
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iridium-based complexes for water oxidation
Julianne M.
Thomsen†
,
Daria L.
Huang†
,
Robert H.
Crabtree
* and
Gary W.
Brudvig
*
Department of Chemistry, Yale University, 225 Prospect St., New Haven, CT 06520, USA. E-mail: robert.crabtree@yale.edu; gary.brudvig@yale.edu
First published on 5th May 2015
Abstract
Organometallic Ir precatalysts have been found to yield homogeneous Ir-based water-oxidation catalysts (WOCs) with very high activity. The Cp*Ir catalyst series can operate under a variety of regimes: it can either act as a homogeneous or a heterogeneous catalyst; it can be driven by chemical, photochemical, or electrochemical methods; and the molecular catalyst can either act in solution or supported as a molecular unit on a variety of solid oxides. In addition to optimizing the various reaction conditions, work has continued to elucidate the catalyst activation mechanism and identify water-oxidation intermediates. This Perspective will describe the development of the Cp*Ir series, their many forms as WOCs, and their ongoing characterization.
 Julianne M. Thomsen | Julianne M. Thomsen received a B.S. in chemistry from Point Loma Nazarene University in San Diego, CA. After receiving her B.S. in 2010, she moved to Yale University to pursue a PhD in chemistry, advised by Gary Brudvig. |
 Daria L. Huang | Daria Huang received a B.S. from Georgetown University where she was a Clare Booth Luce Scholar and studied copper-catalyzed C–H amination under the direction of Professor Timothy Warren. She began her research as an NSF Graduate Research Fellow with Professor Robert Crabtree in 2012, investigating CH and water-oxidation reactions using Cp*Ir complexes. |
 Robert H. Crabtree | Robert H Crabtree, educated at Oxford with Malcolm Green, did his PhD with Joseph Chatt and spent four years in Paris with Hugh Felkin at the CNRS, Gif. He is now Whitehead Professor at Yale and has been an ACS and RSC organometallic chemistry awardee. Early work on catalytic alkane C–H activation and functionalization was followed by work on H2 complexes, dihydrogen bonding, and catalysis for green and energy chemistry. He is a Fellow of the ACS, of the RSC and of the American Academy. |
 Gary W. Brudvig | Gary W. Brudvig received a B.S. in 1976 from the University of Minnesota, a PhD in 1981 from Caltech working with Sunney Chan and was a Miller Postdoctoral Fellow with Ken Sauer at the University of California, Berkeley from 1980 to 1982. He has been on the faculty at Yale University since 1982 where he currently is the Benjamin Silliman Professor of Chemistry, Professor of Molecular Biophysics and Biochemistry, and Director of the Yale Energy Sciences Institute. |
I. Introduction
A. Historical and modern relevance of water oxidation
Water splitting is such a fundamental reaction that it attracted attention soon after water was recognized as a compound of hydrogen and oxygen. As early as 1797, Fourcroy, a collaborator of Lavoisier, reported that their efforts to split water into its elements were only successful for hydrogen production,1 but no reagent could be found to produce oxygen. The report makes it clear that they had already understood that water oxidation (WO) was a vital part of what we now know as photosynthesis and that the release of oxygen from water is driven by sunlight without release of hydrogen. However, it was only much later that the energetic demands of forming O2 from water were understood.2–5 Oxidation of water, which is carried out by photosystem II of green plants, is a four-electron process (eqn (1)) with a thermodynamic potential of 0.82 V vs. NHE at pH = 7.| 2H2O → O2 + 4e− + 4H+ | (1) |
In addition to this large thermodynamic requirement, the formation of O2 is kinetically difficult and in practice requires an additional overpotential to drive the reaction. Thus, it is unsurprising that, although work on WO began early, this problem continues to puzzle modern day investigators.
While early work was driven by academic curiosity, contemporary interest also arises from its relevance to the growing global demand for the generation and storage of renewable energy. Well-known geopolitical and environmental concerns are increasingly driving research efforts to produce carbon-neutral fuels from renewable resources.6,7 Of these resources, only sunlight has enough capacity to fully supply the world's energy needs. Practically, for utilization of sunlight, artificial photosynthesis shows particular promise for the conversion and storage of solar energy in high-energy fuel sources. Artificial photosynthesis can be regarded as the sum of two half-reactions: (1) water oxidation, which provides a source of electrons for (2) the reduction of feedstocks such as CO2 to reduced energy-dense carbon fuels such as methanol or formic acid, or perhaps to H2 (Fig. 1).8 Photons from sunlight are used to generate a charge separation, which drives a water-oxidation catalyst into the high oxidation state necessary to oxidize water and release electrons for the reduction of CO2. As Fourcroy and his contemporaries found, water oxidation is the more difficult half-reaction, and many consider it the bottleneck for development of the field.9 Thus, one of the major challenges for global implementation of artificial photosynthesis is finding competent catalysts for the water-oxidation half-reaction.
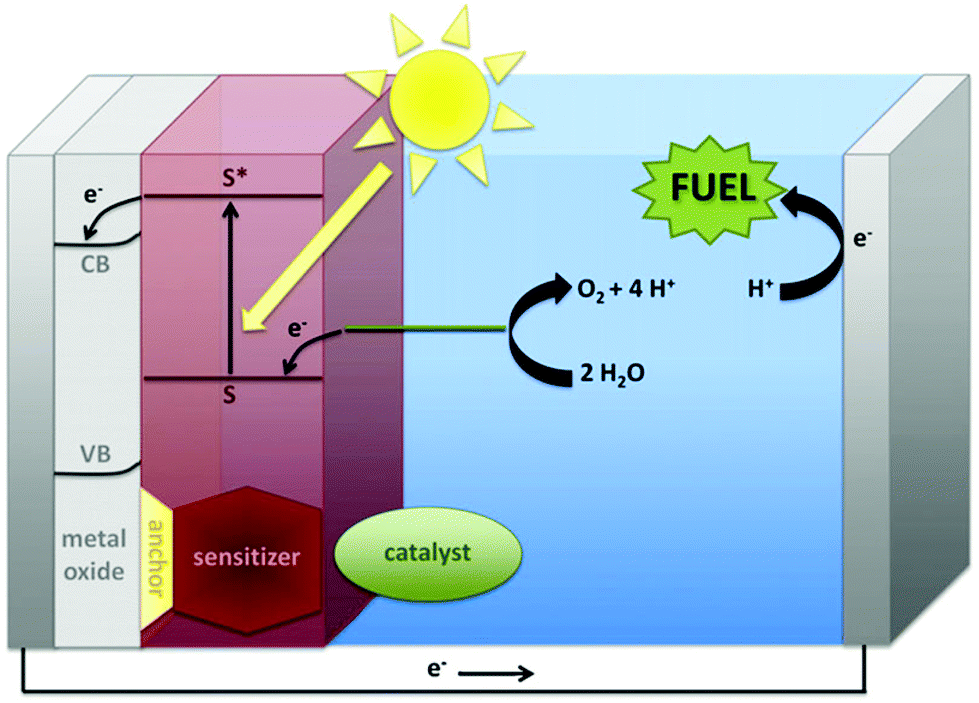 | ||
| Fig. 1 A general schematic for solar fuels formation, showing in more detail the anode half-reaction. Reproduced with permission from ref. 8, copyright 2012 Elsevier. | ||
The best-known and often most robust catalysts contain precious metals, such as platinum and the oxides of iridium and ruthenium. Seminal work by Meyer10 led to the first homogeneous metal-based system—the ruthenium ‘blue dimer’ (Fig. 2, complex 1)—which allowed characterization of the Ru-based water-oxidation intermediates. Mechanistic insights into metal-based water oxidation are vital for improvement of molecular WO catalysts,2 and today homogeneous ruthenium-based water-oxidation catalysts still receive much attention.11–13 However, until recently,14 many of these monomeric and dimeric species suffered from poor catalyst stability, slow rates, and limited turnover numbers (generally <1000).
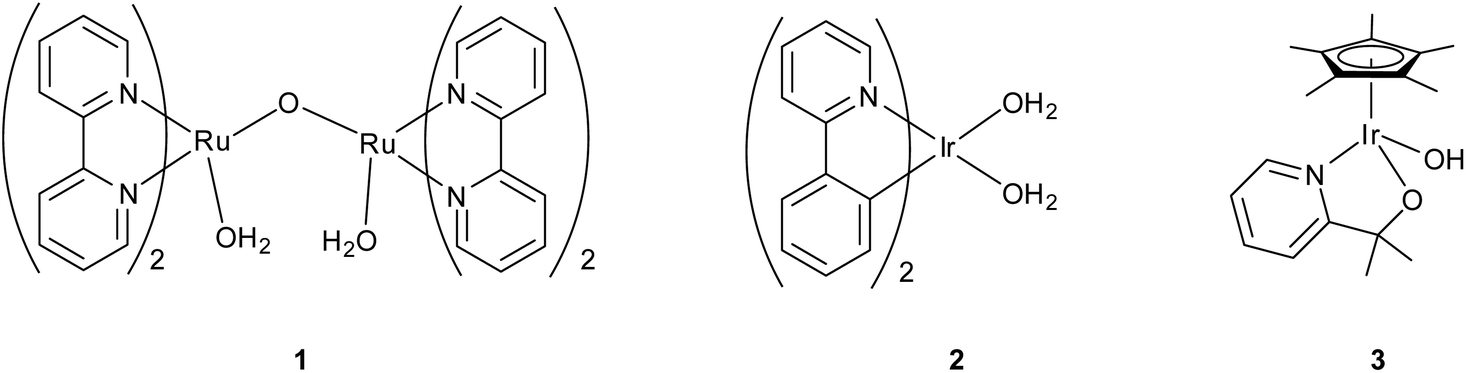 | ||
| Fig. 2 (Left to right) Meyer's “blue dimer,” Bernhard's Ir(phepy) (phepy = 2-phenylpyridine), and our own Cp*Ir(pyalc)OH precatalyst (pyalc = 2-(2′pyridyl)-2-propanoate). | ||
In contrast to these molecular systems, heterogeneous IrO2 was identified as early as the 1970s as an exceptionally effective water-oxidation catalyst (WOC),15–17 with high rates of O2 evolution, good stability and low kinetic barriers. However, the heterogeneous nature of these metal oxides and their incorporation into secondary metallic anodes greatly complicated mechanistic determination and structural characterization of the active site. Thus, it seemed desirable to develop molecular iridium species capable of water oxidation. Indeed in 2008 the first homogeneous iridium WOC was developed,18 and since then the field of homogeneous iridium WOCs has become a very active area of research.13,19 Synthesis of iridium-based catalysts with organic ligands similar to Meyer's “blue dimer” held promise for isolating active WO intermediates, but more study showed that the organometallic complexes were just as mechanistically problematic as their metal oxide counterparts.20–22 More recently, certain organometallic Ir precatalysts have been found to yield homogeneous Ir-based WO catalysts with very high activity.23 The Cp*Ir catalysts can operate under a variety of regimes: they can either act as homogeneous or a heterogeneous catalysts; they can be driven by chemical, photochemical,24,25 or electrochemical20,26–28 methods; and the molecular catalysts can either act in solution23 or supported as molecular units on a variety of solid oxides26 (Fig. 3). In addition to optimizing the various reaction conditions, work has continued to elucidate the catalyst activation mechanism and identify water-oxidation intermediates. The current interest in the area is evident from the number of reviews that have appeared13,29 covering many aspects of iridium-based complexes WO in all forms; this Perspective will describe the development of the Cp*Ir series, their many forms as WOCs, and their ongoing characterization, an area in which we have been active.
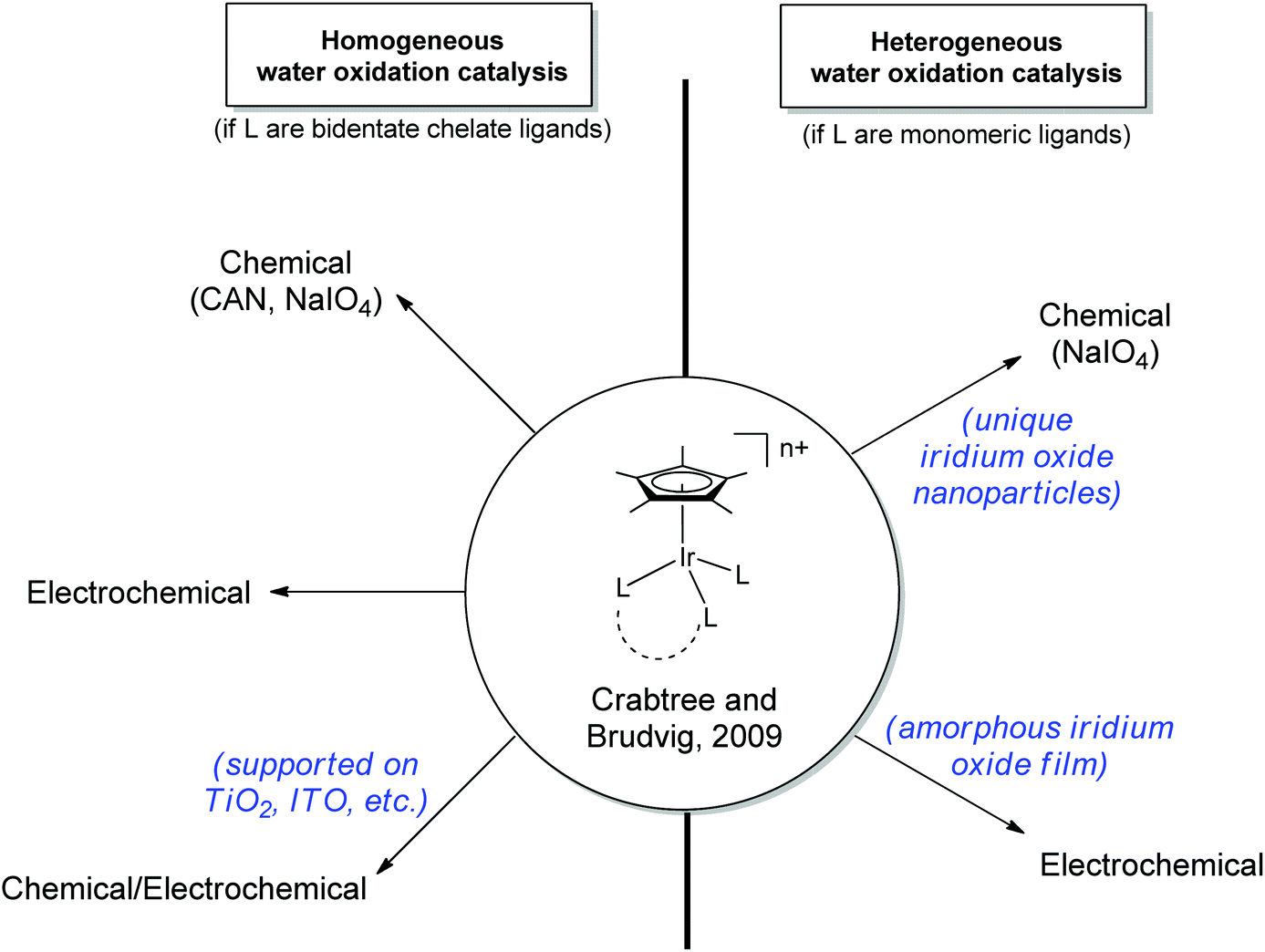 | ||
| Fig. 3 Various regimes under which the Cp*Ir series30 can oxidize water. Ligands (L) may be chelate or monomeric. | ||
B. Overview of Cp*Ir-based water-oxidation catalysis
In 2008, Bernhard and coworkers reported18 WO by an organometallic iridium complex, 2, reminiscent of the monomeric [Ru(bpy)2(H2O)2]2+ (bpy = 2,2′-bipyridine), but 2 showed significantly higher oxygen-evolution rates over a much longer time. Thinking that a high-valent iridium intermediate necessary for catalysis would be stabilized by strong donor ligands, our group made and tested a series of Cp*IrIII(chelate)X catalysts (e.g., 3) that improved on Bernhard's already impressive water-oxidation rates.28,30 Cp* is one of the most strongly donating ligands available, and the commercially available [Cp*IrCl2]2 precursor could be easily modified with a variety of ancillary ligands in minor variants of the original preparation, giving access to a highly tunable series of Cp*Ir complexes. The easy availability of Cp*Ir complexes sparked several other groups to investigate different members of the series, and by now a large library has been investigated.20,22,28,30–39 Iridium-based coordination complexes continue to produce some of the most active and robust WOCs.However, the high activity of iridium has its drawbacks. In terms of mechanistic and structural studies, fast turnover by the Cp*Ir series and other Ir-based WOCs has greatly hindered characterization of any reaction intermediates and determination of the mechanism. Further complicating matters, it soon became apparent that the Cp* units of these catalysts were prone to rapid oxidative degradation.22,40–43 Depending on the presence or absence of suitable ancillary ligands, the Cp*Ir precursors led to homogeneous or heterogeneous WOCs.21,23 As we describe in more detail later, the evidence suggests that the homogeneous catalyst resting states are IrIV oxo-bridged dimers bearing chelate ligands,23 but the heterogeneous catalysts consist either of IrOx nanoparticles or of electrodeposited materials containing [IrOx]y nanoclusters.15,16,21,31,44 All three types of material have proven to be robust WOCs. Later work showed that the homogeneous catalysts, once activated, could easily be supported in molecular form on otherwise inert metal oxide surfaces and remain highly active and robust WOCs.26 Although no characterization of any in-cycle reaction transients has been possible thus far, computational efforts have proposed possible mechanistic pathways.28,30,45–47
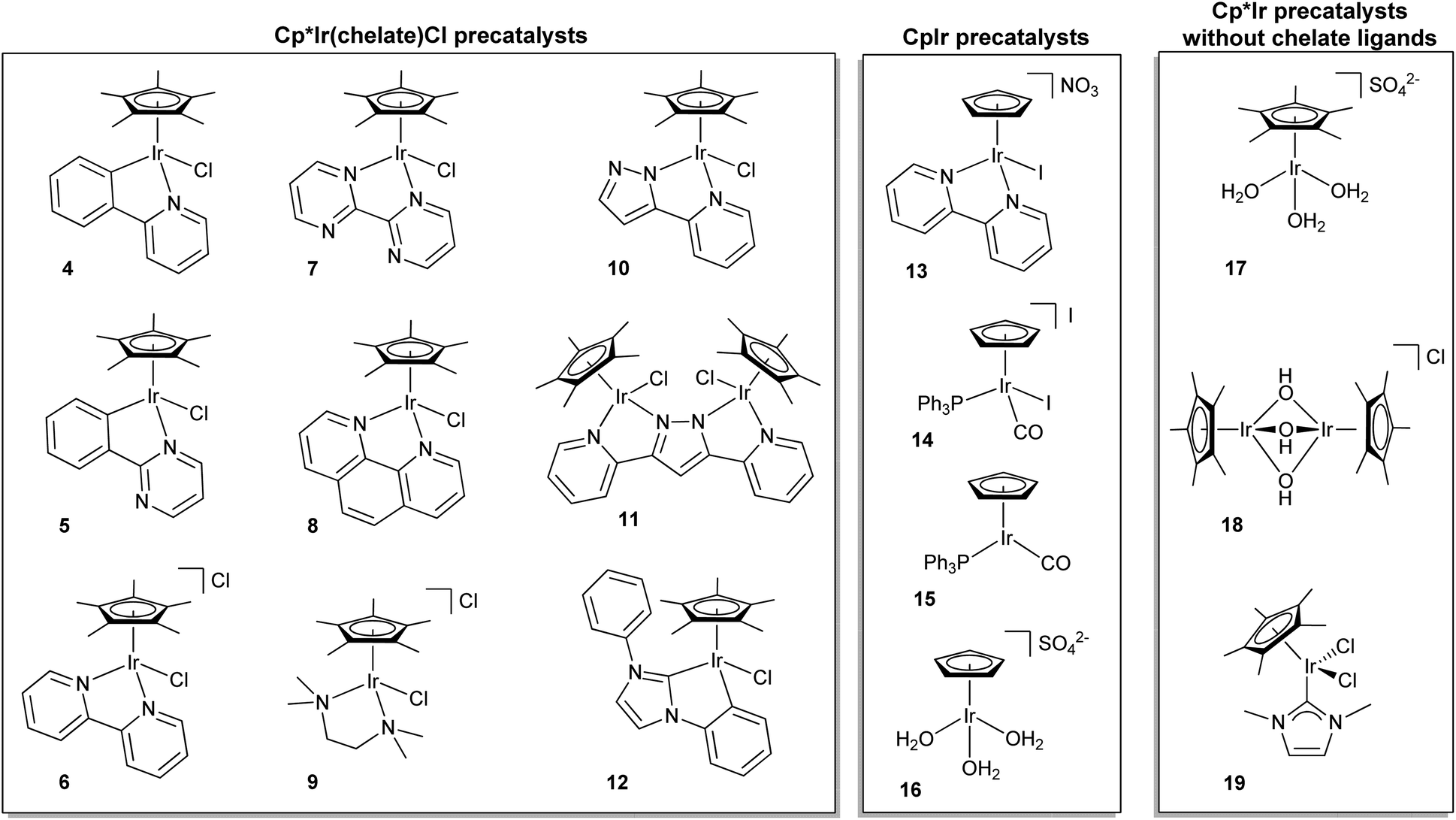 | ||
| Fig. 4 Various iridium-based water-oxidation precatalysts. Cp*Ir(chelate)Cl, CpIr, and Cp*Ir without chelate ligands. | ||
Even before Cp*Ir complexes had been used for water oxidation, it had been indicated in work from the Mayer group that their Cp* moiety was prone to oxidative degradation,40 leading us to test CpIr analogues (13–16). We suspected that because the Cp ligand lacked benzylic hydrogen atoms, it might be more robust than Cp*.28 The CpIr complexes tested did maintain their activity over longer time periods, but they generally showed lower activities per iridium center than their Cp*Ir counterparts. A decisive practical factor was the much more difficult synthesis of the CpIr over the Cp*Ir series. Combined with the lower activity of the Cp complexes, this meant that attention has been concentrated almost exclusively on the Cp* series.
A distinction must be made between chemically- and electrochemically-driven catalysis. In the former case, there can be direct involvement of sacrificial oxidant in the mechanism, for example by transfer of an oxygen atom from the oxidant to the metal, rendering the source of O atoms in product O2 ambiguous. In the latter, electron transfer must occur in one-electron oxidation steps without chemical involvement of the electrode as long as it is suitably oxidation-resistant and water-stable. Thus, water is the only available source of O atoms, and product O2 must be generated by water oxidation.
Some general principles apply to water oxidation by organometallic Ir complexes. The Cp*, Cp or other organometallic placeholder ligands (e.g. cod) are lost by oxidative degradation in an activation step,22,23,42,58 while the O- and N-donor ligands of the Cp*Ir(chelate) series are retained. Depending on the nature of the oxidant and ancillary ligands, activation can result in either a molecular species or a heterogeneous material.21,31 After activation, complexes 3–11 and 13 (all of which bear chelate ligands) form homogeneous coordination complexes which oxidize water at varying rates (Table 1).23 On the other hand, 17–19, having no chelate ligands, give rise to nanoclusters, nanoparticles,21,35 or solid deposits31 that are also catalytically active but are no longer molecular species.
| Compound | Catalyst concentration (μmol L−1) | Oxidant | Oxidant concentration (mmol L−1) | Initial rate (μmol L−1 min−1) | TOF (t.o. min−1) |
|---|---|---|---|---|---|
| a Generally, data analyzed based on first 30 seconds collected using a Clark-type electrode oxygen assay; initial rates are maximum rates; matrix cell is maintained at 25 °C. b Data from Blakemore et al.28 c Data from Hull et al.30 d Calculated from published data. e Data from Graeupner et al.35 f Data from Brewster et al.59 Previous studies have shown that these Cp*Ir(chelate) complexes generally show similar oxidation rates when using CAN, despite the wide range of acidic and basic ligands (4–9, 12) involved. Notable exceptions are 7 and 9, but the decrease in activity in these cases has been suggested to be due to protonation of the chelate ligand and swift loss from the iridium center thereafter; this would lead to heterogeneous iridium species similar to those produced by 17–19, which are generally slower than their homogeneous counterparts. Another interesting point is that bimetallic 11 is significantly slower than 10; this has been attributed to the fact that the Ir-based WO active species do not act cooperatively and the two metals hinder each other's turnover. | |||||
| 4 | 5.0 (±0.1) | CAN | 78 | 49 ± 4 | 10.0 ± 0.9 |
| 5 | 5.4 (±0.1) | CAN | 78 | 92d | 17 |
| 6 | 5.0 (±0.1) | CAN | 78 | 72 ± 3 | 14.4 ± 0.7 |
| 7 | 5.0 (±0.1) | CAN | 78 | 19 ± 2 | 3.9 ± 0.4 |
| 8 | 5.0 (±0.1) | CAN | 78 | 42 ± 2 | 8.4 ± 0.7 |
| 9 | 5.0 (±0.1) | CAN | 78 | 31 ± 3 | 6.3 ± 0.6 |
| 10 | 5.0 (±0.1) | NaIO4 | 10 | 26.4 ± 0.8 | 5.3 ± 0.2d |
| 11 | 5.0 (±0.1) | NaIO4 | 10 | 6.1 ± 0.9 | 1.2 ± 0.2d |
| 12 | 4.5 (±0.1) | CAN | 78 | 36d | 8 |
| 12 | 4.5 (±0.1) | NaIO4 | 5 | ∼63d | 12–16 |
II. Homogeneous regime: the stabilizing effect of chelate ligands on Cp*Ir complexes
A. Oxidative loss of Cp* and characterization of homogeneous catalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ir ratio, similar to the ratios found in water-oxidation work), several equivalents of acetic acid per Ir were formed within minutes of injecting the catalyst into oxidant solution.23 Macchioni and coworkers estimated that about four of the methyl moieties were being converted to acetic acid through oxidative degradation, assuming that the acetic acid was an oxidation product of methyl groups on the Cp*.22 With lower loadings of oxidant, WO and the formation of acetic acid was still seen after treatment with CAN, but formic acid and several other minor products were also observed in the 1H NMR spectra.
:Ir ratio, similar to the ratios found in water-oxidation work), several equivalents of acetic acid per Ir were formed within minutes of injecting the catalyst into oxidant solution.23 Macchioni and coworkers estimated that about four of the methyl moieties were being converted to acetic acid through oxidative degradation, assuming that the acetic acid was an oxidation product of methyl groups on the Cp*.22 With lower loadings of oxidant, WO and the formation of acetic acid was still seen after treatment with CAN, but formic acid and several other minor products were also observed in the 1H NMR spectra.
Through extensive NMR experiments, Macchioni and coworkers characterized several degradation intermediates (Scheme 1).22 Based on these findings, they determined that an iridium center could oxidize its own Cp* methyl groups through a multistep oxidative process in which Cp* degradation and WO are cooperative rather than sequential.58 The Cp*Ir complex loses its labile X ligand and forms an active monomeric oxo-iridium species capable of WO (the mechanism of WO by this monomeric species will be discussed in more depth in Section IIC). However, this species is able to attack C–H bonds as well. O2 produced by WO then coordinates to Ir in an η1- or η2-O2 fashion, and oxidation of this species leads to superoxide formation from the bound O2, 20. The superoxide then initiates oxidation of the Cp* ring, as has been seen before for analogous rhodium complexes.60 Since the same intermediates were observed regardless of chemical oxidant used (CAN,22 NaIO4,61 H2O242), this suggested that the mechanism of activation was not specific to the oxidant employed.58
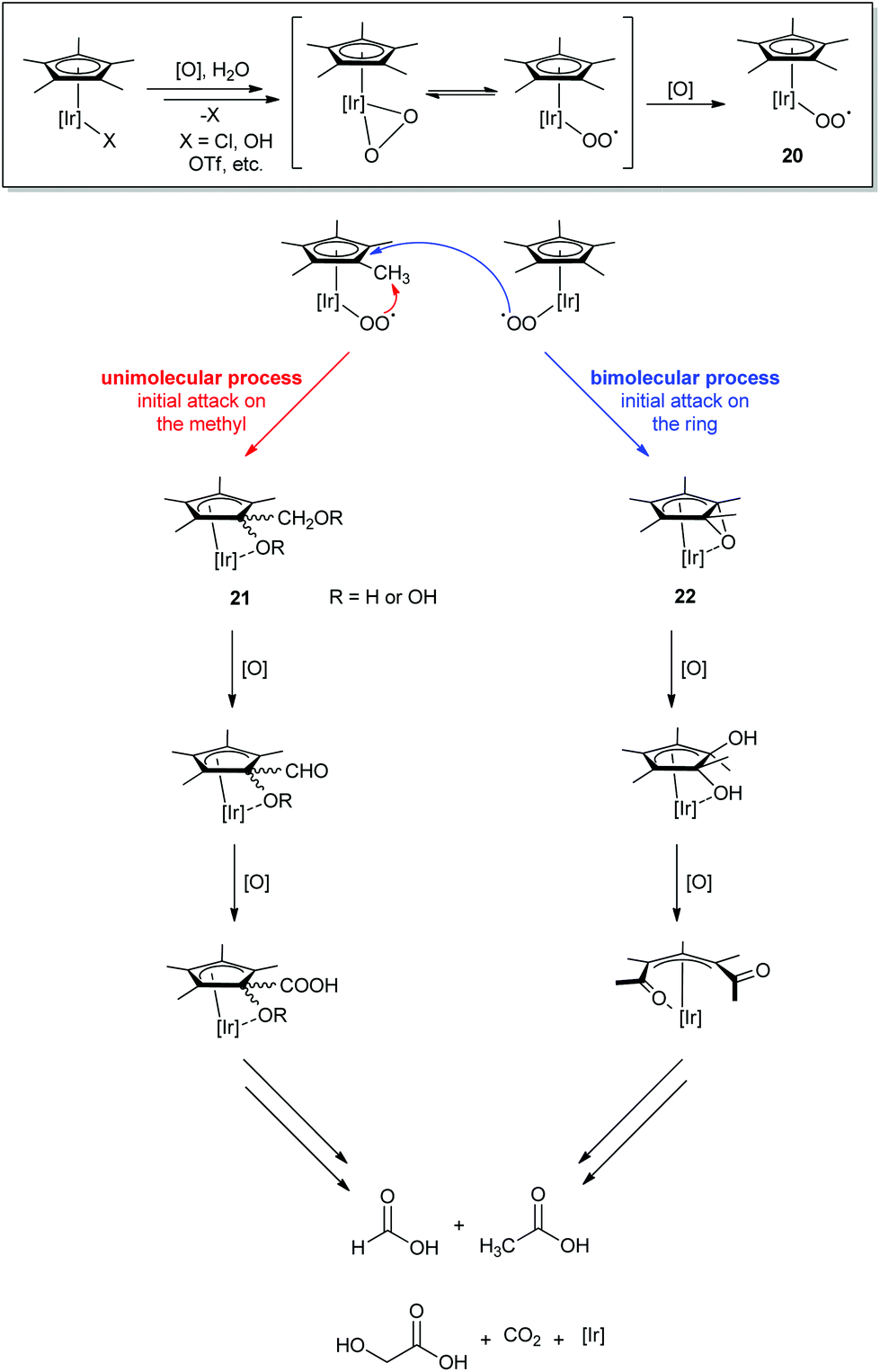 | ||
| Scheme 1 Proposed oxidative degradation pathway of the Cp*.22 | ||
Macchioni and coworkers have reported two intermediates (21 and 22) that suggest that the Cp* is attacked by 20 at two positions: at the H of the methyl22 or at the ring carbon of the Cp*58 (Scheme 1). Alignment of the Ir center and the Cp* ring orbitals would suggest that 20 attacks the methyl position via an intramolecular route. Although it is not clear if 20 inserts O2 in a concerted or stepwise fashion,22 the product has been characterized as an iridium intermediate with a Cp* having an oxidized methyl group and ring carbon. Macchioni et al. proposed that the attack on the ring carbon could also be intramolecular, but because the orbitals of the Ir center do not overlap with the aromatic C as well as the methyl H, it is also possible that another superoxide can attack the ring in a bimolecular fashion. In this case, the superoxide 20 would epoxidize the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the Cp* ring, forming 22.58 Subsequent oxidation would lead to cleavage of the ring to form diacetyl, known to be oxidatively labile. In either the uni- or bimolecular pathway, the Cp* fully degrades into the corresponding ketoacids and CO2, leaving the Ir center and any surviving ancillary ligands (O- and N-donor chelate ligands).
C bond of the Cp* ring, forming 22.58 Subsequent oxidation would lead to cleavage of the ring to form diacetyl, known to be oxidatively labile. In either the uni- or bimolecular pathway, the Cp* fully degrades into the corresponding ketoacids and CO2, leaving the Ir center and any surviving ancillary ligands (O- and N-donor chelate ligands).
While this proposed process explained the source of the organic acids, Macchioni et al. were not able to determine if the iridium species remained homogeneous after the oxidative removal of the Cp*. Work from the Lin group incorporated Cp*Ir(chelate) moieties into metal–organic frameworks (MOFs) (Scheme 2), preventing bimolecular processes through site isolation of the iridium complexes and thus creating a supported “molecular” oxo-iridium species.43 Treating the Ir-MOF with CAN led to loss of Cp*, indicating that Cp* degradation is most likely intramolecular. To test if the Cp* moiety was required for WO, the Ir-MOF was pre-treated with CAN to remove the Cp* moiety before starting a water-oxidation experiment with CAN as the oxidant, and the resulting Ir(chelate) moieties were still able to evolve oxygen at high rates. This work suggested that the likely active species is a monomeric high-valent iridium-oxo species, based on data from XPS, UV-visible, luminescence, and IR spectroscopies. Although multimolecular mechanisms may apply to both ligand degradation and WO for bulk solutions, the Lin group's results demonstrate that even though the Cp* may be lost during WO, the remaining Ir(chelate) species can oxidize water in a molecular fashion. Importantly, this work also demonstrated that Cp* degradation did not necessarily mean formation of heterogeneous iridium oxide particles. Later studies from our group23 suggested that, compared to the monodentate series 17–19, chelate complexes such as 4–12 show enhanced stability and form molecular species capable of water oxidation in solution. Chelation thus prevents nanoparticle formation under oxidative conditions.
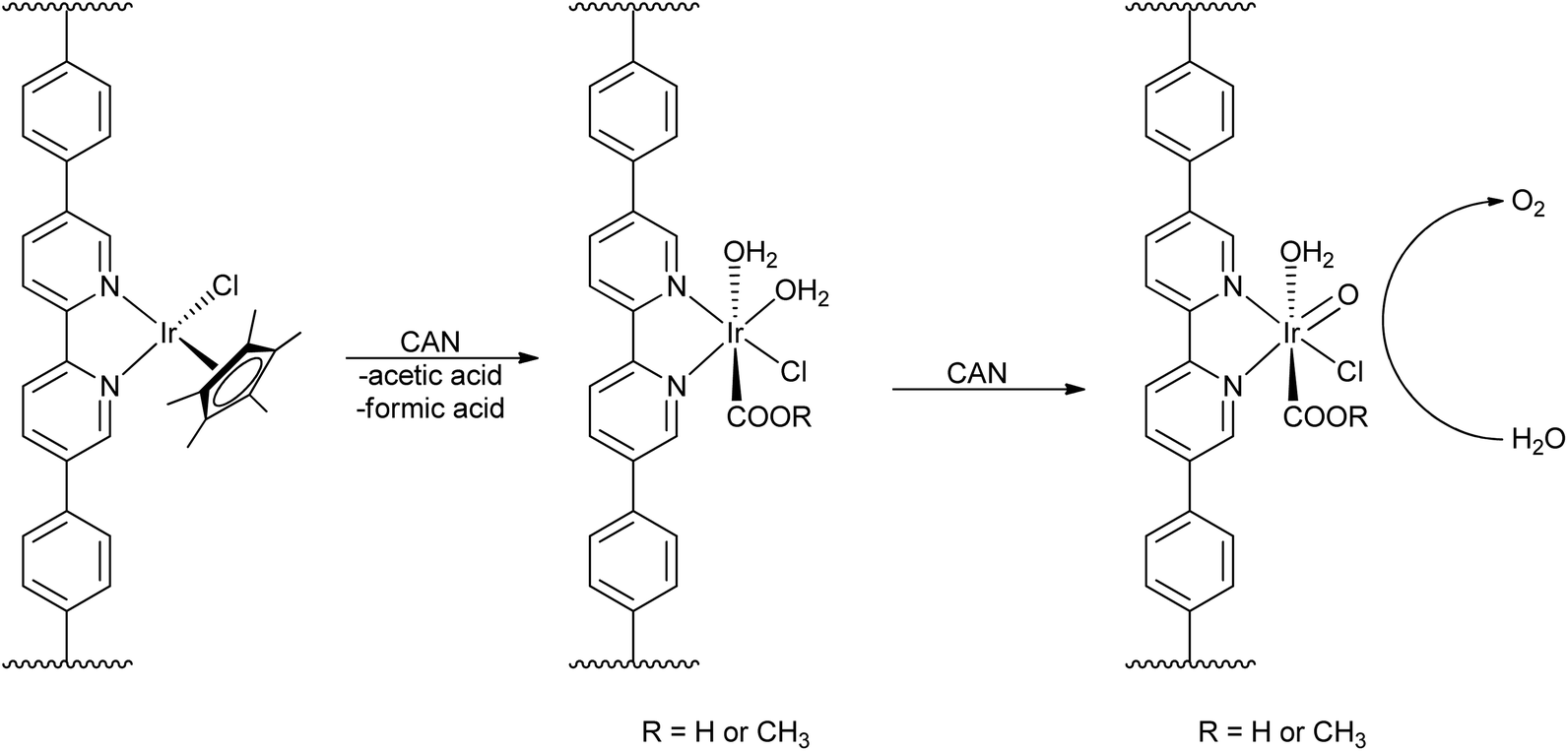 | ||
| Scheme 2 Cp*Ir incorporation into MOFs and oxidation of water by a molecular Ir(chelate) catalyst. Reproduced from ref. 43, with permission from the American Chemical Society. | ||
In the spectroscopic work on the ABS, [Cp*Ir(bpy)OH]BF4, (6-OH), was used23 rather than the chloride complex 6 because chloride oxidation can compete with water oxidation, and the presence of chloride has been observed to contribute to nanoparticle formation.21 Over the first 10 minutes after 6-OH is injected into a solution of oxidant, the solution shows formation of acetic acid by 1H NMR and the rise of a UV-visible absorption at ∼600 nm, which is accompanied by a decrease and ultimate disappearance of the 1H NMR Cp* signal. Cp* loss occurs over the course of a few minutes, but O2 evolution occurs seconds after the injection; therefore, loss of the Cp* may not be necessary for WO to occur through chemical oxidation, although the rate of O2 evolution increases to some extent as the Cp* signal decreases. This suggests that certain Ir(chelate) complexes are competent for WO after loss of Cp*, as suggested by the Lin group. A labile ligand was found to be necessary for both Cp* loss and WO to occur: when the chelate ligands were replaced with tacn (1,4,7-triazacyclononane), a ligand that blocks all the labile aqua sites, the metal became coordinatively saturated and catalytically inactive. The Cp* remained intact by 1H NMR and no WO was observed, confirming that the degradation of the Cp* is not caused by NaIO4 alone. If the Cp* was not required, we thought that other precursors with similar oxidizable placeholder ligands should give access to the catalytic cycle; indeed, IrI(cod)(chelate) precursors oxidized water when driven by NaIO4 with rates similar to their Cp* analogues.23 The ABS had good long-term stability, evident in that adding more oxidant to the same reaction mixture after several hours led to O2 evolution at the same rate as for the first injection.
The chelate ligand, unlike the Cp*, did not appear to degrade or dissociate from the Ir center. MALDI-TOF-MS, TEM-EDX, and XPS showed a 1:1 N:Ir ratio for the ABS isolated from precursor 3, as well as carbon peaks in the NMR spectrum and signals in the mass spectrum indicating that the pyalc ligand was still bonded to Ir. However, the 1H NMR spectrum showed ill-defined ligand peaks and the EPR spectrum showed no signal. Using the Evans method, a weak paramagnetic moment corresponding to ∼0.6 unpaired electrons was estimated. These findings suggested that the ABS is a spin-coupled paramagnetic IrIV dimer or related species. The 17O NMR spectrum showed peaks in regions characteristic for μ-oxo and terminal water ligands in a 1:2 ratio and the resonance Raman spectra were consistent with a bis-μ-oxo core. An IrIV bis-μ-oxo dimer, with each metal having one pyalc chelate ligand was proposed. Each metal would then have two labile sites assigned to 2 H2O ligands. A deprotonation event with a pKa of 5.8 was detected in pH studies using 17O NMR and UV-visible spectroscopy, plausibly assigned to one of the aqua ligands. Optimization by TD-DFT gave 23 as the most energetically stable isomer of the five likely isomers from 3 as the precursor (Scheme 3), though it is of course possible that several isomers coexist in solution. Because the products from both the Cp* and the cod precursors gave similar spectra and WO rates after oxidative activation, it seemed likely that 23 was the activated molecular species for both, after oxidative removal of the organic placeholder ligands. Much like the electrochemically driven deposition of the Blue Layer (as discussed in more detail in Section IIIA),31 the Cp* and cod23 ligands both provided a facile route to a high-valent intermediate from stable precursors. However, though the ABS is easily accessed and quite stable, it has so far proved impossible to crystallize for XRD study in spite of much effort. One possible explanation is that the labile aqua sites pose a problem on concentrating the solution for crystallization, leading to oligomerization due to bridging oxo ligation between dinuclear units. Another possibility is that the IrIV-oxo dimer could exist as several isomers that do not interconvert in solution, hindering crystallization.
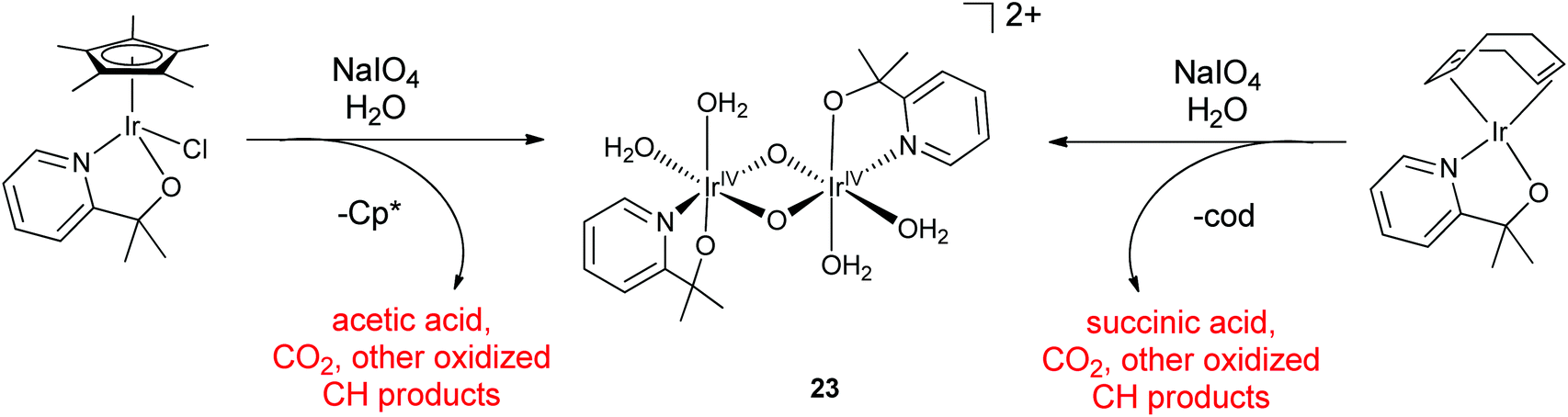 | ||
| Scheme 3 Formation of the proposed IrIV-oxo dimer from Cp*Ir(pyalc)Cl and Ir(cod)(pyalc) precursors using NaIO4. | ||
Shortly after this study was published, a report confirming extensive ligand oxidation of related Cp* complexes appeared from the Templeton group.63 In this report, oxygen atom transfer (OAT) reagents were used with [Cp*Ir(phepy)L]+-type complexes in an attempt to generate terminal IrV oxo species for spectroscopic characterization. As will be discussed in Section IIC, an IrV oxo species is the key intermediate postulated in the mechanism of water-oxidation catalysis by Cp*Ir complexes, and spectroscopic observation of such a species would be a significant advance in elucidation of the catalytic mechanism. Due to electronic repulsion from filled d-orbitals, late transition metals do not typically form terminal oxo species, as the requisite overlap of the metal d-orbitals with oxygen p-orbitals is energetically unfavorable. These authors hypothesized that employing a labile ligand at the iridium center and reacting the complex with OAT reagents would allow concerted 2-electron oxidation to the IrV-oxo species in a single step, which might allow it to be trapped. Two labile, L-type ligands were tested: 3,5-bis-(trifluoromethyl)benzonitrile (NCAr) and styrene (Sty). The counterion used for these studies was the non-coordinating anion tetrakis[3,5-bis(trifluoromethyl)phenyl]borate, or B(ArF)4. The OAT reagents used in the study were dimethyldioxirane (DMDO) and iodosylbenzene. Unfortunately, no IrV-oxo species could be generated, but the authors noticed that the reaction of [Cp*Ir(phepy)(NCAr)][B(ArF)4] with DMDO resulted in O2 evolution. However, repeating the experiment yielded highly inconsistent O2-evolution results, suggesting that their proposed IrIII/IrV mechanism for O2 evolution was not valid. When the authors measured the kinetics of the reaction with DMDO by NMR spectroscopy, signals associated with [Cp*Ir(phepy)(NCAr)][B(ArF)4] disappeared over the course of the reaction, but no identifiable decomposition products were seen in the spectrum. They explained this in terms of the formation of heterogeneous material during the reaction or the generation of paramagnetic species. A related reaction, utilizing [Cp*Ir(phepy)(H2O)][B(ArF)4] for styrene epoxidation driven by iodosylbenzene, resulted in very poor yields of the desired product and the data indicated extensive ligand degradation. They also studied the electrochemistry of [Cp*Ir(phepy)(H2O)][B(ArF)4] in mixed water/ethylene carbonate solution, with phosphate buffer and NaClO4 electrolyte, finding the cyclic voltammograms to be very poorly defined and indicative of ligand degradation. The authors were uncertain if heterogeneous materials or alternative, possibly paramagnetic molecular species had been formed, however, and no studies were carried out to elucidate the nature of the degradation products for any of these reactions. Later work from this group64 showed that a clean molecular product could be formed using a related Cp*Ir complex and alternative OAT reagent at low temperatures, demonstrating that the nature of the ligand has a significant effect on the stability and degradation pathways of this family of complexes.
B. Electrode-driven water-oxidation catalysis
Work on the ABS raised the question of whether the activation process depended on the nature of the oxidant. A 17O NMR experiment comparing CAN and NaIO4 indicated that the same species was formed in either case,23 so the specific nature of the chemical oxidant appeared to be unimportant. However, both CAN and NaIO4 have been implicated in non-innocent electron-transfer pathways, such as oxygen atom transfer,46,65 which introduces complications in elucidating the role of the oxidant in the activation mechanism. Of particular concern for WO driven by NaIO4 was the possibility of direct involvement of IO4− in the mechanism, because isotopic labeling cannot be used to verify the source of O atoms in the product O2 as a result of fast O-atom exchange between IO4− and H2O. Highlighting this concern, a mass spectrometry/DFT study demonstrated that when activation of precursor 19 was driven by NaIO4, the lowest barrier pathway for O–O bond formation showed both oxygen atoms coming IO4− and not from water.46 The proposed mechanism proceeded by O-atom transfer from IO4− to Ir, followed by periodate attack on an intermediate oxoiridium species. ESI-MS signals on the reaction mixture matched the expected Ir–IO3− adducts, formed from the product of periodate reduction. It is significant, however, that the proposed mechanism involved chelation of IO3− to Ir, which would be not be possible with Cp*Ir(chelate) complexes. Furthermore, in light of the large body of evidence that Cp* is degraded rapidly after addition of oxidant, this proposed mechanism may only be operable during the short time following mixing catalyst and oxidant before Cp* loss has occurred. Nevertheless, these results cast doubt on the innocence of IO4− as an oxidant for WO.It was evident from the lack of O2 evolution in control experiments that NaIO4 was not spontaneously disproportionating into IO3− and O2; O2 evolution only began upon injection of Cp*Ir precatalyst. Whether the process catalyzed by the iridium complex was IO4− disproportionation or water oxidation, however, was less clear. Even if WO was dominant, it was still not clear whether or not it could be driven in one-electron steps (such as would be required in a photoelectrochemical cell) with IO4−, because IO4− often acts as a two-electron oxidant.66 Regardless of the oxidant's electron count, there is always the possibility of direct involvement of the oxidant in the catalytic mechanism, where significant association of the catalyst and the oxidant may result in a different pathway than would occur by simple outer-sphere electron transfer.67 If so, a catalyst capable of carrying out WO with an oxidant might still be unable to do so by outer-sphere electron transfer in a photoelectrochemical cell (PEC). Innocent, one-electron oxidation of candidate catalysts is best probed by electrochemistry.68 If the Cp*Ir species were electrocatalytic for O2 evolution in the absence of chemical oxidants, mechanistic ambiguities related to the role of the oxidant could be clarified.
To measure electrode-driven O2 evolution from Cp*Ir species in real time, we coupled electrochemistry with a Clark electrode. To compare these experiments with the analogous experiments with chemical oxidants, we chose a potential of 1.60 V because this is close to the estimated potential of IO4− at the chosen experimental pH of 2.5.69 While a Clark electrode was used to monitor the dissolved O2 levels in a 1 mM solution of 3, a gold working electrode was held at 0 V vs. NHE for a 10-minute period before being stepped to the 1.60 V oxidizing potential. We were surprised to find that at this potential no O2 was measurable by using a Clark electrode over the experimental time frame of 10 minutes (Fig. 5, red line). Intrigued, we wondered if the ABS derived from the Cp*Ir complex and NaIO4 would be electrocatalytic if the unmodified precursor species was not. We thus took a 1 mM solution of 3 and activated it with 100 equivalents of NaIO4, then carried out the same electrochemical experiment coupled with O2 measurement. In contrast with the unmodified precursor, the pre-activated solution resulted in near-immediate evolution of O2 (Fig. 5, blue line). No catalytically active deposits formed on the electrode as determined by the standard “rinse test” as well as by rigorous SEM-EDX examination of the electrode.
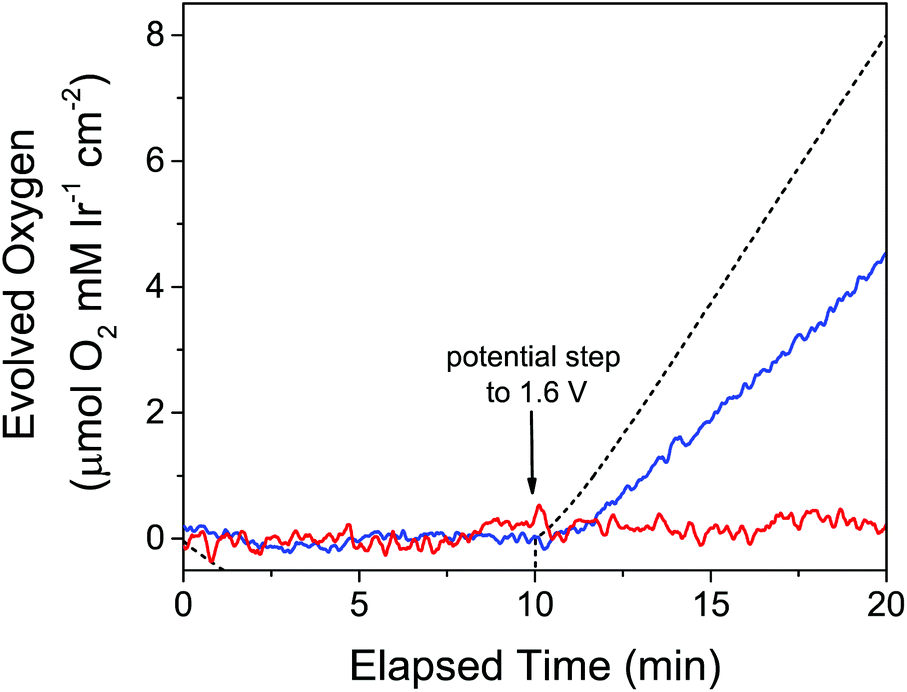 | ||
| Fig. 5 O2 evolution over time in response to applied potential at a gold electrode. The electrode was held at 0 V vs. NHE for the first 10 minutes, followed by a step to 1.60 V vs. NHE for the second 10 minutes. Red: 1 mM 3 in 0.1 M KIO3; blue: 1.2 mM 3 pre-activated with 100 equivalents of NaIO4 (ABS); dashed black: predicted O2 from current passed from preactivated 3. Reprinted from ref. 27 with permission from the American Chemical Society. | ||
The Faradaic efficiency of WO by pre-activated 3 was relatively low, however, at only around 60%, indicating that WO was not the only process occurring at this potential in this solution. Partially oxidized Cp* fragments or residual iodine-containing species from the activation process were some possible causes of oxidation processes potentially responsible for the lowered Faradaic efficiency, but this was difficult to test rigorously due to our inability to unambiguously identify the Cp* oxidation products or change their concentrations. Despite the low Faradaic efficiency, we could still conclude that the ABS derived from 3 pre-activated with NaIO4 was competent for electrocatalytic water oxidation at E = 1.60 V. Because unmodified 3 was catalytically inactive, Cp* loss was evidently necessary to evolve oxygen electrocatalytically. This contrasts with O2 evolution studies with chemical oxidants, despite using the same oxidizing potential.
Therefore, bulk electrolysis of 3 was attempted, using Na2SO4 as the electrolyte to avoid complications from iodine-containing species.9 Gratifyingly, a blue color appeared during bulk electrolysis, though the development of the color took significantly longer by electrochemical methods than for chemical oxidation. However, the appearance of a blue color does not guarantee that the same activated product was formed, as many iridium(IV) oxides are blue (including some generated electrochemically70). Furthermore, slight differences were apparent in the UV-visible spectra (Fig. 6): the λmax corresponding to the blue color seen for the electrolyzed solution (577 nm) was more blue-shifted than the λmax of the solution made by chemical oxidation (608 nm), and the shoulder around 450 nm in the chemically activated solution was absent in the electrochemically activated solution. Microscopy and dynamic light scattering, however, confirmed that the electrochemically formed blue solution contained no nanoparticles. This supported the idea that a molecular species was formed by electrolysis, though we could not conclusively demonstrate that the same species was formed by both activation methods.
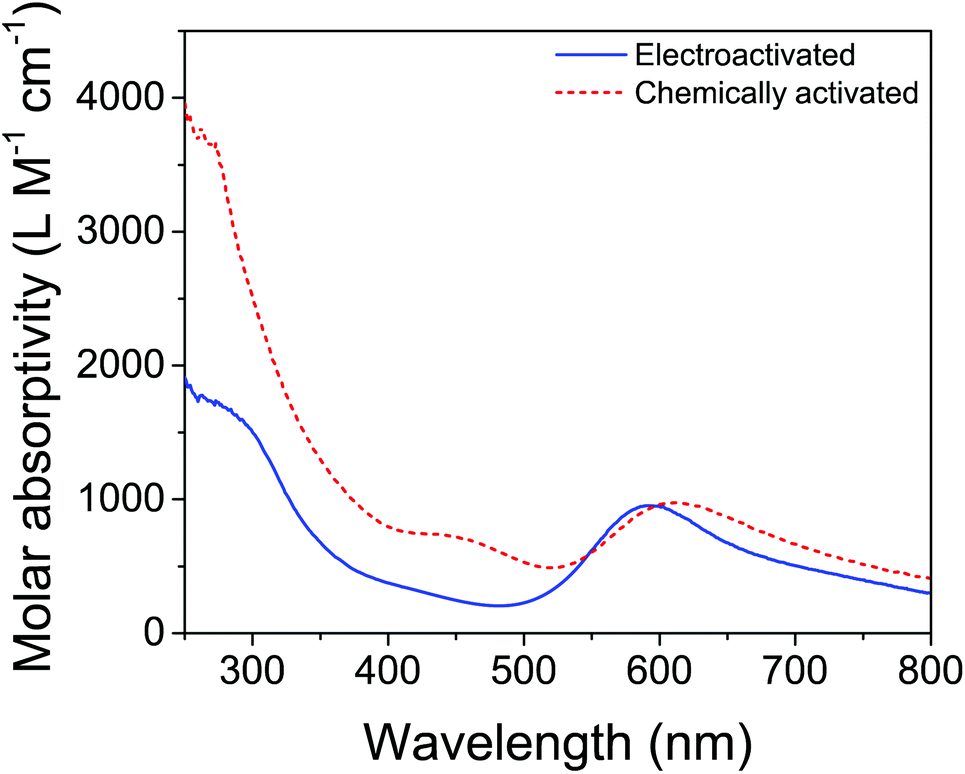 | ||
| Fig. 6 UV-visible spectra of ABS derived from 3 activated by 100 equivalents of NaIO4 (dashed red) and by electrolysis for 36 hours at 1.45 V vs. NHE (solid blue). Reprinted from ref. 27 with permission from the American Chemical Society. | ||
Indirect evidence, including the O2-evolution behavior of the post-electrolysis solution and its spectral features when IO3− was added, supported the notion that a similar activation process took place during electrochemical oxidation as during NaIO4 activation. The electrogenerated ABS was successful in catalyzing electrode-driven O2 evolution. At an overpotential of 345 mV, the Faradaic efficiency approached 100%. Adding IO3− decreased the Faradaic yields, which dropped to ∼60% after adding 150 equivalents of IO3−, and the λmax of the solution shifted to the red with increasing amounts of added IO3−. This indicated that the λmax of the species was sensitive to the anions in solution, and that IO3− coordination resulted in a lower-energy UV-visible transition than did SO42− coordination. Importantly, the electrosynthesized ABS oxidized water at high rates and low overpotential in a solution that had never been exposed to chemical oxidants, and therefore the O2 evolved must be fully sourced from H2O. It also indicated that the byproducts of activation with NaIO4 served as inhibitors of WO, as evidenced by the lower rates and Faradaic yields observed for the NaIO4-derived ABS. Curious to see if the chelate ligand would influence the properties of the ABS, we activated 6-OH by both chemical and electrochemical methods, and compared the resulting O2-evolution activity. As expected, the bipyridine species showed different spectral features and rates of O2 evolution from the standard catalyst, 3, providing evidence that the chelate ligand was retained, as had already been suggested in earlier work by the different rates of WO for 3 and 6-OH with NaIO4.21,23 Taken together, the evidence suggests that molecular iridium species are formed by oxidation of Cp*Ir(chelate) complexes, whether the method of oxidation was chemical or electrochemical, and that these activated molecular species are effective electrocatalysts with high rates and low overpotentials for WO.
A report71 from the groups of Hetterscheid and Koper, in which spectroelectrochemistry was used to evaluate the surface-enhanced Raman spectroscopy (SERS) characteristics of a Cp*Ir complex at applied potentials, appeared shortly before the work on the electrochemical characterization of the ABS. In this work, the electrochemistry of 19-OH, a complex related to 19 in which hydroxo ligands replace both chloro ligands, was examined when 19-OH was immobilized in Nafion films on gold surfaces. The authors reported a high turnover frequency of 2.9 s−1, calculated from current flow at an applied overpotential of 430 mV (1.7 V vs. RHE) and assuming 100% Faradaic efficiency. (This assumption was not verified by quantitative comparison of O2 with current flow, however, and in light of the facile electrochemical oxidation of Cp* in the similar species 3 and 6-OH, it is unclear whether 100% Faradaic efficiency is attained in this case.) SERS was then probed spectroelectrochemically at a series of oxidizing potentials between 0.8 V and 1.4 V vs. RHE, using dropcast 19-OH in the absence of Raman-active Nafion. The authors attributed a feature at 450 cm−1 present at lower potentials (0.8 V–1.3 V) to a bending mode of the Cp*–Ir bond, based on similar features in related metallocene complexes. This feature diminished in intensity with increasing applied potential, while other features at 560 and 730 cm−1 appeared as a result of oxidation. The latter features, appearing at similar wavenumbers to the features attributed to the di-μ-oxo bridge of the ABS, were ascribed to the formation of a similar μ-oxo bridge between monomers of 19-OH, thus resulting in dimerization on the gold surface. Subsequent lowering of the potential led to restoration of the 450 cm−1 feature (though reduced in intensity), which led the authors to conclude that this dimerization process is reversible. It is unclear why the 450 cm−1 feature disappears upon oxidation without loss of Cp*; loss of Cp* from 19-OH would be expected to be irreversible as a result of its rapid oxidative degradation under the oxidizing conditions employed. Furthermore, a proposed dimeric structure is shown in this report that shows two intact Cp* moieties. If Cp* is retained as indicated in the proposed structure, the dimeric species is coordinatively saturated and would be unable to carry out water oxidation. This makes a mono-μ-oxo species, like that recently reported by Ison et al.,72 more plausible. Also, evidence from several other reports38,40,63 suggest that it is possible that the electrochemical current these authors observed was due to Cp* oxidation.
Surprisingly, the ABS formed from 3 and NaIO4 spontaneously chemisorbs to nearly all oxide surfaces, including popular solar fuels substrates such as TiO2 and α-Fe2O3 (hematite). It forms an irreversible, water-stable attachment that is maintained across a broad pH range and seems only limited by the substrate's stable pH range.26 Direct attachment in this way bypasses the often extensive and difficult synthesis and optimization of added anchoring groups. Such spontaneous adsorption behavior of iridium-based materials to conductive substrates has been described before,73,74 but these prior adsorbates were all nanoparticulate. The surface-bound species in our case retains its chelate ligand as shown by the expected 1:1 Ir:N ratio being detected by XPS, and from the STEM-EDX showing a highly conformal coating of the substrate with iridium (Fig. 7).
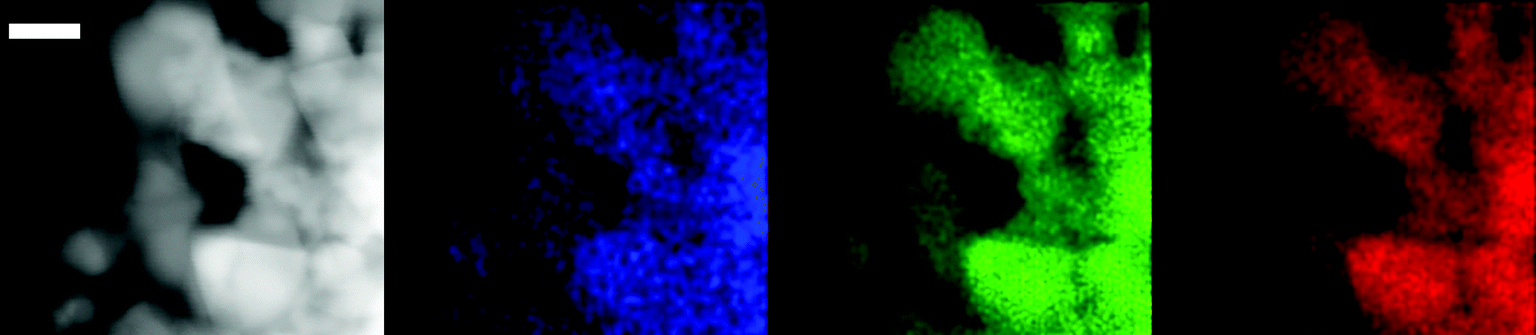 | ||
| Fig. 7 STEM-EDX micrographs showing maps of electrode material as-prepared (left to right): high-angle annular dark field image (HAADF, grey), iridium detected shown in blue, indium in green, and tin in red. The white bar denotes 20 nm. Reproduced from ref. 26. | ||
This conformal coating appears to be a monolayer: this is consistent with the self-limiting adsorption behavior seen in solution over 2 hours, leading to a catalyst loading quantitatively consistent with monolayer formation (Fig. 8). For ease of characterization of the electrochemical behavior of the surface-bound species, we used a conductive nano-ITO substrate. After surface binding, the heterogenized species was analyzed for its water-oxidation activity and stability. The thickness of the nano-ITO substrate influenced the overpotential required to reach the conventional 0.5 mA cm−2 threshold, as would be expected. With thicker films of 10 μm, the overpotential reached the extremely low value of 160 mV for a current density of 0.5 mA cm−2. At slightly higher overpotentials, very high current densities were seen, but more interestingly, these current densities could be maintained over weeks without change. After prolonged electrolysis, XPS analysis revealed the Ir:N ratio to have been fully maintained, suggesting that the ligand had been retained despite the harsh oxidizing conditions. Cyclic voltammetry showed no changes after electrolysis, further supporting this conclusion.
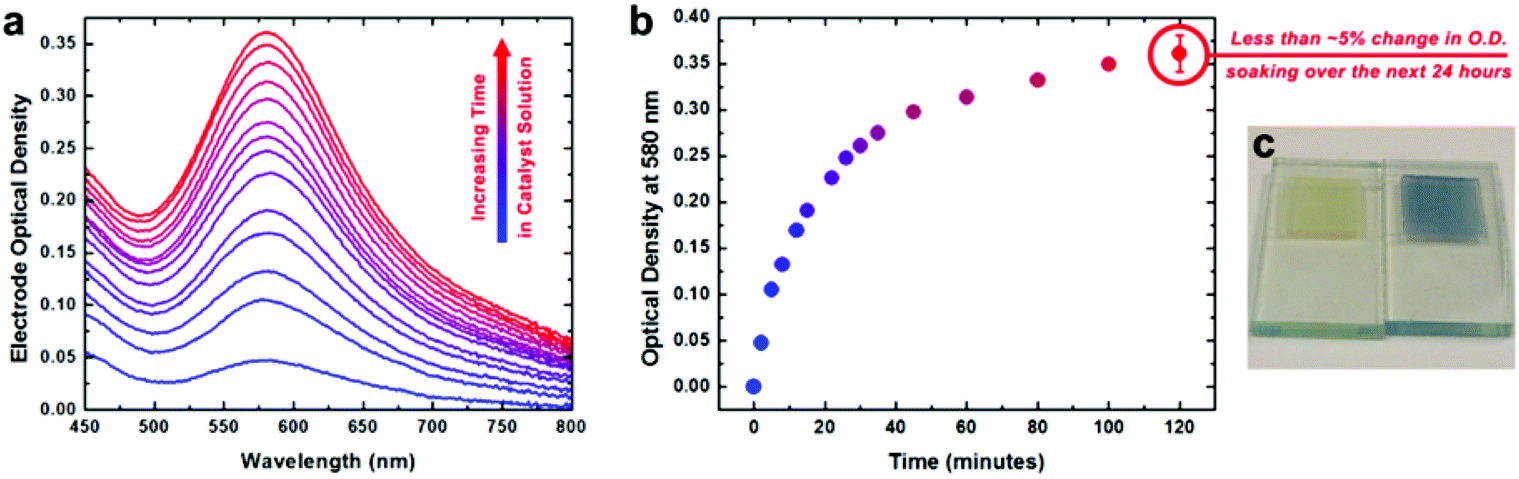 | ||
| Fig. 8 UV-visible spectroscopic and spectroelectrochemical characterization of heterogenized activated 3 on nanoITO electrodes. (a) Optical density spectra of an electrode measured after increasing amounts of time immersed in catalyst solution at room temperature. (b) Increase of optical density at 580 nm for the electrode as a function of immersion time. (c) Photograph of an electrode before (left) and after (right) immersion in catalyst solution for two hours. Reproduced from ref. 26. | ||
O2 evolution reached the very high Faradaic efficiency of 99%. At a modest overpotential of 520 mV, the turnover frequency that resulted, 7.9 s−1, was the highest ever seen for an iridium-based WOC; higher overpotentials are required to reach similar TOFs with the most active prior IrOx species. This means that the ABS derived from 3 not only has unprecedented stability for a molecular WOC, but that it can also be deposited effectively on the metal oxide scaffolds usually considered for PECs, and driven effectively at practical current densities. Activation of precursor 6-OH also led to stable binding of a surface species, but this species showed very different behavior from that derived from 3, again demonstrating that the chelate ligand used affects the properties of the activated catalyst. For its stability, ease of synthesis and surface immobilization, and rapid turnover, the heterogenized ABS is promising for practical applications, and such applications are being explored.
C. Mechanism of water oxidation catalyzed by Cp*Ir(chelate) species
A well-authenticated mechanism for catalysis is not yet available for this family of catalysts, largely due to the poorly resolved nature of the active species and the very high rates, implying short lifetimes for the intermediates. We have so far assumed that the active species for WO by Cp*Ir complexes is an oxo-iridium species. This is consistent with studies in the related field of C–H oxidation using the Cp*Ir catalysts, where C–H bonds are converted to C–O–H with retention of stereochemistry at carbon, consistent with O atom insertion.22,23,26,42,58,61,75–79 However, the exact identity of the proposed oxo-iridium species is not so easy to determine. Whereas the Ru WOCs have been studied in great mechanistic detail, the faster turnover of the Cp*Ir(chelate) complexes has made isolating intermediates very difficult. Nevertheless, through rigorous spectroscopic studies, several oxo-iridium species involved in the WO cycle have been characterized, ranging from those with Cp* still coordinated but partially oxidized22,58 to the fully oxidized IrIV-oxo dimer23 (Fig. 9). For chemical WO studies, the Cp*Ir(chelate) complexes show prompt O2 evolution without any significant lag phase, but the loss of Cp* is observed over several minutes.23 This suggests a variety of Cp*-containing intermediates like 23–27 are capable of oxidizing water and the C–H bonds of the Cp* concurrently, as suggested by Macchioni's “multi-site” hypothesis.58 Each of these species is likely independently active for WO, but likely vary in their effectiveness in catalysis depending on reaction conditions. Based on the electrocatalysis and TOF data, though, the fully oxidized IrIV-oxo dimer form 23 is likely the most highly active form of the catalyst.27 A complete mechanistic cycle has yet to be elucidated, but we will discuss studies in the context of previous Ru WO systems, kinetics, and computational studies carried out on monomeric oxo-iridium species for both WO and C–H oxidation. No computational studies have yet explored the pathways for a di-μ-oxo diiridium species such as 23, but the range of studies so far available can give us some insight into the general mechanistic aspects of the Cp*Ir complexes as WO precatalysts.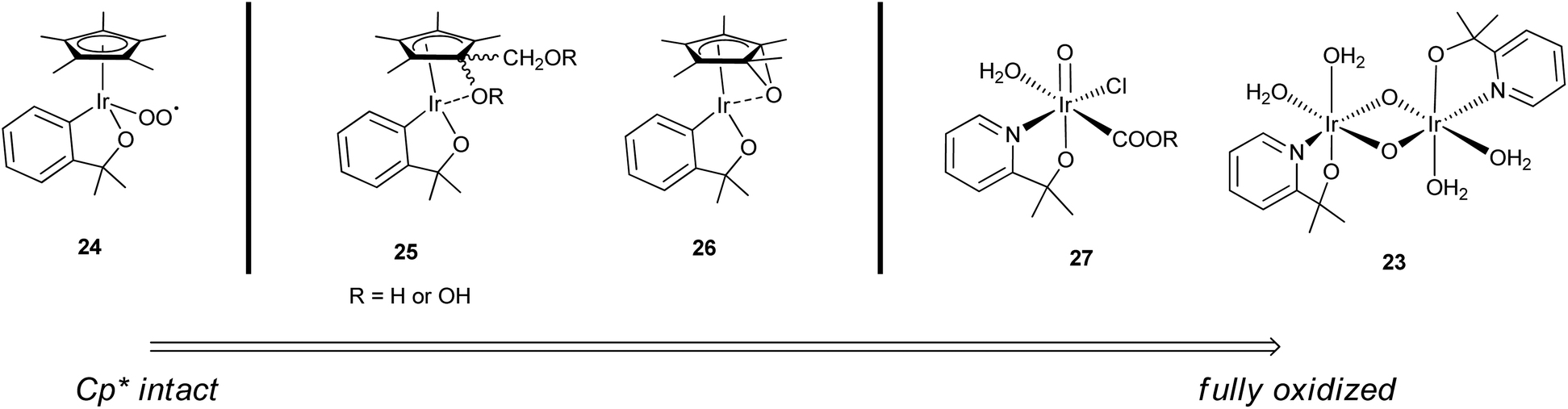 | ||
| Fig. 9 Spectrum of oxidized Cp*Ir complexes predicted to be capable of WOC, based on intermediates characterized by Macchioni (24–26),22,58 Lin (27),43 and Brudvig and Crabtree (23).23 Charge omitted for clarity. | ||
Mechanistic proposals for Ir have mostly been based on studies carried out on analogous Ru systems.80 From these studies, two pathways have been proposed for Ir: water nucleophilic attack (WNA) and radical oxo coupling (ROC) (Fig. 10). Existing mechanistic proposals for Cp*Ir WOCs postulate an IrV-oxo species, 28, as the key intermediate, with water nucleophilic attack (WNA) leading to O–O bond formation, 29.28,30 The high-valent iridium makes the O atom electrophilic enough to enable nucleophilic attack by water, although the water molecule must lose a proton during the process in order to enhance its nucleophilicity. Appropriately basic ancillary ligands could aid in abstraction of protons from water, according to another report,47 in analogy to the effects of bases seen for ruthenium WOCs.2,81,82 Another pathway that has been considered is ROC: this mechanism is usually associated with multi-metallic systems, as two radical metal oxyl species are required to couple together to form the O–O bond. This mechanism is known for some ruthenium dimers. For the postulated active species 23, the oxos are not oriented properly to carry out intramolecular RO; however, it is likely that reaction of 3 with NaIO4 results in many different isomeric forms of the active species, some of which could possibly be oriented appropriately for intramolecular ROC. Bimolecular oxo-coupling pathways are also theoretically possible. However, neither WNA nor ROC has been definitively assigned to any of the Cp*Ir complexes studied so far.
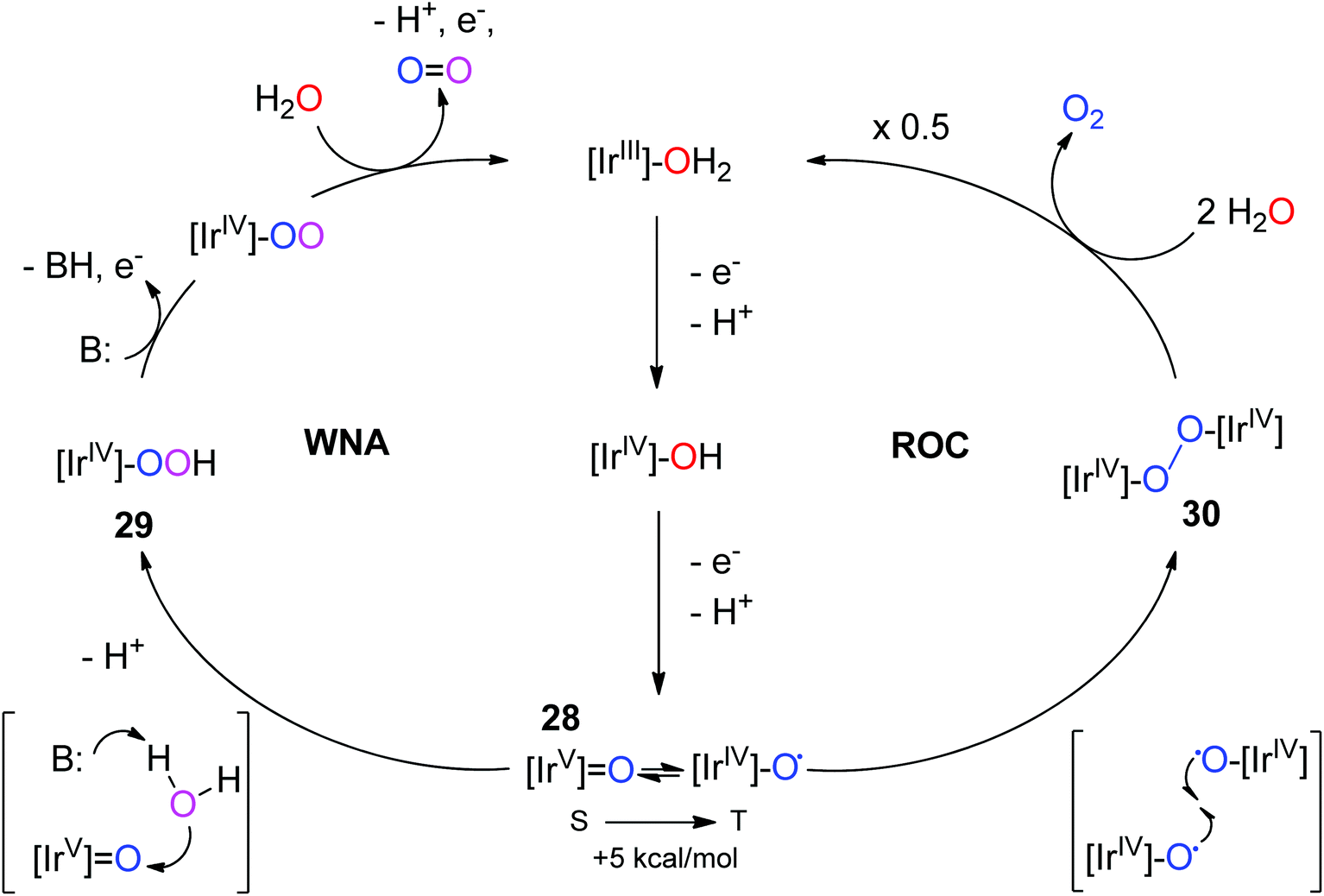 | ||
| Fig. 10 Two proposed pathways for iridium-catalyzed water oxidation. Left: WNA on 28, an electrophilic singlet IrV-oxo species Right: ROC of two IrIV-oxyl species, 30. Chelate ligands and charges are omitted for clarity. Reproduced from ref. 28, with permission from American Chemical Society. | ||
Many groups have studied the kinetics of Cp*Ir-based WOCs in the hope of elucidating the mechanism of action.23,28,35,36,38,39,75,83 The nature of the oxidant used for the study can significantly impact the interpretation of the data.23,46 For example, NaIO4 tends to be a two-electron oxidant, while electrode-driven oxidation is definitely single-electron. CAN is usually a single-electron oxidant as well, but has the added issue that it may directly associate with Ir during catalysis.41,65 The reactions with CAN are usually first-order in catalyst and also in oxidant, but the kinetics are badly behaved when there are no stable chelate ligands present on Ir.28,36,46,57,84,85 The cases of Cp*Ir precursors without chelate ligands are discussed later in Section III, as these precatalysts usually form heterogeneous WOCs with most oxidants and thus show poor kinetic behavior. Additionally, kinetic studies following the decrease in the absorption of CAN, a technique that has been used to calculate rates of WO with these catalysts in the past,22,36,86 is also particularly misleading; it has been found that 20 to 30 equivalents of oxidant are required for activation,41,43,63,87,88 and thus more equivalents of CAN will be consumed than equivalents of O2 released, leading to erroneous rate calculations. With NaIO4, the reaction with 3 is first-order in oxidant, but zero-order in iridium.23 This may mean that initial oxidation of IrIII to IrIV is rate determining, as Macchioni et al.22 concluded for the chelate species 6. Other literature data also indicate that IrIII oxidation to IrIV is often rate limiting for other WOCs,77,78 which does not provide much help in distinguishing WNA from ROC, as both mechanisms require high-valent Ir species and would, therefore, have the same rate-determining step. Furthermore, rate laws vary slightly for different chelate ligands; the bipy species 6, for example, has shown first-order dependence on catalyst concentration in addition to first-order dependence on CAN concentration.28 In all cases, it is unclear how these kinetic data inform mechanism proposals, because the concurrency of oxidative modification into the active species and WO renders all kinetic data inherently ambiguous. To obtain a more accurate picture of the kinetics, studying the fully-activated species would be much more informative; however, because the activation process likely produces multiple species, many of which are probably active for WO, such an analysis would require isolation and characterization of a single active species to be truly meaningful.
Because of the robust nature of the Cp*Ir precursors as WO catalysts, many computational studies have been published examining possible oxidation mechanisms and energetic pathways. However, because the activation phase and the loss of Cp* were not well understood until recently, many recent theoretical models have assumed the Cp* remained attached to the Ir center and have calculated the energies for active species closer in structure to 24 rather than 23. Also, Macchioni's “multi-site” theory58 suggests a variety of intermediates may be able to oxidize water with differing capabilities. Important information can nevertheless be gleaned from current theoretical models with respect to individual aspects of WO: the role of periodate, the contribution of the ligand sets, etc. By examining the general types of monomeric oxo-iridium species, specifically a high-valent mononuclear oxo-iridium (24) with the Cp* and its ancillary ligand still coordinated and a monomeric iridium-oxo with no Cp* but a chelate ligand (27), as well as studies done for C–H oxidation, we can gain insight into the general oxidation pathways of the Cp*Ir precursors.
A computational study has been carried out88 on a species similar to 27, deriving its inspiration from the MOF studies of Lin et al.43 in which a mononuclear Ir species bearing a bipyridine ligand with either formate or acetate bound instead of Cp*moiety.43 The authors investigated the predominant oxidation states involved in the mechanism and concluded that for this species, WO occurs at formal oxidation states much higher than previously thought, with contributions from Ir(VI) and Ir(VII) rather than Ir(V). This study relies on contributions from an acetate ligand bound for proton-coupled electron transfer to get a reasonable reaction barrier. Similarly, calculations from another group suggest that more basic ancillary ligands should greatly accelerate WO, as they have greater donor ability in stabilizing the higher valent Ir intermediates in the absence of Cp*.47 It is unclear how generally applicable these results are in light of the likely dimeric nature of the active species adopted by these catalysts after activation in solution, but this is the first published study that considers the absence of Cp* and is, therefore, interesting to consider in light of the studies of ABS and 23.
Other computational studies examined structures similar to 24, specifically the non-chelate Cp*Ir(NHC) complex 19 reacting with NaIO4 where the Cp* and NHC are still intact during the oxidation cycle.46 They posit a low-energy pathway for O–O bond formation in which both O atoms derive from IO4− rather than water. The same group followed this combined experimental-theoretical study with another theoretical study on the same complex,45 exploring the pathways followed on stepwise oxidation of the complex, without considering an added chemical oxidant. In this study, an intramolecular η2 O2 coordination mode was found to be the most favorable geometry for O–O bond formation. Although precursor 19 does eventually degrade into nanoparticles,35 this computational result suggests possible short-term homogeneous mechanisms, highlighting the O2 bond-formation pathways for species with Cp* and monomeric ancillary ligands still ligated to the Ir center during catalysis.
This coordination mode, however, would not be possible for intact Cp*Ir(chelate) complexes, as it requires two open sites at Ir while Cp* is still ligated to the metal center. However, some experimental studies indicate that oxidation catalysis can occur with Cp* still ligated to an Ir center with an ancillary chelate ligand. Although recent electrocatalytic WO studies with precursor 3 found that Cp* needed to be lost for appreciable O2 evolution to be seen,27 an earlier report on the trifluoroacetate analogue of 320 demonstrated O2 evolution could occur at an electrode without any lag phase, albeit at a very high overpotential (η = ∼900 mV at pH 7.5; the Faradaic yield was not calculated, but appears to be significantly below 100%). This could suggest the potential required to evolve O2 may be higher with the intact Cp* moiety than after its removal, and thus for short times, WO could possibly occur with Cp* still bound. This would imply that intermediates like 24–26 are involved in WO catalysis, although they likely have higher potentials for oxidation than that of the more active species, 23.
Another possible explanation for the much higher overpotential required for an intact precursor comes from the work that has been done on this family of catalysts for C–H oxidation.44,48,76–78 These reactions use Cp*Ir precursors with water as a solvent or co-solvent when oxidants like NaIO4 or CAN are used; other studies use OAT reagents, but these will not be discussed here as they require anhydrous conditions.64 For aqueous C–H oxidation reactions, WO occurs in tandem with C–H oxidation, but the latter is greatly favored, indicating that alkyl C–H bonds are much more reactive than water. Thus, it remains possible that the Cp*Ir precursors can oxidize reactive C–H substrates, such as tetrahydrofuran, in preference to attacking a Cp* ligand—particularly if the oxidative degradation of the Cp* is bimolecular—and thus carry out oxidation catalysis as a high-valent monomeric species with the Cp* still intact. This high-valent Cp*Ir species, similar to 24, could then oxidize both C–H bonds as well as water. If 24 has a higher potential for oxidation than 23, as previously thought, then 24 would more efficiently oxidize C–H bonds. This may explain why, during WO driven by chemical oxidants, Cp* loss occurs over ∼10 minutes, but O2 evolution commences within 1–2 minutes, and it is consistent with mechanistic proposals that oxidation is possible with Cp* still bound. However, whether the Cp*Ir precursors are able to carry out WO catalysis with or without a Cp* is still unclear at present, and only a thorough investigation by both theory and experiment is likely to afford a clear definition of the preferred mechanism.
III. Heterogeneous regime: Cp*Ir complexes without chelate ligands as precursors to heterogeneous catalysts
The most recent work on the Cp*Ir complexes has focused on the homogeneous system, but it was only recently that we could confirm that no Ir NPs were being formed. Shortly after the initial publication on water oxidation by the Cp*Ir complexes,30 work by Grotjahn et al. argued that these WO reactions led to heterogeneous IrOx materials, specifically when driven by CAN.41 They found that TEM of the reaction mixtures showed the formation of iridium-rich particles when as little as 15 equivalents of CAN had been added, with the iridium embedded in a ceria matrix. Although the authors conceded that the Ir-rich particles they observed by TEM were possibly formed as a result of Ir–Ce interactions, this work led to suspicion that the active WOC from Cp*Ir precursors was heterogeneous. As discussed in Section II, later studies from our group21,23 suggested that the chelate ligands on the Cp*Ir complexes are necessary to prevent nanoparticle formation. In contrast, for Cp*Ir complexes having only monodentate ligands, such as 17–19, the kinetic data suggested agglomeration and the formation of heterogeneous materials. Given the well-known stability of IrOx NPs, we focused our efforts on further investigating the homogeneity of Cp*Ir catalysts under oxidizing conditions.A. Detecting heterogeneous material in oxidative conditions
While many techniques exist for assessing the homogeneity of a solution under reducing conditions,55 when we first published the Cp*Ir complexes30 no such techniques had been reported for unambiguous determination of NP formation under highly oxidizing conditions. Additionally, the high salt concentrations and low catalyst loadings in aqueous conditions necessary for both chemical and electrochemical WO limited the effectiveness of traditional techniques for study of heterogeneous materials such as microscopy, kinetic analyses, and catalyst re-isolation. We thus sought out to identify other techniques in order to gain a deeper understanding of our catalyst.We applied the highly sensitive EQCN technique,20 which can detect as little as 10 ng changes in electrode mass in real time. When ramping between oxidizing and reducing potentials, a stepwise electrodeposition pattern was seen. The gain in mass during the oxidative cycle was associated with decomposition of the Cp*Ir complex in solution and deposition of a blue layer of heterogeneous iridium material on the electrode, a deposit that was very active for WO. Rinsing the electrode and screening for activity in plain electrolyte still showed WO and confirmed deposition of an active catalyst on the electrode. Later, in collaboration with Dr Sara Hashmi and Professor Menachem Elimelech of Yale's Chemical Engineering Department, we also used time-resolved DLS to detect nanoparticle formation.21,27,35,78 DLS has the advantage of being a non-destructive method for measuring nanoparticle growth dynamics, average particle size, and particle motion in solution. This powerful technique can monitor the formation of heterogeneous particles on the nanometer scale in operando over the course of several hours with microsecond resolution and a lower detection limit in the 1–2 nm range.
Although we had favored CAN in our initial studies because it has been the historical choice for WO owing to its high oxidizing potential and one-electron preference, it does have some disadvantages that make it unsuitable for DLS. CAN tends to form insoluble ceria particles above pH 3, which would show scattering by DLS that was not a result of iridium complex degradation. CeIV thus requires very low pH, which prevents pH-dependent studies. NPs can easily form upon reduction of CeIV to CeIII, which Grotjahn et al. found can nucleate with Ir.41 CAN is also highly colored; while this has been used to monitor water-oxidation rates and efficiencies, its strong absorption spectrum prevents UV-visible spectroscopic characterization of the working catalyst. CeIII and CeIV solutions are paramagnetic, which can complicate NMR studies. In contrast, NaIO4 is colorless, diamagnetic and soluble from pH 2–7, making it a superior oxidant for use with DLS. A comparison of chemical oxidants for WO is discussed in greater detail elsewhere,69 but after confirming that the Cp*Ir series still produces O2 with similar efficiencies whether CAN or NaIO4 was used, NaIO4 was chosen as the more convenient oxidant for further studies.
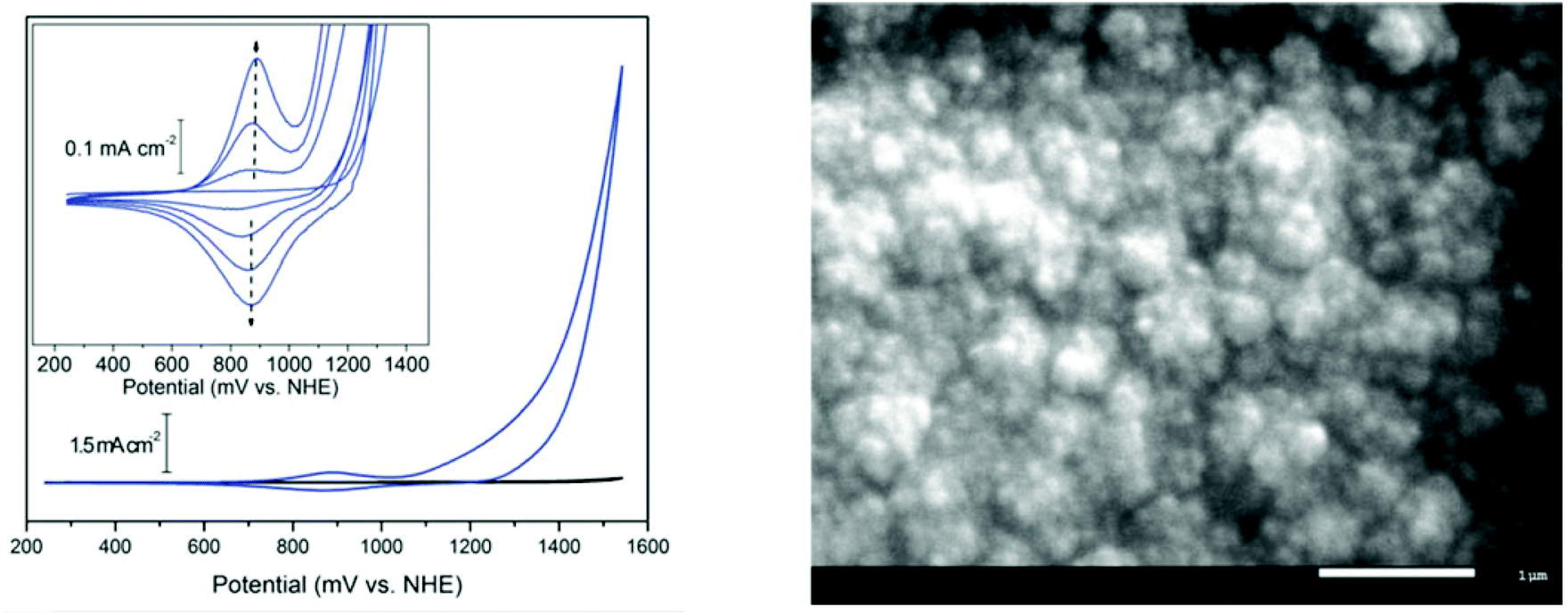 | ||
| Fig. 11 (Left): Cyclic voltammetry of BL (blue) and basal plane graphite background (black) in 0.1 M KNO3 at pH 6 showing the onset of catalytic water oxidation at ∼1.1 V vs. NHE. (Inset): reversible redox-active wave at 0.88 V vs. NHE; the intensity increases for each successive cycle as a result of deposition of BL. (Right): top-view SEM image of BL deposited on an ITO-coated glass substrate. Reproduced from ref. 31, with permission from the Royal Society of Chemistry. | ||
Unlike previous syntheses of hydrous iridium oxide, BL is prepared by unusually mild methods. Traditional preparations usually employ temperatures above 200 °C, powerful oxidants, or specialized equipment.95,98,99 Although the resulting material is often highly active for WO, the harsh conditions make reproducibility difficult. In the case of the BL, starting from 17 or 18, the catalyst loading and formation of the BL on a variety of electrode materials can be finely controlled, easily reproduced, and only require a commercially available potentiostat.31 The BL shows well-defined IR spectroscopic indications for the presence of Cp* C–H bonds at early times; these diminish on continued electrolysis and are replaced by carbonyl bands presumably due to carboxylate formation. In freshly deposited BL, elemental analysis shows 12% carbon from the degradation products of the Cp* is present. After ∼10 hours of bulk electrolysis, much of the carbon is lost, but ∼3% remains even after prolonged electrolysis.32 The catalytic activity of the BL remains unchanged throughout the whole period of carbon loss, which suggests that the organic framework of the catalyst may help deposition of the BL, but is not necessary for WO. A similar blue layer is also formed by oxidation of the CpIr precursor 16, which shows similar catalytic activity to BL but different deposition behavior.20 This is unsurprising, as it is likely that deposition of the active hydrous IrOx nanoclusters from Cp*Ir precursors involves formation of Cp*-derived carboxylate units that could aid in the deposition process by formation of cross-links.
BL shows H/D kinetic isotope effects similar to those of other amorphous iridium oxide materials,33 suggesting the same mechanism of action, yet the BL shows noticeably higher activity. Therefore, the intimate structure of BL has been a topic of great interest, and prior reports have suggested several different possible roles that Cp* plays in BL assembly. Further investigation of BL by IR, X-ray absorption fine structure (XAFS) spectroscopy, high-energy X-ray scattering (HEXS), and X-ray pair distribution function (PDF) analysis suggested that the BL is composed of small (>7.1 Å) iridium-oxo domains.93 Based on computational modeling of an Ir5O22 cluster extracted from crystalline rutile IrO2, the iridium oxo-domains likely contain a mixture of Ir(μ-O3)3Ir and Ir(μ-O2)2Ir substructures (Fig. 12). The small size of the Ir-oxo domains may explain the high activity of the BL, as nearly all the iridium centers are on the surface of the nanocluster and thus potentially available for catalytic reactions with water. The degradation products of the Cp*, likely di- and polyacids, are believed to coordinate to the iridium-oxo clusters and aid in the formation of Ir(μ-O3)3Ir and Ir(μ-O2)2Ir species. The acids are also believed to terminate the growth of the domains before the iridium-oxo centers can continue to grow into extended IrO2 materials. As an amorphous solid, the BL has many more surface sites to perform WO relative to nanoparticulate IrO2 where many of the metal atoms would be buried. Similar ligand templating and termination effects are believed to apply to the well-known cobalt–phosphate WOC of Nocera and coworkers.100 Their cobalt oxide nanocluster is only 11–14 Å in diameter and its small size is attributed to phosphate ligation to Co during assembly. The participation of Cp*-derived acids would also explain the slight mismatch between the modeled Ir(μ-O3)3Ir and Ir(μ-O2)2Ir domains and the BL; sporadic Ir–C bonds would distort the structure from the idealized one derived from crystalline IrO2. Much work is still required to understand the structure of the BL and the role that Cp* degradation plays in the assembly, which will no doubt help elucidate its robust and efficient nature as a WOC.
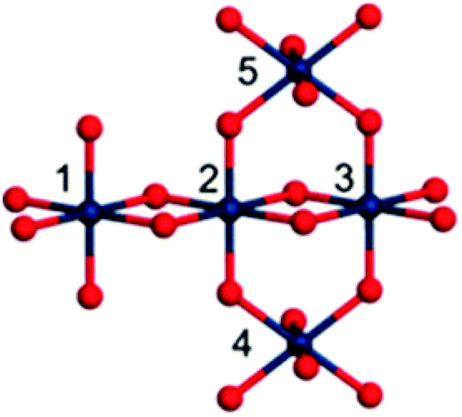 | ||
| Fig. 12 A proposed Ir5O22 cluster from XAS spectroscopy with Ir(μ-O3)3Ir and Ir(μ-O2)2Ir domains. Reproduced from ref. 93, with permission from the Royal Society of Chemistry. | ||
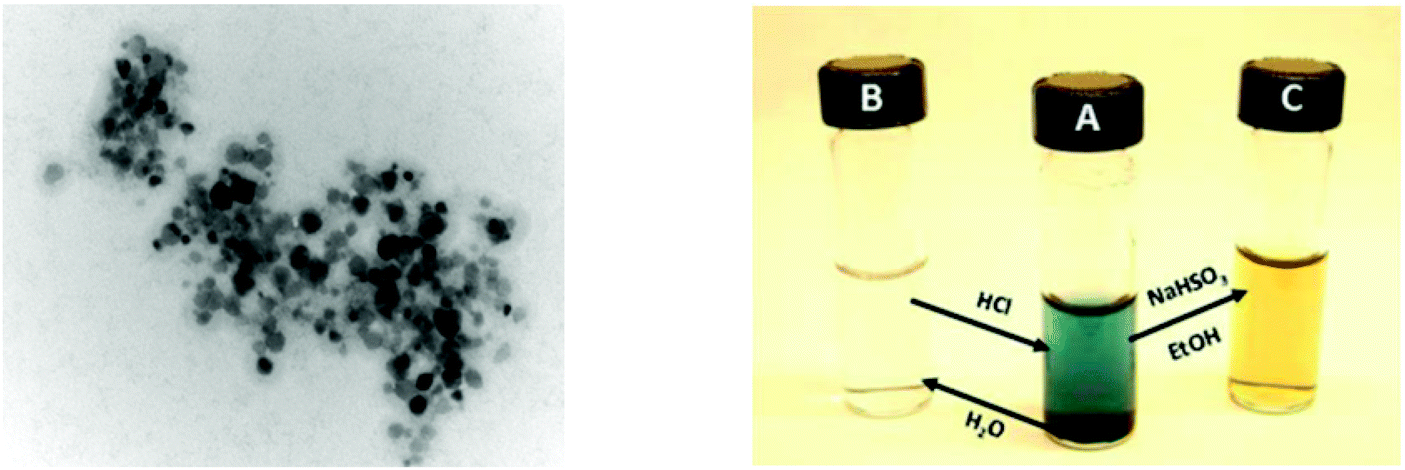 | ||
| Fig. 13 (Left): TEM picture of IrOx NPs derived from 17 at 200000 magnification. (Right): Photo showing conversion of a colloidal solution of NPs from 17 (B) to the deep blue homogeneous solution (A) and the reduced yellow homogeneous solution (C). Reproduced from ref. 21, with permission of the American Chemical Society. | ||
Compound 19 also degrades into NPs under our highly oxidizing conditions despite bearing an N-heterocyclic carbene (NHC) ligand.35 NHCs are often robust in catalysis but they have rarely been exposed to such highly oxidizing conditions; oxidation to the corresponding urea is a likely pathway after detachment from the metal. An earlier report indicated that other Cp*Ir(NHC)Cl2 catalysts were efficient WOCs,85 but our DLS measurements showed formation of particles ∼30 min after injection into an oxidant solution. Much like 17 and 18, both the short-lived homogeneous species and the heterogeneous iridium material are active for WO, as 19 evolves oxygen with reasonable efficiencies before the formation of particles. Of course we cannot exclude the formation of nanoclusters too small for detection by DLS, but such small structures are best considered homogeneous. NHCs are typically among the most donating ligand sets, but 19 does not have drastically improved water-oxidation rates (as a molecular species, before particle formation after 30 minutes) or the longevity observed for equally σ-basic Cp*Ir(phepy)Cl (4). This suggests that the stronger binding power of chelate ligands imparted a stabilizing effect on the Ir species that allows them to remain molecular after Cp* loss.
IV. Summary and outlook
The water-oxidizing chemistry of Cp*Ir complexes critically depends on the presence or absence of chelate ligands (Fig. 14). Oxidation of these pre-catalysts results in irreversible loss of the Cp* moiety en route to a multinuclear active form, which may be homogeneous when chelate ligands are present or heterogeneous otherwise. The presence of chelate ancillary ligands stabilizes the system against agglomeration and particle formation, while in the absence of a chelate ligand, heterogeneous IrOx material is formed. Both the homogeneous and heterogeneous catalysts that are formed from Cp*Ir precursors are highly active for WO, either when driven by a chemical oxidant or electrochemically. The homogeneous species retain their chelate ligands, which leads to tunability in their properties, as shown by the differences in the UV-visible spectral bands as well as rates of activation and O2 evolution. After activation of the chelate series, a di-μ-oxo iridiumIV,IV dimer is formed, having a single chelate ligand per iridium center, with the remaining sites around the iridium atoms occupied by aqua ligands or coordinating electrolytes such as IO3− or SO42− depending on the method of activation.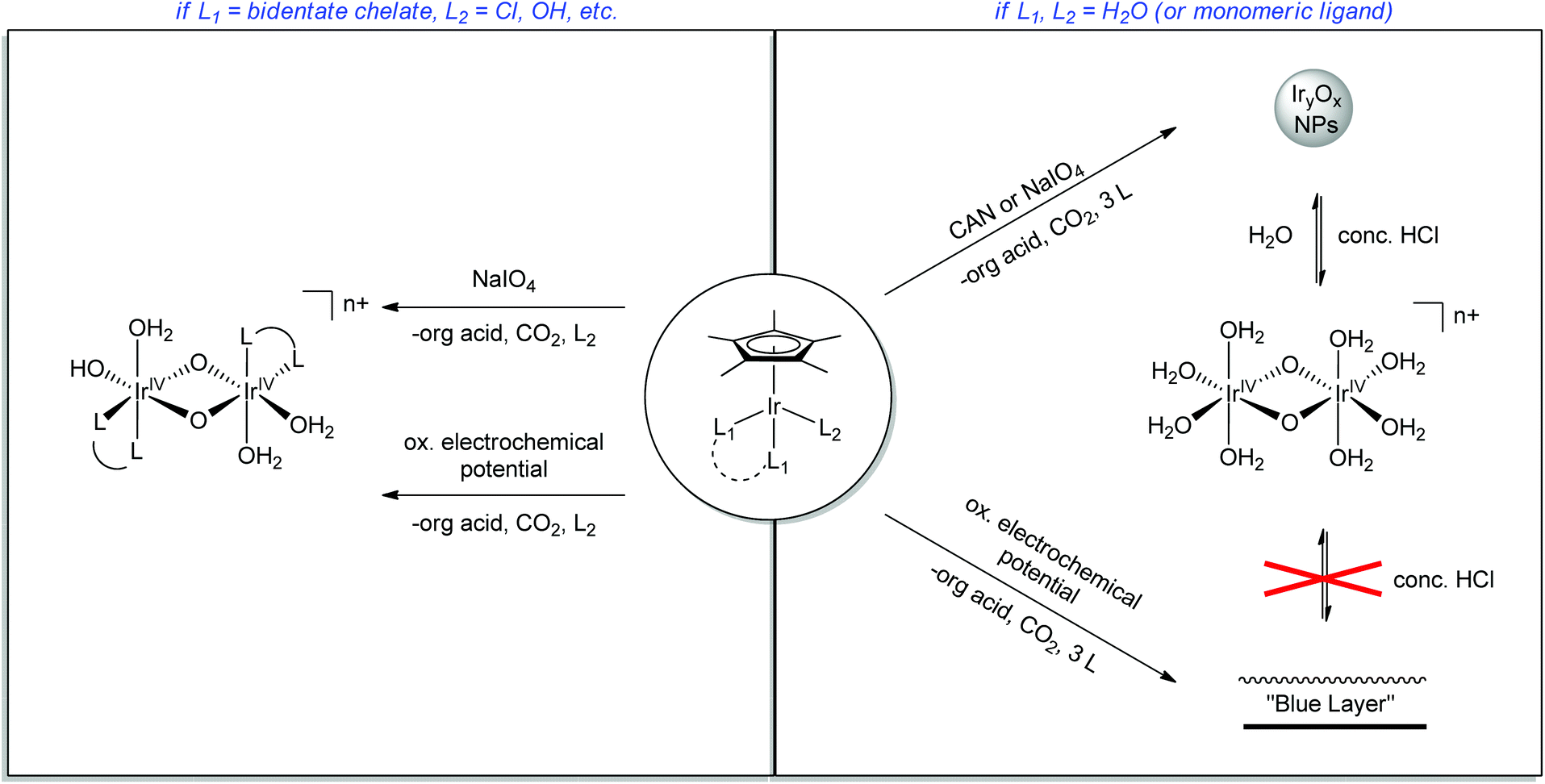 | ||
| Fig. 14 Various regimes of the Cp*Ir complexes as homogeneous catalyst and heterogeneous materials, their production dependent on the method of oxidation. | ||
Because of the multinuclear nature of the active species, the water-oxidizing mechanisms that are favored are probably at least dinuclear. However, at short times, when the Cp* ligands are not yet removed, mononuclear mechanisms may operate. When Cp*Ir(chelate) species are site-isolated in a MOF, they are still active for water oxidation, indicating that mononuclear mechanisms are viable, though Cp* is still lost in these cases. Activation occurs quickly after the addition of chemical oxidants like CAN or NaIO4, but can be carried out by bulk electrolysis as well. Once activated, the highly active WOC that is formed can be easily heterogenized on conductive oxide substrates simply by immersion of the substrate in a solution of activated catalyst. The supported species retains its molecular identity and high activity as an electrocatalyst, and it is stable for millions of turnovers (i.e. weeks) under water-oxidizing conditions on a variety of substrates and a wide range of pH. No discernible degradation occurs and the catalyst remains stably bound to the surface in a monolayer. This shows promise for use in a solar fuels PEC, where high turnover frequency is required to prevent recombination from dominating the electron-transfer processes.
The large body of work on this field has allowed us to identify a wide range of possible catalysts and ways to use them, but there is still much to be explored. The use of the pyalc ligand has enabled the formation of a stable IrIV species with high TOF and low overpotential. The active species formed after Cp* loss from precursors bearing chelate ligands has yet to be unequivocally identified, and work towards the definitive characterization of these species is underway. The identification of the active species would then allow development of methods to synthesize these species directly without going through the Cp* precursors. Such methods would be pivotal in the development of tandem molecular architectures including both light absorber and WOC, such as have been previously studied using the unmodified Cp* precursors.103,104 The catalytic mechanism of the activated species has been difficult to study in light of the extreme difficulty in trapping any mechanistic intermediates and identifying them. Good work has been done on unraveling the mechanism of activation, and there is an open field for computational study of the mechanism of the activated species. Because previous mechanistic work has concentrated on elucidating mononuclear mechanisms, but the active species is most likely dinuclear, this topic is open and could provide insight into why they work so well. Recent discoveries hold promise for successful application of molecular WOCs in photoelectrochemical cells in efforts to design and optimize solar fuels devices.
Acknowledgements
This work was supported as part of the Argonne-Northwestern Solar Energy Research (ANSER) Center, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Award Number DE-SC0001059. D.L.H acknowledges support by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1122492.References
- A.-F. d. c. Fourcroy, Philosophie Chimique, De l'Imprimerie de Cl. Simon, Paris, 1792 Search PubMed.
- F. Liu, J. J. Concepcion, J. W. Jurss, T. Cardolaccia, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2008, 47, 1727 CrossRef CAS PubMed.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163 CrossRef CAS.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724 CrossRef CAS PubMed.
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455 CrossRef CAS PubMed.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729 CrossRef CAS PubMed.
- K. J. Young, L. A. Martini, R. L. Milot, R. C. Snoeberger, V. S. Batista, C. A. Schmuttenmaer, R. H. Crabtree and G. W. Brudvig, Coord. Chem. Rev., 2012, 256, 2503 CrossRef CAS PubMed.
- J. K. Hurst, Science, 2010, 328, 315 CrossRef CAS PubMed.
- S. W. Gersten, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4029 CrossRef CAS.
- B. Limburg, E. Bouwman and S. Bonnet, Coord. Chem. Rev., 2012, 256, 1451 CrossRef CAS PubMed.
- M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21 CrossRef CAS PubMed.
- M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863 Search PubMed.
- L. Duan, F. Bozoglian, S. Mandal, B. Stewart, T. Privalov, A. Llobet and L. Sun, Nat. Chem., 2012, 4, 418 CrossRef CAS PubMed.
- J. Kiwi and M. Grätzel, Angew. Chem., Int. Ed. Engl., 1978, 17, 860 CrossRef PubMed.
- A. Harriman, I. J. Pickering, J. M. Thomas and P. A. Christensen, J. Chem. Soc., Faraday Trans. 1, 1988, 84, 2795 RSC.
- G. Beni, L. M. Schiavone, J. L. Shay, W. C. Dautremont-Smith and B. S. Schneider, Nature, 1979, 282, 281 CrossRef CAS PubMed.
- N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2007, 130, 210 CrossRef PubMed.
- J. A. Woods, S. Bernhard and M. Albrecht, in Molecular Water Oxidation Catalysis, John Wiley & Sons, Ltd, 2014, p. 113 Search PubMed.
- N. D. Schley, J. D. Blakemore, N. K. Subbaiyan, C. D. Incarvito, F. D'Souza, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2011, 133, 10473 CrossRef CAS PubMed.
- U. Hintermair, S. M. Hashmi, M. Elimelech and R. H. Crabtree, J. Am. Chem. Soc., 2012, 134, 9785 CrossRef CAS PubMed.
- A. Savini, P. Belanzoni, G. Bellachioma, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Green Chem., 2011, 13, 3360 RSC.
- U. Hintermair, S. W. Sheehan, A. R. Parent, D. H. Ess, D. T. Richens, P. H. Vaccaro, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2013, 135, 10837 CrossRef CAS PubMed.
- A. Petronilho, M. Rahman, J. A. Woods, H. Al-Sayyed, H. Muller-Bunz, J. M. Don MacElroy, S. Bernhard and M. Albrecht, Dalton Trans., 2012, 41, 13074 RSC.
- L. Badia-Bou, E. Mas-Marza, P. Rodenas, E. M. Barea, F. Fabregat-Santiago, S. Gimenez, E. Peris and J. Bisquert, J. Phys. Chem. C, 2013, 117, 3826 CAS.
- S. W. Sheehan, J. M. Thomsen, U. Hintermair, R. H. Crabtree, G. W. Brudvig and C. A. Schmuttenmaer, Nat. Commun., 2015, 6, 6469 CrossRef PubMed.
- J. M. Thomsen, S. W. Sheehan, S. M. Hashmi, J. Campos, U. Hintermair, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2014, 136, 13826 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, D. Balcells, J. F. Hull, G. W. Olack, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 16017 CrossRef CAS PubMed.
- Molecular Water Oxidation Catalysis: A Key Topic for New Sustainable Energy Conversion Schemes, ed. A. Llobet, John Wiley & Sons, Ltd, Chichester, UK, 2014 Search PubMed.
- J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, G. W. Olack, C. D. Incarvito, G. W. Brudvig and R. H. Crabtree, Chem. Sci., 2011, 2, 94 RSC.
- J. D. Blakemore, M. W. Mara, M. N. Kushner-Lenhoff, N. D. Schley, S. J. Konezny, I. Rivalta, C. F. A. Negre, R. C. Snoeberger, O. Kokhan, J. Huang, A. Stickrath, L. A. Tran, M. L. Parr, L. X. Chen, D. M. Tiede, V. S. Batista, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2013, 52, 1860 CrossRef CAS PubMed.
- J. D. Blakemore, N. D. Schley, M. N. Kushner-Lenhoff, A. M. Winter, F. D'Souza, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2012, 51, 7749 CrossRef CAS PubMed.
- J. Graeupner, T. P. Brewster, J. D. Blakemore, N. D. Schley, J. M. Thomsen, G. W. Brudvig, N. Hazari and R. H. Crabtree, Organometallics, 2012, 31, 7158 CrossRef CAS PubMed.
- J. Graeupner, U. Hintermair, D. L. Huang, J. M. Thomsen, M. Takase, J. Campos, S. M. Hashmi, M. Elimelech, G. W. Brudvig and R. H. Crabtree, Organometallics, 2013, 32, 5384 CrossRef CAS PubMed.
- A. Savini, G. Bellachioma, G. Ciancaleoni, C. Zuccaccia, D. Zuccaccia and A. Macchioni, Chem. Commun., 2010, 46, 9218 RSC.
- A. Savini, A. Bucci, G. Bellachioma, S. Giancola, F. Palomba, L. Rocchigiani, A. Rossi, M. Suriani, C. Zuccaccia and A. Macchioni, J. Organomet. Chem., 2014, 771, 24 CrossRef CAS PubMed.
- J. Depasquale, I. Nieto, L. E. Reuther, C. J. Herbst-Gervasoni, J. J. Paul, V. Mochalin, M. Zeller, C. M. Thomas, A. W. Addison and E. T. Papish, Inorg. Chem., 2013, 52, 9175 CrossRef CAS PubMed.
- D. Hong, M. Murakami, Y. Yamada and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 5708 CAS.
- L. S. Park-Gehrke, J. Freudenthal, W. Kaminsky, A. G. DiPasquale and J. M. Mayer, Dalton Trans., 2009, 1972 RSC.
- D. B. Grotjahn, D. B. Brown, J. K. Martin, D. C. Marelius, M.-C. Abadjian, H. N. Tran, G. Kalyuzhny, K. S. Vecchio, Z. G. Specht, S. A. Cortes-Llamas, V. Miranda-Soto, C. van Niekerk, C. E. Moore and A. L. Rheingold, J. Am. Chem. Soc., 2011, 133, 19024 CrossRef CAS PubMed.
- C. Zuccaccia, G. Bellachioma, S. Bolaño, L. Rocchigiani, A. Savini and A. Macchioni, Eur. J. Inorg. Chem., 2012, 2012, 1462 CrossRef CAS PubMed.
- C. Wang, J.-L. Wang and W. Lin, J. Am. Chem. Soc., 2012, 134, 19895 CrossRef CAS PubMed.
- J. Campos, U. Hintermair, T. P. Brewster, M. K. Takase and R. H. Crabtree, ACS Catal., 2014, 4, 973 CrossRef CAS.
- A. Venturini, A. Barbieri, J. N. H. Reek and D. G. H. Hetterscheid, Chem. – Eur. J., 2014, 20, 5358 CrossRef CAS PubMed.
- D. G. H. Hetterscheid and J. N. H. Reek, Eur. J. Inorg. Chem., 2014, 2014, 742 CrossRef CAS PubMed.
- L. Vilella, P. Vidossich, D. Balcells and A. Lledos, Dalton Trans., 2011, 40, 11241 RSC.
- U. Hintermair, J. Campos, T. P. Brewster, L. M. Pratt, N. D. Schley and R. H. Crabtree, ACS Catal., 2014, 4, 99 CrossRef CAS.
- P. Stonehart, H. A. Kozlowska and B. E. Conway, Potentiodynamic Examination of Electrode Kinetics for Electroactive Adsorbed Species: Applications to the Reduction of Noble Metal Surface Oxides, 1969, vol. 310 Search PubMed.
- S. Gottesfeld and S. Srinivasan, J. Electroanal. Chem. Interfacial Electrochem., 1978, 86, 89 CrossRef CAS.
- T. Pauporté, F. Andolfatto and R. Durand, Electrochim. Acta, 1999, 45, 431 CrossRef.
- R. Crabtree, Acc. Chem. Res., 1979, 12, 331 CrossRef CAS.
- R. H. Crabtree, J. M. Mihelcic and J. M. Quirk, J. Am. Chem. Soc., 1979, 101, 7738 CrossRef CAS.
- R. H. Crabtree, M. F. Mellea, J. M. Mihelcic and J. M. Quirk, J. Am. Chem. Soc., 1982, 104, 107 CrossRef CAS.
- R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed.
- A. Harriman, J. M. Thomas and G. R. Millward, New J. Chem., 1987, 11, 757 CAS.
- H. Junge, N. Marquet, A. Kammer, S. Denurra, M. Bauer, S. Wohlrab, F. Gärtner, M.-M. Pohl, A. Spannenberg, S. Gladiali and M. Beller, Chem. – Eur. J., 2012, 18, 12749 CrossRef CAS PubMed.
- C. Zuccaccia, G. Bellachioma, O. Bortolini, A. Bucci, A. Savini and A. Macchioni, Chem. – Eur. J., 2014, 20, 3446 CrossRef CAS PubMed.
- T. P. Brewster, J. D. Blakemore, N. D. Schley, C. D. Incarvito, N. Hazari, G. W. Brudvig and R. H. Crabtree, Organometallics, 2011, 30, 965 CrossRef CAS.
- D. G. H. Hetterscheid and B. de Bruin, J. Mol. Catal. A: Chem., 2006, 251, 291 CrossRef CAS PubMed.
- A. J. Ingram, A. B. Wolk, C. Flender, J. Zhang, C. J. Johnson, U. Hintermair, R. H. Crabtree, M. A. Johnson and R. N. Zare, Inorg. Chem., 2014, 53, 423 CrossRef CAS PubMed.
- A. R. Parent, J. D. Blakemore, G. W. Brudvig and R. H. Crabtree, Chem. Commun., 2011, 47, 11745 RSC.
- C. R. Turlington, D. P. Harrison, P. S. White, M. Brookhart and J. L. Templeton, Inorg. Chem., 2013, 52, 11351 CrossRef CAS PubMed.
- C. R. Turlington, P. S. White, M. Brookhart and J. L. Templeton, J. Am. Chem. Soc., 2014, 136, 3981 CrossRef CAS PubMed.
- D. J. Wasylenko, C. Ganesamoorthy, M. A. Henderson and C. P. Berlinguette, Inorg. Chem., 2011, 50, 3662 CrossRef CAS PubMed.
- F. R. El-Eziri and Y. Sulfab, Inorg. Chim. Acta, 1977, 25, 15 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 2nd edn, 2001 Search PubMed.
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247 RSC.
- T. Nakagawa, N. S. Bjorge and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 15578 CrossRef CAS PubMed.
- O. Diaz-Morales, T. J. P. Hersbach, D. G. H. Hetterscheid, J. N. H. Reek and M. T. M. Koper, J. Am. Chem. Soc., 2014, 136, 10432 CrossRef CAS PubMed.
- M. C. Lehman, D. R. Pahls, J. M. Meredith, R. D. Sommer, D. M. Heinekey, T. R. Cundari and E. A. Ison, J. Am. Chem. Soc., 2015, 137, 3574 CrossRef CAS PubMed.
- M. Yagi, E. Tomita, S. Sakita, T. Kuwabara and K. Nagai, J. Phys. Chem. B, 2005, 109, 21489 CrossRef CAS PubMed.
- T. Kuwabara, E. Tomita, S. Sakita, D. Hasegawa, K. Sone and M. Yagi, J. Phys. Chem. C, 2008, 112, 3774 CAS.
- A. Savini, A. Bucci, G. Bellachioma, L. Rocchigiani, C. Zuccaccia, A. Llobet and A. Macchioni, Eur. J. Inorg. Chem., 2014, 2014, 690 CrossRef CAS PubMed.
- M. Zhou, N. D. Schley and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 12550 CrossRef CAS PubMed.
- M. Zhou, D. Balcells, A. R. Parent, R. H. Crabtree and O. Eisenstein, ACS Catal., 2011, 2, 208 CrossRef.
- M. Zhou, U. Hintermair, B. G. Hashiguchi, A. R. Parent, S. M. Hashmi, M. Elimelech, R. A. Periana, G. W. Brudvig and R. H. Crabtree, Organometallics, 2013, 32, 957 CrossRef CAS.
- M. C. Lehman, J. B. Gary, P. D. Boyle, M. S. Sanford and E. A. Ison, ACS Catal., 2013, 3, 2304 CrossRef CAS.
- X. Sala, S. Maji, R. Bofill, J. García-Antón, L. Escriche and A. Llobet, Acc. Chem. Res., 2013, 47, 504 CrossRef PubMed.
- J. J. Concepcion, M. K. Tsai, J. T. Muckerman and T. J. Meyer, J. Am. Chem. Soc., 2010, 132, 1545 CrossRef CAS PubMed.
- Y. Tamaki, A. K. Vannucci, C. J. Dares, R. A. Binstead and T. J. Meyer, J. Am. Chem. Soc., 2014, 136, 6854 CrossRef CAS PubMed.
- J. A. Woods, R. Lalrempuia, A. Petronilho, N. D. McDaniel, H. Muller-Bunz, M. Albrecht and S. Bernhard, Energy Environ. Sci., 2014, 7, 2316 CAS.
- N. Marquet, F. Gärtner, S. Losse, M.-M. Pohl, H. Junge and M. Beller, ChemSusChem, 2011, 4, 1598 CrossRef CAS PubMed.
- D. G. H. Hetterscheid and J. N. H. Reek, Chem. Commun., 2011, 47, 2712 RSC.
- I. Corbucci, A. Petronilho, H. Müller-Bunz, L. Rocchigiani, M. Albrecht and A. Macchioni, ACS Catal., 2015, 2714 CrossRef CAS.
- A. Lewandowska-Andralojc, D. E. Polyansky, C. H. Wang, W. H. Wang, Y. Himeda and E. Fujita, Phys. Chem. Chem. Phys., 2014, 16, 11976 RSC.
- R. Z. Liao and P. E. M. Siegbahn, ACS Catal., 2014, 4, 3937 CrossRef CAS.
- Y. Zhao, N. M. Vargas-Barbosa, E. A. Hernandez-Pagan and T. E. Mallouk, Small, 2011, 7, 2087 CrossRef CAS PubMed.
- T. Nakagawa, C. A. Beasley and R. W. Murray, J. Phys. Chem. C, 2009, 113, 12958 CAS.
- K. E. Michaux and R. W. Murray, Langmuir, 2013, 29, 12254 CrossRef CAS PubMed.
- A. A. Gambardella, N. S. Bjorge, V. K. Alspaugh and R. W. Murray, J. Phys. Chem. C, 2011, 115, 21659 CAS.
- J. Huang, J. D. Blakemore, D. Fazi, O. Kokhan, N. D. Schley, R. H. Crabtree, G. W. Brudvig and D. M. Tiede, Phys. Chem. Chem. Phys., 2014, 16, 1814 RSC.
- M. A. Petit and V. Plichon, J. Electroanal. Chem., 1998, 444, 247 CrossRef CAS.
- Y. Kazusuke, Jpn. J. Appl. Phys., 1989, 28, 632 CrossRef.
- P. Steegstra and E. Ahlberg, Electrochim. Acta, 2012, 68, 206 CrossRef CAS PubMed.
- M. N. Kushner-Lenhoff, J. D. Blakemore, N. D. Schley, R. H. Crabtree and G. W. Brudvig, Dalton Trans., 2013, 42, 3617 RSC.
- C. Comninellis and G. P. Vercesi, J. Appl. Electrochem., 1991, 21, 335 CrossRef CAS.
- P. C. S. Hayfield, Platinum Met. Rev., 1998, 42, 116 CAS.
- M. W. Kanan, Y. Surendranath and D. G. Nocera, Chem. Soc. Rev., 2009, 38, 109 RSC.
- S. E. Castillo-Blum, D. T. Richens and A. G. Sykes, J. Chem. Soc., Chem. Commun., 1986, 1120 RSC.
- S. E. Castillo-Blum, D. T. Richens and A. G. Sykes, Inorg. Chem., 1989, 28, 954 CrossRef CAS.
- M. T. Vagnini, A. L. Smeigh, J. D. Blakemore, S. W. Eaton, N. D. Schley, F. D'Souza, R. H. Crabtree, G. W. Brudvig, D. T. Co and M. R. Wasielewski, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15651 CrossRef CAS PubMed.
- M. T. Vagnini, M. W. Mara, M. R. Harpham, J. Huang, M. L. Shelby, L. X. Chen and M. R. Wasielewski, Chem. Sci., 2013, 4, 3863 RSC.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |