
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ZnII and HgII binding to a designed peptide that accommodates different coordination geometries†
Dániel
Szunyogh
a,
Béla
Gyurcsik
ab,
Flemming H.
Larsen
c,
Monika
Stachura
d,
Peter W.
Thulstrup
e,
Lars
Hemmingsen
*e and
Attila
Jancsó
*ab
aMTA-SZTE Bioinorganic Chemistry Research Group, Dóm tér 7, Szeged, H-6720, Hungary
bDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, Szeged, H-6720, Hungary. E-mail: jancso@chem.u-szeged.hu; Fax: (+36) 62544340
cDepartment of Food Science, University of Copenhagen, Rolighedsvej 30, 1958 Frederiksberg C, Denmark
dCERN, 23 Geneva, 1211-Geneva, Switzerland
eDepartment of Chemistry, University of Copenhagen, Universitetsparken 5., 2100 Copenhagen, Denmark. E-mail: lhe@chem.ku.dk; Fax: (+45) 35332398
First published on 26th May 2015
Abstract
Designed metal ion binding peptides offer a variety of applications in both basic science as model systems of more complex metalloproteins, and in biotechnology, e.g. in bioremediation of toxic metal ions, biomining or as artificial enzymes. In this work a peptide (HS: Ac-SCHGDQGSDCSI-NH2) has been specifically designed for binding of both ZnII and HgII, i.e. metal ions with different preferences in terms of coordination number, coordination geometry, and to some extent ligand composition. It is demonstrated that HS accommodates both metal ions, and the first coordination sphere, metal ion exchange between peptides, and speciation are characterized as a function of pH using UV-absorption-, synchrotron radiation CD-, 1H-NMR-, and PAC-spectroscopy as well as potentiometry. HgII binds to the peptide with very high affinity in a {HgS2} coordination geometry, bringing together the two cysteinates close to each end of the peptide in a loop structure. Despite the high affinity, HgII is kinetically labile, exchanging between peptides on the subsecond timescale, as indicated by line broadening in 1H-NMR. The ZnII-HS system displays more complex speciation, involving monomeric species with coordinating cysteinates, histidine, and a solvent water molecule, as well as HS-ZnII-HS complexes. In summary, the HS peptide displays conformational flexibility, contains many typical metal ion binding groups, and is able to accommodate metal ions with different structural and ligand preferences with high affinity. As such, the HS peptide may be a scaffold offering binding of a variety of metal ions, and potentially serve for metal ion sequestration in biotechnological applications.
Introduction
Metal sensor proteins1–5 display high selectivity for both essential and toxic metal ions, as demonstrated by representative members of the MerR family,6,7 such as the CuI-sensing CueR, ZnII-sensing ZntR, and HgII-sensing MerR.8 In this work we have attempted to design a peptide with a broader metal ion binding profile. In a biotechnological perspective, overexpression of such a peptide in suitable bacteria could endow the cells with the capacity to sequester metal ions, including toxic elements, from the environment.9 Additionally, elevated levels of such a peptide could ensure metal ion buffering of the cytosol, allowing the bacterium to survive in harsh conditions of both deprivation and over-exposure to metal ions in the surrounding medium, and serve as an engineered organism with improved properties for biomining and bioremediation.10–12 The template for the design was the CuI binding loop of CueR from V. cholerae, SCPGDQGSDCP. In the related sequence from E. coli, Cu(I) ion is coordinated by two cysteines in a linear coordination geometry.8 The peptide is also expected to possess the capacity to bind the soft HgII ion, due to the thiophilicity of this ion. In order to broaden the metal ion binding profile, and increase the peptide solubility, proline to histidine and proline to serine substitutions were introduced at positions 3 and 11, respectively. The positions of substitutions were chosen to increase ligand-flexibility, and to mimic the presence of His and Ser at these positions in some of the metalloregulatory MerR family members.8 The modifications were expected to promote the coordination of the borderline soft/hard ZnII ion. In a recent study we demonstrated that this designed 12-mer HS peptide (see Scheme 1) forms various species with CdII, including loop structures and metal ion bridged bis-ligand complexes, depending on pH and metal to ligand ratio.13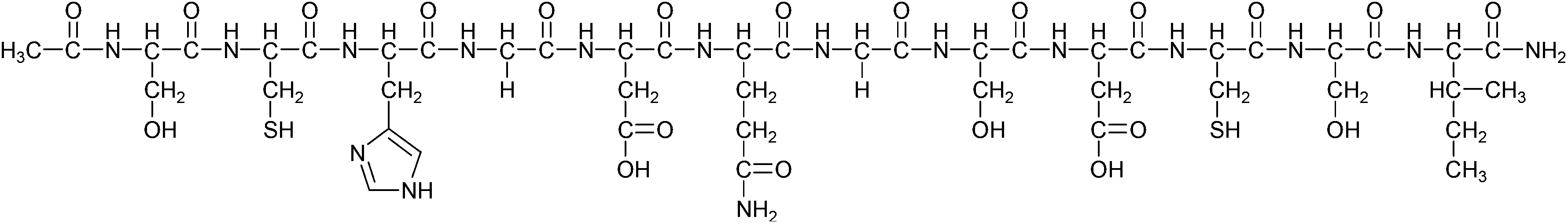 | ||
| Scheme 1 Schematic structure of Ac-SCHGDQGSDCSI-NH2 (HS). | ||
ZnII is rather promiscuous in terms of coordination characteristics as compared to the clearly soft, often two-coordinated HgII. In general, ZnII can easily adopt four-, five- or six-coordinate environments.14 Nevertheless, in zinc-containing enzymes and proteins the most typical coordination number is four.14,15 The preference of ZnII for a tetrahedral coordination geometry in proteins is supported by detailed statistical analyses of crystal structures of zinc-containing proteins deposited in the Protein Data Bank (PDB).16,17 Five- and six-coordinated ZnII centers are typically present due to the complementary coordination of solvent or inhibitor molecules in zinc-containing enzymes.17 Depending on the type of zinc-centers the abundance of Cys and His side chains significantly varies in the donor set patterns (number and type of bound donor groups). At catalytic zinc-centers any three N, O or S donors of Cys, His, Asp and Glu residues bind ZnII in a 4–5 coordinate distorted-tetrahedral or trigonal-bipyramidal geometry, with His being the predominant ligand.18 A water molecule is always found in such centres. His and Asp donors are dominant at the co-catalytic zinc-sites consisting of two or three metal ions in close proximity, two of which are bridged by one of the amino acid side chains or a water molecule.18 Cysteines, however, are not utilized at these motifs. Four protein side chain ligands are bound to ZnII in a tetrahedral or distorted tetrahedral geometry at structural zinc-sites.18 Such a binding mode is characteristic for e.g. the nucleic acid binding zinc finger proteins15 and for the zinc-clusters in metallothioneins.19 In all classes of the structurally diverse zinc fingers20,21 ZnII ions are ligated by a combination of four Cys/His side chain donors, at least two of which are Cys thiolates.15 Thiolate donors, complemented with side chain carboxylates and His-imidazoles, are also typical at the metalloregulatory ZnII binding sites in various zinc sensor proteins, however, coordination number and geometry appears to be more decisive in metal ion selectivity than donor ligand types.15
HgII can tolerate various coordination numbers and geometries, although, six-coordination is much less common than for the other two group 12 metal ions CdII and ZnII.22 Linear two-coordinate, trigonal planar or T-shaped three-coordinate or tetrahedral four-coordinate structures are representative for complexes with monodentate ligands and higher coordination numbers might be accessible mostly with multidentate compounds.22,23 HgII forms complexes with coordination number 2 more commonly than any other metal ion,22 which can be explained by relativistic effects.24 Low coordination numbers are characteristic for complexes formed with thiolates, a class of ligands displaying an outstanding affinity towards the large and soft HgII ion,25 and in biological systems HgII is usually complexed by low molecular weight thiolates or by the Cys side chains of proteins.26 Amongst others, some representative examples are provided by the bacterial mercury resistance systems, e.g. MerP where HgII is bound to a CXXC (X = amino acid other than cysteine) fragment in a typical linear two-coordinate fashion,27 or the metalloregulatory protein MerR where Cys residues from the two protein monomers form a tri-coordinate metal binding site for HgII.28,29 Additionally, distorted tetrahedral HgII coordination environment was reported in a few HgII-substituted proteins.30–32
The substantially different preferences of ZnII and HgII for four- and two-coordinated structures and the negligible role of His side chains in HgII biocoordination prompted us to investigate whether the His residue incorporated in the flexible ligand sequence of HS might have an influence on the binding of either of the two metal ions. In this work we characterize the binding of ZnII and HgII to the peptide in terms of the metal site coordination geometry and exchange dynamics.
Results and discussion
UV absorption and SRCD studies monitoring the formation of thiolate–metal ion bonds and ligand structure
Comparison of pH-dependent series of UV-spectra in the presence of 0, 0.5 and 1.0 equivalent of HgII or ZnII as compared to the ligand provides information on the interaction of the metal ions with donor groups of HS. The occurrence of S− → HgII ligand to metal charge transfer (LMCT) transitions25,33–35 upon the addition HgII to the peptide imply that the cysteine side chain thiolate groups of the ligand are coordinated to HgII already at low pH (see the full spectra in ESI, Fig. S1A–B†). The HgII![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HS 1:1 system shows almost a constant absorbance at λ = 230 nm at pH = 4–11 that is significantly higher than the absorption observed for the ligand in the absence of metal ion between pH ∼ 4.0–9.0 (Fig. 1). This suggests that the thiolate groups of HS are bound to HgII from acidic to alkaline pH. It is important to note that the deprotonation of the cysteine residues of the free peptide between pH ∼ 8–10 is accompanied by the appearance of an n → σ* transition around 230–240 nm characteristic for deprotonated thiols36,37 (Fig. 1 and S2†), and as this is absent for the HgII-HS species, we infer that the {HgS2} coordination geometry is formed at pH lower than 4.
:HS 1:1 system shows almost a constant absorbance at λ = 230 nm at pH = 4–11 that is significantly higher than the absorption observed for the ligand in the absence of metal ion between pH ∼ 4.0–9.0 (Fig. 1). This suggests that the thiolate groups of HS are bound to HgII from acidic to alkaline pH. It is important to note that the deprotonation of the cysteine residues of the free peptide between pH ∼ 8–10 is accompanied by the appearance of an n → σ* transition around 230–240 nm characteristic for deprotonated thiols36,37 (Fig. 1 and S2†), and as this is absent for the HgII-HS species, we infer that the {HgS2} coordination geometry is formed at pH lower than 4.
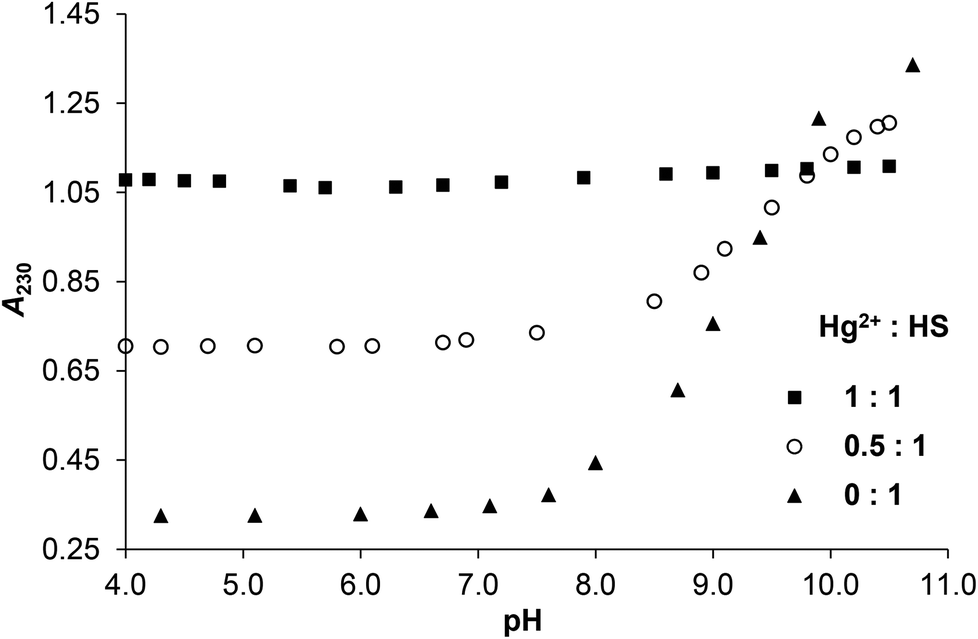 | ||
| Fig. 1 Change of the measured absorbances at 230 nm as a function of pH in the HgII:HS 1:1, 0.5:1 and 0:1 systems (cHS = 1.0 × 10−4 M, I = 0.1 M NaClO4, T = 298 K). | ||
The pH-dependent absorbances detected for the sample containing 0.5 equivalent of HgII compared to HS are in between the values observed for the ligand alone and the HgII–HS 1:1 system at any pH (Fig. 1). This suggests that ∼50% of the cysteine residues are bound to HgII even under acidic conditions and the remaining thiol groups deprotonate in parallel with the free ligand. The spectra recorded in the presence and absence of HgII reflect that the S− → HgII charge transfer transitions are located below λ = 220 nm (ε215 nm ∼ 15900 M−1 cm−1) independently of the pH and metal ion to ligand ratio (see the difference spectra of HgII–HS 1:1 and the free ligand in Fig. S3†). Such high energy LMCT transitions and the observed molar absorbances imply that two thiolates are coordinated to the metal ion, as proposed in previous reports on HgII – oligopeptide model systems.38–42 Three or four HgII-bound thiolates in a trigonal/tetrahedral coordination geometry would result in LMCT peaks or shoulders at lower energies25,31,35,40,42–44 which is not observed here even in the excess of HS over HgII indicating that metal ion bridged species are not formed.
In contrast to HgII, the LMCT band characteristic for S−–ZnII interactions in zinc(II)-bound proteins32–34 and peptides45–49 emerges only above pH ∼ 5.0 in the solutions of ZnII and HS (Fig. 2 and S4A–B†), reflecting the expected, substantially weaker affinity of ZnII towards the ligand. A remarkable spectral change, i.e. a further absorbance increase occurs above pH ∼ 7.5 in the presence of one equivalent ZnII per HS. A similar, but less pronounced spectral change, attributed to the formation of hydroxo mixed ligand species, was also observed in the ZnII-complex of a related 12-mer peptide,50 however, at a higher pH. Thus, the metal bound water appears to display a lower pKa of 8.65 in the ZnII-HS complex, vide infra (potentiometric data).
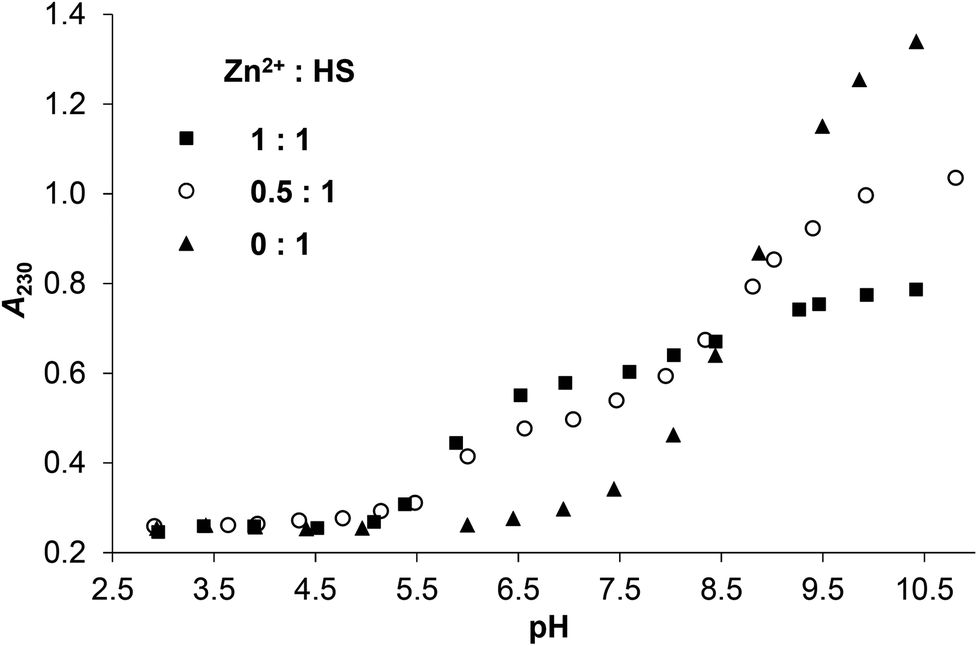 | ||
| Fig. 2 Change of the measured absorbances at 230 nm as a function of pH in the ZnII:HS 1:1, 0.5:1 and 0:1 systems (cHS = 1.0 × 10−4 M, I = 0.1 M NaClO4, T = 298 K). | ||
The A230 nmvs. pH curve obtained for the ZnII–HS 0.5:1 sample runs in between those of the free peptide and the equimolar system in the whole studied pH-range (Fig. 2). The observed profile is closer to that seen in the presence of 1 eq. ZnII between pH 5–9, contrary to the data recorded for HgII. Thus, a more complex speciation must occur for ZnII, with more than half of the thiolates bound to the metal ion at a stoichiometry of 0.5:1 ZnII:HS, indicating the formation of metal bridged species. At high pH, however, the absorbances detected for twofold ligand excess seem to be ca. the averages of those of the free ligand and the equimolar sample (see Fig. 2, S2 and S4†), suggesting similar speciation at any metal ion to ligand ratios.
In order to gain information on the metal ion induced conformational change of the peptide SRCD (synchrotron radiation circular dichroism) spectra were recorded both for HgII and ZnII complexes. Previously we have demonstrated that HS displays a disordered structure with varying levels of transient helicities,13 represented by an intense negative CD-extremum slightly below 200 nm and a less intensive shoulder around 220 nm.34,47,51,52 Addition of HgII to the acidic solution of HS results in a notable decrease of the negative peak at λ ∼ 198 nm while the shoulder is less affected (Fig. 3). A similar type of change was reported to accompany the HgII-coordination of a 18-mer peptide, comprising the metal binding loop of MerP possessing a CAAC motif.53,54 The spectral change was assigned to the folding of the peptide to a thermodynamically (but not necessarily kinetically) stable conformation,53 although the reduction of the negative ellipticity around 200 nm was also observed with other metal ions and two other peptide derivatives with alterations in the metal binding sequence (CCAA and CACA).54 By all accounts, HgII-binding to HS clearly induces a conformational change of the ligand towards a loop structure, presumably similar to the metal-loaded forms of CueR.8 One, however, has to bear in mind that due to the high energy ligand to metal charge transfer bands of the HgII-bound species, CD features of these bands may overlap with the backbone-related CD-effects. This is a known problem in the interpretation of the secondary structures of metalloproteins and metal ion–peptide complexes,34,45,55,56 particularly when relatively small molecules, like the present 12-mer HS peptide, are studied. Distinction of the different contributions may be easier when thiolate to metal ion transitions appear separately at lower energies compared to the peptide backbone bands, like in the tetrahedral {CysS4} type HgII-rubredoxin complex31 or in metallothioneins, where metal induced bands dominate the wavelength region above 220–230 nm.57 Comparison of the SRCD spectra of HS at pH ∼ 2.0 in the presence and absence of HgII (Fig. 3) suggests that any effect of the HgII-binding of the thiolate donors dominate below λ ∼ 210 nm. The increase of pH has practically no further effect on the ellipticity around 198 nm for the HgII-HS complex, however, it slightly influences the lower energy shoulders. This can be assigned to the deprotonation of the Asp and His residues of the peptide inducing modest changes in the backbone of the loop-forming ligand.
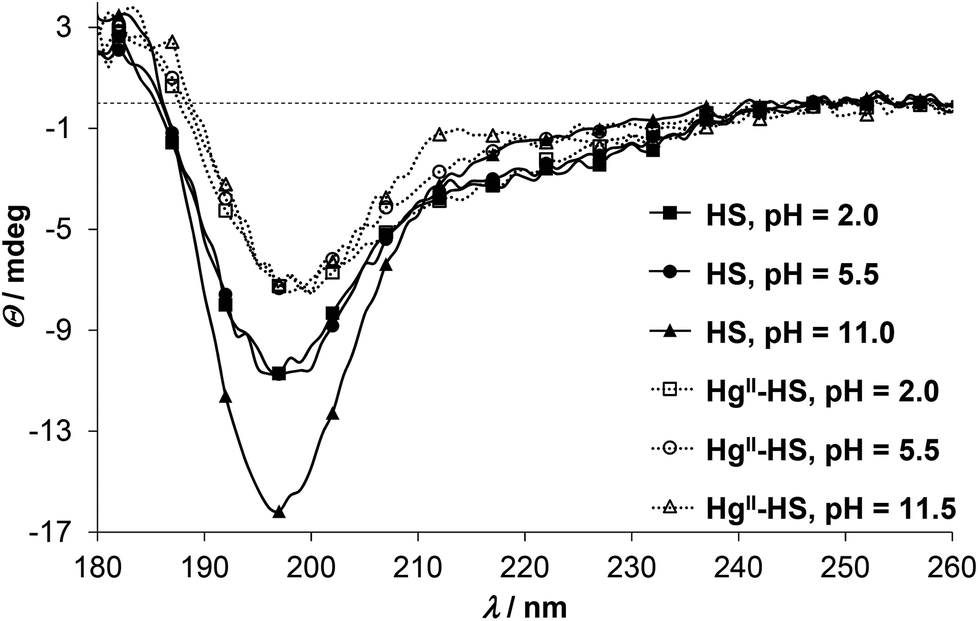 | ||
| Fig. 3 SRCD spectra of HS in the absence (continuous lines with filled markers) and presence of 1.0 eq. of HgII (dotted lines with open markers) at selected pH values (cHS = 1.0 × 10−3 M, I = 0.1 M NaClO4, T = 298 K, l = 0.1 mm). | ||
ZnII has no impact on the SRCD spectra of HS up to pH 5.5 (Fig. 4), which correlates well with the UV-spectra where the LMCT bands emerge above pH ∼ 5. At higher pH, however, the position of the main negative CD-minimum is slightly red-shifted (see spectra at pH = 7.5 and 10.5 on Fig. 4), while the ellipticities around 180 and 230 nm are remarkably increased, as compared to the spectra of the free ligand. As hinted already for HgII, influences of the S−–Zn2+ chromophore and the peptide secondary structure may be superposed in the observed CD-pattern of ZnII-protein/peptide structures.34,55,56 Nevertheless, the direction of the observed changes is rather similar to the ZnII-induced effects on the conformation of a phytochelatin analogue47 and other relatively short oligopeptides52,58 and may suggest an increasing helical content47,52 in the ZnII-bound HS. It was proposed that different coordination properties of metal ions may develop selectivity in the stabilization of the α-helical conformation of 20-mer peptides.58 The fundamentally distinct CD-features of HS in the presence of HgII and ZnII may imply that the different coordination geometry preference of the two metal ions promote large dissimilarity between the HgII- and ZnII-bound structures of the ligand. The characteristic shoulder seen in the spectra of ZnII-HS (Fig. 4 and S5†) starts to develop from ca. pH 6 (data not shown) but increases up to pH 9.5–10. The ZnII:HS ratio dependence of the discussed CD-peak at pH 10.5 reflects a simple equilibrium between the free and ZnII-bound HS (Fig. S5†).
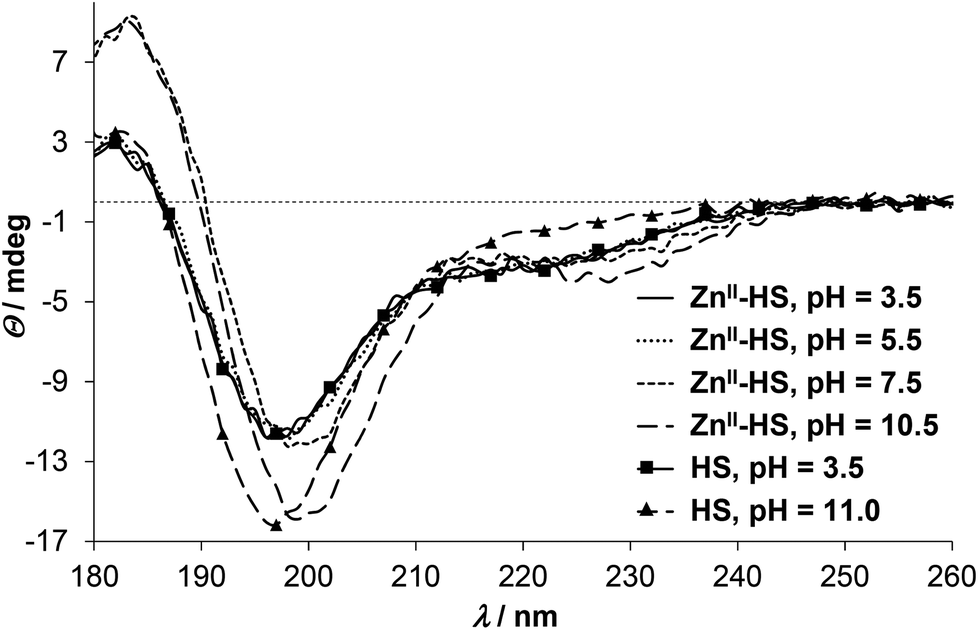 | ||
| Fig. 4 pH-Dependent SRCD spectra recorded in the ZnII–HS 1:1 system. pH = 3.5: continuous line; pH = 5.5: dotted line; pH = 7.5: short dashes; pH = 10.5: long dashes. For comparison, spectra of the free ligand is also shown at pH = 3.5 (continuous line with squares) and 11.0 (long dashes with triangles) (cHS = 1.0 × 10−3 M, I = 0.1 M NaClO4, T = 298 K, l = 0.1 mm). | ||
199mHg PAC spectroscopy for the elucidation of the coordination environment of HgII
The local environment and coordination geometry of HgII was also monitored by 199mHg PAC (perturbed angular correlation of γ-rays) spectroscopy in the presence of one equivalent metal ion at pH = 2.0 and pH = 8.0. The fundamentals of PAC spectroscopy and the interpretation of the parameters obtained by the technique are described in detail in the literature.59 The PAC data may be analyzed with one nuclear quadrupole interaction (NQI) at each pH, and the PAC parameters (νQ, the nuclear quadrupole coupling constant, and η, the so called asymmetry parameter, which is zero for an axially symmetric coordination geometry) for the observed NQIs are collected in Table 1. The fitted νQ and η values are similar at pH 2.0 and 8.0 and comparable to literature data obtained for compounds with two-coordinate {HgS2} structures60,61 (Table 1). The spectrum recorded at low pH is slightly more complex than that obtained at pH ∼ 8.0 as reflected in the lower signal amplitude and the broader and less visible second and third peaks, respectively (Fig. 5). This may suggest the co-existence of a small amount of species with a different structure, nevertheless, the main spectral features, with a support of UV-data, clearly indicate that the major component has a two-coordinate {HgS2} coordination mode.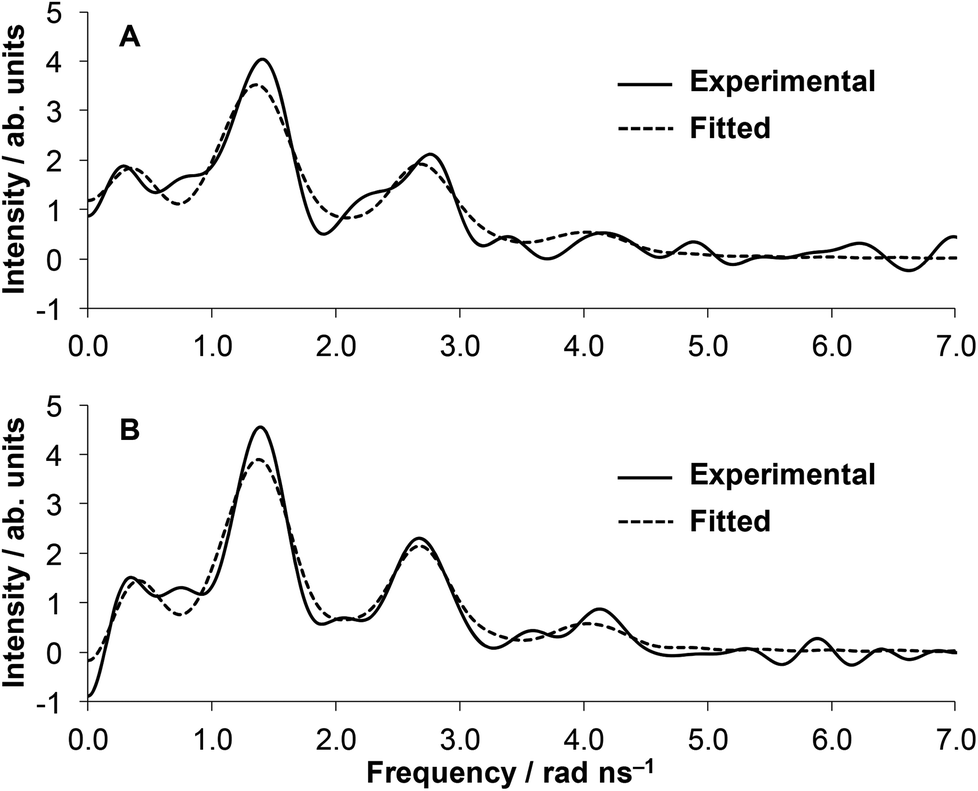 | ||
| Fig. 5 Fourier transformed experimental (solid lines) and fitted (dashed lines) 199mHg PAC data of the HgII:HS 1:1 system at pH = 2.0 (A) and pH = 8.0 (B) (cHgII = cHS = 8.03 × 10−5 M). | ||
:HS and for different HgII–thiolate complexes of known structures
| System/pH | ν Q/GHz | η | Coordination geometry | Ref. |
|---|---|---|---|---|
| HgII-HS 1:1 (pH = 2.0) |
1.43(5) | 0.07(6) | Two-coordinate, 2 thiolates | This work |
| HgII-HS 1:1 (pH = 8.0) |
1.43(1) | 0.13(3) | Two-coordinate, 2 thiolates | This work |
| [Hg(Cysteine)2] | 1.41 | 0.15 | Two-coordinate, 2 thiolates | 60 |
| Ac-Cys-dPro-Pro-Cys-NH2 | 1.42 | 0.19 | Two-coordinate, 2 thiolates | 41 |
| MerA (77 K) | 1.42 | 0.15 | Two-coordinate, 2 thiolates | 61 |
| MerR (77 K) | 1.18 | 0.25 | Three-coordinate, 3 thiolates | 61 |
| Hg-rubredoxin | 0.10 | 0 (fixed) | Four-coordinate, 4 thiolates | 31 |
Potentiometric investigation of distribution and stabilities of the species formed in the ZnII![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :HS system
:HS system
The formation constants (logβ) determined for the proton and ZnII complexes of HS are summarized in Table 2.
β) of the ZnII complexes of HS (estimated errors in parentheses (last digit)) and derived equilibrium data (I = 0.1 M NaClO4, T = 298 K)
| Speciesa | pqr | logβpqr |
pKpqrc, logK2d |
|
|---|---|---|---|---|
|
a logβ values for the protonation processes of HS, re-determined in the present study are: logβ051 = 31.68, logβ041 = 28.33, logβ031 = 24.10, logβ021 = 17.50, logβ011 = 9.06.
b
p,q, and r reflect stoichiometric numbers of the fundamental components the complex species are composed of, as defined in the experimental section.
c pKpqr = logβpqr − logβp(q−1)r.
d logK2 = logβ102 − logβ101.
e H−1 represents an extra deprotonation, beyond the proton releases of the peptide, i.e. deprotonation of a ZnII-bound water molecule.
f NP = number of points.
g FP = fitting parameter representing an average deviation in cm3 between the experimental and fitted data for the full data set (including all evaluated titrations).
|
||||
| [ZnHL]− | 111 | 16.58(4) | pK111 | 5.95 |
| [ZnL]2− | 101 | 10.63(4) | pK101 | 8.65 |
| [ZnH−1L]3−e |
1–11 | 1.98(5) | ||
| [ZnH2L2]4− | 122 | 31.3(2) | pK122 | 7.6 |
| [ZnHL2]5− | 112 | 23.7(1) | pK112 | 8.7 |
| [ZnL2]6− | 102 | 15.0(2) | ||
| NPf | 544 | logK2L |
4.37 | |
| FPg(cm3) | 0.005 | |||
The ligand undergoes five (de)protonation processes in the studied pH-range that were attributed to the carboxylate groups of two Asp residues (pH ∼ 3–5), the imidazole side chain of His (pH ∼ 6–7.2) and the thiol moieties of the two Cys units (pH ∼ 7.8–9.7).13 The deprotonation constants (pKa) of the ligand have been re-determined for the present study and are in a good agreement with those published earlier.13
In Table 2 the species model obtained by best fit of the ZnII:HS system titration curves is presented. Introducing bis-ligand complexes (ZnHxL2) in the model was necessary for the correct description of titration data when HS was used in excess over ZnII (see Experimental). Contrary to this, considering the presence of dinuclear species (Zn2HxL), did not improve the fit of the experimental data neither for the ZnII:HS 0.5:1 and 1:1 samples nor for those containing a two-fold ZnII-excess over the ligand (the latter was evaluated only up to pH ∼ 7).
Complex formation processes start from pH ∼ 4.5 by the appearance of a protonated mono-complex ZnHL as reflected by the calculated species distributions (Fig. 6). A consecutive deprotonation process ZnHL → ZnL + H+ leads to the formation of the parent complex ZnL where all of the dissociable protons of the peptide are already released. The pKa value for this process (= 5.95, see Table 2) is significantly lower than those attributed to the deprotonation processes of the HL and H2L forms of the free ligand (pKHL = 9.06, pKH2L = 8.44) and somewhat below the third pKa of HS (pKH3L = 6.60). This strongly suggests that at least two, but potentially all the three neutral/basic donor groups of the ligand (histidine imidazole and two cysteine thiolates) are bound to ZnII in the ZnL species. Coordination of both cysteines to ZnII in ZnL is also supported by the observed absorbance increase in parallel with the formation of ZnHL/ZnL (A230 traces are overlaid with species distributions calculated for the concentration of UV data, see Fig. S6A–B†).
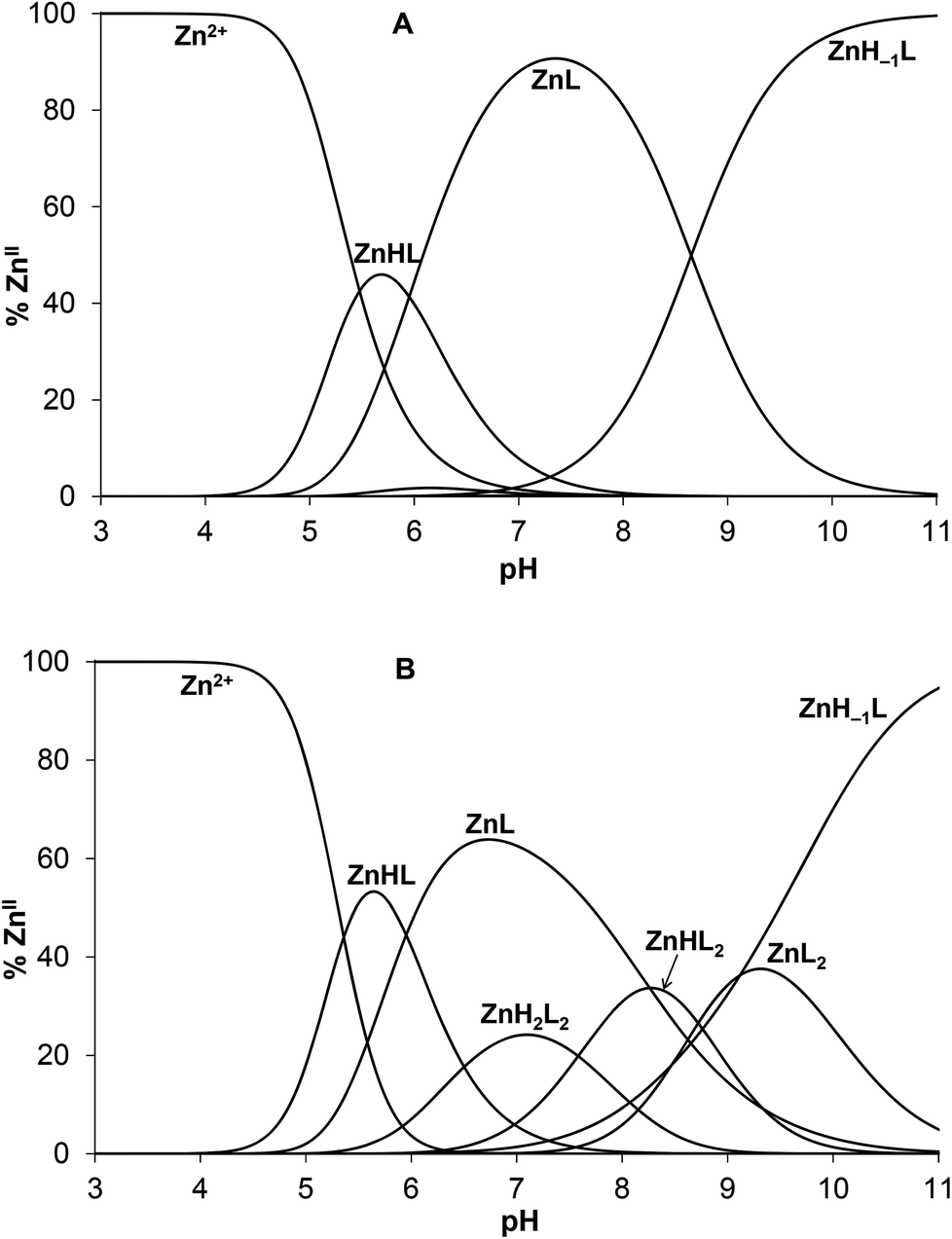 | ||
| Fig. 6 Species distribution diagram for the ZnII:HS 1:1 (A) and 0.5:1 (B) systems (cHS = 1.0 × 10−3 M). The speciation curves for the concentrations applied in the UV experiments are depicted in ESI (Fig. S6A–B†). | ||
The determined stability of ZnL (logK = 10.63) reflects a remarkable affinity of HS to ZnII. This stability constant is, indeed, several orders of magnitude higher than those of the parent ZnII complexes of shorter peptides containing a CXH motif,62 but also surpasses the stabilities of terminally protected tripeptides composed of a CXC sequence,62 in spite of the substantially longer peptide chain and the larger distance between the two Cys residues in HS. Besides, HS has a notably higher affinity to ZnII compared to a similar 12-mer oligopeptide possessing no histidine residues (studied by us, logK = 9.93).50 Although higher stabilities were found for the ZnII complexes of some 10-mer peptides, all of these contained 2–3 histidines in addition to the two cysteine units.63 Thus, the affinity of HS for ZnII falls in range that indicates the coordination of both cysteine and the histidine residues to the metal ion. The ZnII-binding affinity of HS can also be demonstrated by the conditional stability calculated at pH 7.4 and 1:1 metal ion to ligand ratio based on the equations below,
| HqL + Zn ⇄ ZnHqL |
where Zn denotes the free ZnII concentration while HqL and ZnHqL represent the overall concentration of the free and complexed ligands in any protonation states, respectively. The apparent stability constant for the above conditions is Ka = 7.5 × 107 (log
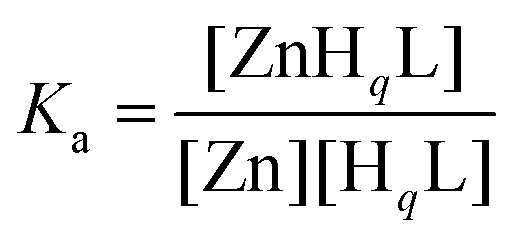 Ka = 7.9) which is in the lower range of affinities reported for various wild-type bacterial ZnII-regulators64 or variants.65
Ka = 7.9) which is in the lower range of affinities reported for various wild-type bacterial ZnII-regulators64 or variants.65
The deprotonation of ZnL above pH ∼ 8 (Fig. 6A) leads to the species ZnH−1L being strongly dominant under alkaline conditions. The observed extra deprotonation is most likely not a ligand-related proton release since the formation of a ZnII–amide bond is a very scarce event in the complexes of ZnII formed with terminally protected peptides.62,66–69 Accordingly, the ZnH−1L composition may represent a species with a deprotonated water ligand, described as Zn(OH)L. The pKa value of the deprotonation process is 8.65 that is ca. 1.7 log units lower than the pKa determined for the same type of proton release of the CdII complex of HS,13 as expected, due to the smaller ionic radius of ZnII as compared to CdII. The deprotonation of the bound H2O occurs also at a somewhat lower pH than in the ZnL complex of a similar ligand containing no His residue in position 3 of the peptide chain (pKa = 9.1150). The {Zn(Cys)2HisH2O/OH−} coordination sphere is also found in horse liver alcohol dehydrogenase (LADH), where the pKa of the metal ion bound water molecule is 9.2 for the native ZnII containing enzyme,70 and 11.0 for the CdII substituted species.71 Interestingly, the pKa of the ZnII-bound water is lower in HS than in LADH. It seems that above neutral pH the histidine of HS significantly influences speciation, the coordination sphere of ZnII and even the peptide structure, as indicated by UV and SRCD data.
Monomeric ZnHL and ZnL complexes dominate in the acidic/neutral pH-range when HS is present in a twofold excess over ZnII (Fig. 6B). As indicated by UV data, metal-bridged bis-ligand species with different protonation states are also formed above pH ∼ 6. Although the determined stabilities of the various bis-complexes do not provide direct information on the binding mode of the ligands, the relatively high pKa value for the ZnHL2 → ZnL2 + H+ process (= 8.7, Table 2) suggests that there are protonated thiol groups in the ZnH2L2 and ZnHL2 species. The stability constant calculated for the binding of the second ligand in ZnL2 (logK2 = 4.37) and the relative stability of the parent mono- and bis-complexes (log(K1/K2) = 6.26) shows a notably weaker binding of the second ligand as compared to the same process in the CdII:HS system (log(K1/K2) = 5.3313) or to the ZnII-binding of the above cited His-free peptide (log(K1/K2) = 5.1450). This finding provides a further support for the important role of histidine in controlling the interaction of ZnII with HS.
1H NMR experiments
Assignment of the 1H NMR resonances of HS and the pH-dependence of the recorded spectra in the absence of metal ions were published previously.13 HgII coordination to the peptide has a strong effect on the resonances of the Cys CβH2 protons (Fig. 7). These signals shift from 2.93 ppm to relatively broad peaks at ∼3.3–3.4 ppm (in an accidental overlap with one of the His CβH2 resonances at pH 4.0–6.0) in the presence of one equivalent of HgII. The significant, ca. 0.4 ppm downfield shift of the Cys CβH2 resonances of the bound ligand, as compared to the same signals of the free HS indicates the binding of both thiolates to the metal ion, as also suggested by UV titrations.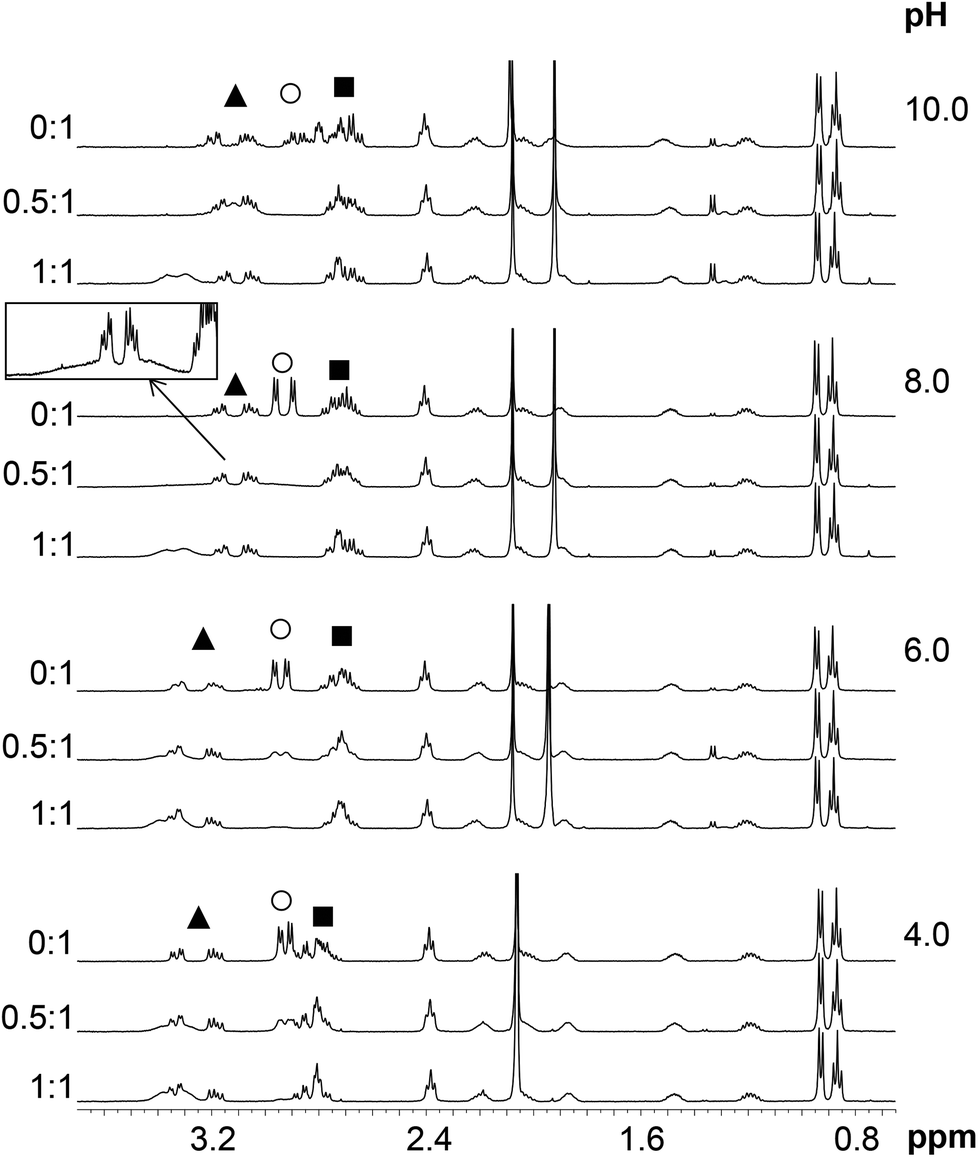 | ||
| Fig. 7 Selected regions of the 1H NMR spectra of HS recorded in the absence and presence of 0.5 and 1.0 eq. of HgII (H2O/D2O = 90:10% v/v, cHS = 1.3 × 10−3 M, T = 298 K). The resonances marked by symbols are: His CβH2: ▲; Cys CβH2: ○; Asp CβH2: ■. The region of δ = 2.7–3.6 ppm from the HgII:HS 0.5:1 spectrum at pH = 8.0 is magnified in the frame. Note that the sharp signals at δ ∼ 1.9 ppm are those of the acetate anion of the added mercury(II) salt which coincide with the acetyl protecting group resonances of HS at pH ∼ 4.0. | ||
Two separate signal sets of the Cys CβH2 protons are observed at pH 4.0–6.0 when HS is in a twofold excess over HgII (Fig. 7) One set is reminiscent of the resonances of the free ligand, whereas the other coincides with those observed in the HgII:HS 1:1 system. Increasing pH to 8.0 results in coalescence of the two signal sets to a very broad bulge-like feature in the range of δ ∼ 2.8–3.4 ppm overlapping with the His CβH2 resonances (Fig. 7). This coalesced signal, with a chemical shift found in between those observed for HgII:HS 1:1 and the free ligand, becomes sharper on increasing pH but is still broad at pH = 10.0. These findings indicate that the ligand exchange rate between the free and bound forms gradually increases from the slow/intermediate to the intermediate/fast time regime in parallel with the deprotonation of the unbound thiol groups of the presumably free ligand being present in the HgII:HS 0.5:1 system.
The exchange rate, kex, between the bound and non-bound ligand forms may be roughly estimated from the observed line-broadening72 at pH 4.0–6.0 which is dominated by slow exchange. The line-broadening, we–w0, occurring for the Cys CβH2 resonances of the free peptide due to the addition of 0.5 eq. HgII is ca. 12 Hz which leads to kex ∼ π × (we − w0)∼ 38 s−1 at pH = 6.0 (we and w0 represent the line width of signals at half height with and without exchange, respectively). The calculation is based on the assumption of a two-site exchange of HS between a specific HgII-peptide bound form and the non-bound form under the applied experimental conditions. kex may also be expressed by a formulae involving the rates of the association and dissociation processes, as follows73
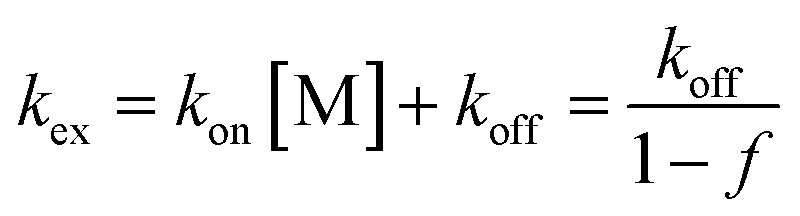
The increase of pH also induces the upfield shift of the resonances of the Asp (CβH2 protons – Fig. 7) and His (CβH2 – Fig. 7 and the Cε1H and Cδ2H protons of the imidazole ring – Fig. 8) reflecting the deprotonation of the side chains of these residues. The chemical shift values are practically independent of the metal ion to ligand ratio at all selected pH values. These findings indicate that the proton releases from the Asp carboxyl groups and the His imidazole moiety are practically unaffected by the presence of HgII and therefore that these groups do not participate in HgII-binding. Nevertheless, coordination of the cysteine residues to HgII has a slight line width increasing effect on the neighbouring Asp side chain resonances under acidic conditions (Fig. 7) and a rather pronounced impact on the His Cε1H and Cδ2H signals in neutral/alkaline solutions (Fig. 8). This shows that although the chemical shifts, apart from those of the cysteines, do not change significantly, the dynamics of the peptide is affected by the binding of HgII.
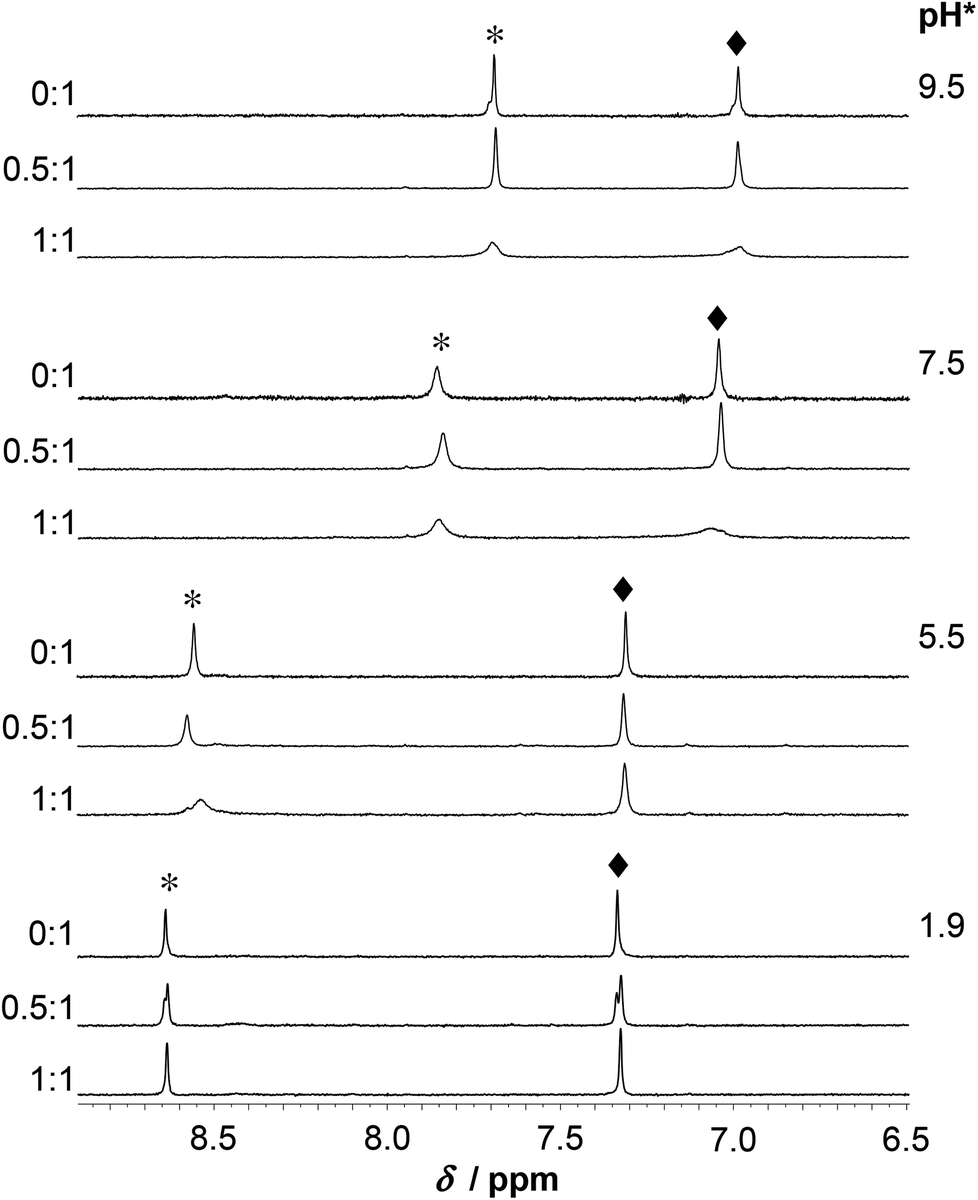 | ||
| Fig. 8 Part of the 1H NMR spectra of the peptide HS recorded at various pH* values in D2O as a function of the HgII to peptide ratio (cHS = 1.3 × 10−3 M, T = 298 K). The symbols denote the Cε1H (*) and Cδ2H (◆) resonances of His. | ||
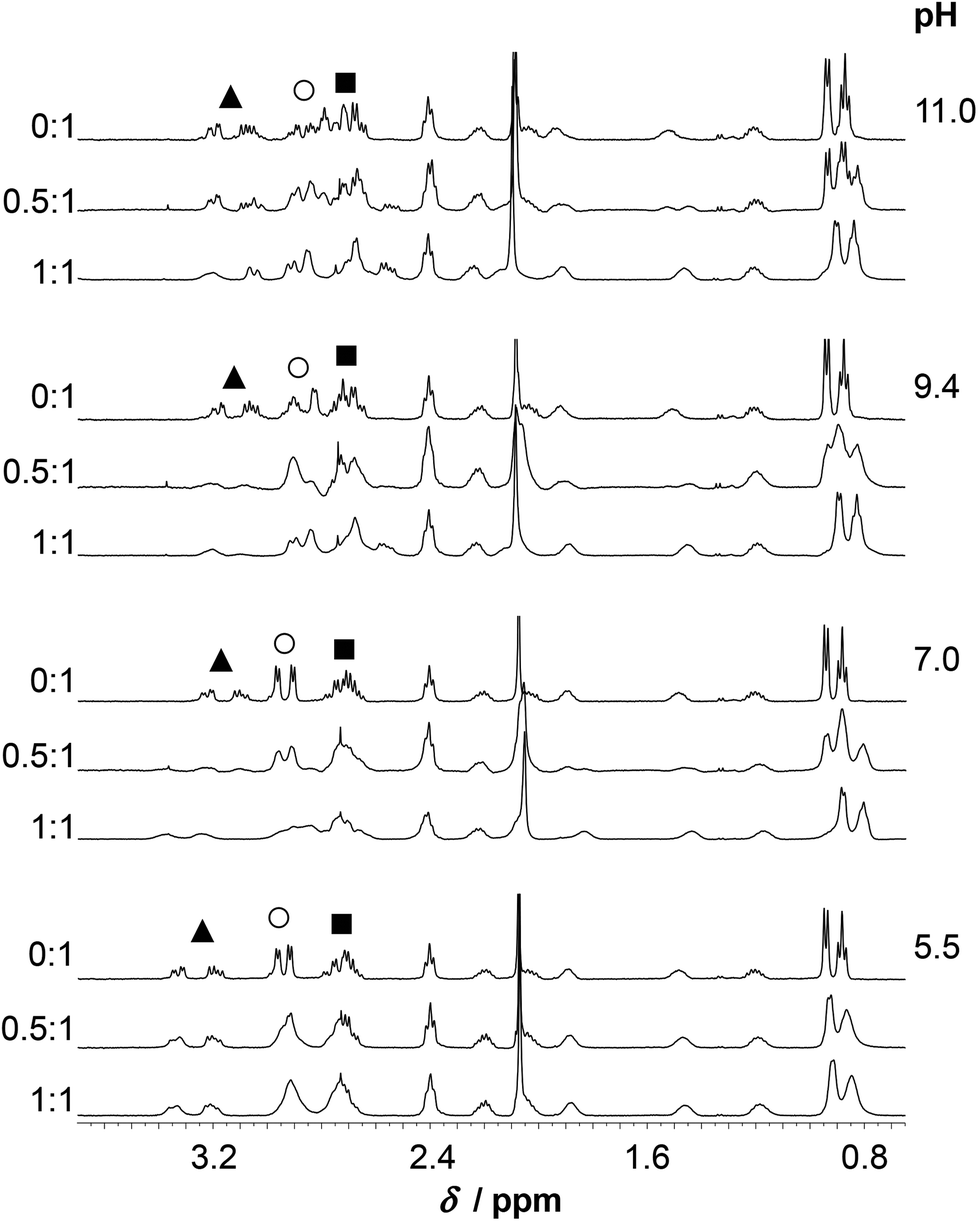
The spectra of HS obtained at pH ∼ 4.4 in the presence and absence of ZnII reflect no differences either in terms of the chemical shifts or the shape of the various 1H-resonances (Fig. S7†). This suggests that, as opposed to HgII, ZnII is not bound to HS under such conditions, which is in agreement with the potentiometric and UV absorption studies, vide supra. At pH ∼ 5.5, however, the presence of ZnII gives rise to pronounced broadening of most resonances. In the presence of 0.5 eq. ZnII this may indicate exchange between the bound and free states of the peptide, but in the fully loaded ZnII:HS system it implies equilibria between conformers falling into the intermediate exchange time regime (ms–s) (Fig. 9–10). At a 1:1 ratio of ZnII and HS the Cε1H and Cδ2H signals of the His imidazole are shifted slightly upfield as compared to the resonances of the free ligand (Fig. 10). At 0.5 eq. of ZnII the chemical shifts of the imidazole ring protons appear in between those of the free HS and the 1:1 system reflecting an equilibrium between the non-bound and metal-bound peptide forms, and fast exchange dynamics for these resonances. The Cε1H and Cδ2H resonances are significantly shifted upfield by a further pH increase (pH 5.5 → 7.0), similarly to the metal ion free solution, which indicates that His-coordination is not completed at pH 5.5. A combined interpretation of the 1H NMR, UV absorption, and potentiometric data at pH ∼ 5.5, (see Fig. 6) leads us to propose co-existing binding isomers of the ZnHL species, with the participation of two Cys-thiolates or one of the Cys-thiolates and the His side chain in metal ion binding.
 | ||
| Fig. 9 Aliphatic region of the 1H NMR spectra of HS recorded in the absence of ZnII and in the ZnII:HS 0.5:1 and 1:1 systems (H2O/D2O = 90:10% v/v, cHS = 1.3 × 10−3 M, T = 298 K). CβH2 resonances of the residues with potential donor groups are indicated by the following symbols: His CβH2: ▲; Cys CβH2: ○; Asp CβH2: ■. | ||
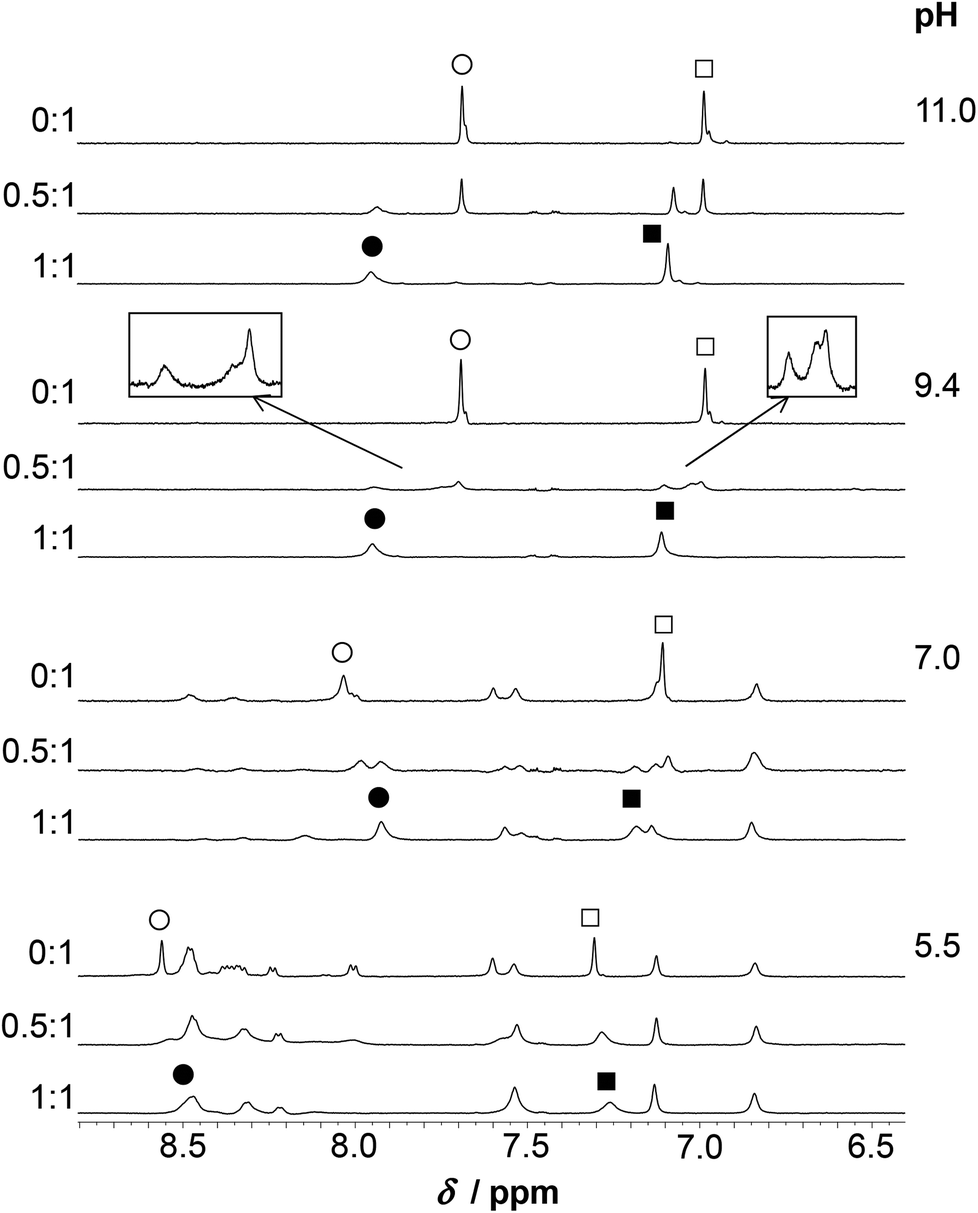 | ||
| Fig. 10 Aromatic/HN region of the 1H NMR spectra of HS recorded in the absence of ZnII and in the ZnII:HS 0.5:1 and 1:1 systems (H2O/D2O = 90:10% v/v, cHS = 1.3 × 10−3 M, T = 298 K). The open circle and square symbols show resonances of the non-bound ligand (Cε1H and Cδ2H, respectively) while the filled symbols mark the same resonances of the bound His residues in the mononuclear species. The regions of δ = 6.9–7.15 and 7.4–8.05 ppm from the ZnII:HS 0.5:1 spectrum at pH ∼ 9.4 are magnified in the frames. | ||
The increase of pH to pH ∼ 7.0 gives rise to a substantial change of the spectral pattern. According to our data, all the metal ions are complexed under such conditions (Fig. 6). Most of the resonances, in addition to those of the Cys residues, display line broadening, in contrast to the signals observed for HgII:HS, where resonances from non-coordinating groups are not affected to the same extent. Thus, ZnII-coordination affects the internal dynamics of the entire peptide on the NMR time scale. Additionally, the ligand exchange dynamics is slowed down to the moderately slow exchange time regime causing the splitting of several 1H resonances (CβH2, Cε1H and Cδ2H of His and all the resonances of Ile) into clearly distinguishable separate signals at a 0.5:1 ZnII:HS ratio (see spectra at pH ≥ 7.0 on Fig. 9–10). The decrease of exchange rate by pH-increase coincides with a remarkable change of the CD-signals (Fig. 4), and occurs in parallel with the formation of the ZnL parent complex. It implies that the participation of several donor groups in metal ion binding leads to a reduced lability of species. At a 1:1 ZnII:HS ratio, the CβH2 protons of the Cys and His residues experience a significant chemical shift change relative to the free ligand, as do the Cε1H and Cδ2H signals of the imidazole ring (Fig. 10). This supports the coordination of the two Cys-thiolates and the His-imidazole groups to ZnII in ZnL, but the poorly resolved spectrum at pH = 7.0 does not provide information on the binding of Asp-carboxylates. As pointed out above, various resonances of the C-terminal Ile residue in the spectral region 0.8–1.0 ppm (CδH3, Cγ2H3) are also strongly affected by metal ion coordination as those of the bound ligand are clearly shifted upfield compared to the ones of the free peptide-like resonances (ZnII:HS 0.5:1, Fig. 9). Analogous spectral features were not observed in the systems of either CdII and HS13 or ZnII and a closely related peptide50 differing only in the His-residue from the presently studied ligand. Thus, while the exact origin of the impact of ZnII-binding on the Ile resonances is not clear, metal ion coordination of the histidine unit very likely plays a key role here.
Based on the observed line broadening of the Cys CβH2 protons at ZnII:HS 0.5:1 (Fig. 9) a similar or slightly lower exchange rate between the bound and non-bound ligands, as compared to HgII:HS, may be predicted. However, the overlap of the various resonances and the complexity of the system (see the distribution curves at pH ∼ 7.0, Fig. 6B) do not allow a deeper discussion. It is, however, an interesting contrast to HgII:HS, that the exchange rate in the presence of ZnII remains relatively slow even at higher pH (see below) approaching the deprotonation-range of the thiol groups of the free ligand. The increased metal ion exchange rate, as observed by the resonances of the Cys CβH2 protons (see Fig. 7) with 0.5 eq. HgII for pH above the pKa of the thiols, imply that the free thiolates take part in the exchange process, and thus that it occurs via an associative mechanism. The low coordination number may be important for this process, as it may allow for coordination of additional thiolates in the equatorial plane. This is analogous to a proposed mechanism of transfer of CuI between proteins,42,75,76 where the metal ion is also found in a structure with two thiolates coordinating. Contrary to this, the metal exchange rate does not change into the fast exchange regime with 0.5 eq. ZnII for pH above the pKa for the thiols, see Fig. 9. This may reflect that the exchange occurs via a dissociative mechanism, although not necessarily via free ZnII, in analogy to the common interpretation of ligand binding reactions for the ZnII aqua ion involving dissociation of coordinated water as the rate determining step.77
As a conclusion, and in line with SRCD data, the simultaneous binding of (at least) three side chain donors induces a more defined ligand structure in the ZnII-bound HS, unlike the loop-like conformation proposed for HgII:HS.
At pH ∼ 9.4 the spectra of the ZnII-containing solutions are still very poorly resolved as the resonances are strongly broadened. Coordination of His to the metal ion in the ZnII:HS 1:1 system is unambiguously demonstrated by the significant downfield shift of the Cε1H and Cδ2H resonances, as compared to the metal ion free sample (Fig. 10). A similar shift was observed and attributed to His-coordination in the CdII-complex of the peptide.13 At least three distinguishable, broad Cδ2H peaks and three Cε1H peaks, albeit less clearly, are observed at 0.5:1 ZnII:peptide ratio (see the enlarged spectrum segments on Fig. 10). One of the Cδ2H and Cε1H peaks appear very close to the free ligand-like signals while the third observed signals have chemical shifts resembling those measured for the 1:1 system. In order to elucidate the processes occurring in the presence of ligand excess above neutral pH, a more detailed series of spectra were recorded at pH ∼ 8. One may follow the evolution of the Cε1H and Cδ2H signals from 0:1 to 1:1 ZnII:peptide ratio on Fig. 11. The series describe the complete transformation of the non-bound ligand to the 1:1 species (mostly ZnL at this pH, see Fig. 6A). The spectra recorded at the intermediate stages, featuring the emergence and transformation of broad peaks, originate from a dynamic exchange between at least three coexisting species (see e.g. the Cε1H resonances at a ZnII:HS ratio of 0.75:1 or the Cδ2H resonances at a ratio of 0.5:1), the free peptide, the fully loaded ZnII-peptide complex, and a species with a plausible 0.5:1 ZnII:HS stoichiometry, i.e. a bis-ligand complex. This provides strong support for the potentiometric data. The species are in slow to intermediate exchange rate relative to the NMR timescale. The emerging signals in the range of δ ∼ 7.0–7.08 and 7.75–7.85 ppm suggests that the His-imidazole moiety of at least one of the two ligands plays a role in ZnII-coordination in some or all of the bis-complexes.
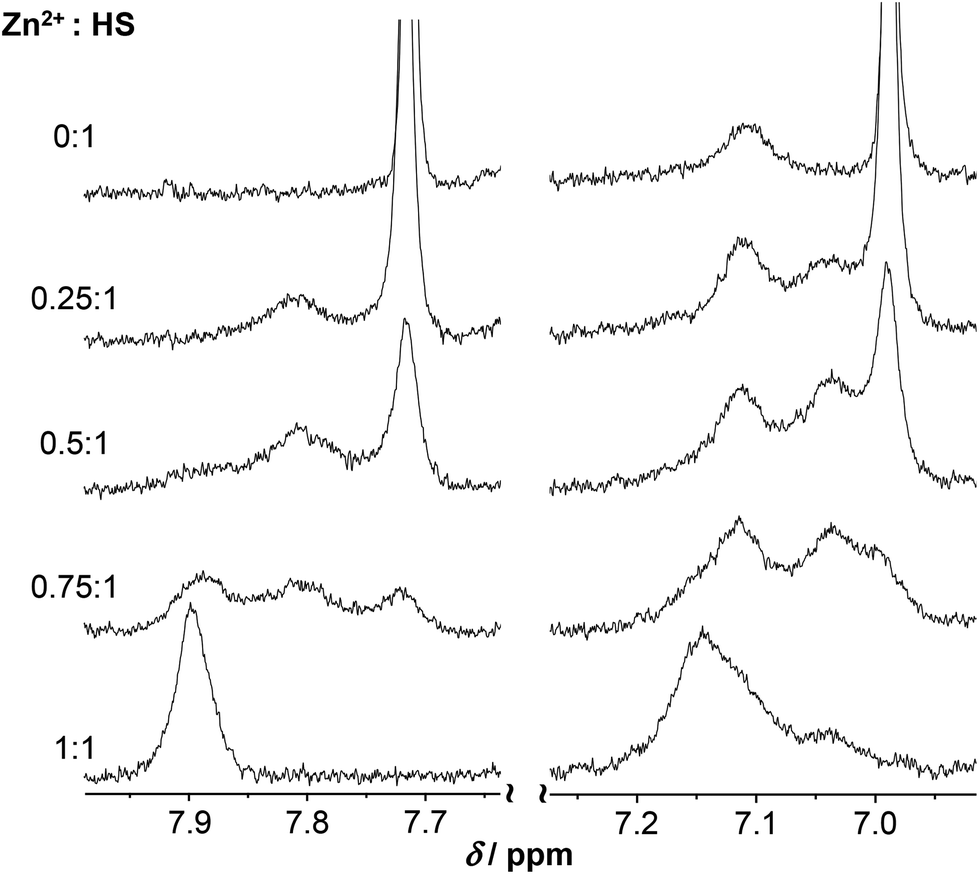 | ||
| Fig. 11 Part of the 1H NMR spectra of HS recorded at pH ∼ 8.0 as a function of the metal ion to peptide ratio (H2O/D2O = 90:10% v/v, cHS = 1.3 × 10−3 M, T = 298 K). The selected regions reveal changes observed on the Cε1H (left) and Cδ2H (right) resonance of the His-imidazole moiety of the ligand. Note that the peak seen at δ ∼ 7.11 ppm on the free HS spectrum is one of the remaining amide resonances still observable at pH ∼ 8.0. | ||
Only two sets of relatively sharp Cε1H and Cδ2H resonances are detected at pH ∼ 11 in the ZnII:HS 0.5:1 system (Fig. 10) which is in excellent correlation with the equilibrium and UV results, i.e. the presence of only the ZnH−1L complex and free ligand. Besides, the notable chemical shift changes observed on the spectra of ZnII:HS 1:1 from pH ∼ 7.0 up to 11.0 (see e.g. the range of δ ∼ 3.5–2.5 ppm (Fig. 9) or the Cδ2H signals (Fig. 10), clearly reflect the conversion of the ZnL parent complex to ZnH−1L.
Conclusion
The 12-mer HS peptide, inspired by the C-terminal metal ion binding domain of a bacterial metalloregulator CueR, is shown to efficiently bind HgII and ZnII, two metal ions with significantly different coordination preferences. HgII is demonstrated to form a loop-like structure in a {HgS2} coordination fashion via binding to the two cysteinates of the ligand, but there is no sign for the participation of any other side chain donors in HgII-coordination. The kinetic lability of HgII is manifested in line broadening on the 1H NMR spectra affecting mostly the resonances of the bound Cys residues and those of the neighbouring units. In contrast to HgII, ZnII dictates the peptide to a more structured form in its ZnL complex via binding to at least three side chain donors, the two Cys thiolates and the His imidazole. Indeed, the SRCD spectra above neutral pH might reflect an increasing helical content in the ZnII-bound HS, although a contribution of the thiolate–metal ion chromophore to the observed CD-pattern may also be present in the same wavelength range. In addition to monomeric species, bis-ligand HS-ZnII-HS complexes are also formed, unlike with HgII. The line broadening in 1H-NMR is pronounced for most of the resonances, indicating that exchange dynamics between different conformers occurs on the NMR time scale (ms–s) and that, in contrast to HgII, ZnII-coordination notably affects the internal dynamics of the entire peptide chain. The results obtained demonstrate that the conformational and coordination flexibility allows HS to adopt diverse structures, favoured by different metal ions, which is a property that may be utilized for metal ion sequestration in practical applications. Experimental studies on the interaction of a flexible peptide like HS with metal ions are a challenge as speciation may be diverse and the system dynamic. In the span between coordination compounds to metalloproteins, dynamic systems like HS, may yield insight into the underlying mechanisms of metal ion exchange that are necessary to account for transport and distribution of essential trace elements in biological systems. In a more fundamental perspective, also the potential role of metal ions for protein folding (and misfolding) through transient binding may also be elucidated by interrogating such peptide–metal ion interactions.Experimental
Materials
The investigated peptide N-acetyl-Ser-Cys-His-Gly-Asp-Gln-Gly-Ser-Asp-Cys-Ser-Ile-NH2 (Ac-SCHGDQGSDCSI-NH2, HS) was synthesized, as described earlier.13 Chemicals and solvents were obtained from Sigma-Aldrich and used without further purification unless otherwise described. The solutions of Zn(ClO4)2·nH2O, Hg(OAc)2 (Aldrich) were standardized complexometrically78 while precise weights of high purity HgCl2 (Aldrich) was used to prepare metal ion stock solutions. pH-Metric titrations were performed with NaOH (Aldrich) solutions standardized using potassium hydrogen phthalate (Sigma-Aldrich).Electronic absorption and SRCD measurements
UV–Visible (UV–Vis) spectra were measured on a Shimadzu UV-3600 UV-VIS-NIR spectrophotometer using a cell with 1 cm optical pathlength. Concentration of the ligand was 1.0 × 10−4 M and the metal ion concentration varied between 5.0 × 10−5 and 2.0 × 10−4 M.The synchrotron radiation CD (SRCD) spectra of the free ligand and the metal complexes were recorded at the SRCD facility at the CD1 beamline on the storage ring ASTRID at the Institute for Storage Ring Facilities (ISA), University of Aarhus, Denmark.79,80 All spectra were recorded with 1 nm steps and a dwell time of 2 s per step, using 0.1 mm quartz cells (SUPRASIL, Hellma GmbH, Germany), for the wavelength range of 175–260 nm. The substances were initially dissolved in 1.0 × 10−2 M HCl in order to avoid the eventual oxidation process. The pH of the samples (cpeptide = 1.0 × 10−3 M) were adjusted by adding the appropriate amount of NaOH solution. From the raw spectra the water baseline was subtracted and spectra were normalized to 1.0 × 10−3 M peptide concentration (to eliminate the effect of dilution).
Perturbed angular correlation of γ-rays
All perturbed angular correlation (PAC) experiments were performed in ISOLDE/CERN with a setup using six BaF2 detectors keeping the samples at a temperature of 1 °C. Production and purification of the radioactive 199mHg is described in the literature.81 The 199mHg solution (150 μL) was mixed with solutions of nonradioactive mercury(II) chloride, sodium perchlorate and buffer if needed. TRIS and CAPS buffers were used for adjusting the pH of samples to pH ∼ 8.0 and 10, respectively. The peptide was dissolved in 0.01 M perchloric acid and amounts of this stock solution were added to the buffered HgII-containing solutions to reach the desired final concentrations. Finally, sucrose was added to 55% w/w. The pH of the solutions was adjusted with NaOH and HClO4. In order to avoid contamination of the samples, small volumes were taken for pH measurements. The temperature dependence of the pH in the TRIS/CAPS-buffered solutions was taken into account and pH-values were corrected to 1 °C.82 The buffers and the peptide stock solutions were purged with argon. The final volume of the samples was 210 μL with cpeptide = cHgII = 8.03 × 10−5 M and cbuffer = 1.60 × 10−2 M.NMR experiments
1H NMR measurements were performed on a Bruker Avance DRX 500 spectrometer operating at 500.132 MHz. The spectra were recorded at T = 298 K in a mixture of H2O/D2O = 90:10% v/v and in a few cases in pure D2O applying the zgpr or zgcppr pulse sequences in order to presaturate the H2O/HDO resonances. In a typical sample the concentration of the peptide was 1.3 × 10−3 M. The chemical shifts were referenced to TSP-d4 at 0.0 ppm. Spectra were recorded using a recycle delay of 5 s, an acquisition time of 1.64 s, a spectral width of 5 or 10 kHz and 64–128 scans. In D2O, the pH* (pH-meter reading uncorrected by the deuterium effect) was adjusted to the desired values with NaOD. The recorded spectra were processed by the ACD/Spectrus Processor software.83
pH-Potentiometric measurements
The protonation and coordination equilibria were investigated in aqueous solutions (I = 0.1 M NaClO4, and T = 298.0 ± 0.1 K) under argon atmosphere with a special care to avoid the oxidation of the peptide. The potentiometric titrations were carried out by an automatic titration set including a PC controlled Dosimat 665 (Metrohm) autoburette, an Orion 710A precision digital pH-meter equipped with an Orion 8103BNUWP Ross Ultra semi micro pH electrode (165 × 6 mm). Conversion of the relative mV values as pH-meter readings to hydrogen ion concentrations was done as described earlier.50 The protonation and complex formation processes were characterized by the following general equilibrium process:| pM + qH + rL ⇄ MpHqLr |
where M denotes the metal ion, L the deprotonated ligand molecule, and H the protons. Charges have been omitted for simplicity but can be easily calculated taking into account the composition of the fully protonated dodecapeptide (H5L+). Please note, that this simplified notion is used generally throughout the text and on the figures. The corresponding formation constants (βMpHqLr ≡ βpqr) were calculated using the PSEQUAD computer program.84 The protonation constants were determined from 3–4 independent titrations (70–80 data points per titration), with a peptide concentration of 1.0 × 10−3 M. The complex formation constants were evaluated from 8 independent titrations (70–80 data points per titration). The applied ratio of ZnII and the ligand was 0.5
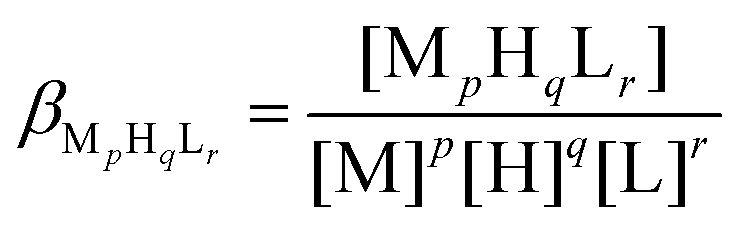 :1, 1:1 and 2:1 with the ZnII concentration varied between 5.2 × 10−4 and 2.04 × 10−3 M. Due to precipitation in the presence of metal ion excess in alkaline pH-range, titration data for the ZnII:HS 2:1 samples were evaluated only up to pH 7.1. (The individual fitting parameter of titrations performed with ligand excess dropped by ca. 40% when considering differently protonated bis-ligand species (ZnHxL2) besides monomeric ones, and accordingly such species were also included in the final model.
:1, 1:1 and 2:1 with the ZnII concentration varied between 5.2 × 10−4 and 2.04 × 10−3 M. Due to precipitation in the presence of metal ion excess in alkaline pH-range, titration data for the ZnII:HS 2:1 samples were evaluated only up to pH 7.1. (The individual fitting parameter of titrations performed with ligand excess dropped by ca. 40% when considering differently protonated bis-ligand species (ZnHxL2) besides monomeric ones, and accordingly such species were also included in the final model.
Acknowledgements
AJ wishes to thank the financial support of the János Bolyai Research Grant from the Hungarian Academy of Sciences. The research leading to the SRCD results has received funding from the European Community's Seventh Framework CALIPSO Programme (FP7/2007–2013, grant No. 312284). LH was supported by ISOLDE/CERN by the beam time grant IS488.References
- D. P. Giedroc and A. I. Arunkumara, Dalton Trans., 2007, 3107 RSC.
- Z. Ma, F. E. Jacobsen and D. P. Giedroc, Chem. Rev., 2009, 109, 4644 CrossRef CAS PubMed.
- K. J. Waldron, J. C. Rutherford, D. Ford and N. J. Robinson, Nature, 2009, 460, 823 CrossRef CAS PubMed.
- K. J. Waldron and N. J. Robinson, Nat. Rev. Microbiol., 2009, 6, 25 CrossRef PubMed.
- B. Zambelli, F. Musiani and S. Ciurli, Metal Ion-Mediated DNA-Protein Interactions in Interplay between Metal Ions and Nucleic Acids, Metal Ions in Life Sciences 10, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Springer, Netherlands, 2012, pp. 135–170 Search PubMed.
- N. L. Brown, J. V. Stoyanov, S. P. Kidd and J. L. Hobman, FEMS Microbiol. Rev., 2003, 27, 145 CrossRef CAS.
- J. L. Hobman, J. Wilkie and N. L. Brown, BioMetals, 2005, 18, 429 CrossRef CAS PubMed.
- A. Changela, K. Chen, Y. Xue, J. Holschen, C. E. Outten, T. V. O'Halloran and A. Mondragon, Science, 2003, 301, 1383 CrossRef CAS PubMed.
- M. Mejáre and l. Bülow, Trends Biotechnol., 2001, 19, 67 CrossRef.
- M. Valls and V. de Lorenzo, FEMS Microbiol. Rev., 2002, 26, 327 CrossRef CAS PubMed.
- K. Kuroda and M. Ueda, Curr. Opin. Biotechnol., 2011, 22, 427 CrossRef CAS PubMed.
- K. D. Brune and T. S. Bayer, Front. Microbiol., 2012, 3, 1 Search PubMed.
- A. Jancso, D. Szunyogh, F. H. Larsen, P. W. Thulstrup, N. J. Christensen, B. Gyurcsik and L. Hemmingsen, Metallomics, 2011, 3, 1331 RSC.
- D. S. Auld, Zinc enzymes, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd., 2011, DOI:10.1002/9781119951438.eibc0241.
- M. A. Pennella and D. P. Giedroc, Zinc: DNA Binding Proteins, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd., 2011, DOI:10.1002/9781119951438.eibc0240.
- I. Dokmanic, M. Sikic and S. Tomic, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 257 CAS.
- M. Laitaoja, J. Valjakka and J. Jänis, Inorg. Chem., 2013, 52, 10983 CrossRef CAS PubMed.
- D. S. Auld, BioMetals, 2001, 14, 271 CrossRef CAS.
- M. J. Stillman, Coord. Chem. Rev., 1995, 144, 461 CrossRef CAS.
- S. S. Krishna, I. Majumdar and N. V. Grishin, Nucleic Acids Res., 2003, 31, 532 CrossRef CAS PubMed.
- R. Gamsjaeger, C. K. Liew, F. E. Loughlin, M. Crossley and J. P. Mackay, Trends Biochem. Sci., 2007, 32, 63 CrossRef CAS PubMed.
- D. C. Bebout, Mercury: Inorganic & Coordination Chemistry, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd., 2011, DOI:10.1002/9781119951438.eibc0124.
- A. Manceau and K. L. Nagy, Dalton Trans., 2008, 1421 RSC.
- M. Kaupp and H. G. von Schnering, Inorg. Chem., 1994, 33, 2555 CrossRef CAS.
- J. G. Wright, M. J. Natan, F. M. MacDonnell, D. M. Ralston and T. V. O'Halloran, Mercury(II)-Thiolate chemistry and the mechanism of the heavy metal biosensor MerR, in Progress in Inorganic Chemistry: Bioinorganic Chemistry, ed. S. J. Lippard, John Wiley & Sons, New York, 1990, vol. 38, pp. 323–412 Search PubMed.
- D. L. Huffman, L. M. Utschig and T. V. O'Halloran, Mercury-responsive gene regulation and mercury-199 as a probe of protein structure, in Metal Ions in Biological Systems, ed. A. Sigel and H. Sigel, Marcel Dekker, New York, 1997, vol. 34, pp. 503–526 Search PubMed.
- R. A. Steele and S. J. Opella, Biochemistry, 1997, 36, 6885 CrossRef CAS PubMed.
- J. G. Wright, H.-T. Tsang, J. E. Penner-Hahn and T. V. O'Halloran, J. Am. Chem. Soc., 1990, 112, 2434 CrossRef CAS.
- J. D. Helmann, B. T. Ballard and C. T. Walsh, Science, 1990, 247, 946 CAS.
- L. M. Utschig, T. Baynard, C. Strong and T. V. O'Halloran, Inorg. Chem., 1997, 36, 2926 CrossRef CAS.
- P. Faller, B. Ctortecka, W. Tröger, T. Butz and M. Vasák, J. Biol. Inorg. Chem., 2000, 5, 393 CrossRef CAS.
- U. Heinz, L. Hemmingsen, M. Kiefer and H.-W. Adolph, Chem. – Eur. J., 2009, 15, 7350 CrossRef CAS PubMed.
- M. Vasak, J. H. Kagi and H. A. Hill, Biochemistry, 1981, 20, 2852 CrossRef CAS.
- J. H. Kägi, M. Vasák, K. Lerch, D. E. Gilg, P. Hunziker, W. R. Bernhard and M. Good, Environ. Health Perspect., 1984, 54, 93 CrossRef.
- L. M. Utschig, J. G. Wright and T. V. O'Halloran, Biochemical and Spectroscopic Probes of Hg(II) Coordination Environments in Proteins, in Methods in Enzymology, ed. B. Vallee and J. Riordan, Academic Press, Inc., San Diego, CA, 1993, vol. 226, pp 71–97 Search PubMed.
- N. A. Rey, O. W. Howarth and E. C. Pereira-Maia, J. Inorg. Biochem., 2004, 98, 1151 CrossRef CAS PubMed.
- A. Kolozsi, A. Lakatos, G. Galbács, A.Ø. Madsen, E. Larsen and B. Gyurcsik, Inorg. Chem., 2008, 47, 3832 CrossRef CAS PubMed.
- G. R. Dieckmann, D. K. McRorie, D. L. Tierney, L. M. Utschig, C. P. Singer, T. V. O'Halloran, J. E. Penner-Hahn, W. F. DeGrado and V. L. Pecoraro, J. Am. Chem. Soc., 1997, 119, 6195 CrossRef CAS.
- P. Rousselot-Pailley, O. Seneque, C. Lebrun, S. Crouzy, D. Boturyn, P. Dumy, M. Ferrand and P. Delangle, Inorg. Chem., 2006, 45, 5510 CrossRef CAS PubMed.
- A. M. Pujol, C. Lebrun, C. Gateau, A. Manceau and P. Delangle, Eur. J. Inorg. Chem., 2012, 3835 CrossRef CAS PubMed.
- S. Pires, J. Habjanic, M. Sezer, C. M. Soares, L. Hemmingsen and O. Iranzo, Inorg. Chem., 2012, 51, 11339 CrossRef CAS PubMed.
- M. Łuczkowski, B. A. Zeider, A. V. H. Hinz, M. Stachura, S. Chakraborty, L. Hemmingsen, D. L. Huffman and V. L. Pecoraro, Chem. – Eur. J., 2013, 19, 9042 CrossRef PubMed.
- O. Iranzo, D. Ghosh and V. L. Pecoraro, Inorg. Chem., 2006, 45, 9959 CrossRef CAS PubMed.
- M. Łuczkowski, M. Stachura, V. Schirf, B. Demeler, L. Hemmingsen and V. L. Pecoraro, Inorg. Chem., 2008, 47, 10875 CrossRef PubMed.
- E.-D. Ciuculescu, Y. Mekmouche and P. Faller, Chem. – Eur. J., 2005, 11, 903 CrossRef CAS PubMed.
- U. Heinz, M. Kiefer, A. Tholey and H.-W. Adolph, J. Biol. Chem., 2005, 280, 3197 CrossRef CAS PubMed.
- Y. Cheng, Y.-B. Yan and J. Liu, J. Inorg. Biochem., 2005, 99, 1952 CrossRef CAS PubMed.
- R. Kobayashi and E. Yoshimura, Biol. Trace Elem. Res., 2006, 114, 313 CrossRef CAS.
- T. Kochanczyk, P. Jakimowicz and A. Krezel, Chem. Commun., 2013, 49, 1312 RSC.
- A. Jancso, B. Gyurcsik, E. Mesterhazy and R. Berkecz, J. Inorg. Biochem., 2013, 126, 96 CrossRef CAS PubMed.
- N. Greenfield and G. D. Fasman, Biochemistry, 1969, 8, 4108 CrossRef CAS.
- A. Lombardi, D. Marasco, O. Maglio, L. Di Costanzo, F. Nastri and V. Pavone, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 11922 CrossRef CAS PubMed.
- G. Veglia, F. Porcelli, T. DeSilva, A. Prantner and S. J. Opella, J. Am. Chem. Soc., 2000, 122, 2389 CrossRef CAS.
- T. M. DeSilva, G. Veglia, F. Porcelli, A. M. Prantner and S. J. Opella, Biopolymers, 2002, 64, 189 CrossRef CAS PubMed.
- L. A. Basile and J. E. Coleman, Protein Sci., 1992, 1, 617 CrossRef CAS PubMed.
- M. Koch, S. Bhattacharya, T. Kehl, M. Gimona, M. Vašák, W. Chazin, C. W. Heizmann, P. M. H. Kroneck and G. Fritz, Biochim. Biophys. Acta, 2007, 1773, 457 CrossRef CAS PubMed.
- W. Lu and M. J. Stillman, J. Am. Chem. Soc., 1993, 115, 3291 CrossRef CAS.
- M. R. Ghadiri and C. Choi, J. Am. Chem. Soc., 1990, 112, 1630 CrossRef CAS.
- L. Hemmingsen, K. N. Sas and E. Danielsen, Chem. Rev., 2004, 104, 4027 CrossRef CAS PubMed.
- T. Butz, W. Tröger, T. Pöhlmann and O. Nuyken, Z. Naturforsch., 1992, 47a, 85 Search PubMed.
- W. Tröger, Hyperfine Interact., 1999, 120/121, 117 CrossRef.
- P. Gockel, M. Gelinsky, R. Vogler and H. Vahrenkamp, Inorg. Chim. Acta, 1998, 272, 115 CrossRef CAS.
- M. Rowinska-Zyrek, D. Witkowska, S. Bielinska, W. Kamysz and H. Kozlowski, Dalton Trans., 2011, 40, 5604 RSC.
- H. Reyes-Caballero, G. C. Campanello and D. P. Giedroc, Biophys. Chem., 2011, 156, 103 CrossRef CAS PubMed.
- M. A. Pennella, A. I. Arunkumar and D. P. Giedroc, J. Mol. Biol., 2006, 356, 1124 CrossRef CAS PubMed.
- C. Migliorinia, E. Porciatti, M. Luczkowski and D. Valensina, Coord. Chem. Rev., 2012, 256, 352 CrossRef PubMed.
- K. Kulon, D. Woźniak, K. Wegner, Z. Grzonka and H. Kozłowski, J. Inorg. Biochem., 2007, 101, 1699 CrossRef CAS PubMed.
- C. Kállay, K. Ősz, A. Dávid, Z. Valastyán, G. Malandrinos, N. Hadjiliadis and I. Sóvágó, Dalton Trans., 2007, 4040 RSC.
- C. Kállay, K. Várnagy, G. Malandrinos, N. Hadjiliadis, D. Sanna and I. Sóvágó, Inorg. Chim. Acta, 2009, 362, 935 CrossRef PubMed.
- P. Andersson, J. Kvassman, A. Lindström, B. Oldén and G. Pettersson, Eur. J. Biochem., 1980, 108, 303 CrossRef CAS PubMed.
- L. Hemmingsen, R. Bauer, M. J. Bjerrum, M. Zeppezauer, H. W. Adolph, G. Formicka and E. Cedergren-Zeppezauer, Biochemistry, 1995, 34, 7145 CrossRef CAS.
- K. L. Bren, Nuclear Magnetic Resonance (NMR) Spectroscopy of Metallobiomolecules, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd., 2011, DOI:10.1002/9781119951438.eibc0296.
- M. Ringkjøbing Jensen, M. A. S. Hass, D. F. Hansen and J. J. Led, Cell. Mol. Life Sci., 2007, 64, 1085 CrossRef PubMed.
- P. Atkins and J. de Paula, Reaction dynamics, in Physical Chemistry, W. H. Freeman and Company, New York, 9th edn, 2010, ch. 22, pp. 831–875 Search PubMed.
- R. A. Pufahl, C. P. Singer, K. L. Peariso, S.-J. Lin, P. J. Schmidt, C. J. Fahrni, V. Cizewski Culotta, J. E. Penner-Hahn and T. V. O'Halloran, Science, 1997, 278, 853 CrossRef CAS.
- A. K. Boal and A. C. Rosenzweig, Chem. Rev., 2009, 109, 4760 CrossRef CAS PubMed.
- D. T. Richens, The Chemistry of Aqua Ions: Synthesis, Structure and Reactivity: A Tour Through the Periodic Table of the Elements, John Wiley & Sons, Chichester, 1997 Search PubMed.
- R. Pribil, Applied Complexometry, ed. R.A. Chalmers, Pergamon Press, Oxford, UK, 1982 Search PubMed.
- A. J. Miles, R. W. Janes, A. Brown, D. T. Clarke, J. C. Sutherland, Y. Tao, B. A. Wallace and S. V. Hoffmann, J. Synchrotron Radiat., 2008, 15, 420 CrossRef CAS PubMed.
- A. J. Miles, S. V. Hoffmann, Y. Tao, R. W. Janes and B. A. Wallace, Spectroscopy, 2007, 21, 245 CrossRef CAS.
- O. Iranzo, P. W. Thulstrup, S. B. Ryu, L. Hemmingsen and V. L. Pecoraro, Chem. – Eur. J., 2007, 13, 9178 CrossRef CAS PubMed.
- R. J. Beynon and J. S. Easterby, Buffer Solutions, Taylor & Francis, UK, 2003 Search PubMed.
- ACD/Spectrus Processor, version 2012 (Build 61851, 24 Jan 2013), Advanced Chemistry Development, Inc., Toronto, ON, Canada, http://www.acdlabs.com, 2012 Search PubMed.
- L. Zékány, I. Nagypál and G. Peintler, PSEQUAD for Chemical Equilibria, Technical Software Distributors, Baltimore, MD, 1991 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5dt00945f |
| This journal is © The Royal Society of Chemistry 2015 |