
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFluorescent benzene-centered mono-, bis- and tris-triazapentadiene–boron complexes†‡
Christoph
Glotzbach
a,
Nadine
Gödeke
a,
Roland
Fröhlich
a,
Constantin-Gabriel
Daniliuc
a,
Shohei
Saito
b,
Shigehiro
Yamaguchi
b and
Ernst-Ulrich
Würthwein
*a
aOrganisch-Chemisches Institut, Westfälische Wilhelms-Universität, Corrensstr. 40, 48149 Münster, Germany. E-mail: wurthwe@uni-muenster.de
bInstitute of Transformative Bio-molecules, and Department of Chemistry, Graduate School of Science, Nagoya University, Furo, Chikusa, Nagoya 464-8602, Japan
First published on 29th April 2015
Abstract
A series of novel benzene centered mono-, bis- and tris-1,3,5-triazapentadiene ligands 6a–e was synthesized and investigated with respect to their reactivity towards triphenylborane. The resulting blue-fluorescent boron complexes 14a–e with a six-membered ring chelate structure show excellent thermal and chemical stability. All title compounds were completely characterized including X-ray diffraction studies for 14a–c and 14e. Whereas the absorption spectra of all three classes of compounds are similar, the fluorescence spectra show distinct differences. Thus, the emission spectra of 14a,b show Stokes shifts of 4100–6700 cm−1 with low quantum yields both in solution and in the solid state. However, the more bulky compounds 14c–e show markedly larger molar extinction coefficients and smaller bathochromic shifts compared to 14a,b. For all compounds, we observe significantly more intense red-shifted fluorescence in the solid state compared to that in dichloromethane solutions. For the interpretation of the absorption properties TD-DFT studies were performed based on DFT geometry optimizations.
Introduction
Although 1,3,5-triazapentadienes are known since 1907,1–4 relatively little is known about their coordination chemistry compared to their carbon analogues β-diimine- and the well-known β-diketone ligands (Scheme 1).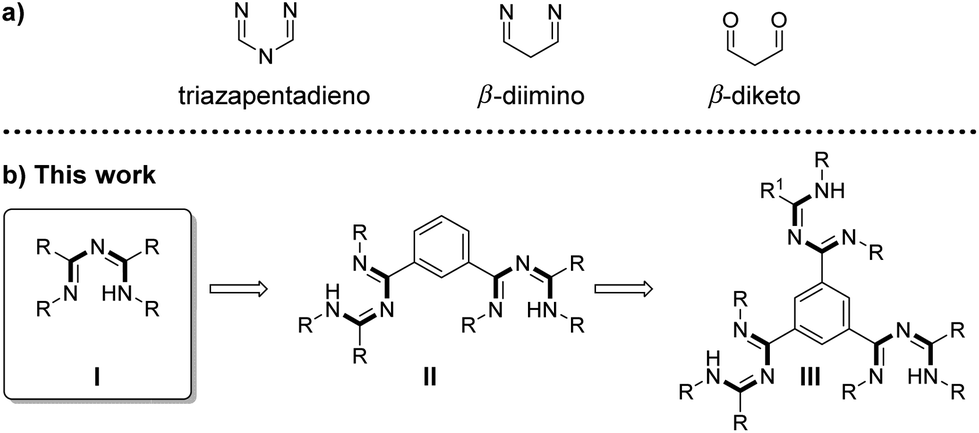 | ||
| Scheme 1 (a) Backbone of a 1,3,5-triazapentadiene ligand and its carbon analogues. (b) Extension of the monotriazapentadiene (I) to the benzene-centered bis- (II) and tris-triazapentadiene (III). | ||
1,3,5-Triazapentadienes are mostly used as ligands for transition metal ions,5,6 whereas coordination to main group elements, such as Al,7 Ga,8 Tl9 and boron, is currently underrepresented in the literature. Especially boron seems of interest to us, since boron–nitrogen compounds are known to exhibit intense fluorescence properties10,11 and are also used in modern applications like OLEDs.12 In 1962 the first neutral 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 triazapentadiene–boron complex was synthesized by Milks et al.13 and, a decade later, Mikhailov et al. started to intensively study the synthesis of similar complexes.14 Since then, only a small number of research groups including ours15–18 have studied this class of neutral triazapentadiene–boron complexes. Species with such compositions and structural properties have not yet received intense attention, with some aza-boron-dipyridomethenes and aza-boron-diquinomethene systems showing the closest similarity to our compounds.19 In most of the studies, the photophysical properties are either not mentioned or are only of minor interest. This background encouraged us to look into this matter more closely.
:1 triazapentadiene–boron complex was synthesized by Milks et al.13 and, a decade later, Mikhailov et al. started to intensively study the synthesis of similar complexes.14 Since then, only a small number of research groups including ours15–18 have studied this class of neutral triazapentadiene–boron complexes. Species with such compositions and structural properties have not yet received intense attention, with some aza-boron-dipyridomethenes and aza-boron-diquinomethene systems showing the closest similarity to our compounds.19 In most of the studies, the photophysical properties are either not mentioned or are only of minor interest. This background encouraged us to look into this matter more closely.
In this article we describe (a) the synthesis and full structural characterization of a series of novel benzene-centered mono-, bis- and tris-1,3,5-triazapentadiene ligands and their di-phenylboron complexes featuring an increasing number of possible electronic and steric interactions, (b) their photophysical properties in solution and in the crystalline state as a function of the size of the complexes, which are (c) supported by extensive quantum chemical calculations for a better understanding of their electronic structures in the ground state and in the excited state. We were especially interested to know whether the accumulation of up to three boron complexes at the periphery of a benzene moiety exhibits intense electronic interactions of these photochemically active subgroups.
Results and discussion
Synthesis
Mono-triazapentadiene ligands 6a,b: the synthesis of the hitherto unknown secondary triazapentadiene ligand 6a, which bears two hydrogen atoms at the terminal nitrogen atom, was achieved by following a procedure developed by Heße et al.,4,5 whereas the novel tertiary triazapentadiene ligand 6b (with one hydrogen atom at the terminal nitrogen atom) was synthesized by adapting a procedure of Häger et al. (Scheme 2).16 | ||
| Scheme 2 Synthetic pathways for secondary (6a) and tertiary (6b) mono-triazapentadiene ligands. | ||
Bis- and tris-triazapentadiene ligands 6c–e: the procedure used for 6b is also the basis for the preparation of the bis-triazapentadienes 6c–d20 and the novel three-armed tris-triazapentadiene ligand 6e (Scheme 3).
 | ||
| Scheme 3 Synthesis of (a) bis- (6c, d) and (b) tris-triazapentadiene ligands 6e. | ||
Because of the incomplete reaction and its distinct basic nature the free ligand 6e is quite difficult to isolate. Therefore we decided to use the crude product 6e for further reactions since we know from experience that the triazapentadiene boron compounds are readily purified by either recrystallization or via column chromatography on alumina oxide.15,21
Boron complexes 14a–e: the novel boron complexes derived from these ligands were synthesized by heating a solution of the respective triazapentadiene ligands 6a–e with 1 to 3 equivalents of triphenylborane in toluene under reflux for 5 hours (Scheme 4).
 | ||
| Scheme 4 Synthesis of the triazapentadiene–boron complexes 14a–e. | ||
After column chromatography on neutral alumina oxide, boron complexes 14a–e featuring up to seven, thirteen and nineteen cyclic units in close chemical neighbourhood were obtained as yellow solids in yields from 47% to 77%. Recrystallization afforded crystalline solids, which have melting points in the range of 125–205 °C and show excellent thermal and chemical stability.
With these boron compounds in hand, a closer look into the structural properties and the influence of steric and electronic effects on the photophysical properties of the triazapentadiene boron complexes were the next aims of this study.
X-Ray structures
Single crystal X-ray diffraction analyses were carried out for 14a–c and 14e (Fig. 1; for 14a, see Fig. S4, ESI‡). For all three types of compounds substantial deviations from planarity are observed for the heterocyclic units due to the steric properties of the aryl substituents at the B, N, and C-positions and due to the sp3-hybridized boron atom (the dihedral angle C–N–B–N amounts to 43–51°). Furthermore the peripheral aryl substituents are strongly tilted with respect to the heterocycle (34–54° for the C-bound aryl groups, 44–64° for the N-bound aryl groups). Individual differences of the tilt angles may be due to the nature of the respective substitution patterns as well as due to crystal packing effects in the solid state. In general, our new boron compounds 14a–c and 14e may be described more or less as propeller shaped molecules, which was postulated to be a prerequisite to display the AIE effect in the solid state (see below).22 | ||
| Fig. 1 ORTEP plots (thermal ellipsoids with 50% probability) of the molecular structures of 14b, c and e. For detailed structural data for compounds 14a–c, and 14e see the ESI.‡ | ||
Photophysical properties
Dichloromethane solutions of the boron complexes 14a,b absorb light between λabs = 250–450 nm with 14a having its maximum at λabs,max = 263 nm and 14b at λabs,max = 307 nm. In both cases the longest absorption maximum occurs at λabs = 353 nm in the form of a broad shoulder with molar extinction coefficients of ε353 nm = 1000 M−1 cm−1 L for 14a and ε353 nm = 1300 M−1 cm−1 L for 14b. They also exhibit blue emission in the range of λem = 413–487 nm (Fig. 2, Table 1). In each case three bands are observed for 14a at λem = 413 nm, 453 nm (fluorescence maximum) and 479 nm (very broad), for 14b at λem = 414 nm (fluorescence maximum), 446 nm and 487 nm (very broad). This leads to Stokes shifts of Δν = 4100 cm−1 (14a) and Δν = 4200 cm−1 (14b). The absolute quantum yields of the emissions are below one percent (ΦF < 0.01). This is consistent with the observation that the emission of 14a and 14b cannot be detected by the naked eye as shown in Fig. 2. | ||
| Fig. 2 (a) Photograph of compound 14a in solution and (b) the normalized absorption and emission spectra of 14a in CH2Cl2. (c) Normalized absorption and emission spectra of 14b in CH2Cl2 and (d) the photograph of compound 14b in solution. Irradiation was performed with a UV lamp with a wavelength of 366 nm. | ||
| Compound | λ abs /nm | ε max /M−1 cm−1 L | λ em /nm | Δνd/cm−1 | Φ F |
|---|---|---|---|---|---|
| a Longest wavelength absorption maxima recorded in dichloromethane using concentrations of about 10−5 M. b Molar extinction coefficient at the longest-wavelength absorption maximum. c Fluorescence maxima. Wavelengths are given for solution/solid. d Stokes shift in cm−1 as calculated from the longest wavelength absorption λabs and the shortest wavelength emission λem data (compare text and Fig. 2 and 3). e Absolute fluorescence quantum yield determined by a calibrated integration sphere system. Quantum yields are given for the solution/solid state. | |||||
| 14a | 353 | 1000 | 453/600 | 4100 | <0.01/0.02 |
| 14b | 353 | 1300 | 414/590 | 4200 | <0.01/0.03 |
| 14c | 365 | 19000 |
542/560 | 6100 | <0.01/0.03 |
| 14d | 365 | 19000 |
539/560 | 6100 | <0.01/0.04 |
| 14e | 363 | 26700 |
517/560 | 6700 | <0.01/0.02 |
Interestingly, introducing an electron-donating group such as the p-methoxyphenyl group at the triazapentadiene unit (14b) has only a minor influence on the photophysical properties. As seen from the UV-spectra, the exchange of a single substituent at one of the many aryl groups has only a very little effect on the spectroscopic properties. The background for choosing different substitution patterns is mainly to demonstrate the synthetic versatility of our experimental method.
By increasing the steric bulk of this class of compounds by introducing a second (14c,d) and a third (14e) triazapentadiene boron unit, we attempted to decrease the conformational freedom of rotations of the ancillary substituents, thus making the possibility for radiative decay from the excited state dominant, which would result in an enhanced emission compared to the mono triazapentadiene boron compounds 14a,b. These additional triazapentadiene boron units of compounds 14c–e led in solution to relatively small bathochromic shifts but a strong increase of the molar extinction coefficients ε of the absorption (14c,d: λabs = 365 nm, ε365 nm = 19000 M−1 cm−1 L; 14e: λabs = 363 nm, ε363 nm = 26700 M−1 cm−1 L; Table 1), which goes in line with the higher number of chromophores per volume in the bis- and tris-triazapentadiene boron complexes compared to the mono-compounds, resulting in more electronic transitions in the UV/vis-spectra. Similarly, the fluorescence maxima (14c: λem,max = 542 nm; 14d: λem,max = 539 nm; 14e: λem,max = 517 nm) are red-shifted compared to the mono triazapentadiene boron complexes 14a,b (Fig. 3, Table 1). The Stokes shifts of these three compounds 14c–e are significantly larger compared to those of 14a,b. However, there is no enhancement of the absolute quantum yields, which are still below one percent (ΦF < 0.01). Thus, neither the differing substituent effects (14b, 14d) nor the enhanced steric bulk (14e) result in a strong increase of the fluorescence of the complexes 14 in solution.
 | ||
| Fig. 3 (a) Absorption spectra of 14c, d and (b) their normalized emission spectra. (c) Absorption spectrum of tris-triazapentadiene boron complex 14e and (d) its normalized emission spectrum (10−5 M solutions in CH2Cl2). | ||
In contrast to the fluorescence of the solutions, the fluorescence of the crystalline materials 14a–e is clearly visible to the naked eye upon irradiating the sample with a handheld UV-lamp as shown exemplarily in Fig. 4 for complexes 14a,c and e.
 | ||
| Fig. 4 Photographs of crystalline 14a, 14c and 14eunder (a) daylight and (b) in a dark room under UV irradiation. Irradiation was performed with a UV lamp with a wavelength of 366 nm. (c) Normalized emission spectra of 14a, 14c, and 14e in the solid state. | ||
The solid state emissions of the mono triazapentadiene boron complexes 14a and 14b are detected at λem = 600 nm and λem = 590 nm, respectively. The bis-triazapentadiene boron complexes 14c,d and the tris-triazapentadiene boron compound 14e show their emission maximum in the solid state at the same wavelength (λem = 560 nm). Thus, all the emissions of the crystalline compounds are significantly red-shifted. While, as stated above, the quantum yields for 14a–e in solution are below one percent, the absolute quantum yields in the crystalline state are at ΦF = 0.02 for 14a, ΦF = 0.03 for 14b and 14c, ΦF = 0.04 for 14d and ΦF = 0.02 for 14e (Table 1).
This interesting behaviour of compounds 14a–e indicates different emission properties in solution compared to those in the solid state. The formation of excimers and the aggregation induced emission effect (AIE), as first described in 2001 by the research group of B. Z. Tang,22 are possible explanations for these phenomena, which have to be studied in more detail before a conclusive interpretation can be made. Certainly, steric bulk and therefore hindered rotation of the aryl substituents are possible reasons which have to be considered.23
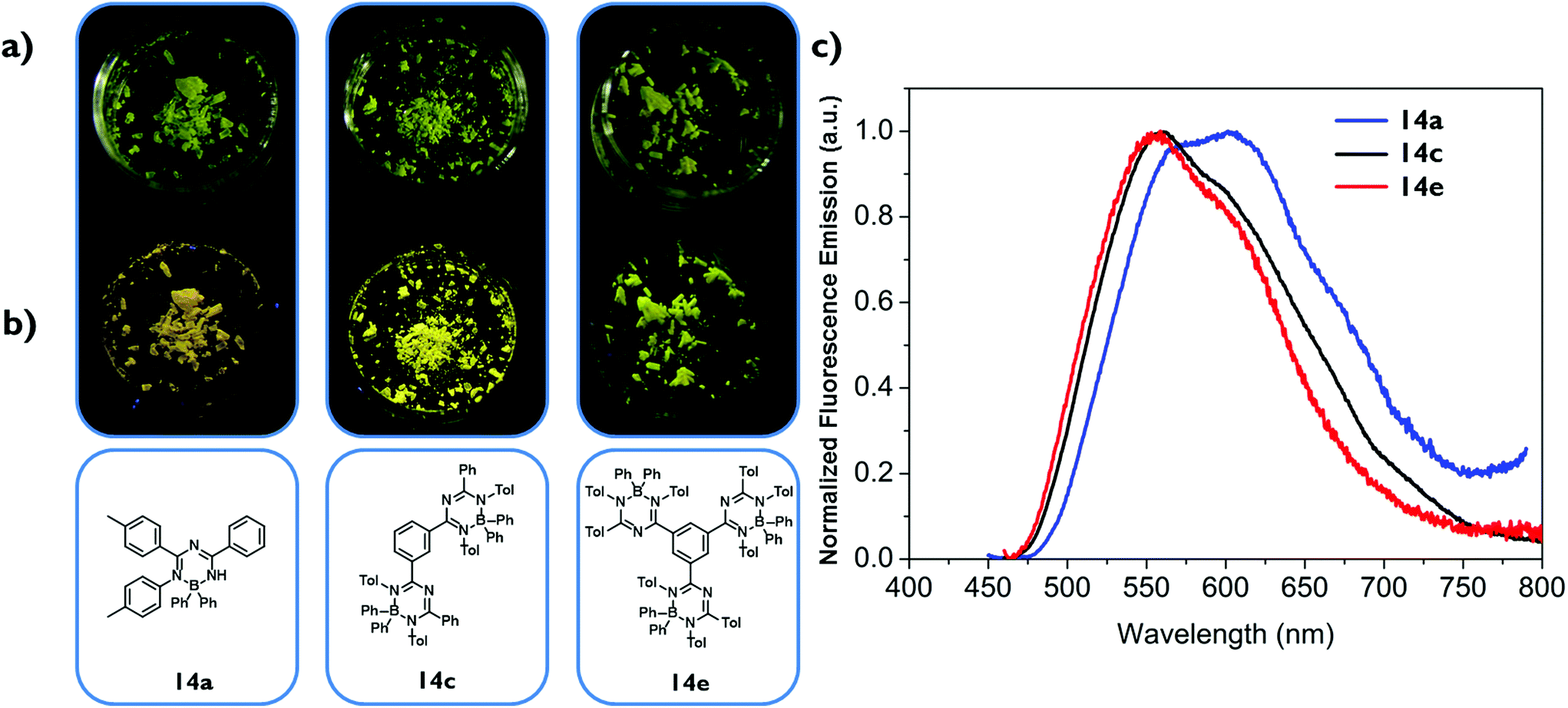
To demonstrate the different emission properties in solution and in the solid state by an additional qualitative experiment, compound 14a was dissolved in acetonitrile and water was added successively (Fig. 5).24 As stated above, the acetonitrile solution of 14a is not emitting light visible to the naked eye upon UV irradiation, but after addition of water 14a precipitates due to its poor solubility in water and starts to emit yellow light. Worth noting is the considerable red-shift in the photoluminescence spectra of 14a from λem = 453 nm for a solution in pure acetonitrile to λem = 550 nm for a suspension in a 1:1 acetonitrile/water mixture, which is possibly due to aggregation resulting in the formation of excimers. One should keep in mind that these spectra are to be treated qualitatively, since scatter light or other detection errors caused by particles are likely to occur.
 | ||
| Fig. 5 Images of an acetonitrile/water (1:0–1:1) solution of 14a during UV irradiation (left). Normalized photoluminescence spectrum of 14a in a 1:0 (CH3CN/H2O) solution and a 1:1 (CH3CN/H2O) suspension (right). Irradiation was performed with a UV lamp with a wavelength of 366 nm. | ||
Quantum chemical-DFT calculations
To investigate the electronic structures of the mono-, bis- and tris-triazapentadiene boron compounds, DFT geometry optimizations of 14a–c and 14e at the B3LYP25/def2-TZVP level using the GAUSSIAN 09 package of programs26 for the gas phase without consideration of solvent effects were performed using the X-ray data as starting geometries. Subsequently, to study the light absorption of these molecules, excited singlet states and the respective transitions were evaluated using TD-DFT calculations considering each of 20 states (Gaussian 09: td = (root = 1, nstates = 20); see the ESI‡).27 The calculated and experimental UV/vis spectra including the oscillator strengths of the excited singlet states and the relevant Kohn–Sham molecular orbitals of 14a, 14c and 14e are shown in Fig. 6–9. Additional data are given in the ESI.‡ | ||
| Fig. 6 The experimental (black, y1-axis) and calculated (blue) UV/VIS spectra of 14a with the associated oscillator strength (y2-axis) (left). Transitions involving the excited states S1, S2, S3 and S4 and the relevant Kohn–Sham orbitals (right). For more detailed information see the ESI.‡ | ||
 | ||
| Fig. 7 The experimental (black, y1-axis) and calculated (blue) UV/VIS spectra of 14c with the associated oscillator strength (y2-axis) (upper left). Kohn–Sham molecular orbitals of 14c. The degree of degeneracy of the energy levels is clearly visible within the HOMO/HOMO−1, HOMO−2/HOMO−3 and HOMO−4/HOMO−5 pairs. For more detailed information, see the ESI.‡ | ||
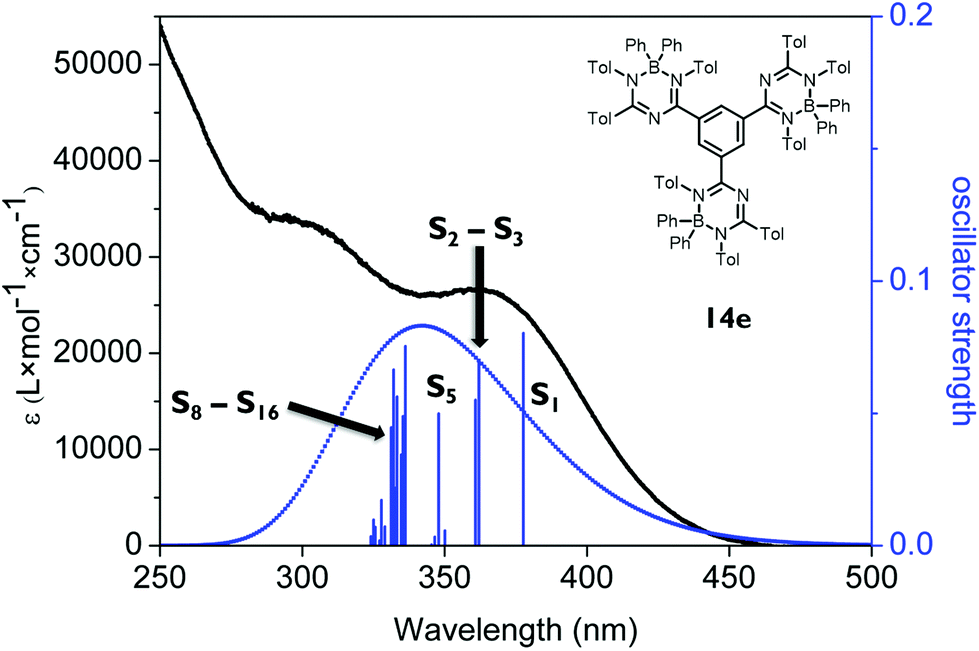 | ||
| Fig. 8 The experimental UV/VIS spectrum (black, y1-axis) and the calculated excitation wavelengths of states S1–S20 for 14e with the associated oscillator strength (y2-axis). | ||
 | ||
| Fig. 9 Kohn–Sham molecular orbital pictures of compound 14e. | ||
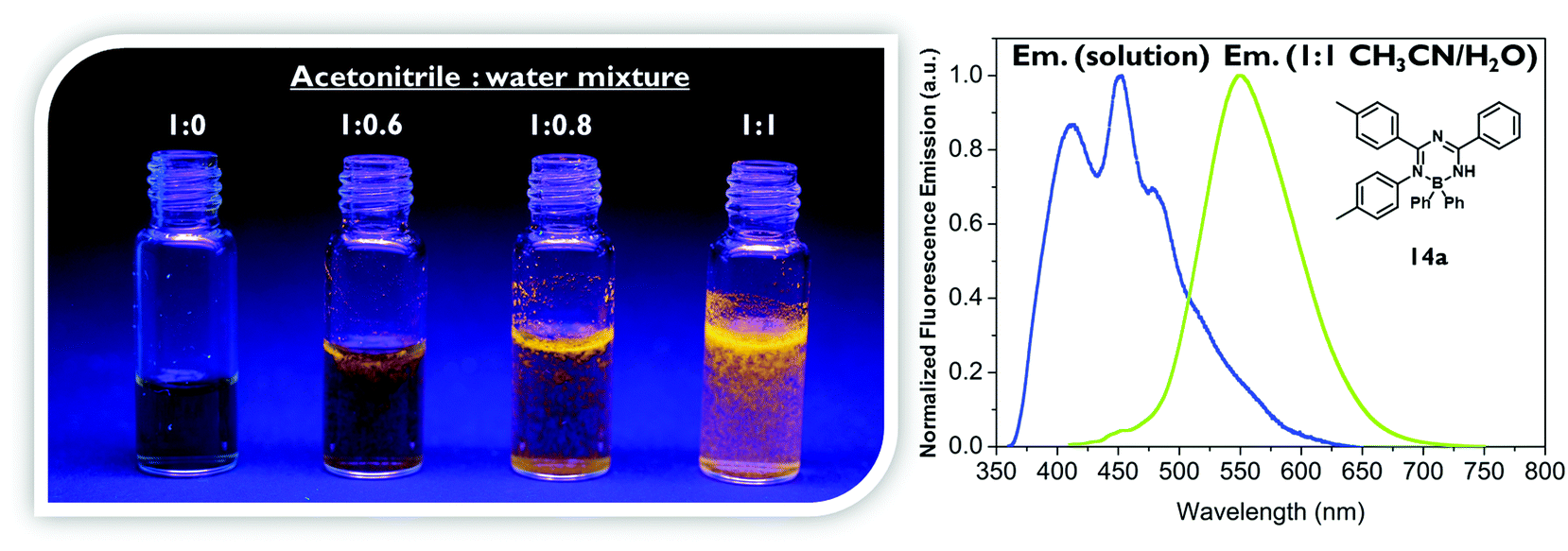
For the mono-1,3,5-triazapentadiene–boron complex 14a, the calculated UV/vis absorption spectrum matches quite well with the experimental one (Fig. 6). The absorption band at the longer wavelength, λabs,calc = 343 nm (λabs,exp = 353 nm), is described by the transitions involving S1, S2, S3 and S4 from the occupied molecular orbitals located mostly at the aryl substituents of the boron atom (HOMO−1, HOMO−2, HOMO−3) and at the nitrogen lone pairs as well as at the substituents of the triazapentadiene backbone (HOMO, HOMO−4) to the LUMO. Thus, the photochemistry of this system might be traced back to intramolecular charge transfer processes (ICT). The most intense absorption band at λabs,calc = 273 nm (λabs,exp = 263 nm) is predominantly described by the π–π* transitions involving the states S7, S9 and S13, which can exclusively be traced back to excitations from occupied molecular orbitals at the terminal substituents and the electron pairs at the nitrogen atoms of the triazapentadiene backbone (see the ESI‡) into the LUMO which is concentrated at the triazapentadiene boron chelate.
For the bis-1,3,5-triazapentadiene–boron complex 14c the experimentally observed absorption band at λabs,exp = 359 nm is reproduced by the calculations with a value of λabs,calc = 365 nm (Fig. 7). We assign it mainly to π–π* transitions involving the states S1 (HOMO−1 → LUMO, HOMO → LUMO, HOMO → LUMO+1), S2 (HOMO → LUMO; HOMO−1 → LUMO) and S5 (HOMO−3 → LUMO; (HOMO−4 → LUMO) (see the ESI‡).
The absorption spectrum of the tris-1,3,5-triazapentadiene–boron complex 14e (Fig. 8) is qualitatively also similar to the spectra of 14a and 14c (Fig. 7). The calculated and the experimental spectra show their longest wavelength absorption at λabs,exp = 363 nm and λabs,calc = 345 nm, which is represented by calculated transitions involving the states S1 (λabs,calc = 378 nm), S2 (λabs,calc = 362 nm), S3 (λabs,calc = 361 nm) and S5 (λabs,calc = 348 nm) involving usual π–π* excitations from the three distinctly different orbitals HOMO, HOMO−1 and HOMO−2 located at the individual triazapentadiene subunits into their LUMO counterparts. The latter are characterized by participation of one (LUMO), two (LUMO+1, LUMO+3, LUMO+4) and three (LUMO+2) triazapentadiene boron units, which are electronically separated from each other. The absorption band at lower wavelength at λabs,exp = 298 nm is only partially represented by the accumulation of transitions involving the states S8–S20 (λabs,calc = 335 nm) concerning occupied orbitals at the peripheral aryl substituents of the triazapentadiene boron units (e.g. HOMO−3, HOMO−4, HOMO−5) and the LUMO, LUMO+1 and LUMO+2 located at the three triazapentadiene–boron cores of the molecule (Fig. 9) (see the ESI‡ for additional Kohn–Sham molecular orbitals and calculation details). Not unexpectedly we noticed that the calculated gas phase absorption features of 14c and 14e do not match well with the experimental results at short wavelength when compared to 14a and 14b. Certainly, steric bulk and consequently more possible conformations and the lack of the solvent in the calculations are reasons for this observation.
As the experimental and calculated data clearly indicate, the mono-, the bis- and the tris-triazapentadiene boron compounds behave unexpectedly similarly with respect to their light absorption properties. This is indicated by the spectra and the orbital pictures and also by the similar HOMO/LUMO gaps which vary only in the range of ΔEHOMO–LUMO = 3.75–3.93 eV. Thus, due to the heavy steric hindrance, all three systems exhibit only limited delocalization of electrons throughout the molecule. In contrast, as shown above, the fluorescence properties (emission maxima and Stokes shifts) of these three types of boron compounds differ significantly, especially in the solid state. This indicates the importance of steric properties caused by intra- and intermolecular interactions.
Experimental section
General procedures
Compounds 1–3, 5 and 7 are commercially available. The bis-triazapentadiene ligands 6c and 6d including their precursors (11c–13c, 11d–13d) were synthesized following a literature procedure.20 Melting points are uncorrected. Nuclear magnetic resonance spectroscopy (1H, 13C, 11B, 19F) was performed on superconductive spectrometers: 300 MHz (1H: 300.1 MHz, 13C: 75.5 MHz, 19F: 282.4 MHz, 11B: 96.3 MHz), 400 MHz (1H: 400.1 MHz, 13C: 100.6 MHz), 500 MHz (1H: 499.9 MHz, 13C: 125.7 MHz, 19F: 470.3 MHz, 11B: 160.4 MHz) and 600 MHz (1H: 599.8 MHz, 13C: 150.8 MHz, 19F: 564.4 MHz, 11B: 192.4 MHz). 1H NMR and 13C NMR: chemical shifts δ are given relative to TMS and referenced to the solvent signal. 19F NMR: chemical shifts δ are given relative to CFCl3 (external reference), 11B NMR: chemical shifts δ are given relative to BF3·OEt2 (external reference). All signals in the 1H NMR and 13C spectra were assigned on the basis of relative intensities, coupling constants, and GCOSY, GHSQC, GHMBC, 1D-COSY and 1D-NOE experiments. Mass spectra were recorded on a micrOTOF using electron spray ionization. UV/VIS absorption spectra were recorded with a resolution of 0.2 nm to 0.5 nm. If not stated otherwise, dilute solutions with concentrations about 10−5 M in a 1 cm square quartz cell in degassed dichloromethane were used. Fluorescence lifetimes were measured with a Picosecond Fluorescence Measurement System equipped with a picosecond light pulser (excitation wavelength 377 nm with a repetition rate of 10 Hz). Absolute fluorescence quantum yields were determined with a calibrated integrating sphere system. X-Ray diffraction (for more information please see the ESI‡): Data sets were collected with a Nonius KappaCCD diffractometer or a Rigaku CCD diffractometer with a rotating anode generator. The programs used: data collection, COLLECT (R. W. W. Hooft, Bruker AXS, 2008, Delft, The Netherlands) and CrystalClear (Rigaku Corp., 2000); data reduction Denzo-SMN (Z. Otwinowski, W. Minor, Methods Enzymol., 1997, 276, 307–326); absorption correction, Denzo (Z. Otwinowski, D. Borek, W. Majewski, W. Minor, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2003, 59, 228–234); structure solution SHELXS-97 (G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467–473); structure refinement SHELXL-97 (G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122) and graphics, XP (Bruker AXS, 2000). R-values are given for the observed reflections, and wR2 values are given for all reflections. Supplementary crystallographic data for 14a, 14b, 14c and 14e CCDC 1011607–1011610 are given in the ESI.‡Synthesis of the starting materials and the mono- and tris-triazapentadiene ligands
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 147.7, 141.1, 134.4 (Cq), 132.5, 131.1, 129.6, 129.2, 128.8, 128.6, 128.1, 126.9, 121.4 (CAr), 21.3, 20.7 (CH3) ppm. Anal. Calcd for C22H21N3: C, 80.71; H, 6.46; N, 12.83. Found: C, 80.68; H, 6.40; N, 12.88.
N), 144.2, 142.1, 140.5, 136.8 (Cq), 136.6 (CAr), 135.3, 134.7, 134.5, 133.0 (Cq), 132.9, 131.0, 130.1, 129.4, 129.3, 129.1, 128.9, 127.3, 126.9, 113.6 (CAr), 55.5 (–OCH3), 21.6, 21.1, 21.0, 20.8 (CH3) ppm. HRMS (ESI): calcd for C32H34N3O ([M + H]+) 476.2696, observed 476.2695.
N), 158.5 (N–CN), 156.6 (Cq–OCH3), 147.5, 140.7, 133.7, 131.8, 130.0 (Cq), 129.2, 128.9, 128.1, 124.0, 121.7, 113.7 (CAr), 63.0 (H3CCH2O–), 55.4 (–OCH3), 21.6, 21.1 (–CH3), 14.5 (H3CCH2O–) ppm. HRMS (ESI): calcd for C24H27N2O2 ([M + H]+) 387.2067, observed 387.2067.
O), 136.5, 135.6, 133.0 (Cq), 129.7, 129.2, 120.4 (CAr), 20.6 (–CH3) ppm. HRMS (ESI): calcd for C30H27N3O3Na ([M + Na]+) 500.1945, observed 500.1945.
:1 mixture yielding 12e as a yellow crystalline compound in 71% (12.2 g) yield: mp 133 °C. 1H NMR (300 MHz; CD2Cl2): δ 9.08 (s, 3H, ArH), 7.36–7.13 (m, 6H, ArH), 7.12–6.93 (m, 6H, ArH), 2.40 (s, 9H, –CH3) ppm. 13C NMR (75 MHz; CD2Cl2): δ 145.0 (–(Cl–)CN), 141.2, 137.2, 136.2 (Cq), 133.3, 130.1, 121.1 (CAr), 21.3 (–CH3) ppm.
N), 147.6, 142.2, 137.4, 132.7 (Cq), 129.4, 129.3, 128.3, 122.1 (CAr), 63.7 (–O–CH2CH3), 21.7, 21.1 (–CH3), 14.6 (–O–CH2CH3) ppm. HRMS (ESI): calcd for C60H61N6O3 ([M + H]+) 913.4800, observed 913.4806.
N), 147.7, 141.1, 134.4 (Cq), 132.5, 131.1, 129.6, 129.2, 128.8, 128.6, 128.1, 126.9, 121.4 (CAr), 21.3, 20.7 (CH3) ppm. Anal. Calcd for C22H21N3: C, 80.71; H, 6.46; N, 12.83. Found: C, 80.68; H, 6.40; N, 12.88.
N), 144.2, 142.1, 140.5, 136.8 (Cq), 136.6 (CAr), 135.3, 134.7, 134.5, 133.0 (Cq), 132.9, 131.0, 130.1, 129.4, 129.3, 129.1, 128.9, 127.3, 126.9, 113.6 (CAr), 55.5 (–OCH3), 21.6, 21.1, 21.0, 20.8 (CH3) ppm. HRMS (ESI): calcd for C32H34N3O ([M + H]+) 476.2696, observed 476.2695.
N), 158.5 (N–CN), 156.6 (Cq–OCH3), 147.5, 140.7, 133.7, 131.8, 130.0 (Cq), 129.2, 128.9, 128.1, 124.0, 121.7, 113.7 (CAr), 63.0 (H3CCH2O–), 55.4 (–OCH3), 21.6, 21.1 (–CH3), 14.5 (H3CCH2O–) ppm. HRMS (ESI): calcd for C24H27N2O2 ([M + H]+) 387.2067, observed 387.2067.
O), 136.5, 135.6, 133.0 (Cq), 129.7, 129.2, 120.4 (CAr), 20.6 (–CH3) ppm. HRMS (ESI): calcd for C30H27N3O3Na ([M + Na]+) 500.1945, observed 500.1945.
:1 mixture yielding 12e as a yellow crystalline compound in 71% (12.2 g) yield: mp 133 °C. 1H NMR (300 MHz; CD2Cl2): δ 9.08 (s, 3H, ArH), 7.36–7.13 (m, 6H, ArH), 7.12–6.93 (m, 6H, ArH), 2.40 (s, 9H, –CH3) ppm. 13C NMR (75 MHz; CD2Cl2): δ 145.0 (–(Cl–)CN), 141.2, 137.2, 136.2 (Cq), 133.3, 130.1, 121.1 (CAr), 21.3 (–CH3) ppm.
N), 147.6, 142.2, 137.4, 132.7 (Cq), 129.4, 129.3, 128.3, 122.1 (CAr), 63.7 (–O–CH2CH3), 21.7, 21.1 (–CH3), 14.6 (–O–CH2CH3) ppm. HRMS (ESI): calcd for C60H61N6O3 ([M + H]+) 913.4800, observed 913.4806.
Synthesis of boron compounds 14a–14e based on mono-, bis- and tris-triazapentadiene ligands 6
N), 143.1, 140.3, 135.9, 135.7 (Cq), 134.9 (CAr), 134.6 (Cq), 133.1, 132.3, 131.3, 129.2, 129.1, 129.0, 127.0 (CAr), 21.5, 21.0 (–CH3) ppm. 11B NMR (96 MHz; C6D6): δ 0.4 (s, Bquart, ν1/2 ∼ 910 Hz) ppm HRMS (ESI): calcd for C34H31N3B ([M + H]+) 492.2611, observed 492.2603. UV/VIS (dichloromethane): λabs (shape, ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) , ε) = 353 nm (sh, 28329 cm−1, 1000 L mol−1 cm−1), 263 nm (p, 38023 cm−1, 4400 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 479 nm (p, 20876 cm−1), 453 nm (p, 22075 cm−1), 412 nm (p, 24272 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%.
N), 162.7, 143.4, 142.8, 140.7 (Cq), 136.4 (CAr), 136.2, 136.0, 135.6, 135.3 (Cq), 133.5, 131.8, 130.1, 129.5, 129.1, 129.2, 127.0, 126.6, 114.0 (CAr), 55.0 (–OCH3), 21.5, 21.4, 21.1, 21.0 (–CH3) ppm. 11B NMR (96 MHz; C6D6): δ 2.9 (s, Bquart, ν1/2 ∼ 960 Hz) ppm. HRMS (ESI): calcd for C44H43N3OB ([M + H]+) 640.3501, observed 640.3507. UV/VIS (dichloromethane): λabs (shape, , ε) = 353 nm (sh, 28329 cm−1, 1300 L mol−1 cm−1), 307 nm (p, 35573 cm−1, 1800 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 494 nm (sh, 20243 cm−1), 447 nm (p, 22371 cm−1), 414 nm (p, 24155 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%. Absolute quantum yield (crystalline): ΦF = 3%.
Cq–Cq–CH–Cq), 7.63–7.59 (m, 4H, ArH), 7.55–7.50 (m, 8H, B–ArH), 7.41 (dd, JHH = 7.8 Hz, JHH = 1.9 Hz, 2H, –NCq–Cq–CH–CH–CH–Cq), 7.35–7.31 (m, 2H, ArH), 7.29–7.23 (m, 4H, ArH), 7.20–7.10 (m, 12H, B–ArH), 6.99 (t, JHH = 7.8 Hz, 1H, –NCq–Cq–CH–CH–CH–Cq), 6.69 (ddt, JHH = 8.9, 7.4, 0.8 Hz, 8H, ArH), 6.65–6.60 (m, 4H, ArH), 6.51–6.46 (m, 4H, ArH), 2.13 (s, 6H, –CH3), 2.11 (s, 6H, –CH3) ppm. 13C NMR (150 MHz, CD2Cl2): δ 166.9, 165.9 (CN), 147.9 (B–Cq), 142.1, 142.0, 137.5, 136.9 (Cq), 135.9, 135.8 (Cq–CH3), 135.3 (B–CAr), 133.4, 131.7 (CAr,centralBenzene), 130.7, 130.1, 128.8, 128.7, 128.5, 128.4, 128.1 (CAr), 127.2 (CAr,centralBenzene), 127.0, 126.1 (B–CAr), 21.0, 20.9 (–CH3) ppm. 11B NMR (192 MHz, CD2Cl2): δ 2.4 (s, Bquart, ν1/2 ∼ 740 Hz) ppm. HRMS (ESI): calcd for C74H63B2N6 ([M + H]+) 1057.5316, observed 1057.5307. UV/VIS (dichloromethane): λabs (shape, , ε) = 365 nm (p, 27397 cm−1, 19000 L mol−1 cm−1), 297 nm (sh, 33670 cm−1, 20700 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 538 nm (p, 18587 cm−1), 469 nm (p, 21322 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%. Absolute quantum yield (crystalline): ΦF = 3%.
Cq–Cq–CH–Cq), 7.60–7.55 (m, 4H, ArH), 7.52–7.47 (m, 8H, B–ArH), 7.39–7.33 (m, 4H, ArH), 7.31–7.24 (m, 4H, ArH), 7.19–7.07 (m, 12H, B–ArH), 6.98 (td, JHH = 7.7 Hz, 5JHH = 0.5 Hz, 1H, –NCq–Cq–CH–CH–CH–Cq), 6.84–6.78 (m, 4H, ArH), 6.70–6.65 (m, 4H ArH), 6.65–6.61 (m, 4H, ArH), 6.47–6.41 (m, 4H, ArH), 2.11 (s, 6H, –CH3) ppm. 13C NMR (150 MHz, CD2Cl2): δ 167.3, 166.4 (CN), 147.7 (B–Cq), 143.7, 141.9, 137.2, 137.0 (Cq), 136.4 (Cq–CH3), 135.4 (B–CAr), 133.6, 132.0 (CAr,centralBenzene), 131.7 (Cq–Cl), 130.9, 130.7, 130.3, 129.0, 128.7, 128.5, 128.2 (CAr), 127.5 (CAr,centralBenzene), 127.4, 126.6 (B–CAr), 21.2 (–CH3) ppm. 11B NMR (192 MHz, CD2Cl2): δ 2.6 (s, Bquart, ν1/2 ∼ 600 Hz) ppm. HRMS (ESI): calcd for C72H57B2Cl2N6 ([M + H]+) 1097.4224, observed 1097.4219. UV/VIS (dichloromethane): λabs (shape, , ε) = 365 nm (p, 27397 cm−1, 19000 L mol−1 cm−1), 297 nm (sh, 33670 cm−1, 22800 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 538 nm (p, 18587 cm−1), 469 nm (p, 21322 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%. Absolute quantum yield (crystalline): ΦF = 4%.
N), 147.7 (B–Cq), 142.4, 141.4, 140.8 (Cq), 136.1 (Cq,centralBenzene), 135.9, 135.7 (Cq–CH3), 135.4 (B–CAr), 134.5 (Cq–CH3), 133.3 (CAr,centralBenzene), 130.8, 129.2, 128.7, 128.4, 128.2 (CAr), 127.0, 126.1 (B–CAr), 21.6, 21.1, 20.9 (–CH3) ppm. 11B NMR (192 MHz, CD2Cl2): δ −2.8 (s br, Bquart, ν1/2 ∼ 3000 Hz) ppm. HRMS (ESI): calcd for C111H97B3N9 ([M + H]+) 1588.8151, observed 1588.8167. UV/VIS (dichloromethane): λabs (shape, , ε) = 363 nm (p, 27548 cm−1, 26700 L mol−1 cm−1), 298 nm (p, 33557 cm−1, 33400 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 517 nm (p, 19342 cm−1), 479 nm (sh, 20877 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%. Absolute quantum yield (crystalline): ΦF = 2%.
, ε) = 353 nm (sh, 28329 cm−1, 1000 L mol−1 cm−1), 263 nm (p, 38023 cm−1, 4400 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 479 nm (p, 20876 cm−1), 453 nm (p, 22075 cm−1), 412 nm (p, 24272 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%.
N), 162.7, 143.4, 142.8, 140.7 (Cq), 136.4 (CAr), 136.2, 136.0, 135.6, 135.3 (Cq), 133.5, 131.8, 130.1, 129.5, 129.1, 129.2, 127.0, 126.6, 114.0 (CAr), 55.0 (–OCH3), 21.5, 21.4, 21.1, 21.0 (–CH3) ppm. 11B NMR (96 MHz; C6D6): δ 2.9 (s, Bquart, ν1/2 ∼ 960 Hz) ppm. HRMS (ESI): calcd for C44H43N3OB ([M + H]+) 640.3501, observed 640.3507. UV/VIS (dichloromethane): λabs (shape, , ε) = 353 nm (sh, 28329 cm−1, 1300 L mol−1 cm−1), 307 nm (p, 35573 cm−1, 1800 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 494 nm (sh, 20243 cm−1), 447 nm (p, 22371 cm−1), 414 nm (p, 24155 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%. Absolute quantum yield (crystalline): ΦF = 3%.
Cq–Cq–CH–Cq), 7.63–7.59 (m, 4H, ArH), 7.55–7.50 (m, 8H, B–ArH), 7.41 (dd, JHH = 7.8 Hz, JHH = 1.9 Hz, 2H, –NCq–Cq–CH–CH–CH–Cq), 7.35–7.31 (m, 2H, ArH), 7.29–7.23 (m, 4H, ArH), 7.20–7.10 (m, 12H, B–ArH), 6.99 (t, JHH = 7.8 Hz, 1H, –NCq–Cq–CH–CH–CH–Cq), 6.69 (ddt, JHH = 8.9, 7.4, 0.8 Hz, 8H, ArH), 6.65–6.60 (m, 4H, ArH), 6.51–6.46 (m, 4H, ArH), 2.13 (s, 6H, –CH3), 2.11 (s, 6H, –CH3) ppm. 13C NMR (150 MHz, CD2Cl2): δ 166.9, 165.9 (CN), 147.9 (B–Cq), 142.1, 142.0, 137.5, 136.9 (Cq), 135.9, 135.8 (Cq–CH3), 135.3 (B–CAr), 133.4, 131.7 (CAr,centralBenzene), 130.7, 130.1, 128.8, 128.7, 128.5, 128.4, 128.1 (CAr), 127.2 (CAr,centralBenzene), 127.0, 126.1 (B–CAr), 21.0, 20.9 (–CH3) ppm. 11B NMR (192 MHz, CD2Cl2): δ 2.4 (s, Bquart, ν1/2 ∼ 740 Hz) ppm. HRMS (ESI): calcd for C74H63B2N6 ([M + H]+) 1057.5316, observed 1057.5307. UV/VIS (dichloromethane): λabs (shape, , ε) = 365 nm (p, 27397 cm−1, 19000 L mol−1 cm−1), 297 nm (sh, 33670 cm−1, 20700 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 538 nm (p, 18587 cm−1), 469 nm (p, 21322 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%. Absolute quantum yield (crystalline): ΦF = 3%.
Cq–Cq–CH–Cq), 7.60–7.55 (m, 4H, ArH), 7.52–7.47 (m, 8H, B–ArH), 7.39–7.33 (m, 4H, ArH), 7.31–7.24 (m, 4H, ArH), 7.19–7.07 (m, 12H, B–ArH), 6.98 (td, JHH = 7.7 Hz, 5JHH = 0.5 Hz, 1H, –NCq–Cq–CH–CH–CH–Cq), 6.84–6.78 (m, 4H, ArH), 6.70–6.65 (m, 4H ArH), 6.65–6.61 (m, 4H, ArH), 6.47–6.41 (m, 4H, ArH), 2.11 (s, 6H, –CH3) ppm. 13C NMR (150 MHz, CD2Cl2): δ 167.3, 166.4 (CN), 147.7 (B–Cq), 143.7, 141.9, 137.2, 137.0 (Cq), 136.4 (Cq–CH3), 135.4 (B–CAr), 133.6, 132.0 (CAr,centralBenzene), 131.7 (Cq–Cl), 130.9, 130.7, 130.3, 129.0, 128.7, 128.5, 128.2 (CAr), 127.5 (CAr,centralBenzene), 127.4, 126.6 (B–CAr), 21.2 (–CH3) ppm. 11B NMR (192 MHz, CD2Cl2): δ 2.6 (s, Bquart, ν1/2 ∼ 600 Hz) ppm. HRMS (ESI): calcd for C72H57B2Cl2N6 ([M + H]+) 1097.4224, observed 1097.4219. UV/VIS (dichloromethane): λabs (shape, , ε) = 365 nm (p, 27397 cm−1, 19000 L mol−1 cm−1), 297 nm (sh, 33670 cm−1, 22800 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 538 nm (p, 18587 cm−1), 469 nm (p, 21322 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%. Absolute quantum yield (crystalline): ΦF = 4%.
N), 147.7 (B–Cq), 142.4, 141.4, 140.8 (Cq), 136.1 (Cq,centralBenzene), 135.9, 135.7 (Cq–CH3), 135.4 (B–CAr), 134.5 (Cq–CH3), 133.3 (CAr,centralBenzene), 130.8, 129.2, 128.7, 128.4, 128.2 (CAr), 127.0, 126.1 (B–CAr), 21.6, 21.1, 20.9 (–CH3) ppm. 11B NMR (192 MHz, CD2Cl2): δ −2.8 (s br, Bquart, ν1/2 ∼ 3000 Hz) ppm. HRMS (ESI): calcd for C111H97B3N9 ([M + H]+) 1588.8151, observed 1588.8167. UV/VIS (dichloromethane): λabs (shape, , ε) = 363 nm (p, 27548 cm−1, 26700 L mol−1 cm−1), 298 nm (p, 33557 cm−1, 33400 L mol−1 cm−1). Fluorescence (dichloromethane): λem (shape, ) = 517 nm (p, 19342 cm−1), 479 nm (sh, 20877 cm−1). Absolute quantum yield (dichloromethane): ΦF = <1%. Absolute quantum yield (crystalline): ΦF = 2%.
Conclusions
In summary, we have described the modular synthesis and full structural characterization including X-ray diffraction analysis of two novel mono-1,3,5-triazapentadiene–boron complexes 14a,b, two hitherto unknown bis-1,3,5-triazapentadiene–boron complexes 14c,d and the first tris-triazapentadiene boron complex 14e. In order to evaluate their photophysical properties all compounds were investigated by absorption and emission spectroscopy in solution as well as in the solid state. The experimentally observed relatively similar absorption properties in solution of 14a,c,e were interpreted on the basis of quantum chemical TD-DFT calculations as a result of only limited electron delocalization involving the central benzene ring and the neighboring triazapentadiene boron systems due to the specific steric properties. The inspection of the relevant Kohn–Sham molecular orbitals indicates that the photochemistry of all these systems 14 is based on intramolecular charge transfer processes (ICT). In contrast, the emission properties of the three classes of compounds differ significantly with respect to emission maxima and Stokes shifts. Quite interestingly, whereas the fluorescence in solution of the compounds 14a–e is not visible to the naked eye, bright red-shifted emission was seen for these compounds in the solid state and, qualitatively, in an acetonitrile–water suspension. Most likely, steric bulk hindering the rotation of the substituents of these compounds in the solid state is responsible for these emission properties.Acknowledgements
Financial support from the Deutsche Forschungsgemeinschaft (IRTG 1143 Münster-Nagoya and SFB 858) is gratefully acknowledged. This work was partly supported by JSPS Core-to-Core Program, A. Advanced Research Networks (S.Y.).References
- H. Ley and F. Müller, Ber. Dtsch. Chem. Ges., 1907, 40, 2950–2958 CrossRef.
- J. A. Elvidge and N. R. Barot, Imidines and diamidides (1,3,5-Triazapentadienes) Patai, The chemistry of functional groups, Suppl. A. The chemistry of doubly bonded functional groups part 2, Wiley, London, 1977, ch. 13 Search PubMed.
- F. C. Cooper, M. W. Partridge and F. W. Short, J. Chem. Soc., 1951, 391–404 RSC.
- N. Heße, R. Fröhlich, B. Wibbeling and E.-U. Würthwein, Eur. J. Org. Chem., 2006, 3923–3937 CrossRef.
- N. Heße, R. Fröhlich, I. Humelnicu and E.-U. Würthwein, Eur. J. Inorg. Chem., 2005, 2189–2197 CrossRef.
- (a) H. V. R. Dias, J. A. Flores, M. Pellei, B. Morresi, G. G. Lobbia, S. Singh, Y. Kobayashi, M. Yousufuddin and C. Santini, Dalton Trans., 2011, 40, 8569–8580 RSC; (b) M. N. Kopylovich and A. J. L. Pombeiro, Coord. Chem. Rev., 2011, 255, 339–355 CrossRef CAS; (c) J. Barker and M. Kilner, J. Chem. Soc., Dalton Trans., 1989, 837–841 RSC; (d) F. Liu, X. Qiao, M. Wang, M. Zhou, H. Tong, D. Gup and D. Liu, Polyhedron, 2013, 52, 539–544 Search PubMed; (e) A. Igashira-Kamiyama, T. Kajiwara, T. Konno and T. Ito, Inorg. Chem., 2006, 45, 6460–6466 CrossRef CAS PubMed; (f) V. A. K. Adiraju, J. A. Flores, M. Yousufuddin and H. V. R. Dias, Organometallics, 2012, 31, 7925–7932 CrossRef and references therein (g) N. Q. Shixaliyev, A. M. Maharramov, A. V. Gurbanov, V. G. Nenajdenko, V. M. Muzalevskiy, K. T. Mahmudov and M. N. Kopylovich, Catal. Today, 2013, 217, 76–79 CrossRef CAS and references therein.
- (a) K. Bakthavachalam and N. D. Reddy, Organometallics, 2013, 32, 3174–3184 CrossRef CAS; (b) J. Masuda and D. W. Stephan, Dalton Trans., 2006, 2089–2097 RSC.
- D. R. Aris, J. Barker, P. R. Phillips, N. W. Alcock and M. G. H. Wallbridge, J. Chem. Soc., Dalton Trans., 1997, 909–910 RSC.
- H. V. R. Dias, S. Singh and T. R. Cundari, Angew. Chem., Int. Ed., 2005, 44, 4907–4910 CrossRef CAS PubMed.
- (a) A. Treibs and F.-H. Kreuzer, Liebigs Ann. Chem., 1968, 718, 208–223 CrossRef CAS; (b) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed; (c) Q.-D. Liu, M. S. Mudadu, R. Thummel, Y. Tao and S. Wang, Adv. Funct. Mater., 2005, 15, 143–154 CrossRef CAS; (d) J. A. Araneda, W. E. Piers, B. Heyne, M. Parvez and R. McDonald, Angew. Chem., Int. Ed., 2011, 50, 12214–12217 CrossRef CAS PubMed.
- D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS PubMed.
- (a) Y.-L. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263–12274 CrossRef CAS PubMed; (b) G. E. Morse and T. P. Bender, ACS Appl. Mater. Interfaces, 2012, 4, 5055–5068 CrossRef CAS PubMed; (c) Z. M. Hudson and S. Wang, Dalton Trans., 2011, 40, 7805–7816 RSC.
- J. R. Milks, G. W. Kennerly and J. H. Polevy, J. Am. Chem. Soc., 1962, 84, 2529–2534 CrossRef CAS.
- (a) V. A. Dorokhov, B. M. Zolotarev, O. S. Chizhov and B. M. Mikhailov, Izv. Akad. Nauk. SSSR, Ser. Khim., 1977, 7, 1587–1593 Search PubMed; (b) B. M. Mikhailov, V. A. Dorokhov and V. I. Seredenko, Izv. Akad. Nauk. SSSR, Ser. Khim., 1977, 6, 1385–1390 Search PubMed; (c) B. M. Mikhailov, Pure Appl. Chem., 1977, 49, 749–764 CrossRef CAS; (d) O. G. Boldyreva, V. A. Dorokhov and B. M. Mikhailov, Izv. Akad. Nauk. SSSR, Ser. Khim., 1985, 2, 428–430 Search PubMed.
- (a) K. B. Anderson, R. A. Franich, H. W. Kroese, R. Meder and C. E. F. Rickard, Polyhedron, 1995, 14, 1149–1153 CrossRef CAS; (b) G. Sathyamoorthi, M.-L. Soong, T. W. Ross and J. H. Boyer, Heteroat. Chem., 1993, 4, 603–608 CrossRef CAS.
- I. Häger, R. Fröhlich and E.-U. Würthwein, Eur. J. Org. Chem., 2009, 2415–2428 CrossRef.
- (a) M. A. Dureen and D. W. Stephan, J. Am. Chem. Soc., 2010, 123, 13559–13568 CrossRef PubMed; (b) D. Zhao, G. Li, D. Wu, X. Qin, P. Neuhaus, Y. Cheng, S. Yang, Z. Lu, X. Pu, C. Long and J. You, Angew. Chem., Int. Ed., 2013, 52, 13676–13680 CrossRef CAS PubMed.
- (a) B. Neue, R. Fröhlich, B. Wibbeling, A. Fukazawa, A. Wakamiya, S. Yamaguchi and E.-U. Würthwein, J. Org. Chem., 2012, 77, 2176–2184 CrossRef CAS PubMed; (b) B. Neue, A. Wakamiya, R. Fröhlich, B. Wibbeling, S. Yamaguchi and E.-U. Würthwein, J. Org. Chem., 2013, 78, 11747–11755 CrossRef CAS PubMed.
- (a) D. Wang, R. Liu, C. Chen, S. Wang, J. Chang, C. Wu, H. Zhu and E. R. Waclawik, Dyes Pigm., 2013, 99, 240–249 CrossRef CAS; (b) Y. Deng, Y.-y. Cheng, H. Liu, J. Mack, H. Lu and L.-g. Zhu, Tetrahedron Lett., 2014, 55, 3792–3796 CrossRef CAS.
- J. I. Clodt, C. Wigbers, R. Reiermann, R. Fröhlich and E.-U. Würthwein, Eur. J. Org. Chem., 2011, 3197–3209 CrossRef CAS.
- C. Glotzbach, U. Kauscher, J. Voskuhl, N. S. Kehr, M. C. A. Stuart, R. Fröhlich, H. J. Galla, B. J. Ravoo, K. Nagura, S. Saito, S. Yamaguchi and E.-U. Würthwein, J. Org. Chem., 2013, 78, 4410–4418 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 36, 1740–1741 RSC.
- (a) C. Kitamura, Y. Tanigawa, T. Kobayashi, H. Naito, H. Kurata and T. Kawase, Tetrahedron, 2012, 68, 1688–1694 CrossRef CAS; (b) A. Dreuw, J. Plötner, L. Lorenz, J. Wachtveitl, J. E. Djanhan, J. Brüning, T. Metz, M. Bolte and M. U. Schmidt, Angew. Chem., Int. Ed., 2005, 44, 7783–7786 CrossRef CAS PubMed; (c) H. Langhals, T. Potrawa, H. Nöth and G. Linti, Angew. Chem., Int. Ed. Engl., 1989, 28, 478–480 CrossRef.
- (a) J. Chen, C. C. W. Law, J. W. Y. Lan, Y. Dong, S. M. F. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS; (b) G. Yu, S. Yin, Y. Liu, J. Chen, X. Xu, X. Sun, D. Ma, X. Zhan, Q. Peng, Z. Shuai, B. Z. Tang, D. Zhu, W. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335–6346 CrossRef CAS PubMed; (c) Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 44, 4332–4353 RSC.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS; (c) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS; (b) R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS.
- D. Hellwinkel, F. Laemmerzahl and G. Hofmann, Chem. Ber., 1983, 116, 3375–3405 CrossRef CAS.
- G. Tarzia, P. Schiatti, D. Selva, D. Favara and S. Ceriani, Eur. J. Med. Chem., 1976, 11, 263–270 CAS.
- H. Nakajima, M. Takahashi and Y. Kimura, Macromol. Mater. Eng., 2010, 295, 460–468 CAS.
- M. V. Zatsepina, T. V. Artamonova and G. I. Koldobskii, Russ. J. Org. Chem., 2008, 44, 577–581 CrossRef CAS.
- R. Kupfer, M. Nagel, E.-U. Würthwein and R. Allmann, Chem. Ber., 1985, 118, 3089–3104 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Prof. Dr Paul von Ragué Schleyer. |
| ‡ Electronic supplementary information (ESI) available: Spectroscopic, crystallographic and quantum chemical details. CCDC 1011607–1011610. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00908a |
| This journal is © The Royal Society of Chemistry 2015 |