrich and S. J. Elliott, Biochemistry, 2012, 51, 974–985 CrossRef CAS PubMed;
(c) M. Hoffmann, J. Seidel and O. Einsle, J. Mol. Biol., 2009, 393, 951–965 CrossRef CAS PubMed;
(d) A. Echalier, T. Brittain, J. Wright, S. Boycheva, G. B. Mortuza, V. Fülöp and N. J. Watmough, Biochemistry, 2008, 47, 1947–1956 CrossRef CAS PubMed;
(e) G. W. Pettigrew, A. Echalier and S. R. Pauleta, J. Inorg. Biochem., 2006, 100, 551–567 CrossRef CAS PubMed;
(f) A. Brige, D. Leys, T. E. Meyer, M. A. Cusanovich and J. J. Van Beeumen, Biochemistry, 2002, 41, 4827–4836 CrossRef CAS PubMed;
(g) A. Kadziola and S. Larsen, Structure, 1997, 5, 203–216 CrossRef CAS;
(h) P. M. Matias, J. Morais, A. V. Coelho, R. Meijers, A. Gonzalez, A. W. Thompson, L. Sieker, J. LeGall and M. A. Carrondo, J. Biol. Inorg. Chem., 1997, 2, 507–514 CrossRef CAS.
-
(a) F. M. Stone and C. B. Coulter, J. Gen. Physiol., 1932, 15, 629–639 CrossRef CAS;
(b) L. Smith, J. Biol. Chem., 1959, 234, 1571–1574 CAS;
(c) P. M. Matias, A. V. Coelho, F. M. Valente, D. Placido, J. LeGall, A. V. Xavier, I. A. Pereira and M. A. Carrondo, J. Biol. Chem., 2002, 277, 47907–47916 CrossRef CAS PubMed.
-
(a) O. Einsle, A. Messerschmidt, P. Stach, G. P. Bourenkov, H. D. Bartunik, R. Huber and P. M. H. Kroneck, Nature, 1999, 400, 476–480 CrossRef CAS PubMed;
(b) T. M. Iverson, D. M. Arciero, A. B. Hooper and D. C. Rees, J. Biol. Inorg. Chem., 2001, 6, 390–397 CrossRef CAS;
(c) D. Heitmann and O. Einsle, Biochemistry, 2005, 44, 12411–12419 CrossRef CAS PubMed;
(d) H. R. Gibson, C. G. Mowat, C. S. Miles, B.-R. Li, D. Leys, G. A. Reid and S. K. Chapman, Biochemistry, 2006, 45, 6363–6371 CrossRef CAS PubMed;
(e) R. S. Hartshorne, B. N. Jepson, T. A. Clarke, S. J. Field, J. Fredrickson, J. Zachara, L. Shi, J. N. Butt and D. J. Richardson, J. Biol. Inorg. Chem., 2007, 12, 1083–1094 CrossRef CAS PubMed;
(f) D. Leys, T. E. Meyer, A. S. Tsapin, K. H. Nealson, M. A. Cusanovich and J. J. Van Beeumen, J. Biol. Chem., 2002, 277, 35703–35711 CrossRef CAS PubMed;
(g) M. V. Pattarkine, J. J. Tanner, C. A. Bottoms, Y.-H. Lee and J. D. Wall, J. Mol. Biol., 2006, 358, 1314–1327 CrossRef CAS PubMed;
(h) I. Bento, V. H. Teixeira, A. M. Baptista, C. M. Soares, P. M. Matias and M. A. Carrondo, J. Biol. Chem., 2003, 278, 36455–36469 CrossRef CAS PubMed.
-
(a) S. Brahma, S. A. Ikbal and S. P. Rath, Inorg. Chim. Acta, 2011, 372, 62–70 CrossRef CAS PubMed;
(b) S. Dey and S. P. Rath, Dalton Trans., 2014, 43, 2301–2314 RSC.
-
(a) S. Bhowmik, S. K. Ghosh and S. P. Rath, Chem. Commun., 2011, 47, 4790–4792 RSC;
(b) S. K. Ghosh, R. Patra and S. P. Rath, Inorg. Chem., 2010, 49, 3449–3460 CrossRef CAS PubMed.
-
(a) S. Bhowmik, S. Dey, D. Sahoo and S. P. Rath, Chem. – Eur. J., 2013, 19, 13732–13744 CrossRef CAS PubMed;
(b) S. Bhowmik, D. Sil, R. Patra and S. P. Rath, J. Chem. Sci., 2011, 123, 827–837 CrossRef CAS.
- D. Sil, F. S. T. Khan and S. P. Rath, Inorg. Chem., 2014, 53, 11925–11936 CrossRef CAS PubMed.
-
(a) S. K. Ghosh, R. Patra and S. P. Rath, Inorg. Chim. Acta, 2010, 363, 2791–2799 CrossRef CAS PubMed;
(b) S. K. Ghosh, R. Patra and S. P. Rath, Inorg. Chem., 2008, 47, 10196–10198 CrossRef CAS PubMed.
-
(a) S. K. Ghosh, S. Bhowmik, D. Sil and S. P. Rath, Chem. – Eur. J., 2013, 19, 17846–17859 CrossRef CAS PubMed;
(b) S. Bhowmik, S. K. Ghosh, S. Layek, H. C. Verma and S. P. Rath, Chem. – Eur. J., 2012, 18, 13025–13037 CrossRef CAS PubMed;
(c) S. K. Ghosh and S. P. Rath, J. Am. Chem. Soc., 2010, 132, 17983–17985 CrossRef CAS PubMed;
(d) M. A. Sainna, D. Sil, D. Sahoo, B. Martin, S. P. Rath, P. Comba and S. P. de Visser, Inorg. Chem., 2015, 54, 1919–1930 CrossRef CAS PubMed.
-
(a) S. Brahma, S. A. Ikbal and S. P. Rath, Inorg. Chem., 2014, 53, 49–62 CrossRef CAS PubMed;
(b) S. Brahma, S. A. Ikbal, A. Dhamija and S. P. Rath, Inorg. Chem., 2014, 53, 2381–2395 CrossRef CAS PubMed;
(c) S. A. Ikbal, S. Brahma and S. P. Rath, Chem. Commun., 2014, 50, 14037–14040 RSC;
(d) S. A. Ikbal, S. Brahma and S. P. Rath, Chem. Commun., 2015, 51, 895–898 RSC;
(e) S. Brahma, S. A. Ikbal, S. Dey and S. P. Rath, Chem. Commun., 2012, 48, 4070–4072 RSC.
-
(a) A. Chaudhary and S. P. Rath, Chem. – Eur. J., 2012, 18, 7404–7417 CrossRef CAS PubMed;
(b) A. Chaudhary and S. P. Rath, Chem. – Eur. J., 2011, 17, 11478–11487 CrossRef CAS PubMed;
(c) P. Mondal, A. Chaudhary and S. P. Rath, Dalton Trans., 2013, 42, 12381–12394 RSC;
(d) P. Mondal and S. P. Rath, Isr. J. Chem., 2015, 55 DOI:10.1002/ijch.201500014;
(e) A. Chaudhary, R. Patra and S. P. Rath, Indian J. Chem., 2011, 50A, 1436–1442 CAS;
(f) A. Chaudhary, S. A. Ikbal, S. Brahma and S. P. Rath, Polyhedron, 2013, 52, 761–769 CrossRef CAS PubMed;
(g) S. A. Ikbal, S. Brahma, A. Dhamija and S. P. Rath, J. Chem. Sci., 2014, 126, 1451–1461 CrossRef.
- D. Arnold, A. W. Johnson and M. Winter, J. Chem. Soc., Perkin Trans. 1, 1977, 1643–1647 RSC.
-
(a) J. P. Collman, P. S. Wagenknecht and J. E. Hutchison, Angew. Chem., Int. Ed. Engl., 1994, 33, 1537–1554 CrossRef PubMed;
(b) J. Rosenthal and D. G. Nocera, Acc. Chem. Res., 2007, 40, 543–553 CrossRef CAS PubMed;
(c) P. D. Harvey, C. Stern, C. P. Gros and R. Guilard, Coord. Chem. Rev., 2007, 251, 401–428 CrossRef CAS PubMed.
-
(a) V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, Tetrahedron Lett., 1999, 40, 5051–5054 CrossRef CAS;
(b) V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, J. Phys. Chem. B, 1999, 103, 5151–5156 CrossRef CAS;
(c) K.-I. Sugiura, G. V. Ponomarev, S. Okubo, A. Tajiri and Y. Sakata, Bull. Chem. Soc. Jpn., 1997, 70, 1115–1123 CrossRef CAS.
- M. Kasha, H. R. Rawls and M. A. E. Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- I. Fujii, V. V. Borovkov and Y. Inoue, Anal. Sci., 2006, 22, x77–x78 CrossRef CAS.
-
(a) G. A. Hembury, V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, Chem. Lett., 2003, 32, 428–429 CrossRef CAS;
(b) V. Borokov, J. M. Lintuluoto and Y. Inoue, Helv. Chim. Acta, 1999, 82, 919–934 CrossRef.
-
(a) R. Weiss, A. Gold and J. Terner, Chem. Rev., 2006, 106, 2550–2579 CrossRef CAS PubMed;
(b) M. Nakamura, Coord. Chem. Rev., 2006, 250, 2271–2294 CrossRef CAS PubMed;
(c) D. Sahoo, M. G. Quesne, S. P. de Visser and S. P. Rath, Angew. Chem., Int. Ed., 2015, 54, 4796–4800 CrossRef CAS PubMed;
(d) R. Patra, D. Sahoo, S. Dey, D. Sil and S. P. Rath, Inorg. Chem., 2012, 51, 11294–11305 CrossRef CAS PubMed;
(e) R. Patra, S. Bhowmik, S. K. Ghosh and S. P. Rath, Dalton Trans., 2010, 39, 5795–5806 RSC;
(f) R. Patra, A. Chaudhary, S. K. Ghosh and S. P. Rath, Inorg. Chem., 2010, 49, 2057–2067 CrossRef CAS PubMed;
(g) R. Patra and S. P. Rath, Inorg. Chem. Commun., 2009, 515–519 CrossRef CAS PubMed;
(h) R. Patra, A. Chaudhary, S. K. Ghosh and S. P. Rath, Inorg. Chem., 2008, 47, 8324–8335 CrossRef CAS PubMed.
-
(a) H. Masuda, T. Taga, K. Osaki, H. Sugimoto, Z. Yoshida and H. Ogoshi, Inorg. Chem., 1980, 19, 950–955 CrossRef CAS;
(b) N. Xu, D. R. Powell and G. B. Richter-Addo, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1366 CAS;
(c) A. D. Boersma and H. M. Goff, Inorg. Chem., 1982, 21, 581–586 CrossRef CAS.
- C. A. Reed and F. Guiset, J. Am. Chem. Soc., 1996, 118, 3281–3282 CrossRef CAS.
-
(a) D. Kanamori, Y. Yamada, A. Onoda, T. Okamura, S. Adachi, H. Yamamoto and N. Ueyama, Inorg. Chim. Acta, 2005, 358, 331–338 CrossRef CAS PubMed;
(b) A. M. Helms, W. D. Jones and G. L. McLendon, J. Coord. Chem., 1991, 23, 351–359 CrossRef CAS PubMed;
(c) N. Ueyama, N. Nishikawa, Y. Yamada, T. Okamura and A. Nakamura, Inorg. Chim. Acta, 1998, 283, 91–97 CrossRef CAS;
(d) M. P. Byrn, C. J. Curtis, Y. Hsiou, S. I. Khan, P. A. Sawin, S. K. Tendick, A. Terzis and C. E. Strouse, J. Am. Chem. Soc., 1993, 115, 9480–9497 CrossRef CAS.
- K. M. Miller and C. E. Strouse, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1984, 40, 1324–1327 CrossRef.
-
(a)
Cytochrome P450: Structure, Mechanism, and Biochemistry, ed. P. R. Ortiz de Montellano, Kluwer Academic/Plenum Publishers, New York, 3rd edn, 2005 Search PubMed;
(b) I. G. Denisov, T. M. Makris, S. G. Sligar and I. Schlichting, Chem. Rev., 2005, 105, 2253–2277 CrossRef CAS PubMed.
- T. E. Clement, D. J. Nurco and K. M. Smith, Inorg. Chem., 1998, 37, 1150–1160 CrossRef CAS PubMed.
- B. Cheng, J. D. Hobbs, P. G. Debrunner, J. Erlebacher, J. A. Shelnutt and W. R. Scheidt, Inorg. Chem., 1995, 34, 102–110 CrossRef CAS.
-
(a) J. Rosenthal, T. D. Luckett, J. M. Hodgkiss and D. G. Nocera, J. Am. Chem. Soc., 2006, 128, 6546–6547 CrossRef CAS PubMed;
(b) J. Rosenthal, J. Bachman, J. L. Dempsey, A. J. Esswein, T. G. Gray, J. M. Hodgkiss, D. R. Manke, T. D. Luckett, B. J. Pistorio, A. S. Veige and D. G. Nocera, Coord. Chem. Rev., 2005, 249, 1316–1326 CrossRef CAS PubMed.
-
(a) D. R. Evans and C. A. Reed, J. Am. Chem. Soc., 2000, 122, 4660–4667 CrossRef CAS;
(b) D. R. Evans, R. S. Mathur, K. Heerwegh, C. A. Reed and Z. Xie, Angew. Chem., Int. Ed. Engl., 1997, 36, 1335–1337 CrossRef CAS PubMed;
(c) W. R. Scheidt, B. Cheng, M. K. Safo, F. Cukiernik, J.-C. Marchon and P. G. Duebrunner, J. Am. Chem. Soc., 1992, 114, 4420–4421 CrossRef CAS.
Footnote |
| † Dedicated to Professor Animesh Chakravorty on the occasion of his 80th birthday. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence


















 ) and thiophenolato iron(III)monoporphyrins (
) and thiophenolato iron(III)monoporphyrins ( ).
).