
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and structure of Ag(I), Pd(II), Rh(I), Ru(II) and Au(I) NHC-complexes with a pendant Lewis acidic boronic ester moiety†
Momar
Toure
,
Olivier
Chuzel
* and
Jean-Luc
Parrain
*
Aix-Marseille Université, Centrale Marseille, CNRS, iSm2 UMR 7313, 13397, Marseille, France. E-mail: olivier.chuzel@univ-amu.fr; jl.parrain@univ-amu.fr
First published on 24th March 2015
Abstract
Bifunctional Ag(I), Pd(II), Rh(I), Ru(II) and Au(I) complexes containing a NHC ligand and a pendant trivalent boron moiety have been synthesized in high yields. Fine-tuned reaction conditions were used to prevent potential ligand self-quenching or polymerization due to the eventual co-existence in situ of free NHC (Lewis base) and boronic ester (Lewis acid) in the same molecule.
Boron-centered ligands have relatively large structural and electronic diversity allowing for different coordination modes to a metal center, as found, in borane, boryl, borylene or boride complexes.1 In some particular cases of Lewis-basic metals, the Lewis-acidic trivalent boron atom can be coordinated to the metal center by a dative bond, becoming a σ-acceptor Z-type ligand,2 mainly with borane derivatives,3 which was first authenticated in 1999 by Hill et al. through the isolation of a metallaboratrane.4 Complexes featuring this supported M–B Z-type interactions are derived from the B–H activation of hydrido tris(methimazolyl)borates and related systems, or are obtained directly from ambiphilic phosphine borane ligands5 where the boron atom moiety is structurally maintained close to the metal center.
There are rare examples of bidentate or bifunctional ligands containing a boronic ester moiety that are reported in the literature,6–9 and the only observed transition metal–Z interactions with the boronic ester moiety disclosed so far are the bridging borane–boryl compounds.10 Because the structural feature of these bifunctional ligands does not allow the boronic ester moiety to coordinate to the metal center, we designed a more flexible bifunctional ligand allowing the possibility of such an interaction. To increase the interaction, a strong σ-donor N-heterocyclic carbene (NHC) ligand was chosen to enhance the Lewis base character of the transition metal. Synthesis of such NHC-boron transition metal complexes was highly challenging due to the necessary generation of a coexisting free NHC (Lewis base) and not the hindered boron derivative (Lewis acid) in situ (Scheme 1).
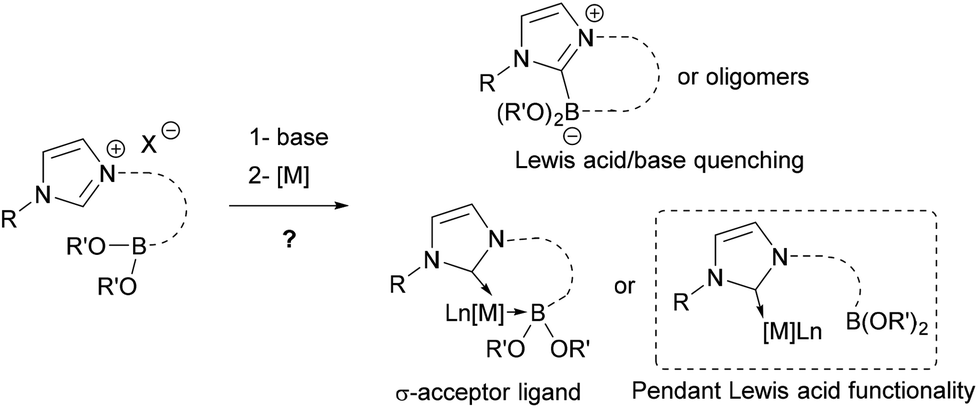 | ||
| Scheme 1 Possible NHC/boronic ester/metal combined product pathways. | ||
Here, we report the synthesis and the characterization of the original stable bifunctional NHC-boronic ester silver, palladium, rhodium, ruthenium and gold complexes. Despite several modifications, the boronic ester moiety remained pendant and no Z-type interaction to the metal center was observed in this series of complexes (Scheme 1).
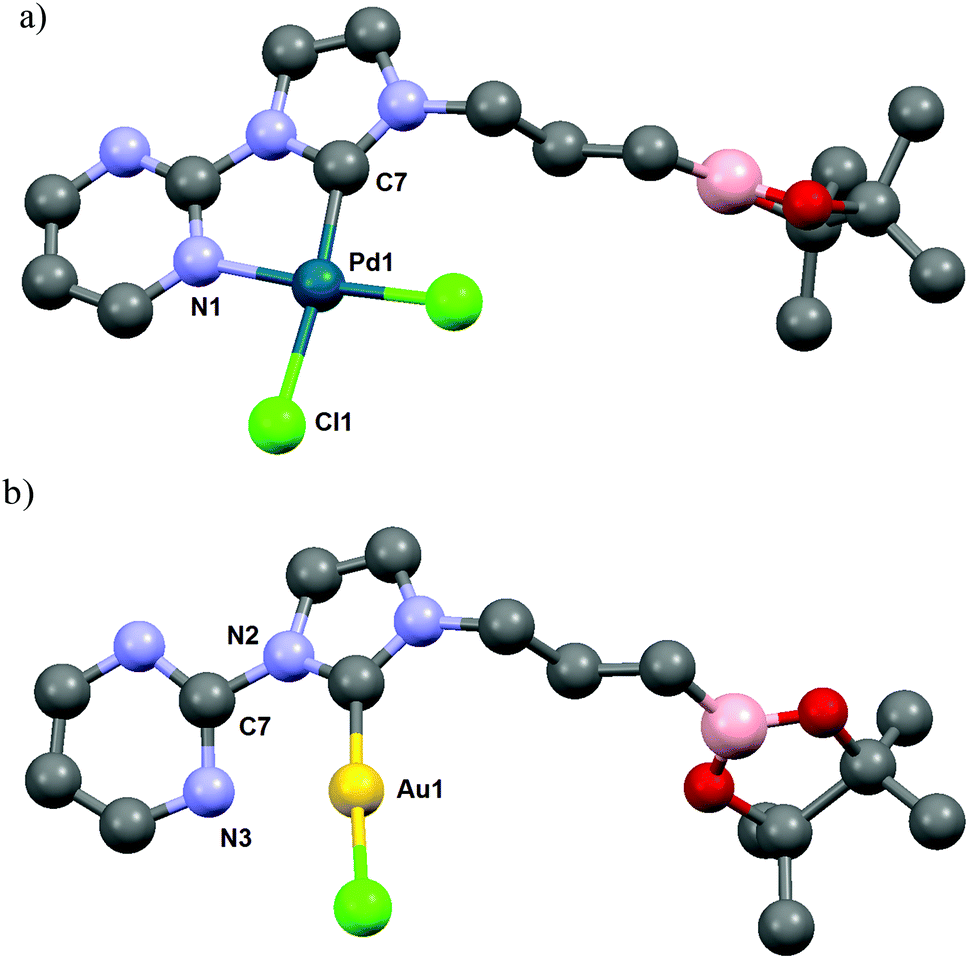
In the initial attempts, NHC-boron salts were obtained by reacting phenyl-substituted imidazole 1a with the bromopropyl dioxaborolane 2 that was prepared in one step through hydroboration of allylbromide.11 Attempts to prepare the complex 5a directly from imidazolium salt 1a and classic bases such as n-BuLi,12 KHMDS, NaH or t-BuOK did not succeed and only degradation of the starting material was observed. No self-quenching resulting in cyclic NHC-boranes, polymerization processes or a frustrated Lewis pair was detected. To generate the carbenoid species and avoid decomposition, we decided to use silver oxide as a soft base. Gratifyingly the reaction between Ag2O and the imidazolium salt 1a gave the desired cationic bis-NHC silver complex 4a in quantitative yield, demonstrating the importance of the silver oxide mediated approach (Scheme 2). The structure was confirmed by NMR and mass spectrometry analysis (ESI, MeOH, m/z for [M − AgBr2]+ 731.4). Subsequent treatment of 4a with 2.5 equiv. of PdCl2(CH3CN)2 at room temperature in dichloromethane afforded binuclear dichloride complexes 5a in 84% yield. Transmetallation of the silver complex with other transition metals was also investigated. The reactivity of 4a towards [Rh(η4-cod)Cl]2 or AuCl metallic sources afforded the stable neutral complexes 6a and 7a in 74% and 87% yield respectively (Fig. 1). Negligible difference in chemical shifts of the boron atom in 11B NMR (128 MHz, CDCl3) for imidazolium salts 3a (33.3 ppm) and all complexes prepared (33.3–34.0 ppm) showed that the boron moiety was not ligated to the metal center and remained pendant. This initial evidence was further confirmed for 5a, 6a and 7a in the solid state by X-ray diffraction studies (Fig. 2).
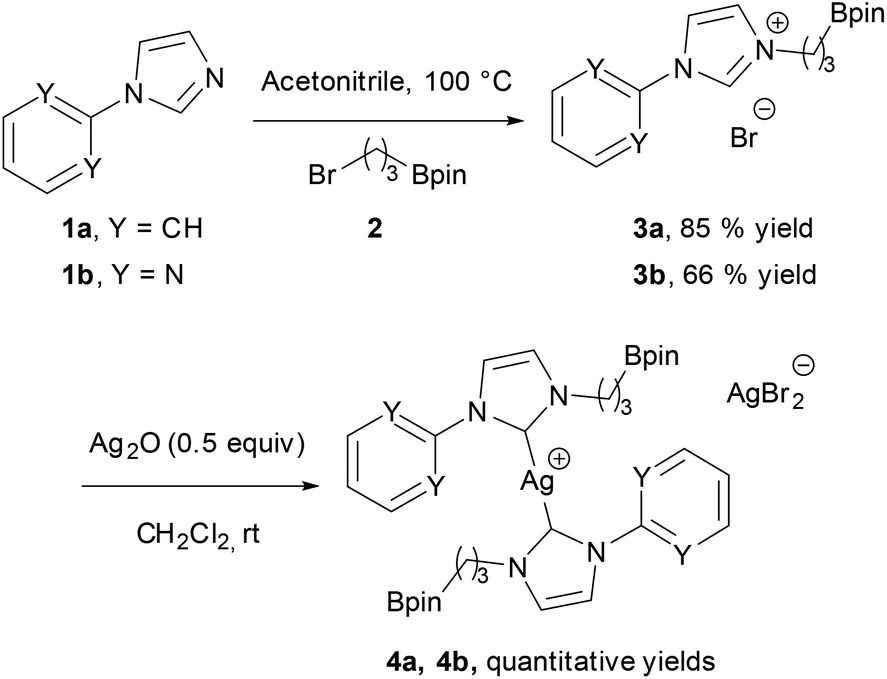 | ||
| Scheme 2 Synthesis of Ag-NHC boronic ester complexes. | ||
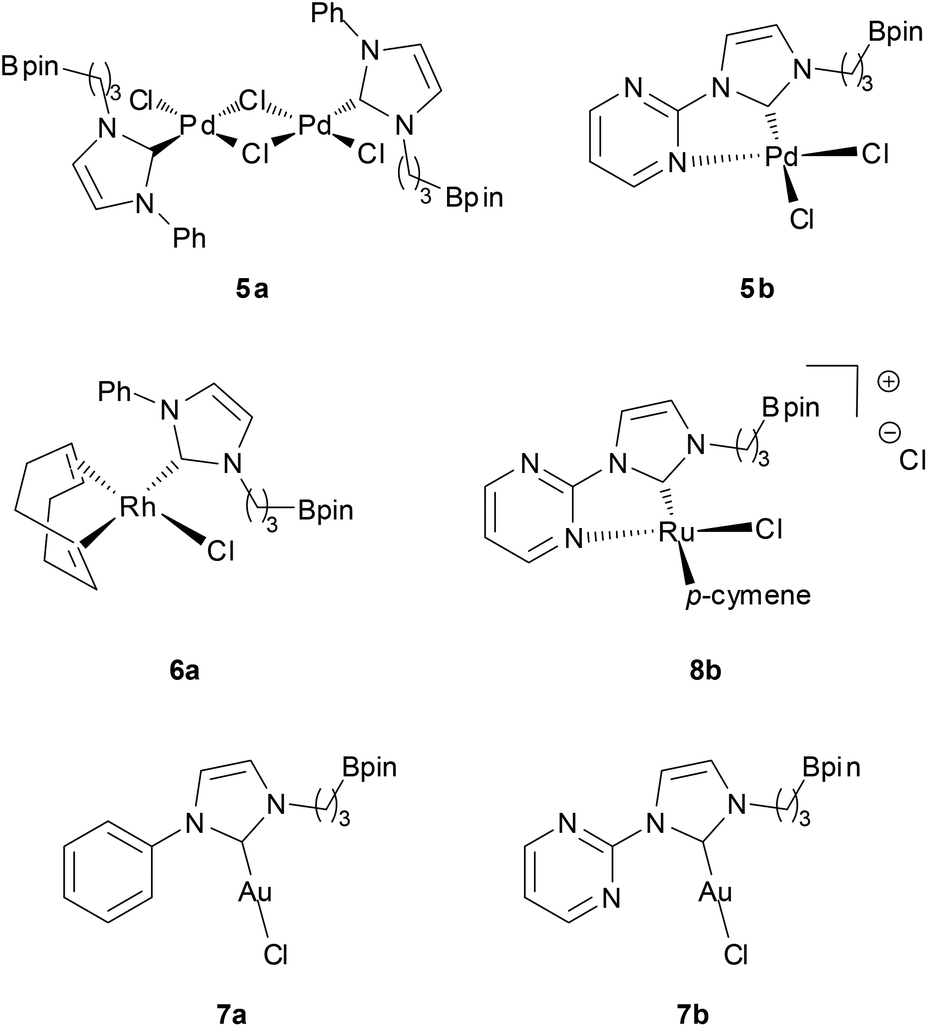 | ||
| Fig. 1 Structures of palladium, rhodium, gold and ruthenium NHC-boronic ester complexes. | ||
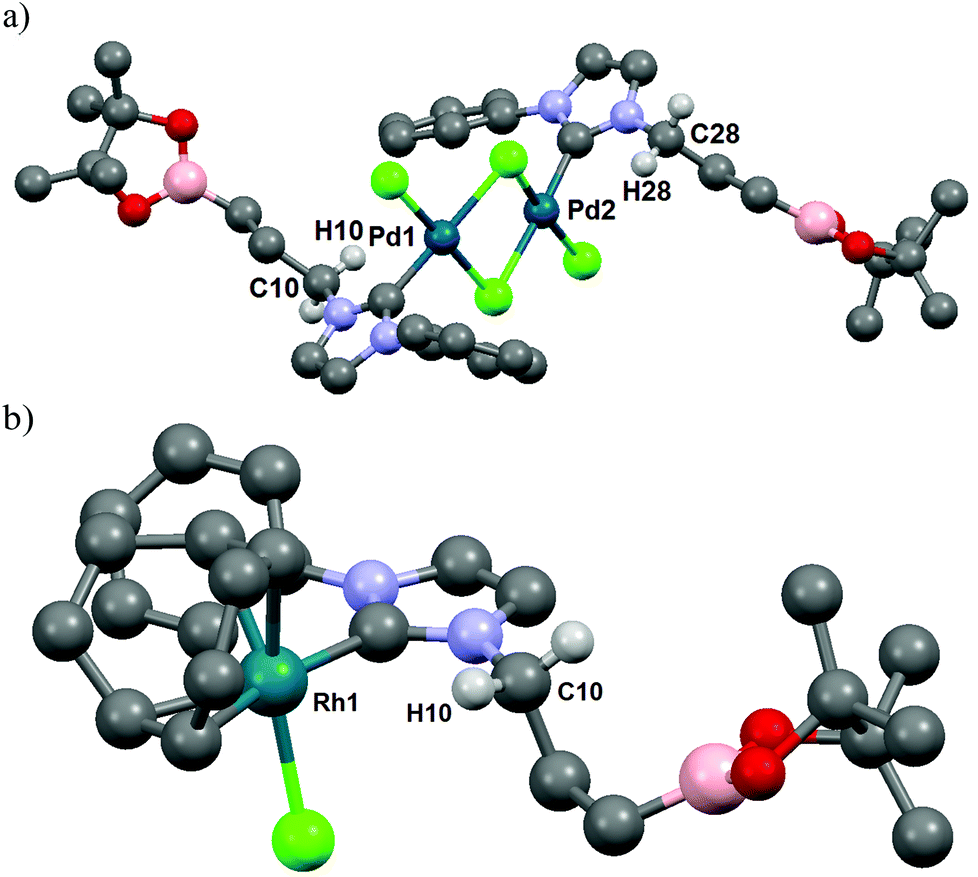 | ||
| Fig. 2 ORTEP diagrams of (a) [PdCl2NHC]2 complex 5a; (b) [RhClNHC] complex 6a are depicted with thermal ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. | ||
Interestingly, for complexes 5a and 6a the existence of hydrogen bonding or anagostic interactions (three-centers/four-electrons) between the metal and the hydrogen atom of the methylene group in α to the NHC was suspected by 1H NMR (400 MHz, CDCl3). Hydrogen atoms of this CH2 group were equivalent both in 3a (4.57 ppm, 2H, t, 3J = 7.3 Hz) and 4a (4.13 ppm, 2H, t, 3J = 7.3 Hz), but a downfield shift of these protons in an unresolved broad signal appeared for 5a at room temperature (4.40–4.95 ppm, 2H, m). Upon cooling to 243 K in CD2Cl2 solution, this signal splits into two distinct sets attributed respectively to the hydrogen atom participating in an interaction with the palladium atom (4.57–4.91 ppm, 1H, m) and to the one that is not (4.27–4.57 ppm, 1H, m) (see ESI†). In the case of rhodium complex 6a the 1H NMR signal of one of these two protons is strongly shifted downfield and these protons appeared as two distinct sets of broad signals at room temperature (4.32 and 4.89 ppm for non-coordinated and coordinated hydrogen atoms respectively) (see ESI†). The downfield chemical shift of one of these protons indicated a clearer rhodium–H interaction than those observed for complex 5a, typical of metal–hydrogen bonding or anagostic interactions. The existence of such interactions in 5a and 6a was confirmed in the solid state by an X-ray diffraction study (Fig. 2) and their structural parameters (Pd1–H10 2.8203(4) Å, Pd1–C10 3.338(6) Å, Pd1–H10–C10 114.31(3)°; Pd2–H28 2.8580(5) Å, Pd2–C28 3.346(8) Å, Pd2–H28–C28 112.08(4)°; (Rh1–H10) 2.9297(2) Å, Rh1–C10 = 3.449(3) Å and angle (Rh1–H10–C10) = 114.65(2)°) are in good agreement with those of anagostic interactions in transition metal d8 complexes (lit.14d(M–H) ≈ 2.3–2.9 Å, M–H–C ≈ 110–170°). The origin of these interactions is still under debate and may involve donation of filled dz2 or dxz/yz orbitals of the metal center into the C–H σ* orbital.15 In order to obtain a more detailed understanding of the involved orbitals, a Natural Bond Orbital (NBO)16 analysis was performed (see ESI† for details). Two distinct interactions were found, one involving the sp3 C–H orbitals and the antibonding rhodium s-orbital and a second interaction between the antibonding sp3 C–H with a rhodium lone pair with second order perturbation energies, E2P = 2.94 and 2.44 kJ mol−1, respectively. Moreover, the hydrogen atom close to the rhodium atom has a slightly higher natural charge of 0.222 than the other hydrogen atoms bonded by the same carbon atom (0.204). To conclude, the M–H bond distance and the M–H–C angle values and the very low E2P (<5 kJ mol−1) strongly suggest the establishment of a very weak hydrogen bond,17 probably led to the alkyl chain conformation. Such an interaction was not observed in the corresponding silver or gold complexes 4a and 7a, respectively.
The presence of privileged conformers having equivalent (4a, 7a) or non-equivalent (5a, 6a) protons bearing the methylene group α to the NHC is of interest. In the last case, this effect is strongly pronounced in complex 6a and to estimate the participation of the rhodium complex geometry and/or the boronic ester moiety in such preferential conformation the analogous rhodium complex 11 (Scheme 3) was synthesized. It was observed in 11 that these two protons were downfield shifted and not equivalent, but the effect was not as important as observed in 6a (see ESI†). Clearly, the boronic ester moiety has an effect on the conformation of the alkyl chain, leading to a rhodium complex 6a more stabilized by an additional C–H hydrogen interaction. This could generate a difference in the reactivity of 6a and 11 complexes (see further).
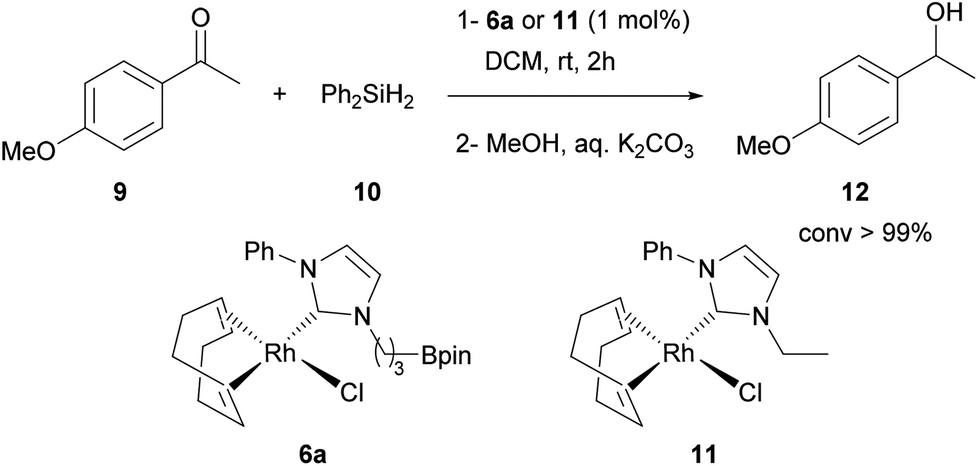 | ||
| Scheme 3 Catalytic hydrosilylation of ketone 9 with analogous rhodium-NHC complexes 6a and 11. | ||
Furthermore, to increase the Lewis basicity at the metallic center and the possibility of Z-interaction to the boron atom, a donor pyrimidine hemilabile ligand was incorporated at the N-position of the NHC instead of the phenyl group (Scheme 2). 1H NMR and 13C NMR spectra of palladium dichloride complex 5b showed clearly three non-equivalent pyrimidine hydrogens and carbon atoms with a downfield shift (δH > 1) of one aromatic hydrogen atom in the α position to the nitrogen atom. As expected, these observations clearly indicate bidentate coordination of the metal to the NHC and one of the nitrogen atoms of the pyrimidine moiety. In the solid state, complex 5b features a pseudo square planar geometry with a Pd1–N1 distance of 2.049(2) Å and N1–Pd1–C7 and N1–Pd1–Cl1 angles of 80.18(1)° and 93.47(7)°, respectively, which confirms the spectroscopic NMR data (Fig. 3). Unfortunately, attempts to synthesize the rhodium complex analogue from [Rh(η4-cod)Cl]2 gave an undetermined complex mixture.
 | ||
| Fig. 3 ORTEP diagrams of (a) [PdCl2NHC] complex 5b; (b) [AuClNHC] complex 7b are depicted with thermal ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. | ||
Alternatively, the treatment of 4b with [Ru(p-cymene)Cl2]2 gave quantitatively the corresponding 18-electron ruthenium(II) complex 8b. The cationic nature of the complex was correlated by high-resolution mass spectrometry (ESI, MeOH, m/z 585.1742 [M − Cl]+). Unfortunately, all attempts to crystallize complex 8b failed, but the NMR spectroscopic data, as seen previously in the complex 5b, provided evidence for the bidentate nature of the NHC-pyrimidine ligand as well as the pendant nature of the boron moiety (11B NMR, 128 MHz, in CDCl3, 34.0 ppm). As expected, reaction of 4b with an AuCl gold source gave the linear geometric complex 7b, where the pyrimidine aromatic ring was twisted to hold the gold metal center far from the nitrogen atom of the pyrimidine (Au1–N3, 3.118(1) Å, Au1–N3–C7–N2 dihedral angle, −34.91(1)°). As previously observed in this series of complexes, NMR data and X-ray characterization revealed again the pendant character of the boron moiety in the complex 7b (11B NMR, 128 MHz, in CDCl3, 33.8 ppm) (Fig. 3).
Despite some evidence in the literature of transition metal–Z interactions with the boronic ester moiety,10 or a related reaction intermediate found in the borylation reaction mechanism,13 such interactions were not observed in this series of flexible complexes, owing to a less buttress structure that does not allow potential recruitment of the boron atom moiety close to the metal center. Interestingly, these stable complexes exhibit a Lewis acidic pendant boronic ester moiety that could serve as an additional binding site in bifunctional catalysis. An initial 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equivalent mixture of complex 5a and Et3N was analyzed by 11B NMR (128 MHz, CDCl3) showing a complete upfield chemical shift of the boron atom (from 33.5 to 2.85 ppm) corresponding to a borate species that revealed an unambiguous covalent B–N bond. This information gave us confidence to use the boron atom in these complexes as an additional electrophilic activating site. In this context, stability and reactivity of complex 6a were evaluated in a preliminary hydrosilylation reaction and 6a was found to be a very efficient catalyst (conv. >99% by 1H NMR) (Scheme 3). However, the analogous complex 11 gave the same results in terms of kinetics (see ESI†), meaning that the boronic part of the ligand seems to behave only as a spectator in this reaction. Nevertheless, the use of the stable palladium, rhodium, ruthenium and gold complexes described here as potential catalysts for the synergistic activation of reaction partners will be explored soon, as well as extension to structurally analogous more electrophilic NHC-borane ligands.
:1 equivalent mixture of complex 5a and Et3N was analyzed by 11B NMR (128 MHz, CDCl3) showing a complete upfield chemical shift of the boron atom (from 33.5 to 2.85 ppm) corresponding to a borate species that revealed an unambiguous covalent B–N bond. This information gave us confidence to use the boron atom in these complexes as an additional electrophilic activating site. In this context, stability and reactivity of complex 6a were evaluated in a preliminary hydrosilylation reaction and 6a was found to be a very efficient catalyst (conv. >99% by 1H NMR) (Scheme 3). However, the analogous complex 11 gave the same results in terms of kinetics (see ESI†), meaning that the boronic part of the ligand seems to behave only as a spectator in this reaction. Nevertheless, the use of the stable palladium, rhodium, ruthenium and gold complexes described here as potential catalysts for the synergistic activation of reaction partners will be explored soon, as well as extension to structurally analogous more electrophilic NHC-borane ligands.
Conclusions
In summary, we have reported the synthesis and characterization of novel bifunctional NHC-boronic ester ligands and their corresponding silver(I), palladium(II), rhodium(I), ruthenium(II) and gold(I) complexes.18 It was observed that only the use of Ag2O as a base allowed us to obtain these complexes in very good yields, and without either a self-quenching or a polymerization process taking place during the generation of the in situ Lewis base (NHC) and Lewis acid (boronic ester). It was confirmed by 11B NMR and X-ray diffraction that the boron moiety is not coordinated to the metal center and remained pendant. Complexes bearing pendant trivalent borane derivatives are of interest19 and could be an alternative to enhance potential electrophilic activation of a substrate. Reported examples are essentially built on metallocene shape systems,20 but many of them must be stabilized in situ as borate derivatives,21 to avoid potential intramolecular rearrangement.22 The stable complexes developed here would let us confidently use them in bifunctional catalysis in the cooperative activation of small molecules.9 These hypotheses will be examined in further detail in due course.Acknowledgements
This work was supported by grant from the French Research Ministry to M.T., Aix-Marseille Université, CNRS UMR 7313. We thank M. Giorgi (http://www.spectropole.fr) for the X-ray diffraction studies.Notes and references
- H. Braunschweig, C. Kollann and D. Rais, Angew. Chem., Int. Ed., 2006, 45, 5254 CrossRef CAS PubMed; H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924 CrossRef PubMed.
- H. Braunschweig and R. D. Dewhurst, Dalton Trans., 2011, 40, 549 RSC; A. Amgoune and D. Bourissou, Chem. Commun., 2011, 47, 859 RSC; H. Kameo and H. Nakazawa, Chem. – Asian J., 2013, 8, 1720 CrossRef CAS PubMed; G. R. Owen, Chem. Soc. Rev., 2012, 41, 3535 RSC; M. Sircoglou, S. Bontemps, M. Mercy, N. Saffon, M. Takahashi, G. Bouhadir, L. Maron and D. Bourissou, Angew. Chem., Int. Ed., 2007, 46, 8583 CrossRef PubMed; A. F. Hill, Organometallics, 2006, 25, 4741 CrossRef; G. Parkin, Organometallics, 2006, 25, 4744 CrossRef; F.-G. Fontaine, J. Boudreau and M.-H. Thibeault, Eur. J. Inorg. Chem., 2008, 5439 CrossRef.
- Selected references: F. Inagaki, C. Matsumoto, Y. Okada, N. Maruyama and C. Mukai, Angew. Chem., Int. Ed., 2015, 54, 818 CrossRef CAS PubMed; W. H. Harman, T.-P. Lin and J. C. Peters, Angew. Chem., Int. Ed., 2014, 53, 1081 CrossRef PubMed; M.-E. Moret and J. C. Peters, Angew. Chem., Int. Ed., 2011, 50, 2063 CrossRef PubMed; H. Fong, M.-E. Moret, Y. Lee and J. C. Peters, Organometallics, 2013, 32, 3053 CrossRef PubMed; W. H. Harman and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 5080 CrossRef PubMed; S. N. MacMillan, W. H. Harman and J. C. Peters, Chem. Sci., 2014, 5, 590 RSC; M. Sircoglou, S. Bontemps, G. Bouhadir, N. Saffon, K. Miqueu, W. Gu, M. Mercy, C.-H. Chen, B. M. Foxman, L. Maron, O. V. Ozerov and D. Bourissou, J. Am. Chem. Soc., 2008, 130, 16729 CrossRef PubMed; M.-E. Moret, Y. Lee and J. C. Peters, J. Am. Chem. Soc., 2011, 133, 18118 CrossRef PubMed; D. L. M. Suess, C. Tsay and J. C. Peters, J. Am. Chem. Soc., 2012, 134, 14158 CrossRef PubMed; H. Kameo, Y. Hashimoto and H. Nakazawa, Organometallics, 2012, 31, 3155 CrossRef; M. Sircoglou, S. Bontemps, M. Mercy, K. Miqueu, S. Ladeira, N. Saffon, L. Maron, G. Bouhadir and D. Bourissou, Inorg. Chem., 2010, 49, 3983 CrossRef PubMed; B. E. Cowie and D. J. H. Emslie, Chem. – Eur. J., 2014, 20, 16899 CrossRef PubMed; W. A. Gunderson, D. L. M. Suess, H. Fong, X. Wang, C. M. Hoffmann, G. E. Cutsail III, J. C. Peters and B. M. Hoffman, J. Am. Chem. Soc., 2014, 136, 14998 CrossRef PubMed.
- A. F. Hill, G. R. Owen, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1999, 38, 2759 CrossRef CAS.
- G. Bouhadir, A. Amgoune, D. Bourissou and I. R. Crossley, in Advances in Organometallic Chemistry, ed. A. F. Hill and M. J. Fink, Elsevier: Academic Press, 2010, vol. 8, p. 1 CrossRef CAS PubMed; S. Bontemps, G. Bouhadir, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2006, 128, 12056 CrossRef CAS PubMed.
- A. Börner, J. Ward, K. Kortus and H. B. Kagan, Tetrahedron: Asymmetry, 1993, 4, 2219 CrossRef CAS; L. B. Fields and E. N. Jacobsen, Tetrahedron: Asymmetry, 1993, 4, 2229 CrossRef.
- S. Chikkali, S. Magens, D. Gudat, M. Nieger, I. Hartenbach and T. Schleid, Eur. J. Inorg. Chem., 2008, 2207 CrossRef CAS.
- J. Navarro, O. Torres, M. Martín and E. Sola, J. Am. Chem. Soc., 2011, 133, 9738 CrossRef CAS PubMed.
- O. Tutusaus, C. Ni and N. K. Szymczak, J. Am. Chem. Soc., 2013, 135, 3403 CrossRef CAS PubMed.
- D. Curtis, M. J. G. Lesley, N. C. Norman, A. G. Orpen and J. Starbuck, J. Chem. Soc., Dalton Trans., 1999, 1687 RSC; S. A. Westcott, T. B. Marder, R. T. Baker, R. L. Harlow, J. C. Calabrese, K. C. Lam and Z. Lin, Polyhedron, 2004, 23, 2665 CrossRef CAS; R. S. Anju, D. K. Roy, K. Geetharani, B. Mondal, B. Vargheseb and S. Ghosh, Dalton Trans., 2013, 42, 12828 RSC.
- G. A. Molander, C. S. Yun, M. Ribagorda and B. Biolatto, J. Org. Chem., 2003, 68, 5534 CrossRef CAS PubMed.
- Wang et al. have synthesized a NHC-borane platinum complex but the tethered and rigid structure of the free ligand does not allow self-quenching, see: Z. M. Hudson, C. Sun, M. G. Helander, Y.-L. Chang, Z.-H. Lu and S. Wang, J. Am. Chem. Soc., 2012, 134, 13930 CrossRef CAS PubMed ; By this way, a more flexible structure led to the corresponding Lewis pair ions, see: Y.-L. Rao, L. D. Chen, N. J. Mosey and S. Wang, J. Am. Chem. Soc., 2012, 134, 11026 CrossRef PubMed.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; Q. Li, C. W. Liskey and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 8755 CrossRef PubMed.
- D. Braga, F. Grepioni and E. Tedesco, Organometallics, 1997, 16, 1846 CrossRef CAS; M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef PubMed; A. Mukhopadhyay and S. Pal, Eur. J. Inorg. Chem., 2006, 4879 CrossRef; R. Angamuthu, L. L. Gelauff, M. A. Siegler, A. L. Spek and E. Bouwman, Chem. Commun., 2009, 2700 RSC; A. T. Çolak, O. Z. Yesilel and O. Büyükgüngör, J. Mol. Struct., 2011, 991, 68 CrossRef; N. Singh and A. J. Elias, Dalton Trans., 2011, 40, 4882 RSC; A. G. Jarvis, P. E. Sehnal, S. E. Bajwa, A. C. Whitwood, X. Zhang, M. S. Cheung, Z. Lin and I. J. S. Fairlamb, Chem. – Eur. J., 2013, 19, 6034 CrossRef PubMed; M. G. D. Holaday, G. Tarafdar, A. Kumar, M. L. P. Reddy and A. Srinivasan, Dalton Trans., 2014, 43, 7699 RSC; H. V. Huynh, L. R. Wong and P. S. Ng, Organometallics, 2008, 27, 2231 CrossRef.
- W. Yao, O. Eisenstein and R. H. Crabtree, Inorg. Chim. Acta, 1997, 254, 105 CrossRef CAS; Y. Zhang, J. C. Lewis, R. Bergman, J. A. Ellman and E. Oldfield, Organometallics, 2006, 25, 3515 CrossRef.
- E. D. Glendening, C. R. Landis and F. Weinhold, Natural bond orbital methods, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 1–42 CrossRef CAS; F. Weinhold and C. R. Landis, Natural bond orbitals and extensions of localized bonding concepts, Chem. Educ.: Res. Pract. Eur., 2001, 2, 91–104 RSC; F. Weinhold and C. R. Landis, Valency and Bonding – a natural bond donor-acceptor perspective, Cambridge University Press, 2005 Search PubMed.
- J. Saßmannshausen, Dalton Trans., 2012, 41, 1919 RSC.
- To our knowledge, only one metallic complex featuring bifunctional NHC-boronic ester was reported, see ref. 8.
- For bifunctional complexes with pendant borane features, see: W.-J. Xu, S.-J. Liu, X.-Y. Zhao, S. Sun, S. Cheng, T.-C. Ma, H.-B. Sun, Q. Zhao and W. Huang, Chem. – Eur. J., 2010, 16, 7125 CrossRef CAS PubMed; J. Vergnaud, M. Grellier, G. Bouhadir, L. Vendier, S. Sabo- Etienne and D. Bourissou, Organometallics, 2008, 27, 1140 CrossRef; T. G. Ostapowicz, C. Merkens, M. Hölscher, J. Klankermayer and W. Leitner, J. Am. Chem. Soc., 2013, 135, 2104 CrossRef PubMed; A. J. M. Miller, J. A. Labinger and J. E. Bercaw, J. Am. Chem. Soc., 2008, 130, 11874 CrossRef PubMed; A. J. M. Miller, J. A. Labinger and J. E. Bercaw, Organometallics, 2010, 29, 4499 CrossRef; H. Braunschweig, R. Dirk and B. Ganter, J. Organomet. Chem., 1997, 545–546, 257 CrossRef.
- For bifunctional complexes based on a metallocene–borane pendant shape, see: R. E. v. H. Spence and W. E. Piers, Organometallics, 1995, 14, 4617 CrossRef CAS; R. S. Rojas, B. C. Peoples, A. R. Cabrera, M. Valderrama, R. Fröhlich, G. Kehr, G. Erker, T. Wiegand and H. Eckert, Organometallics, 2011, 30, 6372 CrossRef; P. A. Deck, T. S. Fisher and J. S. Downey, Organometallics, 1997, 16, 1193 CrossRef; A. J. Ashe III, X. Fang and J. W. Kampf, Organometallics, 1999, 18, 2288 CrossRef; H. Braunschweig, R. Dirk, M. Müller, P. Nguyen, R. Resendes, D. P. Gates and I. Manners, Angew. Chem., Int. Ed., 1997, 36, 2338 CrossRef; A. Appel, F. Jäkle, T. Priermeier, R. Schmid and M. Wagner, Organometallics, 1996, 15, 1188 CrossRef; J. Chen, K. Venkatasubbaiah, T. Pakkirisamy, A. Doshi, A. Yusupov, Y. Patel, R. A. Lalancette and F. Jäkle, Chem. – Eur. J., 2010, 16, 8861 CrossRef PubMed; G. E. Herberich, A. Fisher and D. Wiebelhaus, Organometallics, 1996, 15, 3106 CrossRef; S. A. Larkin, J. T. Golden, P. J. Shapiro, G. P. A. Yap, D. M. J. Foo and A. L. Rheingold, Organometallics, 1996, 15, 2393 CrossRef.
- M. Hill, G. Erker, G. Kehr, R. Fröhlich and O. Kataeva, J. Am. Chem. Soc., 2004, 126, 11046 CrossRef CAS PubMed; C. Herrmann, G. Kehr, R. Fröhlich and G. Erker, Organometallics, 2008, 27, 2328 CrossRef; M. Emmert, G. Kehr, R. Fröhlich and G. Erker, Chem. – Eur. J., 2009, 15, 8124 CrossRef PubMed; S. J. Lancaster, S. Al-Benna, M. Thornton-Pett and M. Bochmann, Organometallics, 2000, 19, 1599 CrossRef.
- M. Hill, G. Kehr, G. Erker, O. Kataeva and R. Fröhlich, Chem. Commun., 2004, 1020 RSC; C. Herrmann, G. Kehr, R. Fröhlich and G. Erker, Eur. J. Inorg. Chem., 2008, 2273 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectroscopic data, detailed calculations and CIF for complexes 5a, 5b, 6a, 7a and 7b. CCDC 864270, 880506, 880508, 1023303 and 1023304. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00814j |
| This journal is © The Royal Society of Chemistry 2015 |