DOI:
10.1039/C5DT00942A
(Paper)
Dalton Trans., 2015,
44, 9230-9240
Copper mediated decyano decarboxylative coupling of cyanoacetate ligands: Pesci versus Lewis acid mechanism†
Received
9th March 2015
, Accepted 10th April 2015
First published on 24th April 2015
Abstract
A combination of gas-phase ion trap multistage mass spectrometry (MSn) experiments and density functional theory (DFT) calculations have been used to examine the mechanisms of the sequential decomposition reactions of copper cyanoacetate anions, [(NCCH2CO2)2Cu]−, introduced into the gas-phase via electrospray ionization. Gas phase IR spectroscopy, used to probe the coordination mode of the cyanoacetate ligands, revealed that the initial precursor ions are bound to the Cu via the carboxylate, [NCCH2CO2CuO2CCH2CN], 1. Multistage collision-induced dissociation (CID) of 1 gave sequential losses of CO2 and ethene. DFT calculations suggest that the lowest energy pathways for sequential decarboxylation involve Lewis acid mechanisms in which the binding of the cyanoacetate ligand sequentially rearranges from O to N: [NCCH2CO2CuO2CCH2CN]− → [NCCH2CO2CuNCCH2CO2]− → [NCCH2CO2CuNCCH2]− + CO2 and [NCCH2CO2CuNCCH2]− → [O2CCH2CNCuNCCH2]− → [CH2CNCuNCCH2]− + CO2. Loss of ethene involves sequential rearrangement of the binding of the cyanomethyl carbanion ligands from N to C: [CH2CNCuNCCH2]− → [NCCH2CuNCCH2]− → [NCCH2CuCH2CN]−. CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 loss then proceeds via a 1,2-dyotropic rearrangement to form [NCCuCH2CH2CN]− followed by β-cyanide transfer. This study highlights the rich mechanistic possibilities for metal mediated decarboxylation reactions involving ambidentate carboxylate ligands.
CH2 loss then proceeds via a 1,2-dyotropic rearrangement to form [NCCuCH2CH2CN]− followed by β-cyanide transfer. This study highlights the rich mechanistic possibilities for metal mediated decarboxylation reactions involving ambidentate carboxylate ligands.
Introduction
The search for new types of metal catalyzed C–C bond-coupling reactions continues, with an active research front being the development of alternatives to stoichiometric organometallic cross coupling reagents.1 Metal catalyzed decarboxylative coupling reactions represent an attractive way of creating carbon frameworks.2,3 The most commonly reported new synthetic methods involving metal catalyzed decarboxylation reactions are those involving sp2 hybridized aromatic carboxylic acids2g and sp hybridized alkynoic acids.2j Fewer metal catalyzed decarboxylation reactions of sp3 hybridized carboxylic acids have been noted.4 An exception is α-cyanoacetic acid and its derivatives (Scheme 1).5 As early as 1974, Tsuda et al. reported that Cu(I) cyanoacetate underwent decarboxylation to form cyanomethylcopper, which could be used in C–C bond coupling reactions.5a More recently catalytic variants have been reported, including palladium catalyzed decarboxylative coupling of cyanoacetate salts with aryl halides5b,c and in the copper catalyzed decarboxylative alkynation of quaternary α-cyanoacetate salts.5d
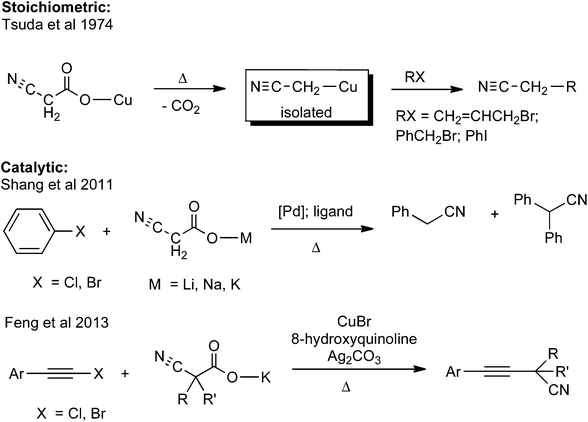 |
| | Scheme 1 Metal mediated decarboxylative coupling reactions of cyanoacetates.5 | |
One of the fascinating aspects of metal catalyzed decarboxylative coupling reactions is that the decarboxylation step can proceed via a number of different mechanisms, especially for ambidentate carboxylate ligands such as the cyanoacetate anion.6–13 These include: decarboxylation of the free anion (eqn (1));6 electron transfer induced free radical decarboxylation (eqn (2));7 the Pesci reaction8 in which decarboxylation of the O bound carboxylate9 directly yields an organometallic intermediate10 (eqn (3)); a Lewis acid decarboxylation reaction11 in which the metal acts as a Lewis acid and decarboxylation occurs via binding through the cyano group (eqn (4)).12,13 The last 2 mechanisms have been proposed for copper(I) catalyzed decarboxylation of cyanoacetic acid.14–16
| | 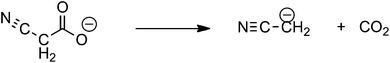 | (1) |
| | 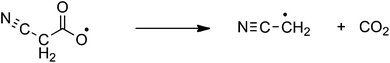 | (2) |
| | 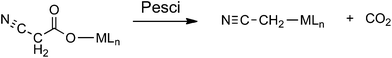 | (3) |
| |  | (4) |
Gas phase studies, in which multistage mass spectrometry experiments (MSn)17 are used to form organometallic ions, [RML]−/+ (ref. 18 and 19), via collision induced dissociation (CID) of metal carboxylate precursors, [RCO2ML]−/+, have proven to be a useful way of surveying the role of the metal, carboxylate, and auxiliary ligand in promoting known types of C–C bond coupling reactions as well as in exploring new classes of reactions.17c For example, we have discovered a new copper mediated reaction involving dehydro decarboxylative coupling of acetate ligands (eqn (5)).18k CID on the copper acetate anions, [CH3CO2CuO2CCH3]−, formed via electrospray ionization (ESI), proceeds via the sequential losses of CO2 (eqn (6) and (7)) and ethene. DFT calculations revealed that the mechanisms for this reaction involved a novel 1,2-dyotropic rearrangement20 (eqn (8)) followed by β-hydride transfer (eqn (9)).
| | | 2CH3CO2− → CH2CH2 + 2CO2 + 2H− | (5) |
| | | [CH3CO2CuO2CCH3]− → [CH3CO2CuCH3]− + CO2 | (6) |
| | | [CH3CO2CuCH3]− → [CH3CuCH3]− + CO2 | (7) |
| | | [CH3CuCH3]− → [HCuCH2CH3]− | (8) |
| | | [HCuCH2CH3]− → [HCuH]− + CH2CH2 | (9) |
A combination of DFT calculations and gas phase IR spectroscopy21 was first used to determine the binding modes22,23 of cyanoacetate ligands to Cu(I), Scheme 2. We then used MSn experiments and DFT calculations to examine the sequential decomposition reactions of cyanoacetate ligands coordinated to copper(I). DFT calculations were carried out to establish whether the mechanism for sequential decarboxylation proceeds via the Pesci reaction (eqn (3))8 or via a Lewis acid pathway (eqn (4)).11
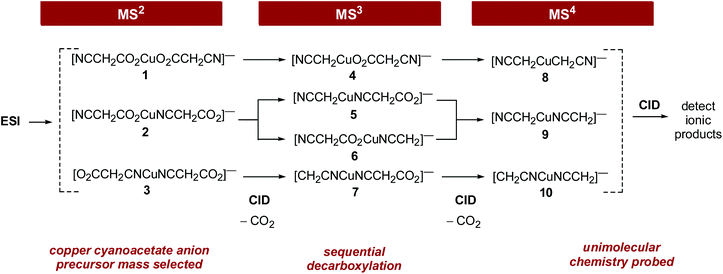 |
| | Scheme 2 Gas-phase sequential decarboxylation and subsequent unimolecular chemistry using multistage mass spectrometry experiments. Note the potential involvement of different isomers in each of the CID steps. These isomers arise from different binding modes of the cyanoacetate ligands to the Cu(I) center (isomers 1–3 and 4–7) and/or cyanomethyl anion ligands to the Cu(I) center (isomers 4–7 and 8–10). | |
Experimental section
Reagents
Copper(II) acetate and cyanoacetic acid were obtained from Aldrich and methanol was AR grade obtained from Merck. All chemicals were used without further purification.
Gas phase IR spectroscopy
Vibrational spectra were obtained using a Fourier transform ion cyclotron resonance spectrometer (Bruker 7T FT-ICR) equipped with an ESI source, and coupled with the tuneable IR free-electron laser (FEL) of CLIO (Centre Laser Infrarouge d'Orsay) producing mid-infrared radiation in the 900–1800 cm−1 range.24 Desired mass-selected ions are trapped in a ∼5 cm long hexapole ion-trap contained within a collision cell where ions are collisionally cooled using a flow of high-purity argon buffer gas. They are then pulse extracted to the ICR cell where they are irradiated with several25 IR macropulses delivered at 25 Hz. Upon resonant vibrational excitation, dissociation of the selected ion can be monitored via its fragment peaks. The most abundant fragments of [(NCCH2CO2)2Cu]− with decreasing relative abundance are fragment ions of m/z 187 and 143, corresponding to losses of CO2 and 2CO2, respectively. The IR action spectra were derived by plotting the IRMPD efficiency against the photon energy. A scaling factor of 0.98 was applied to the calculated B3-LYP/6-31+G(d) harmonic frequencies in the 900–1800 cm−1 region. Each calculated band was convoluted assuming a Gaussian shape with full width at half maximum of 20 cm−1.
Mass spectrometry
Mass spectra were recorded using a Thermo Scientific (Bremen, Germany) LTQ FT hybrid mass spectrometer equipped with a Finnigan ESI (electrospray ionization) source. This instrument consists of a linear ion trap (LTQ) coupled to a Fourier-transform ion cyclotron resonance (FT-ICR) mass spectrometer.25 Copper(II) acetate was dissolved in methanol together with cyanoacetic acid in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 molar ratio, with a final concentration of 1 mM.26 These solutions were transferred to the electrospray source via a syringe pump at a rate of 5 μl min−1. Typical electrospray source conditions with needle potentials of 3.5–5.0 kV and a heated capillary temperature of 200–300 °C were used. The desired precursor ion was mass selected with a window of 7 and subjected to CID the following typical conditions: activation energy between 25% and 35%; activation (Q), 0.25, and activation time 30 ms prior to ejection from the ion-trap and detection. The copper isotope pattern (63Cu 69.2%, 65Cu 30.8%) as well as high resolution mass spectrometry experiments were used to identify copper containing species.
:10 molar ratio, with a final concentration of 1 mM.26 These solutions were transferred to the electrospray source via a syringe pump at a rate of 5 μl min−1. Typical electrospray source conditions with needle potentials of 3.5–5.0 kV and a heated capillary temperature of 200–300 °C were used. The desired precursor ion was mass selected with a window of 7 and subjected to CID the following typical conditions: activation energy between 25% and 35%; activation (Q), 0.25, and activation time 30 ms prior to ejection from the ion-trap and detection. The copper isotope pattern (63Cu 69.2%, 65Cu 30.8%) as well as high resolution mass spectrometry experiments were used to identify copper containing species.
DFT calculations
The Gaussian 09 package27 was used to carry out theoretical calculations aimed at providing insights into the binding modes of the cyanoacetate ligands to Cu(I) and their fragmentation mechanisms. Geometry optimizations were conducted using the B3LYP DFT method.28 The Stuttgart Dresden effective core potential (SDD) and basis set was used for copper and the 6-31+G(d) all electron basis set for carbon, nitrogen and oxygen.29 This method and basis set, which we designate hereafter as B3LYP/SDD6-31+G(d), was specifically chosen as it has been proven to reasonably describe the gas phase chemistry of copper carboxylates and metallates.18f,i,k,n,o
All transition states (TS) were characterized by the presence of a single imaginary frequency and intrinsic reaction coordinates (IRC) were carried out to ensure smooth connection of reactants and products. Geometry optimizations in methanol solvent were carried out using Tomasi's Polarizable Continuum Model (PCM).30
Results and discussion section
Binding mode of cyanoacetate to Cu(I)
ESI/MS of a methanolic solutions of copper(II) acetate and cyanoacetic acid gave rise to a series of ions including an ion corresponding to Cu(I) coordinated to two cyanoacetate ligands.26 Similar ions have been previously observed for methanolic solutions of copper(II) acetate with other aliphatic and aromatic carboxylic acids.18f We have used DFT calculations to examine the structures and relative energies of the three isomers of Cu(I) coordinated to two cyanoacetate ligands (1, 2 and 3 of Scheme 2) in methanol and in the gas phase. For each of these isomers a range of conformations of the cyanoacetate ligands was considered in the gas phase and the most stable conformer of each isomer was then reoptimized using a methanol solvent continuum model. Fig. 1 shows the structures of these isomers in the solution and gas phase, while Table 1 lists their relative energies. Structures of higher energy gas-phase conformers are given in ESI Fig. S1–S3.†
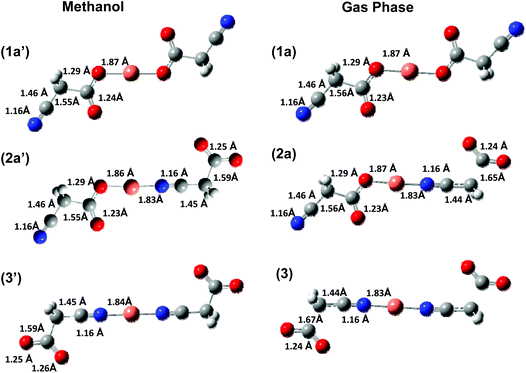 |
| | Fig. 1 DFT calculated (B3LYP/SDD6-31+G(d)) structures of isomers 1a; 2a; and 3 in methanol solution ((1a′), (2a′) and (3′) respectively) and in the gas phase ((1a), (2a) and (3) respectively). Bond lengths are in Angstroms. | |
Table 1 DFT calculated (B3LYP/SDD6-31+G(d)) relative energies of isomeric structures in which cyanoacetate binds via either the O,O, O,N or N,N atoms to the copper in methanol and the gas phase. Energies are reported in eV and are relative to the case where O,O bind to the copper in methanol (isomer 1a). In addition, relative stabilities of the isomers in the gas phase are given in parentheses
| Medium |
Isomer 1a |
Isomer 2a |
Isomer 3 |
|
The higher energies highlight the solvent stabilization effect.
|
| Methanol |
0 |
0.13 |
0.33 |
| Gas phasea |
2.08 (0) |
2.43 (0.35) |
2.95 (0.87) |
Comparison of the gas-phase and solution-phase complexes of the linkage isomers 1, 2 and 3 (Scheme 2) reveals that the nitrogen-bound isomers are higher in energy than the oxygen-bound in both the gas phase and the condensed phase (Table 1). Thus the oxygen-bound linkage isomer 1 should be considered the major parent complex isolated from the ESI/MS experiments. The structural features of the lowest energy O,O bound isomer are almost identical in both the gas (1a) and condensed (1a′) phases, with only small changes to key bond lengths. In contrast, for the cyanoacetate ligand(s) bound through the N atom in isomers 2 and 3, there is a significant stretching of the C–C bond length on moving from solution to the gas phase (Fig. 1). Finally, comparison of the differences in the gas versus condensed phase energies of isomers 2a and 3 relative to 1a further highlights the stabilizing effect of the solvent on the N coordinated cyanoacetate ligand.
To confirm that 1 is isolated upon ESI/MS of copper solutions of cyanoacetate, we have carried out gas phase IR spectroscopy experiments (Fig. 2) since the C–O stretch of carboxylates in metal complexes are known to be sensitive to the binding mode to the metal centre in both the condensed22 and gas23 phases. The symmetrical isomers 1 and 3 are predicted to only have 3 active IR modes in the IR range used in our experiments (900–1800 cm−1). These correspond to the asymmetric combinations of the C–O stretching, CH2 wagging and CO stretching for 1 (Fig. 2b, red lines) and for 3 (Fig. 2d, green lines). The asymmetrical isomer 2 is predicted to have 6 active IR modes in the IR range used in our experiments, corresponding to two C–O stretching, CH2 wagging and CO stretching (Fig. 2c, blue lines). The experimental IR spectrum only contains 3 bands (Fig. 2a), thereby ruling out isomer 2. The examination of the overlap of the experimental bands (grey lines) with those predicted for 1 and 3 reveals that the best match between experiment and theory is for 1, with the relative error for the three bands being on the order of 1%. These observations are consistent with solution phase IR spectra of mononuclear O-bound copper cyano acetate phosphine complexes for which the acetate moiety was found to have bands in the 1339–1379 and 1611–1640 cm−1 regions.9
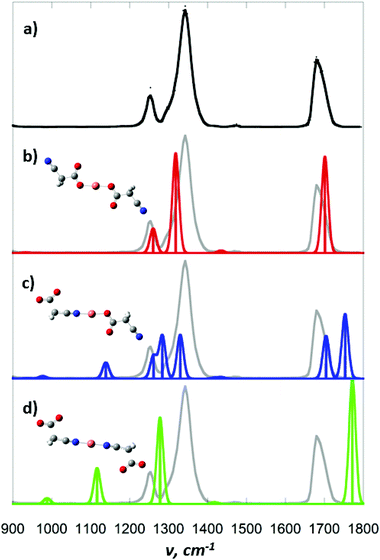 |
| | Fig. 2 Gas phase infrared spectra of [NCCH2CO2CuO2CCH2CN]−: (a) experimental; DFT calculated (B3LYP/SDD6-31+G(d)) spectra for isomer (b) 1a; (c) 2a; and (d) 3. | |
Multistage CID of [NCCH2CO2CuO2CCH2CN]−
Low energy collision-induced dissociation (CID) experiments were carried out on [NCCH2CO2CuO2CCH2CN]− in order to establish its gas phase unimolecular reactivity and to compare it to that of the parent acetate.18f,k In the first stage of CID, CO2 loss is observed from the parent ion [NCCH2CO2CuO2CCH2CN]− (m/z 231) (Fig. 3a) to generate [NCCH2CO2CuO2CCH2CN–CO2]− (m/z 187) and a further secondary loss of another CO2 generating [NCCH2CO2CuO2CCH2CN–2CO2]− (m/z 143).
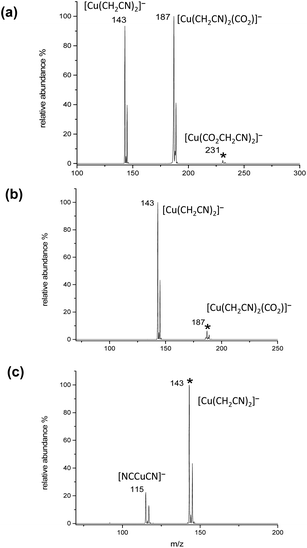 |
| | Fig. 3 Gas phase ion trap MSn mass spectra showing the collision induced dissociation of: (a) [(NCCH2CO2)2Cu]−, m/z 231; (b) [(NCCH2CO2)2Cu–CO2]−, m/z 187; (c) [(NCCH2CO2)2Cu–2CO2]−, m/z 143. The mass selected precursor ion is denoted with an *. | |
Mass selection of the initial product ([NCCH2CO2CuO2CCH2CN–CO2]−) followed by CID, gives rise to a second decarboxylation reaction (Fig. 3b). The now formed doubly decarboxylated product ([NCCH2CO2CuO2CCH2CN–2CO2]−) undergoes loss of ethene to generate [NCCuCN]− (m/z 115) upon CID (Fig. 3c). All of these reactions are directly related to those observed for the parent acetate, [CH3CO2CuO2CCH3]−, (eqn (6)–(9)) and the sequence of double decarboxylation followed by loss of ethene corresponds to the copper promoted decyano decarboxylative coupling of two cyanoacetate ligands (eqn (10)).
| | | 2NCCH2CO2− → CH2CH2 + 2CO2 + 2CN− | (10) |
DFT calculated mechanisms for copper(I)-mediated decarboxylation of cyanoacetate
As noted in the introduction, a key issue regarding the decomposition mechanism of cyanoacetate ligands is the structure of the parent carboxylate complex. In particular, since it can either bind to the metal via an oxygen or a nitrogen atom, the cyanoacetate ligand is potentially ambidentate. Potential mechanistic pathways for the double decarboxylation of [NCCH2CO2CuO2CCH2CN]− are summarized in Scheme 3, where the energies are presented relative to the most stable starting isomer 1a. CID in an ion trap is akin to an “on/off” switch, the product of the first decarboxylation step is collisionally cooled back to the temperature of the helium bath before it is mass selected and subjected to a new stage of CID. Thus the energies of the key species associated with the first decarboxylation step are presented relative to the starting isomer 1a (Fig. 4), while those associated with the second decarboxylation step are presented relative to the isomer 6a (Fig. 5).
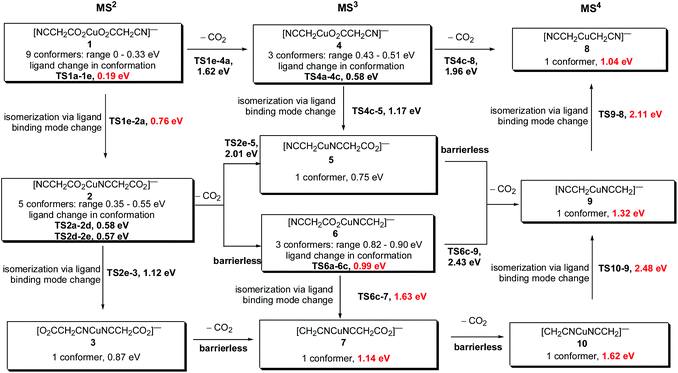 |
| | Scheme 3 Summary of DFT calculated energetics associated with double decarboxylation proceeding via isomers 1, 2 and 3. The most likely pathway under the low energy CID conditions of the ion trap is highlighted in bold red. The potential energy surfaces for the first stage and second stage of decarboxylation are given in Fig. 4 and Fig. 5, while the potential energy surfaces for the rearrangement from structure [CH2CNCuNCCH2]− to [NCCH2CuCH2CN]− are given in Fig. 6. All energies are given relative to 1a, the most stable conformation of the most stable starting isomer 1. | |
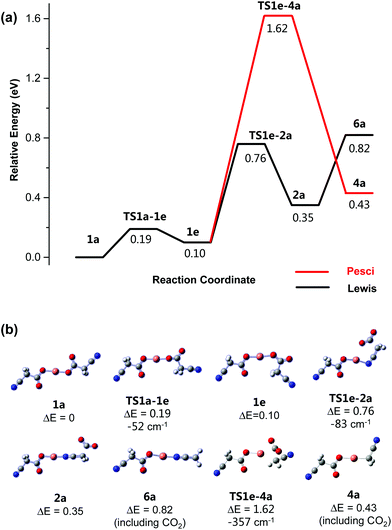 |
| | Fig. 4 (a) Calculated B3LYP/SDD6-31+G(d) energy diagram (eV) for CID of [NCCH2CO2CuO2CCH2CN]− undergoing first decarboxylation. The red line shows the possible Pesci pathway. (b) Structures of key minima and transition states. All energies are relative to the precursor structure 1a. The imaginary (negative) frequency associated with each transition state is given in units of cm−1. | |
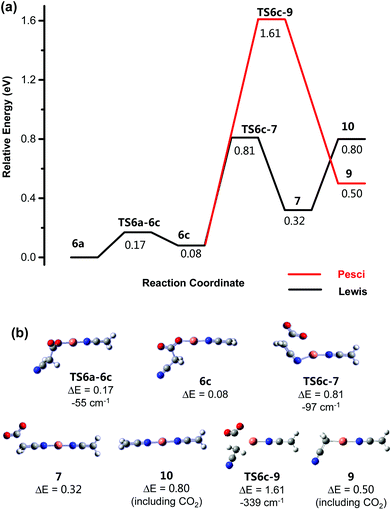 |
| | Fig. 5 Calculated B3LYP/SDD6-31+G(d) energy diagram (eV) for CID of [NCCH2CO2CuNCCH2]− undergoing decarboxylation. The red line shows the possible Pesci pathway. (b) Structures of key minima and transition states. All energies are relative to the precursor structure 6a. The imaginary (negative) frequency associated with each transition state is given in units of cm−1. | |
The first decarboxylation step starts from the most stable precursor, 1a, which rearranges first to the less stable structure, 1e, via a transition state, TS1a-1e, requiring an energy of 0.19 eV (Fig. 4). Isomer 1e then undergoes an intramolecular rearrangement, which changes the coordination mode of one of the cyanoacetate ligands from O to N bound, via a second transition state, TS1e-2a, requiring 0.76 eV. Isomer 2a undergoes a barrierless loss of CO2via a Lewis acid mechanism to form the decarboxylated product, [NCCH2CO2CuNCCH2]−, 6a. The direct Pesci mechanism for decarboxylation of 1e to form the organometallic 4a proceeds viaTS1e-4a, which is 1.62 eV higher in energy than the entrance channel (1a). While the energy for this decarboxylation in slightly less than that required for decarboxylation of [CH3CO2Cu2OCCH3]−,18f it is 0.8 eV higher than the highest energy requirement for the Lewis acid mechanism. Hence, this mechanism is unlikely to occur under the low energy CID conditions used in the ion trap.
The lowest energy pathway for the second decarboxylation, proceeding from [NCCH2CO2CuNCCH2]−, 6a, also involves change in conformation viaTS6a-6c, requiring 0.17 eV, to form, 6c, followed by a change in the coordination mode of the remaining cyanoacetate ligand from O to N bound, to form 7via transition state TS6c-7, which requires an energy input of 0.81 eV above that of the starting structure 6a (Fig. 5). Subsequently, 7 loses CO2 to form the doubly decarboxylated product, [CH2CNCuNCCH2]−, 10via a Lewis acid mechanism.
Decarboxylation via the Pesci mechanism occurs via the transition state TS6c-9, which is calculated to be 1.61 eV higher in energy than the starting product 6a and 0.8 eV higher than the highest energy requirement for the Lewis acid mechanism. Hence, this mechanism is also unlikely to occur under the ion trap conditions used.
How do our results compare to previous studies on the DFT calculated mechanisms for metal mediated decarboxylation of ambidentate carboxylate ligands? The Lewis acid mechanism is also predicted to be preferred in the palladium promoted decarboxylation of cyano acetate16b and in the reverse reaction of CuNCH2 with CO2.31 A recent study by Uggerud and coworkers has used DFT calculations to suggest a change in binding of the ambidentate nicotinate ligand to a Cu(II) centre from O bound carboxylate to N bound followed by decarboxylation via a Lewis acid mechanism.32
DFT calculated mechanisms for the loss of ethene: isomerization, 1,2-dyotropic rearrangement and β-cyanide transfer
The observation of ethene loss suggests that the N bound isomer 10 formed upon double decarboxylation needs to undergo sequential rearrangement of the binding of the cyanomethyl carbanion ligands from N to C to give the organometallate, 8, which can then undergo a dyotropic rearrangement analogous to that observed for [CH3CuCH3]− (Scheme 4B). Such intramolecular interconversion of C- and N-bound isomers of transition-metal cyanocarbanions have been reported previously33 and based on DFT calculations a “metal-sliding mechanism” mechanism has been proposed (Scheme 4A).
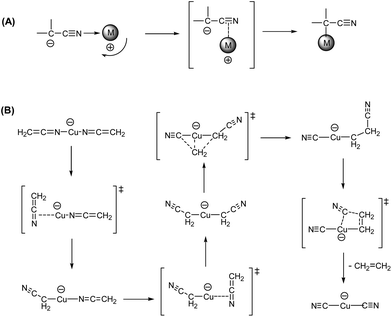 |
| | Scheme 4 (A) Previously proposed intramolecular “metal-sliding mechanism” for N- to C-isomerization of cyano carbanions in transition metal complexes;33b (B) sequential rearrangement reactions of the N bound isomer [CH2CNCuNCCH2]−, 10 to the organometallate, [NCCH2CuCH2CN]−, 8, which then undergoes a dyotropic rearrangement. | |
DFT calculations were used to explore, amongst others, the rearrangement processes shown in Scheme 4B and the results of these studies are highlighted for the N- to C-bound isomerisation reactions and the 1,2-dyotropic rearrangement. Since these reactions all occur during the same CID experiment, the energies of the key species associated with these reactions are presented relative to 10 (Fig. 6 and 7).
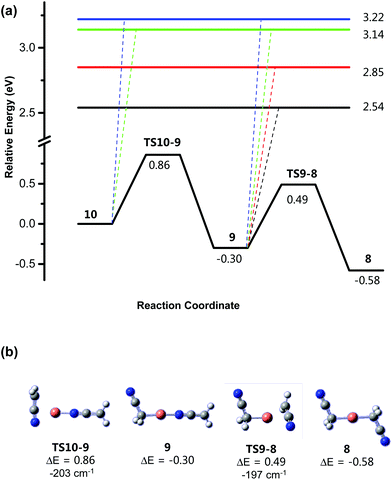 |
| | Fig. 6 (a) Calculated B3LYP/SDD6-31+G(d) (eV) rearrangement from structure [CH2CNCuNCCH2]− (10) to [NCCH2CuCH2CN]− (8). (b) Structures of key minima and transition states. All energies are relative to the precursor structure 1a. The energy calculated includes both of the CO2 lost in Fig. 4 and 5. The horizontal lines represent the energy for the barrierless losses of: [NCCH2]− to form [CuCH2CN], 2.54 eV; [NCCH2]˙ to form [CuCH2CN]˙−, 2.85 eV; [CH2CN]− to form [CuNCCH2], 3.14 eV; [CH2CN]˙ to form [CuNCCH2]˙−, 3.22 eV. The imaginary (negative) frequency associated with each transition state is given in units of cm−1. | |
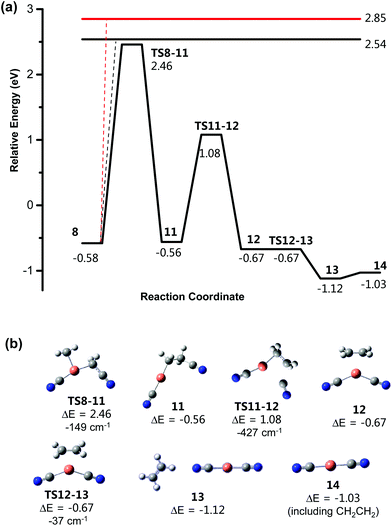 |
| | Fig. 7 (a) Calculated B3LYP/SDD6-31+G(d) energy diagram (eV) for CID of [NCCH2CuCH2CN]− undergoing a 1,2-dyotropic rearrangement with subsequent loss of ethene. (b) Structures of key minima and transition states. All energies are relative to the structure 8. The horizontal lines represent the energy for barrierless loss of: [NCCH2]− to form [CuCH2CN], 2.54 eV; and [NCCH2]˙ to form [CuCH2CN]˙−, 2.85 eV. The imaginary (negative) frequency associated with each transition state is given in units of cm−1. | |
Fig. 6 reveals that structure 10 can undergo a N,C bond rearrangement to form the more stable N,C linkage isomer, 9, via a transition state TS10-9, requiring 0.86 eV of energy relative to 10. Isomer 9 can then undergo another N,C bond rearrangement to form the more stable C,C linkage isomer, 8, via a transition state TS9-8 requiring 0.49 eV of energy input above that of the starting structure (10).
The potential energy diagram for the loss of ethene proceeding via a dyotropic rearrangement followed by β-cyanide transfer is shown in Fig. 7. As described above, 8 is initially formed via rearrangement from 10. The dyotropic rearrangement proceeds over a relatively high energy (3.04 eV higher than 8) transition state (TS8-11), which involves insertion of CH2 from one of the cyanomethyl ligand into the Cu–C bond of the other cyanomethyl ligand with concomitant transfer of the CN group to the copper centre. The product [NCCuCH2CH2CN]−, 11, then undergoes a β-cyanide transfer via the lower energy transition state (TS11-12) to form intermediate 12, which consists of ethene bound to the copper centre of [NCCuCN]−via a π electron interaction, and which is more stable than the starting organocuprate by 0.09 eV. Loss of ethene via transition state (TS12-13) gives stable complex 13, which is 0.54 eV lower in energy than 8 and 0.45 eV lower than 12. Subsequent loss of ethene to form [NCCuCN]− (14) only requires 0.09 eV.34 Overall the reaction of 8 to form ethene and [NCCuCN]− is exothermic by 0.45 eV.
Finally, the possible homolytic or heterolytic cleavage of Cu–N or Cu–C bonds in [CH2CNCuNCCH2]− (10), [NCCH2CuNCCH2]− (9) and [NCCH2CuCH2CN]− (8) was also considered (eqn (11)–(18)). The endothermicities for these processes, which are presumed to be barrierless, are given as coloured horizontal lines in Fig. 6 and 7. For the rearrangement of [CH2CNCuNCCH2]− (10) to [NCCH2CuNCCH2]− (9) and then [NCCH2CuCH2CN]− (8), the energies for the homolytic or heterolytic cleavage reactions (eqn (11)–(18)) are considerably higher than the barriers for rearrangement viaTS10-9 and TS9-8 and are therefore unlikely to be competitive with these rearrangement reactions (Fig. 6). For the dyotropic rearrangement of [NCCH2CuCH2CN]− (8) followed by β-cyanide transfer and loss of ethene, the energy for the homolytic cleavage reaction (eqn (18)) is considerably higher than the barrier for the dyotropic rearrangement and is therefore unlikely to occur under the low energy CID conditions of the ion trap, consistent with the fact that the [CuCH2CN]˙− product is not observed experimentally (Fig. 3c). The energy for the heterolytic cleavage reaction (eqn (17)) is slightly higher than the barrier for the dyotropic rearrangement, however we are unable to observe whether CH2CN− is formed experimentally due to the low mass cutoff of the ion trap.
| | | [CH2CNCuNCCH2]− → CH2CN− + [CuNCCH2] | (11) |
| | | [CH2CNCuNCCH2]− → [CuNCCH2]˙− + CH2CN˙ | (12) |
| | | [NCCH2CuNCCH2]− → CH2CN− + [CuNCCH2] | (13) |
| | | [NCCH2CuNCCH2]− → [CuNCCH2]˙− + CH2CN˙ | (14) |
| | | [NCCH2CuNCCH2]− → CH2CN− + [CuCH2CN] | (15) |
| | | [NCCH2CuNCCH2]− → [CuCH2CN]˙− + CH2CN˙ | (16) |
| | | [NCCH2CuCH2CN]− → CH2CN− + [CuCH2CN] | (17) |
| | | [NCCH2CuCH2CN]− → [CuCH2CN]˙− + CH2CN˙ | (18) |
Conclusions
Our previous study on the fragmentation reactions of trifluoro acetate ligands coordinated to a Cu(I) centre suggested the possibility of isomerization of the complex via a change in the coordination mode. Here we have examined the gas phase fragmentation of cyanaoacetate ligands coordinated to a Cu(I) centre. The precursor ion [NCCH2CO2CuO2CCH2CN]− was determined to be the O-bound isomer, 1, via gas-phase IR spectroscopy combined with DFT calculations. The DFT calculations suggest that 1 is the most stable isomer in both the gas-phase and in a model methanol medium (where explicit solvent molecules are not considered). [NCCH2CO2CuO2CCH2CN]− undergoes sequential losses of CO2, CO2 and ethene. DFT calculations highlight that the mechanisms for these losses are amongst the most complex that we have encountered for the fragmentation of coordinated carboxylate ligands. Starting from 1, the direct sequential Pesci decarboxylation reactions of both O coordinated cyanaoacetate ligands is predicted to be significantly higher in energy than the Lewis acid mechanisms. The latter require conformational changes of the cyanaoacetate ligands followed by an intramolecular isomerization to the N bound ligand and then barrierless loss of CO2. Extrusion of ethene requires formation of the organometallate, [NCCH2CuCH2CN]−, 8, via sequential “copper-sliding mechanism” for the N- to C-isomerization of both coordinated cyano carbanions in the N bound isomer [CH2CNCuNCCH2]−, 10. 8 then undergoes a unimolecular C–C bond forming reaction via a dyotropic rearrangement. It seems possible that copper(I) catalyzed decarboxylation reactions of cyanoacetate in solution may involve Lewis acid pathways and that the resultant complexes may isomerize into organometallic complexes.35
Acknowledgements
We thank the ARC for financial support via grant DP110108344 (to RAJO and GNK). GNK thank CLIO for an access grant and UPSud for a visiting scientist fellowship. Financial support from the National FT-ICR network (FR 3624 CNRS) for conducting the research is gratefully acknowledged. The authors gratefully acknowledge the generous allocation of computing time from the Melbourne University high performance computing Facility (Edward).
References
-
(a) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417 CrossRef CAS PubMed;
(b) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236 CrossRef CAS PubMed.
- For reviews on the development of synthetic methods using metal catalyzed decarboxylation see:
(a) L. J. Gooßen, K. Gooßen, N. Rodriguez, M. Blanchot, C. Linder and B. Zimmermann, Pure Appl. Chem., 2008, 80, 1725 CrossRef;
(b) L. J. Gooßen, N. Rodriguez and K. Gooßen, Angew. Chem., Int. Ed., 2008, 47, 3100 CrossRef PubMed;
(c) L. J. Goossen, F. Collet and K. Goossen, Isr. J. Chem., 2010, 50, 617 CrossRef CAS;
(d) J. D. Weaver, A. Recio, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846 CrossRef CAS PubMed;
(e) N. Rodriguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030 RSC;
(f) R. Shang and L. Liu, Sci. China: Chem., 2011, 54, 1670 CrossRef CAS;
(g) J. Cornella and I. Larrosa, Synthesis, 2012, 653 CAS;
(h) W. I. Dzik, P. P. Lange and L. J. Gooßen, Chem. Sci., 2012, 3, 2671 RSC;
(i) L. J. Gooßen and K. Gooßen, Top. Organomet. Chem., 2013, 44, 121 CrossRef;
(j) K. Park and S. Lee, RSC Adv., 2013, 3, 14165 RSC.
- Decarboxylation has also been used to synthesize organometallics in solution. For reviews, see:
(a) G. B. Deacon, Organomet. Chem. Rev., Sect. A, 1970, 5, 355 CAS;
(b) G. B. Deacon, S. J. Faulks and G. N. Pain, Adv. Organomet. Chem., 1986, 25, 237 CrossRef CAS.
- S. B. Lang, K. M. O'Nele and J. A. Tunge, J. Am. Chem. Soc., 2014, 136, 13606 CrossRef CAS PubMed.
-
(a) T. Tsuda, T. Nakatsuka, T. Hirayama and T. Saegusa, J. Chem. Soc., Chem. Commun., 1974, 557 RSC;
(b) R. Shang, D.-S. Ji, L. Chu, Y. Fu and L. Liu, Angew. Chem., Int. Ed., 2011, 50, 4470 CrossRef CAS PubMed;
(c) P. W. Yeung, K. H. Chung and F. Y. Kwong, Org. Lett., 2011, 13, 2912 CrossRef CAS PubMed;
(d) Y.-S. Feng, Z.-Q. Xu, L. Mao, F.-F. Zhang and H.-J. Xu, Org. Lett., 2013, 15, 1472 CrossRef CAS PubMed.
- For decarboxylation of cyanoacetate in the gas phase:
(a) S. T. Graul and R. R. Squires, J. Am. Chem. Soc., 1990, 112, 2517 CrossRef CAS; in solution:
(b) A. J. Belsky, P. G. Maiella and T. B. Brill, J. Phys. Chem. A, 1999, 103, 4253 CrossRef CAS.
- R. G. Johnson and R. K. Ingham, Chem. Rev., 1956, 56, 219 CrossRef CAS.
- L. Pesci, Atti R. Accad. Naz. Lincei, 1901, 10(I), 362 CAS . For an English abstract, see: L. Pesci, J. Chem. Soc., Abstr., 1901, 80, 576 Search PubMed.
- For X-ray structures of metal complexes in which cyanoacetate is bound to the metal centre via O, see: D. J. Darensbourg, M. W. Holtcamp, E. M. Longridge, B. Khandelwal, K. K. Klausmeyer and J. H. Reibenspies, J. Am. Chem. Soc., 1995, 117, 318 CrossRef CAS.
- For X-ray structures of organometallic complexes of deprotonated acetonitrile, see:
(a) D. J. Darensbourg, J. A. Joyce and A. Rheingold, Organometallics, 1991, 10, 3407 CrossRef CAS;
(b) A. M. Oertel, V. Ritleng, M. J. Chetcuti and L. F. Veiros, J. Am. Chem. Soc., 2010, 132, 13588 CrossRef CAS PubMed;
(c) D. Emeljanenko, A. Peters, V. Vitske, E. Kaifer and H.-J. Himmel, Eur. J. Inorg. Chem., 2010, 30, 4783 CrossRef;
(d) J. Tehranchi, P. J. Donoghue, C. J. Cramer and W. B. Tolman, Eur. J. Inorg. Chem., 2013, 22–23, 4077 CrossRef PubMed.
- For reviews on Lewis acid decarboxylation see:
(a) A. Liu and H. Zhang, Biochemistry, 2006, 45, 10407 CrossRef CAS PubMed;
(b) F. Jordan and H. Patel, ACS Catal., 2013, 3, 1601 CrossRef CAS PubMed.
- For X-ray structures of metal complexes in which cyanoacetate is bound to a Cu centre via N, see: V. V. Oliinik, B. M. Mykhalichko, D. Schollmeier and M. G. Mys'kiv, Russ. J. Coord. Chem., 1997, 23, 352 CAS.
- For X-ray structures of metal complexes of deprotonated acetonitrile in which binding is via N, see: D. Emeljanenko, A. Peters, V. Vitske, E. Kaifer and H.-J. Himmel, Eur. J. Inorg. Chem., 2010, 4783 CrossRef CAS.
- An alternative mechanism involving an O-coordinated cyanoacetate ligand as well as N coordinated cyanoacetic acids has been described: D. J. Darensbourg, J. A. Chojnacki and E. V. Atnip, J. Am. Chem. Soc., 1993, 115, 4675 CrossRef CAS.
- For literature in which the organometallic pathway has been suggested see: T. Tsuda, Y. Chujo and T. Saegusa, J. Am. Chem. Soc., 1978, 100, 630 CrossRef CAS.
- For literature in which the Lewis acid pathway has been suggested see:
(a)
ref. 10a; ;
(b) Y. Y. Jiang, Y. Fu and L. Liu, Sci. China: Chem., 2012, 55, 2057 CrossRef CAS.
-
(a) R. A. J. O'Hair, Chem. Commun., 2006, 1469 RSC;
(b)
R. A. J. O'Hair, in MS Investigations of Reactive Intermediates in Solution, ed. L. S. Santos, Wiley-VCH, Weinheim, 2010, ch. 6, pp. 199–227 Search PubMed;
(c) N. J. Rijs and R. A. J. O'Hair, Acc. Chem. Res., 2015, 48, 329 CrossRef PubMed.
- For organometallic anions see:
(a) S. M. Bachrach, M. Hare and S. R. Kass, J. Am. Chem. Soc., 1998, 120, 12646 CrossRef CAS;
(b) R. A. J. O'Hair, Chem. Commun., 2002, 20 RSC;
(c) P. F. James and R. A. J. O'Hair, Org. Lett., 2004, 6, 2761 CrossRef CAS PubMed;
(d) R. A. J. O'Hair, A. K. Vrkic and P. F. James, J. Am. Chem. Soc., 2004, 126, 12173 CrossRef PubMed;
(e) A. P. Jacob, P. F. James and R. A. J. O'Hair, Int. J. Mass Spectrom., 2006, 45, 255–256 Search PubMed;
(f) N. J. Rijs, G. N. Khairallah, T. Waters and R. A. J. O'Hair, J. Am. Chem. Soc., 2008, 130, 1069 CrossRef CAS PubMed;
(g) C. C. L. Thum, G. N. Khairallah and R. A. J. O'Hair, Angew. Chem., Int. Ed., 2008, 47, 9118 CrossRef CAS PubMed;
(h) N. J. Rijs and R. A. J. O'Hair, Organometallics, 2009, 29, 2684 CrossRef;
(i) G. N. Khairallah, C. Thum and R. A. J. O'Hair, Organometallics, 2009, 28, 5002 CrossRef CAS;
(j) N. J. Rijs, B. F. Yates and R. A. J. O'Hair, Chem. – Eur. J., 2010, 16, 2674 CrossRef CAS PubMed;
(k) N. J. Rijs and R. A. J. O'Hair, Organometallics, 2010, 29, 2282 CrossRef CAS;
(l) N. J. Rijs, G. B. Sanvido, G. N. Khairallah and R. A. J. O'Hair, Dalton Trans., 2010, 39, 8655 RSC;
(m) N. J. Rijs, N. Yoshikai, E. Nakamura and R. A. J. O'Hair, J. Am. Chem. Soc., 2012, 134, 2569 CrossRef CAS PubMed;
(n) N. J. Rijs and R. A. J. O'Hair, Dalton Trans., 2012, 41, 3395 RSC;
(o) L. O. Sraj, G. N. Khairallah, G. da Silva and R. A. J. O'Hair, Organometallics, 2012, 31, 1801 CrossRef CAS;
(p) M. I. S. Röhr, J. Petersen, C. Brunet, R. Antoine, M. Broyer, P. Dugourd, V. Bonačić-Koutecký, R. A. J. O'Hair and R. Mitrić, J. Phys. Chem. Lett., 2012, 3, 1197 CrossRef PubMed;
(q) N. J. Rijs and R. A. J. O'Hair, Organometallics, 2012, 31, 8012 CrossRef CAS;
(r) K. L. Vikse, G. N. Khairallah and R. A. J. O'Hair, Organometallics, 2012, 31, 7467 CrossRef CAS;
(s) K. L. Vikse, G. N. Khairallah, J. S. McIndoe and R. A. J. O'Hair, Dalton Trans., 2013, 42, 6440 RSC;
(t) G. N. Khairallah, C. C. L. Thum, D. Lesage, J.-C. Tabet and R. A. J. O'Hair, Organometallics, 2013, 32, 2319 CrossRef CAS;
(u) N. J. Rijs, N. Yoshikai, E. Nakamura and R. A. J. O'Hair, J. Org. Chem., 2014, 79, 1320 CrossRef CAS PubMed;
(v) M. Leeming, G. N. Khairallah, S. Osburn, K. L. Vikse and R. A. J. O'Hair, Aust. J. Chem., 2014, 67, 701 CrossRef CAS;
(w) J. Li, G. N. Khairallah and R. A. J. O'Hair, Organometallics, 2015, 34, 488 CrossRef CAS.
- For organometallic cations see:
(a) A. Fiedler, D. Schröder, W. Zummack and H. Schwarz, Inorg. Chim. Acta, 1997, 259, 227 CrossRef CAS;
(b) R. A. J. O'Hair, T. Waters and B. Cao, Angew. Chem., Int. Ed., 2007, 46, 7048 CrossRef PubMed;
(c) G. N. Khairallah, T. Waters and R. A. J. O'Hair, Dalton Trans., 2009, 2832 RSC;
(d) G. N. Khairallah, J. H. Yoo and R. A. J. O'Hair, Organometallics, 2010, 29, 1238 CrossRef CAS;
(e) A. B. Attygale, C.-C. Chan, F. U. Axe and M. Bolgar, J. Mass Spectrom., 2010, 45, 72 Search PubMed;
(f) C.-C. Chan, F. U. Axe, M. Bolgar and A. B. Attygale, J. Mass Spectrom., 2010, 45, 1130 CrossRef CAS PubMed;
(g) B. Butschke and H. Schwarz, Organometallics, 2010, 29, 6002 CrossRef CAS;
(h) M. Woolley, G. N. Khairallah, P. S. Donnely and R. A. J. O'Hair, Rapid Commun. Mass Spectrom., 2011, 25, 2083 CrossRef CAS PubMed;
(i) M. G. Leeming, G. N. Khairallah, G. da Silva and R. A. J. O'Hair, Organometallics, 2011, 30, 4297 CrossRef CAS;
(j) C. Brunet, R. Antoine, M. Broyer, P. Dugourd, A. Kulesza, J. Petersen, M. I. S. Röhr, R. Mitrić, V. Bonačić-Koutecký and R. A. J. O'Hair, J. Phys. Chem. A, 2011, 115, 9120 CrossRef CAS PubMed;
(k) G. N. Khairallah, C. M. Williams, S. Chow and R. A. J. O'Hair, Dalton Trans., 2013, 42, 9462–9467 RSC;
(l) G. N. Khairallah, K. A. Saleeba, S. Chow, W. Eger, C. M. Williams and R. A. J. O'Hair, Int. J. Mass Spectrom., 2013, 354–355, 229 CrossRef CAS;
(m) M. J. Woolley, G. N. Khairallah, P. S. Donnelly, B. F. Yates and R. A. J. O'Hair, Organometallics, 2013, 32, 6931 CrossRef CAS;
(n) H. Al Sharif, K. L. Vikse, G. N. Khairallah and R. A. J. O'Hair, Organometallics, 2013, 32, 5416 CrossRef CAS;
(o) M. J. Woolley, G. N. Khairallah, G. R. da Silva, P. S. Donnelly and R. A. J. O'Hair, Organometallics, 2014, 33, 5185 CrossRef CAS;
(p) M. J. Woolley, A. Ariafard, G. N. Khairallah, K. H.-Y. Kwan, P. S. Donnelly, J. M. White, A. J. Canty, B. F. Yates and R. A. J. O'Hair, J. Org. Chem., 2014, 79, 12056 CrossRef CAS PubMed.
- I. Fernandez, F. P. Cossio and M. A. Sierra, Chem. Rev., 2009, 109, 6687 CrossRef CAS PubMed.
-
(a) L. MacAleese and P. Maître, Mass Spectrom. Rev., 2007, 26, 583 CrossRef CAS PubMed;
(b) J. Roithova, Chem. Soc. Rev., 2012, 41, 547 RSC;
(c) K. R. Asmis and J. Sauer, Mass Spectrom. Rev., 2007, 26, 542 CrossRef CAS PubMed.
- G. B. Deacon and R. J. Phillips, Coord. Chem. Rev., 1980, 33, 227 CrossRef CAS.
-
(a) G. S. Groenewold, W. A. de Jong, J. Oomens and M. J. Van Stipdonk, J. Am. Soc. Mass Spectrom., 2010, 21, 719 CrossRef CAS PubMed;
(b) L. Ducháčková, D. Schröder and J. Roithová, Inorg. Chem., 2011, 50, 3153 CrossRef PubMed;
(c) J. Paterová, J. Heyda, P. Jungwirth, C. J. Shaffer, Á. Révész, E. L. Zins and D. Schröder, J. Phys. Chem. A, 2011, 115, 6813 CrossRef PubMed.
- R. Prazeres, F. Glotin, C. Insa, D. A. Jaroszynskiand and J. M. Ortega, Eur. Phys. J. D, 1998, 3, 87 CrossRef CAS.
- L. Feketeová, G. N. Khairallah and R. A. J. O'Hair, Eur. J. Mass Spectrom., 2008, 14, 107 CrossRef PubMed.
- There have been numerous reports on Cu(II) salts giving rise to Cu(I) ions upon ESI/MS. There are several mechanisms by which these redox processes can occur. They may: (i) involve redox changes in solution upon binding of a ligand prior to ESI/MS; (ii) arise during the ESI process itself; (iii) occur via gas-phase dissociation of Cu(II) containing ions after ESI. For lead references that describe each of these scenarios see:
(a) A. Tintaru, L. Charles, P. Milko, J. Roithová and D. Schröder, J. Phys. Org. Chem., 2009, 22, 229 CrossRef CAS;
(b) A. Tsybizova, J. Tarábek, M. Buchta, P. Holý and D. Schröder, Rapid Commun. Mass Spectrom., 2012, 26, 2287 CrossRef CAS PubMed;
(c) A. Tsybizova, B. L. Ryland, N. Tsierkezos, S. S. Stahl, J. Roithová and D. Schröder, Eur. J. Inorg. Chem., 2014, 1407 CrossRef CAS.
-
M. J. Frisch, et al., Gaussian 09, A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
-
(a) M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866 CrossRef CAS;
(b) P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213 CrossRef CAS;
(c) T. Clark, J. Chandrasekhar and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294 CrossRef CAS;
(d) R. Krishnam, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650 CrossRef;
(e) P. M. W. Gill, B. G. Johnson, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1992, 197, 499 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- C.-H. Guo, H.-S. Wu, X.-M. Zhang, J.-Y. Song and X. Zhang, J. Phys. Chem. A, 2009, 113, 6710 CrossRef CAS PubMed.
- H. Dossmann, C. Afonso, J.-C. Tabet and E. Uggerud, Int. J. Mass Spectrom., 2013, 354–355, 165 CrossRef CAS.
-
(a) T. Naota, A. Tannna, S. Kamuro and S.-I. Murahashi, J. Am. Chem. Soc., 2002, 124, 6842 CrossRef CAS PubMed;
(b) T. Naota, A. Tannna, S. Kamuro, M. Hieda, K. Ogata, S.-I. Murahashi and H. Takaya, Chem. – Eur. J., 2008, 14, 2482 CrossRef CAS PubMed.
- For previous gas-phase studies of [NCCuCN]−, see:
(a) I. G. Dance, P. A. W. Dean and K. J. Fisher, Inorg. Chem., 1994, 33, 6261 CrossRef CAS;
(b) X. B. Wang, Y. L. Wang, J. Yang, X. P. Xing, J. Li and L. S. Wang, J. Am. Chem. Soc., 2009, 131, 16368 CrossRef CAS PubMed.
- A reviewer has raised an intriguing observation regarding the major changes in shape and hence volume during the decarboxylation reaction pathways calculated in Fig. 4 and 5. In the condensed phase, this would result in a volume of activation that might be measured through high pressure studies (see for example:
(a) D. R. Stranks, Pure Appl. Chem., 1974, 38, 303 CrossRef CAS). We are unaware of any such studies for Cu(I) mediated decarboxylation in the condensed phase, although the activation volumes of other decarboxylation reactions have been determined:
(b) R. Van Eldik, T. Asano and W. J. Le Noble, Chem. Rev., 1989, 89, 549 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Complete citation for ref. 26. Cartesian coordinates and energies for all calculated species, additional mass spectra, structural comparisons, electron affinity comparisons, level of theory comparisons, additional figures as mentioned in the text (S1–S7). See DOI: 10.1039/c5dt00942a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a