
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduction of hydroxy-functionalised carbaboranyl carboxylic acids and ketones by organolithium reagents†
W.
Neumann
,
M.
Hiller
,
M. B.
Sárosi
,
P.
Lönnecke
and
E.
Hey-Hawkins
*
Universität Leipzig, Institut für Anorganische Chemie, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
First published on 2nd March 2015
Abstract
While the reaction of carbaboranyl carboxylic acids and ketones with organolithium reagents generally leads to cleavage of the exo-polyhedral C–C bond, introduction of a hydroxyl group at the second carbon atom of the cluster enables the reduction of the carbonyl compounds to tertiary alcohols. The proposed mechanism involving the formation of dimeric contact ion pairs was supported by X-ray crystallography and theoretical calculations.
Introduction
Carbonyl-functionalised carbaboranes are versatile building blocks in carbaborane chemistry.1 However, one drawback of these compounds is the high lability of their exo-polyhedral C–C bond. Reaction of ortho-carbaboranyl ketones, carboxylic acids and esters with organolithium reagents mainly results in cleavage of the carbon–carbon bond rather than reduction of the carbonyl group, as the lithiated intermediates are probably destabilised due to the strong electron-withdrawing effect and steric hindrance of the cluster.2 Less cleavage was observed with Grignard reagents, as the less polar O–Mg bonds apparently better stabilise the intermediate alkoxides.2e In contrast, ortho-carbaboranyl aldehydes can be reduced to secondary alcohols by reaction with organolithium reagents with only little C–C bond cleavage.2e,f,3 We have recently reported that carbaboranyl carboxylic acids can also be reduced to the respective tertiary alcohols under such conditions with less exo-polyhedral C–C bond cleavage if a hydroxyl group is present at the second carbon atom of the cluster.2g As the reduction led to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of tertiary alcohol and cleavage product, a reaction mechanism involving the formation of dimeric contact ion pairs was proposed. These dimers may enable intermolecular (intra-aggregate) transfer of an oxygen atom, thereby allowing the direct conversion of the acid to an alcohol instead of the expected ketone. Herein, further investigations into this unique reaction behaviour are presented.
:1 mixture of tertiary alcohol and cleavage product, a reaction mechanism involving the formation of dimeric contact ion pairs was proposed. These dimers may enable intermolecular (intra-aggregate) transfer of an oxygen atom, thereby allowing the direct conversion of the acid to an alcohol instead of the expected ketone. Herein, further investigations into this unique reaction behaviour are presented.
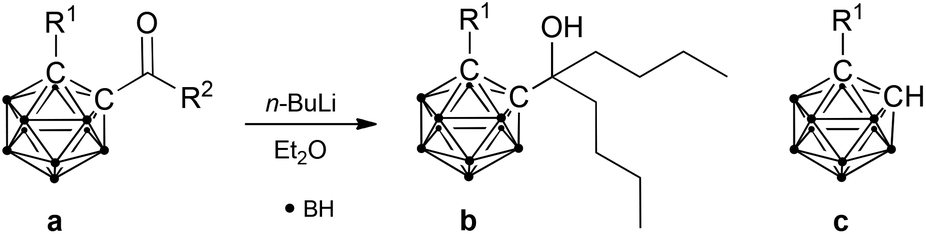
The reduction of carboxylic acids by organolithium reagents typically results in the formation of geminal diolates, which spontaneously rearrange to the corresponding ketones upon acidic work-up. Reduction of the ketones to tertiary alcohols can then be achieved in a second step. In contrast, reduction of 1-hydroxy-1,2-dicarba-closo-dodecaboranyl-2-carboxylic acid (salborin) directly yields tertiary alcohols (reaction II, Table 1).2g The formation of an intermediate ketone after intermolecular oxygen transfer has been proposed. To further investigate the formation of such an intermediate as well as the influence of the hydroxyl group on exo-polyhedral C–C bond cleavage, ketones of unsubstituted and hydroxy-functionalised ortho-carbaborane were synthesised and treated with n-BuLi.
Results and discussion
1-Acetyl-1,2-dicarba-closo-dodecaborane was synthesised by treating lithiated ortho-carbaborane with acetyl chloride and separated from the disubstituted derivative by column chromatography (Scheme 1).2d Accordingly, 1-acetoxy-2-acetyl-1,2-dicarba-closo-dodecaborane was prepared from lithiated 1-hydroxy-1,2-dicarba-closo-dodecaborane and acetyl chloride (Scheme 1). For subsequent deprotection of the hydroxyl group, a specific protocol had to be developed. Acetyl-protected alcohols are generally deprotected under alkaline conditions. However, carbaboranes tend to undergo deboronation of the cluster (decapping) under such conditions, and their lability is noticeably enhanced by carbonyl groups.4 The carboxylic acid congeners of the carbaboranyl ketone, salborin and asborin (1-acetoxy-1,2-dicarba-closo-dodecaboranyl-2-carboxylic acid), were shown to be already decapped under neutral conditions in aqueous solution.5 Furthermore, also the exo-polyhedral C–C bond of carbonyl-substituted carbaboranes is prone to cleavage under alkaline conditions.2b,d,6 Therefore, acidic conditions were chosen for hydrolysis of the ester. Although asborin was shown to readily be deacylated already under neutral conditions,5 the acidic environment was also expected to minimise the concurrent decapping of the cluster. However, even the use of acetone, which has been reported to suppress deboronation of carbonyl-functionalised carbaboranes,4 unexpectedly led to rapid decapping of the carbaboranyl ketone. Thus, chloroform was chosen as reaction medium and acetone was only added at the end of the reaction to accelerate deacylation (Scheme 1).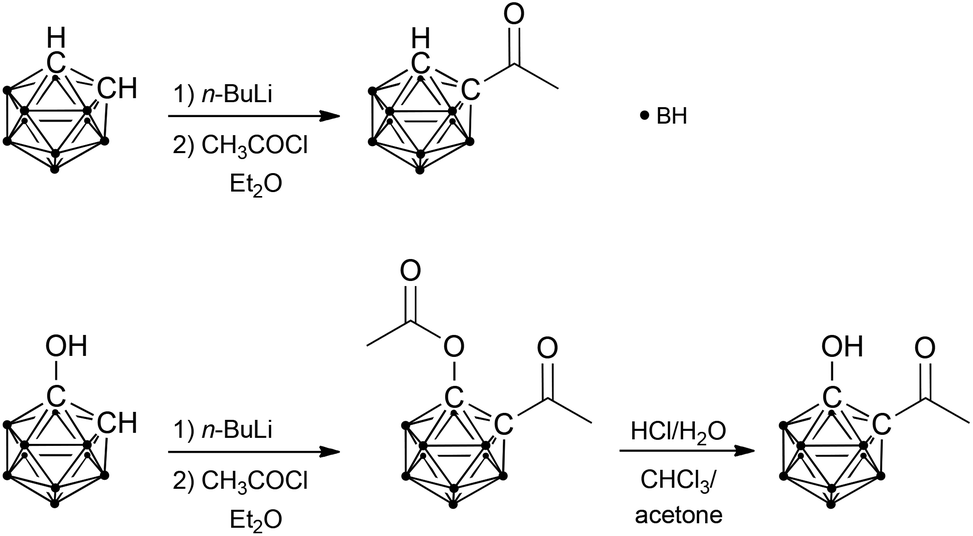 | ||
| Scheme 1 Syntheses of the studied carbaboranyl ketones. | ||
Both ketones were then treated with n-BuLi. As expected and reported for analogous derivatives,2b,e,f the reaction of 1-acetyl-1,2-dicarba-closo-dodecaborane resulted in cleavage of the exo-polyhedral C–C bond yielding unsubstituted ortho-carbaborane (reaction III, Table 1). In contrast, reaction of the hydroxy-functionalised carbaboranyl ketone gave the respective tertiary alcohol in a mixture with the cleavage product, 1-hydroxy-1,2-dicarba-closo-dodecaborane (reaction IV, Table 1). This confirms the previous findings that a hydroxyl group decreases C–C bond cleavage during reduction with organolithium reagents2g and also supports the proposed formation of a ketone-like intermediate during reduction of the corresponding carboxylic acid (salborin).
The previously proposed mechanism involved the formation of dimeric contact ion pairs.2g This dimer formation is now supported by a crystal structure of the lithium salt of salborin (Fig. 1). In the solid state, a centrosymmetric alkoxide-bridged Li2O2 rhomboid is formed in which each Li ion is coordinated by both deprotonated hydroxyl groups and an oxygen atom of a carboxylato group. The tetrahedral coordination sphere of each Li ion is completed by a coordinating water molecule and thus formed exclusively by O atoms. Although generally known as an excellent Lewis base for the complexation of Li ions, the co-crystallised tetramethylethylenediamine (tmeda) molecules are not coordinated at the metal ions but protonated, and the resulting two cations compensate the charge of the dianionic dimeric contact ion pair. Apart from dimer formation, the carbaborane clusters do not interact with other clusters. In contrast, interaction of hydroxyl groups of adjacent clusters of salborin leads to formation of one-dimensional chains of dimers;5 the latter are formed via hydrogen bonds of the carboxylato groups, similar to other carbaboranyl carboxylic acids.5,7 Upon deprotonation, the hydroxyl groups seem to become the pivot of dimer formation.
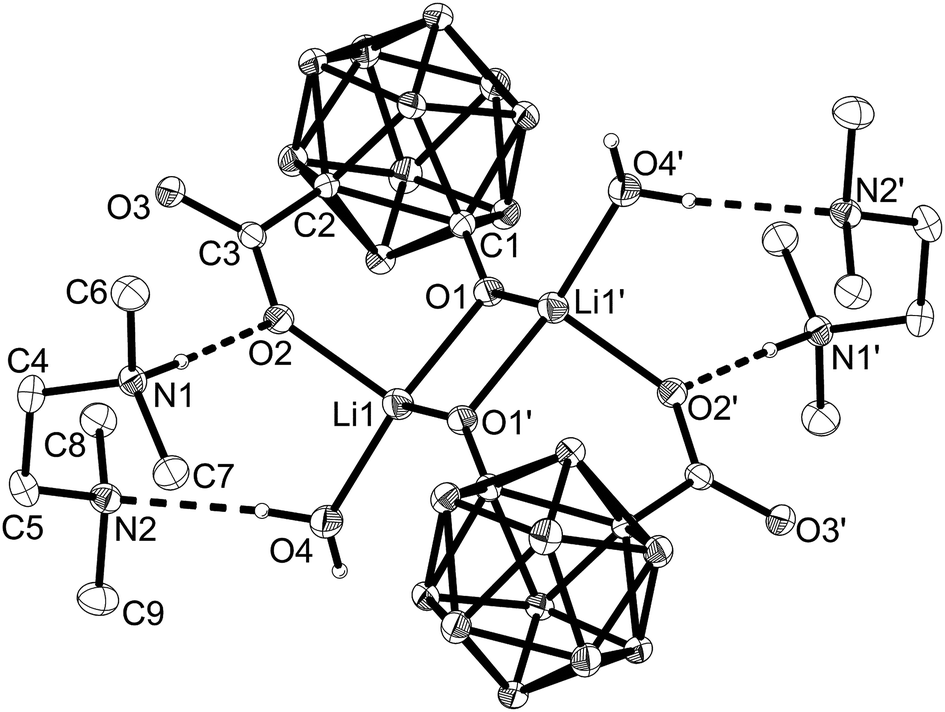 | ||
Fig. 1 ORTEP representation of the crystal structure of the lithium salt of 1-hydroxy-1,2-dicarba-closo-dodecaboranyl-2-carboxylic acid. Thermal ellipsoids are drawn at 50% probability; only selected hydrogen atoms are shown to indicate the direction of hydrogen bonds; all other hydrogen atoms are omitted for clarity. Selected bond lengths (Å): C1–O1 1.277(2), C1–C2 1.867(9), C1–C3 1.527(1), C3–O2 1.263(5), C3–O3 1.239(1). Crystallographic data: C18H58B20Li2N4O8, M = 688.76, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 7.2752(3), b = 11.5518(4), c = 12.3958(5) Å, V = 974.44(7) Å3, Z = 1, T = 130(2) K, 19988 reflections measured, 5958 unique (Rint = 0.0250). Final R indices (all data): R1 = 0.0452, wR2 = 0.0922. Chains along the a axis are formed via intermolecular OH⋯O donor–acceptor bonds. , a = 7.2752(3), b = 11.5518(4), c = 12.3958(5) Å, V = 974.44(7) Å3, Z = 1, T = 130(2) K, 19988 reflections measured, 5958 unique (Rint = 0.0250). Final R indices (all data): R1 = 0.0452, wR2 = 0.0922. Chains along the a axis are formed via intermolecular OH⋯O donor–acceptor bonds. | ||
Deprotonation also leads to a remarkable change in the geometry of the cluster caused by an increase in the C–O exo-dative π bonding of the hydroxyl group.8 Thus, the exo-polyhedral C–O bond (C(1)–O(1) 1.277(2) Å) is significantly shortened compared to salborin (1.374(2) Å;5 Table S1 in the ESI†), with an associated increase in the bond order (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ca. 1.23 Å; C–O ca. 1.43 Å).8a,e Delocalisation of the anionic charge of the oxygen atom into the carbaborane cage results in lengthening of the cluster C–C bond (C(1)–C(2)) concomitant with an opening of the closo geometry. While the C–C bond of salborin (1.660(1) Å) is only slightly longer than that of unsubstituted ortho-carbaborane (1.62 Å),9 the C(1)–C(2) bond in the lithium salt is significantly longer (1.867(9) Å). The B–B bonds remain mainly unaffected by deprotonation (Table S1 in the ESI†), whereas the similar C–O bond lengths of the carboxylato group (C(3)–O(2), C(3)–O(3)) indicate C–O double-bond character.
O ca. 1.23 Å; C–O ca. 1.43 Å).8a,e Delocalisation of the anionic charge of the oxygen atom into the carbaborane cage results in lengthening of the cluster C–C bond (C(1)–C(2)) concomitant with an opening of the closo geometry. While the C–C bond of salborin (1.660(1) Å) is only slightly longer than that of unsubstituted ortho-carbaborane (1.62 Å),9 the C(1)–C(2) bond in the lithium salt is significantly longer (1.867(9) Å). The B–B bonds remain mainly unaffected by deprotonation (Table S1 in the ESI†), whereas the similar C–O bond lengths of the carboxylato group (C(3)–O(2), C(3)–O(3)) indicate C–O double-bond character.
The arrangement of the clusters in the crystal structure of the lithium salt supports the importance of the hydroxyl groups for the unique reaction behaviour. In the reaction mixture, solvent molecules (Et2O) may complete the coordination sphere of the lithium ions in place of the water molecules, and as structures in solution are dynamic, the bridging lithium ions may enable an orientation of both clusters appropriate for the proposed oxygen transfer between the clusters during reduction of the carbaboranyl acid (Scheme 2). Theoretical calculations support the rearrangement of the dimer in solution by revealing similar Gibbs free energies (ΔG) for different dimeric structures (Fig. 2): a centrosymmetric dimer, as found in the crystal structure (A), a C2-symmetric dimer (B), a dimer with a bridging monodentate coordination (μ) of one carboxylato group (C), and a dimer with a bridging bidentate coordination (μ,κ1:κ1) of one carboxylato group (D). Further dimeric structures with chelating carboxylato groups and non-bridging hydroxyl groups would be theoretically possible. However, as only bridging hydroxyl groups were found in the crystal structure, dimers with chelating carboxylato groups were not considered in the theoretical calculations. The modelled dimer C is thermodynamically favoured over the other dimeric structures, and the orientation of the carboxylato groups in this dimer may facilitate the proposed intra-aggregate oxygen transfer.
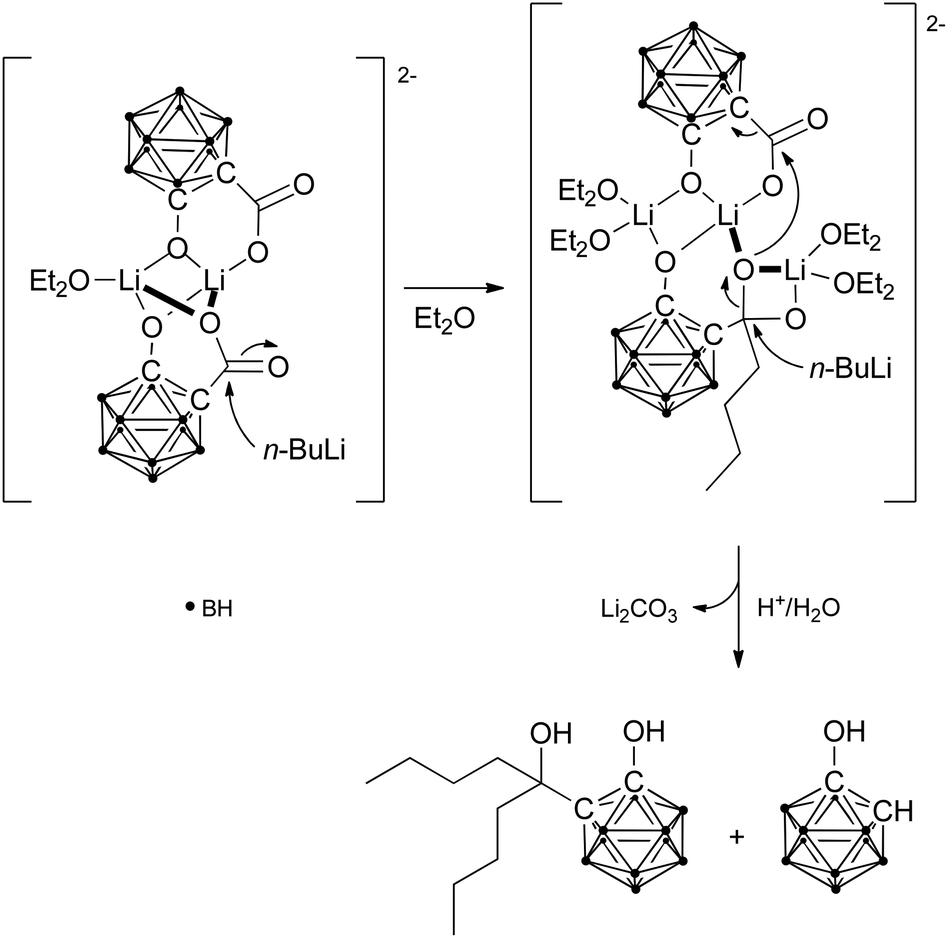 | ||
| Scheme 2 Proposed mechanism for the reduction of 1-hydroxy-1,2-dicarba-closo-dodecaboranyl-2-carboxylic acid by n-BuLi involving an intra-aggregate oxygen transfer. | ||
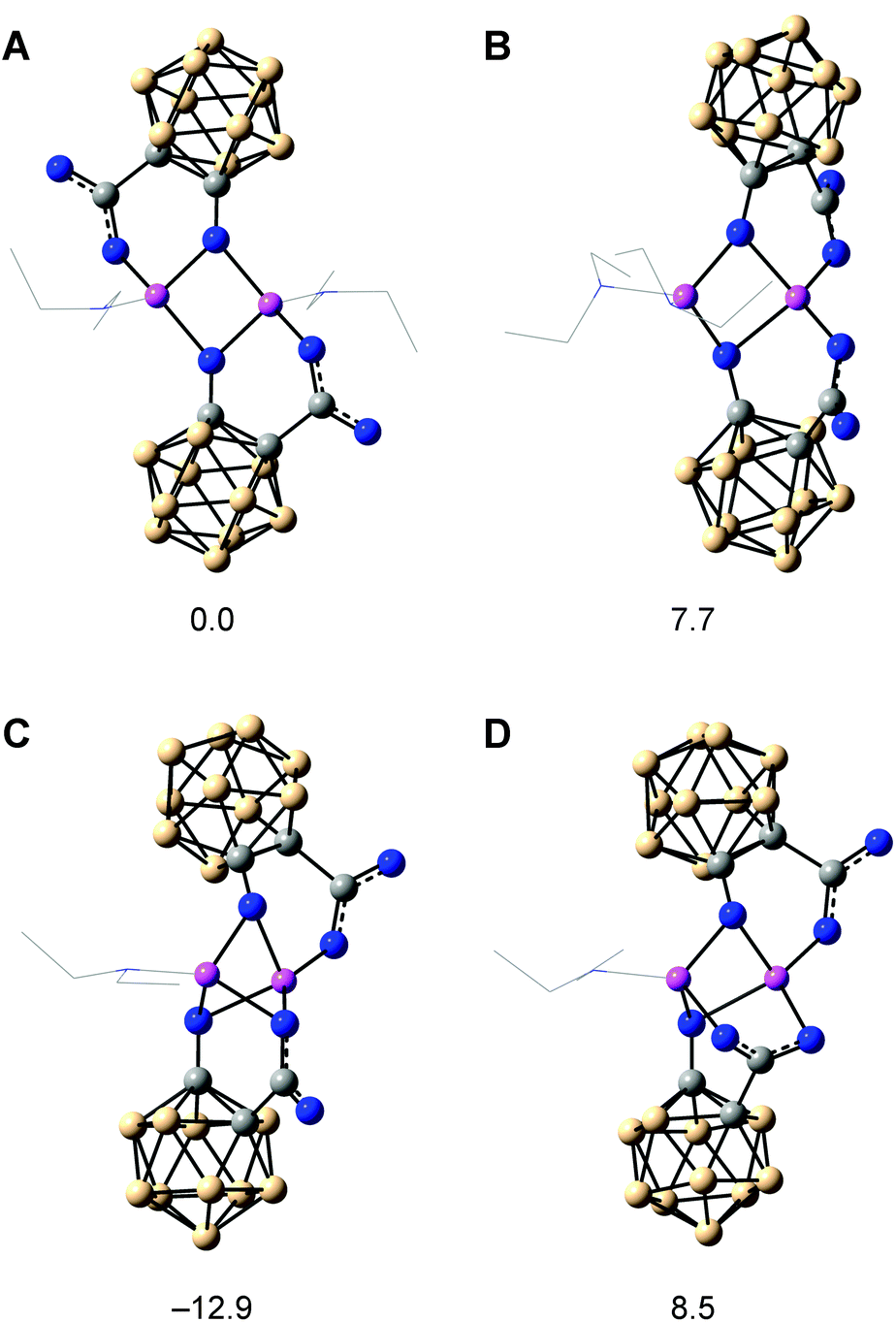 | ||
| Fig. 2 Optimised geometries of the dimers of lithiated 1-hydroxy-1,2-dicarba-closo-dodecaboranyl-2-carboxylic acid. Diethyl ether molecules are shown as a wireframe representation. All geometries are minima, and the relative Gibbs free energies (ΔG) are given below each dimer in kJ mol−1. The calculated free energy of an additional diethyl ether molecule has been included in the ΔG values of C and D. | ||
Nucleophilic attack of n-BuLi may first lead to a ketone-like intermediate (Scheme 2). The attack could either take place at the non-bridging or the bridging carboxylato group of C. An attack at the first could be sterically favoured as a result of less hindrance. However, nucleophilic attack at the bridging carboxylato group could be more favoured due to the higher electrophilicity of the carbon centre. Based on this attack a possible reaction intermediate was calculated (Fig. 3). Furthermore, the carboxylato bridge can be cleaved by the incoming lithium ion, and resulting free coordination sites could be filled by solvent molecules. As geminal diolates are generally unstable, oxygen transfer to the carboxylato group of the other cluster may then occur rapidly with simultaneous attack of the second n-BuLi molecule at this intermediate to generate the tertiary alkoxide and lithium carbonate. The bridging Li coordination of one of the carboxylate oxygen atoms may facilitate its intra-aggregate transfer resulting in the elimination of lithium carbonate.
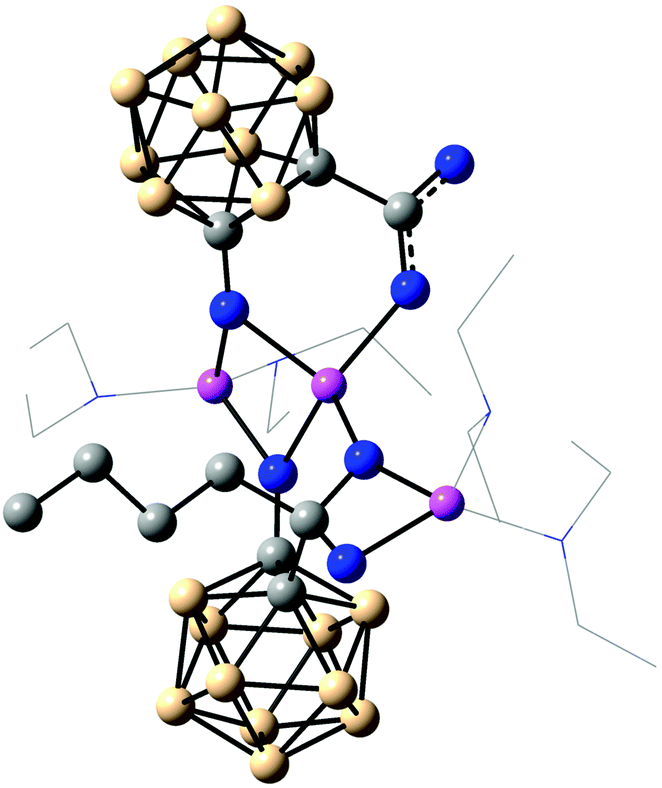 | ||
| Fig. 3 Optimised geometry of a plausible intermediate formed after nucleophilic attack of n-BuLi at lithiated 1-hydroxy-1,2-dicarba-closo-dodecaboranyl-2-carboxylic acid (dimer C). Diethyl ether molecules are shown as a wireframe representation. | ||
Besides facilitating oxygen transfer from the geminal diolate, Li coordination by the alkoxide and the deprotonated hydroxyl group may also stabilise the intermediates and thereby suppress cleavage of the exo-polyhedral C–C bond during reduction of the acid (reaction II) or the ketone (reaction IV) and explain the different ratios between tertiary alcohol and cleavage product (Table 1). Furthermore, the exo-dative π bonding of the deprotonated hydroxyl group increases the electron density within the cluster and may thus also contribute to stabilisation of the intermediate alkoxides.
The importance of the hydroxyl group for the formation of tertiary alcohols was further corroborated by replacing the hydroxyl with an amino group. Similar to oxygen, nitrogen bases strongly coordinate at lithium cations (HSAB principle)10 and also show exo-dative π bonding.8e Thus, an amino group at the carbaborane cluster may enable an analogous reaction sequence. However, as primary amines readily react with carbonyl groups and attack the carbaborane cluster resulting in deboronation,11 a protecting group had to be used. The tert-butyloxycarbonyl (Boc) group was chosen as a rather small group which is stable under alkaline conditions, also in the presence of n-BuLi, and can be directly introduced during the synthesis of the carbaboranyl amine from an intermediate isocyanate.12 Carboxylation of the Boc-protected carbaboranyl amine has already been reported.12 However, reaction with n-BuLi did not yield the tertiary alcohol but only led to cleavage of the exo-polyhedral C–C bond (reaction V, Table 1). This behaviour resembles the previously reported decarboxylation of 1-methoxy-1,2-dicarba-closo-dodecaboranyl-2-carboxylic acid (reaction VI, Table 1)2g and indicates that the Boc group probably impedes coordination of lithium ions in a bridging manner.
Conclusions
Similar to 1-hydroxy-1,2-dicarba-closo-dodecaboranyl-2-carboxylic acid, reaction of the corresponding ketone with n-BuLi led to formation of a tertiary alcohol. This supports the proposed mechanism of reduction of the carboxylic acid proceeding via a ketone-like intermediate, thereby directly yielding the tertiary alcohol upon work-up. Formation of the proposed dimeric contact ion pairs was supported by a crystal structure of the lithium salt of salborin. The observed centrosymmetric Li2O2 arrangement with bridging deprotonated hydroxyl groups may enable intra-aggregate oxygen transfer during reduction of the acid. The proposed mechanism was also supported by theoretical calculations. Furthermore, the observed intramolecular coordination of lithium by the deprotonated hydroxyl and carbonyl group of the cluster may stabilise the intermediate alkoxides and reduce exo-polyhedral C–C bond cleavage during reduction of the acid and ketone. In addition, the strong exo-dative π-bonding of the deprotonated hydroxyl group may further stabilise the intermediate alkoxides. The importance of the hydroxyl group for this unique reactivity was further confirmed by reactions of derivatives without a second functional group (R = H) or with a bulky functional group (R = OMe, NHBoc) hindering coordination at the lithium cation, thus resulting in quantitative cleavage of the exo-polyhedral C–C bond upon treatment with n-BuLi.Experimental
General
Reactions were carried out under a nitrogen atmosphere in anhydrous solvents, which were purified with an MBRAUN Solvent Purification System MB SPS-800 Series. Chemicals were used as purchased. Thin-layer chromatography (TLC) was performed on pre-coated glass plates (0.25 mm, silica gel 60 F254); visualisation of carbaborane compounds on TLC plates was achieved by treatment with a solution of PdCl2 (1% in MeOH) and gentle heating. Column chromatography was carried out with silica gel (0.035–0.070 mm, 60 Å). NMR spectra were recorded on Bruker AVANCE DRX 400 and AVANCE III HD 400 spectrometers. The chemical shifts of 1H, 11B and 13C NMR spectra are reported in parts per million at 400.13, 128.38, and 100.63 MHz, respectively, with tetramethylsilane as internal standard and referencing to the unified scale.13 Mass spectra were recorded on an ESQUIRE 3000 Plus Bruker-Daltonics ESI spectrometer.Synthetic procedures
:1) to yield a pale yellow oil (0.05 g, 8%): 1H NMR (CDCl3): δ = 1.40–3.10 (10H, br, C2B10H10), 2.43 (3H, s, CH3), 4.16 (1H, s, CH). 11B{1H} NMR (CDCl3): δ = −13.2 (2B, s), −12.8 (2B, s), −11.8 (2B, s), −8.7 (2B, s), −2.5 (2B, s). MS (ESI(−), acetone): m/z (%) 175.2 (100) [M − B]−; the observed isotopic pattern was in agreement with the calculated one.
:1) to yield a colourless oil (0.02 g, 13%): 1H NMR (CDCl3): δ = 1.30–3.60 (10H, br, C2B10H10), 2.10 (3H, s, CH3), 2.47 (3H, s, CH3). 11B NMR (CDCl3): δ = −13.5 (2B, d, 1JB,H = 184 Hz), −12.0 (2B, d, 1JB,H = 159 Hz), −10.8 (4B, d, 1JB,H = 168 Hz), −7.8 (1B, d, 1JB,H = 150 Hz), −3.2 (1B, d, 1JB,H = 154 Hz). 13C{1H} NMR (CDCl3): δ = 20.9 (CH3), 28.8 (CH3), 79.8 (Ccluster), 96.3 (Ccluster), 165.2 (CO), 189.7 (CO). MS (ESI(−), acetone): m/z (%) 234.1 (100) [M − B]−, 191.1 (30) [M − B − COCH3]−; the observed isotopic patterns were in agreement with the calculated ones.
1-Acetoxy-2-acetyl-1,2-dicarba-closo-dodecaborane(12) (0.25 mg) was dissolved in CHCl3 (35 mL). HCl (37% w/w, 3.5 mL) was added and the reaction mixture was stirred for 2.5 d at room temperature. Acetone (10 mL) was added and the reaction mixture was stirred for another 5 h. The phases were separated and the aqueous phase was extracted with n-hexane. The combined organic phases were dried over MgSO4, filtered and the solvent was removed under reduced pressure. Unconsumed starting material was separated from the product by column chromatography (n-hexane–ethyl acetate 10:1 → 0:1) to yield an orange oil (0.05 g, 24%): 1H NMR (CDCl3): δ = 1.30–3.00 (10H, br, C2B10H10), 2.51 (3H, s, COCH3), 2.65 (1H, br s, OH). 11B{1H} NMR (CDCl3): δ = −18.1 (1B, s), −14.5 (2B, s), −11.8 (5B, br), −9.5 (1B, s), −3.8 (1B, s). MS (ESI(−), CH3CN): m/z (%) 201.1 (100) [M − H]−; the observed isotopic pattern was in agreement with the calculated one.
1-[(tert-Butyloxycarbonyl)amino]-1,2-dicarba-closo-dodecabo-rane(12) (0.10 g, 0.4 mmol) was dissolved in Et2O (15 mL) and the solution was cooled to 0 °C. n-BuLi (0.64 mL, 1.5 M in n-hexane, 2.5 equiv.) was added and the solution was stirred for 90 min at 0 °C. The solvent was removed, and the residue was washed with n-hexane (3× 15 mL) and redissolved in Et2O (40 mL). CO2 was passed through the solution for 10 min at 0 °C. N2 was passed through the solution for 10 min at 0 °C to remove any dissolved CO2; then n-BuLi (0.64 mL, 1.5 M in n-hexane, 2.5 equiv.) was added. The solution was stirred for 1 h at room temperature. The solvent was removed under reduced pressure; aqueous HCl (30 mL, 1 M) was added and the solution was stirred for 1 h at room temperature. The solution was extracted with n-hexane and the combined organic phases were washed with water, dried over MgSO4, filtered and all volatile material was removed under reduced pressure. The product crystallised as colourless, needle-like crystals, which were analysed by 1H, 11B NMR spectroscopy and MS and were identified as 1-[(tert-butyloxycarbonyl)amino]-1,2-dicarba-closo-dodecaborane(12).
Crystallography
Theoretical calculations
All calculations have been carried out with the Gaussian 09 programme package19 at the M06-2X/6-31+G(d,p) level of theory20 using a pruned (99590) grid. Explicit solvent molecules (diethyl ether) have been included wherever appropriate and the SMD solvent model (diethyl ether)21 has been used for all calculations.
Acknowledgements
Financial support from the Fonds der Chemischen Industrie (FCI, doctoral grant to W.N.) and the Graduate School “Leipzig School of Natural Sciences – Building with Molecules and Nano-objects (BuildMoNa)” is gratefully acknowledged.Notes and references
- R. N. Grimes, Carboranes, Elsevier, Amsterdam, 2nd edn, 2011 Search PubMed.
- (a) L. I. Zakharkin, Dokl. Akad. Nauk SSSR, 1965, 162, 817 CAS; (b) V. I. Stanko, A. I. Klimova, Y. A. Chapovskii and T. P. Klimova, Zh. Obshch. Khim., 1966, 36, 1779 CAS; (c) L. I. Zakharkin and A. I. L'vov, J. Organomet. Chem., 1966, 5, 313 CrossRef CAS; (d) L. I. Zakharkin and A. I. L'vov, Zh. Obshch. Khim., 1967, 37, 1217 CAS; (e) A. I. L'vov and L. I. Zakharkin, Izv. Akad. Nauk. SSSR, Ser. Khim., 1967, 2653 Search PubMed; (f) R. N. Grimes, Carboranes, Elsevier, Amsterdam, 2nd edn, 2011, ch. 9.9, pp. 427–430 and ch. 9.10, pp. 436–437 Search PubMed; (g) W. Neumann, M. Hiller, P. Lönnecke and E. Hey-Hawkins, Dalton Trans., 2014, 43, 4935 RSC.
- (a) L. I. Zakharkin and A. I. L'vov, Zh. Obshch. Khim., 1967, 37, 742 CAS; (b) V. I. Stanko, V. A. Brattsev, N. E. Al'perovich and N. S. Titova, Zh. Obshch. Khim., 1968, 38, 1056 CAS; (c) J. L. Maurer, F. Berchier, A. J. Serino, C. B. Knobler and M. F. Hawthorne, J. Org. Chem., 1990, 55, 838 CrossRef CAS.
- J. J. Schaeck and S. B. Kahl, Inorg. Chem., 1999, 38, 204 CrossRef CAS.
- M. Scholz, G. N. Kaluđerović, H. Kommera, R. Paschke, J. Will, W. S. Sheldrick and E. Hey-Hawkins, Eur. J. Med. Chem., 2011, 46, 1131 CrossRef CAS PubMed.
- (a) V. I. Stanko, T. V. Klimova and I. P. Beletskaya, Dokl. Akad. Nauk SSSR, Ser. Khim., 1974, 216, 329 CAS; (b) V. I. Stanko and T. V. Klimova, Zh. Obshch. Khim., 1977, 47, 2017 CAS; (c) D. A. Brown, H. M. Colquhoun, J. A. Daniels, J. A. H. MacBride, I. R. Stephenson and K. Wade, J. Mater. Chem., 1992, 2, 793 RSC; (d) H. Nakamura, K. Aoyagi and Y. Yamamoto, J. Org. Chem., 1997, 62, 780 CrossRef CAS; (e) H. Nakamura, K. Aoyagi and Y. Yamamoto, J. Organomet. Chem., 1999, 574, 107 CrossRef CAS.
- (a) A. J. Welch, U. Venkatasubramanian, G. M. Rosair, D. Ellis and D. J. Donohoe, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 1295 CAS; (b) U. Venkatasubramanian, D. Ellis, G. M. Rosair and A. J. Welch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, o559 Search PubMed; (c) U. Venkatasubramanian, D. Ellis, G. M. Rosair and A. J. Welch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, o586 Search PubMed; (d) U. Venkatasubramanian, D. J. Donohoe, D. Ellis, B. T. Giles, S. A. Macgregor, S. Robertson, G. M. Rosair, A. J. Welch, A. S. Batsanov, L. A. Boyd, R. C. B. Copley, M. A. Fox, J. A. K. Howard and K. Wade, Polyhedron, 2004, 23, 629 CrossRef CAS PubMed.
- (a) D. A. Brown, W. Clegg, H. M. Colquhoun, J. A. Daniels, I. R. Stephenson and K. Wade, J. Chem. Soc., Chem. Commun., 1987, 889 RSC; (b) R. Coult, M. A. Fox, W. R. Gill and K. Wade, Polyhedron, 1992, 11, 2717 CrossRef CAS; (c) I. Zharov, A. Saxena, J. Michl and R. D. Miller, Inorg. Chem., 1997, 36, 6033 CrossRef CAS PubMed; (d) J. Llop, C. Viñas, J. M. Oliva, F. Teixidor, M. A. Flores, R. Kivekas and R. Sillanpää, J. Organomet. Chem., 2002, 657, 232 CrossRef CAS; (e) L. A. Boyd, W. Clegg, R. C. B. Copley, M. G. Davidson, M. A. Fox, T. G. Hibbert, J. A. K. Howard, A. Mackinnon, R. J. Peace and K. Wade, Dalton Trans., 2004, 2786 RSC.
- (a) M. G. Davidson, T. G. Hibbert, J. A. K. Howard, A. Mackinnon and K. Wade, Chem. Commun., 1996, 2285 RSC; (b) R. J. Blanch, M. Williams, G. D. Fallon, M. G. Gardiner, R. Kaddour and C. L. Raston, Angew. Chem., Int. Ed., 1997, 36, 504 CrossRef CAS; (c) M. J. Hardie and C. L. Raston, CrystEngComm, 2001, 3, 162 RSC; (d) M. A. Fox and A. K. Hughes, Coord. Chem. Rev., 2004, 248, 457 CrossRef CAS PubMed; (e) A. R. Turner, H. E. Robertson, K. B. Borisenko, D. W. H. Rankin and M. A. Fox, Dalton Trans., 2005, 1310 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533 CrossRef CAS.
- L. I. Zakharkin and V. N. Kalinin, Tetrahedron Lett., 1965, 6, 407 CrossRef.
- R. A. Kasar, G. M. Knudsen and S. B. Kahl, Inorg. Chem., 1999, 38, 2936 CrossRef CAS PubMed.
- R. K. Harris, E. D. Becker, S. M. Cabral de Menezes, R. Goodfellow and P. Granger, Pure Appl. Chem., 2001, 73, 1795 CrossRef CAS.
- E. Hey-Hawkins and M. Scholz, Ger. Appl., DE 102007026701 A1, 2008 Search PubMed; E. Hey-Hawkins and M. Scholz, PCT Int. Appl., WO 2008145733 A2, 2008 Search PubMed.
- K. Ohta, T. Goto, H. Yamazaki, F. Pichierri and Y. Endo, Inorg. Chem., 2007, 46, 3966 CrossRef CAS PubMed.
- CrysAlis Pro, Agilent Technologies, Version 1.171.36.28 Search PubMed.
- SIR92: A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- SHELXL-2013: G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS; (b) Y. Zhao and D. Truhlar, Theor. Chem. Acc., 2008, 119, 525 CrossRef CAS PubMed; (c) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157 CrossRef CAS PubMed; (d) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed; (e) R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Further crystal data. CCDC 1039317. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00761e |
| This journal is © The Royal Society of Chemistry 2015 |