
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Syntheses and reductions of C-dimesitylboryl-1,2-dicarba-closo-dodecaboranes†‡
Jan
Kahlert
a,
Lena
Böhling
a,
Andreas
Brockhinke
a,
Hans-Georg
Stammler
a,
Beate
Neumann
a,
Louis M.
Rendina
b,
Paul J.
Low
c,
Lothar
Weber
*a and
Mark A.
Fox
 *d
*d
aFakultät für Chemie der Universität Bielefeld, 33615 Bielefeld, Germany. E-mail: lothar.weber@uni-bielefeld.de
bSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia
cSchool of Chemistry and Biochemistry, University of Western Australia, 35 Stirling Highway, Crawley, Perth 6009, Australia
dDepartment of Chemistry, Durham University, Durham DH1 3LE, UK. E-mail: m.a.fox@durham.ac.uk
First published on 24th April 2015
Abstract
Two C-dimesitylboryl-1,2-dicarba-closo-dodecaboranes, 1-(BMes2)-2-R-1,2-C2B10H10 (1, R = H, 2, R = Ph), were synthesised by lithiation of 1,2-dicarba-closo-dodecaborane and 1-phenyl-1,2-dicarba-closo-dodecaborane, respectively, with n-butyllithium and subsequent reaction with fluorodimesitylborane. These novel compounds were structurally characterised by X-ray crystallography. Compounds 1 and 2 are hydrolysed on prolonged exposure to air to give mesitylene and boronic acids 1-(B(OH)2)-2-R-1,2-C2B10H10 (3, R = H, 4, R = Ph respectively). Addition of fluoride anions to 1 and 2 resulted in boryl-carborane bond cleavage to give dimesitylborinic acid HOBMes2. UV absorption bands at 318–333 nm were observed for 1 and 2 corresponding to local π–π*-transitions within the dimesitylboryl groups while visible emissions at 541–664 nm with Stokes shifts of 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 920–16170 cm−1 were attributed to intramolecular charge transfer transitions between the mesityl and cluster groups. Compound 2 was shown by cyclic voltammetry to form a stable dianion on reduction. NMR spectra for the dianion [2]2− were recorded from solutions generated by reductions of 2 with alkali metals and compared with NMR spectra from reductions of 1,2-diphenyl-ortho-carborane 5. On the basis of observed and computed 11B NMR shifts, these nido-dianions contain bowl-shaped cluster geometries. The carborane is viewed as the electron-acceptor and the mesityl group is the electron-donor in C-dimesitylboryl-1,2-dicarba-closo-dodecaboranes.
920–16170 cm−1 were attributed to intramolecular charge transfer transitions between the mesityl and cluster groups. Compound 2 was shown by cyclic voltammetry to form a stable dianion on reduction. NMR spectra for the dianion [2]2− were recorded from solutions generated by reductions of 2 with alkali metals and compared with NMR spectra from reductions of 1,2-diphenyl-ortho-carborane 5. On the basis of observed and computed 11B NMR shifts, these nido-dianions contain bowl-shaped cluster geometries. The carborane is viewed as the electron-acceptor and the mesityl group is the electron-donor in C-dimesitylboryl-1,2-dicarba-closo-dodecaboranes.
Introduction
Tri-coordinate boron compounds have been intensely investigated in the past two decades in view of potential applications as functional materials.1 The most widely employed functional moiety containing a tri-coordinate boron atom is the dimesitylboryl group (BMes2; Mes = 2,4,6-Me3C6H2) in which the unsaturated boron centre is kinetically stabilised by steric shielding of the mesityl groups. The empty pz-orbital at the boron atom can interact with the π-system of attached organic skeletons which leads to a narrowing of the HOMO–LUMO-gap (HLG) by lowering the LUMO energy. Indeed, the π-acceptor strength of the BMes2 group is similar to those of cyano-2 and nitro-3 groups. These electronic characteristics have led to organic materials containing BMes2 units finding application as electron-transporting materials in opto-electronic devices.4,5 Compounds containing BMes2 can be strongly fluorescent and thus have been used in organic light emitting diodes (OLEDs).5,6 Moreover, the ability of the boron atom to form selectively covalent adducts with small anions has led to applications of these compounds as colorimetric and luminescent sensors for fluoride7–9 and cyanide.10,11Other boron-containing compounds that have gained considerable interest in the last seven years in the field of opto-electronic materials are derivatives of the dicarba-closo-dodecaborane isomers (1,2-, 1,7- and 1,12-C2B10H12 which are ortho-, meta- and para-carborane, respectively).12,13 Due to their delocalised σ-electron systems (‘3D aromaticity’), these clusters possess high thermal and chemical stabilities.14 The ortho-carborane is a unique electron-acceptor when connected to a donor at one or both cluster carbon atoms (at C1 and/or C2 in 1,2-C2B10H12) due to the elasticity of the cluster C1–C2 bond.15–17 The ortho-carborane unit thus can play an active role as the acceptor in donor–acceptor molecules (dyads). Photoexcitation of such dyads induces a charge transfer from an organic scaffold to the carborane cluster which either led to luminescence quenching18 or to charge transfer (CT) emissions or both depending on the solvents used19–21 and whether the materials were investigated as solids.22–28 Compounds with both BMes2 and ortho-carboranyl groups are known with para-phenylene bridges linking both units.29,30 In these systems, the ortho-carboranyl group served as a strongly inductive electron-withdrawing group as the cluster increased the Lewis acidity of the triarylborane as found by fluoride ion titrations compared to the triarylborane without the cluster attached.29 Another compound containing both BMes2 and ortho-carboranyl group was reported with an ethylene bridge linking both units.31
Of the few ortho-carboranes with tri-coordinate boron substituents at their carbon atoms reported,24–26,32–36 only C-benzodiazaborolyl-ortho-carboranes have been investigated regarding their photophysical, electrochemical and spectroelectrochemical properties.25 However, the benzodiazaborolyl group generally acts as a π-donor and is therefore electronically quite distinct from the BMes2 moiety. In order to better understand the photophysical and electrochemical properties of ortho-carboranes with boryl groups, studies with compounds containing a tri-coordinate boron π-acceptor would be appealing and allow one to determine more precisely the electronic interplay between the carborane and tri-coordinate boron electron-withdrawing units. Therefore we present herein, the syntheses and crystal structures of two ortho-carborane derivatives 1 and 2 with a BMes2 group at one of the cage carbon atoms and their photophysical and electrochemical properties (Fig. 1). The geometry of the reduced species of 2 was also determined by a combination of 11B NMR spectroscopy and GIAO-NMR DFT computations.
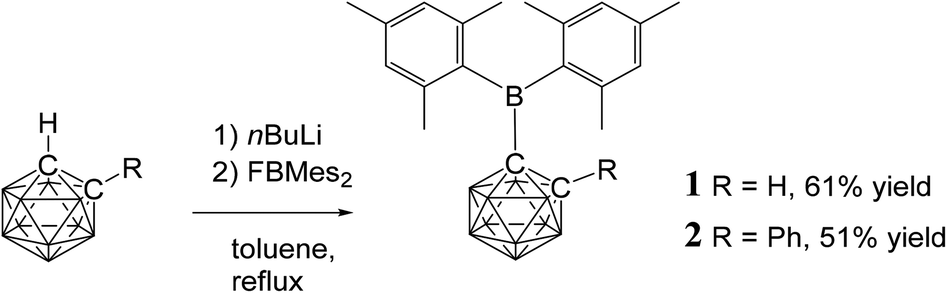 | ||
| Fig. 1 Syntheses of the novel C-dimesitylboryl-ortho-carboranes 1 and 2. | ||
Results and discussion
Syntheses and characterisation of 1 and 2
Compounds 1 and 2 were synthesised by reaction of fluorodimesitylborane with the corresponding C-lithiocarborane, generated in situ by metallation of ortho-carborane and 1-phenyl-ortho-carborane, in boiling toluene (Fig. 1). The elevated temperatures proved to be essential as no conversion was observed at ambient temperature. Purification was achieved by aqueous work-up and the target compounds were isolated in moderate yields by crystallisation from n-hexane/dichloromethane mixtures. The elemental analytical result for 1 was significantly lower (0.8% for carbon) than the calculated value. Similar discrepancies have been noted elsewhere for related compounds.33 It is possible that the formation of boron carbide during the combustion analysis may adversely affect the values obtained.Signals in the 11B{1H} NMR spectra of 1 and 2 between 3.7 and −12.9 ppm confirm the presence of the ortho-carborane clusters. The 11B peaks at 78.9 ppm (1) and 80.4 ppm (2) are assigned to the BMes2 groups and are very broad compared to the peaks corresponding to the cluster. The 11B chemical shifts of the Mes2B groups in 1 and 2 are virtually identical to trimesitylborane (79.2 ppm) and phenyldimesitylborane (79.3 ppm).37 Thus, the ortho-carboranyl groups influence the chemical shift of the tri-coordinate boron atom of the BMes2 unit in the same manner as a phenyl or mesityl substituent.
Hydrolyses of 1 and 2
Since aqueous work-ups were used in the preparation of 1 and 2 these C-boryl-ortho-carboranes are considered to be water-stable – unlike many reported C-boryl-ortho-carboranes that could not be isolated pure due to facile hydrolysis.32 The 1H and 11B NMR spectra for 1 and 2 in deuterated chloroform solutions containing excess water also showed no changes. The sterics of the mesityl and the carboranyl groups appear to prevent facile hydrolysis of the boron atom in 1 and 2.Solids of 1 and 2 do, however, hydrolyse on prolonged exposure to air (complete conversion after three weeks for 1 and eighteen months for 2) to give mesitylene and the new carboranylboronic acids, 3 and 4 (Fig. 2). While these acids could not be obtained pure, they were identified by multinuclear NMR spectroscopy and mass spectrometry. The observed cleavage of the B–C(mesityl) bond in the process is not without precedent. It has been shown elsewhere that mesitylene is formed from the reaction of dimesitylborinic acid, Mes2BOH, with trimethylaluminium.38 The initial steps in these air-induced hydrolyses of 1 and 2 probably involve cleavage of the B–C(mesityl) bonds by oxygen (as in the B–C(phenyl) bond cleavage reaction of triphenylboron by oxygen39) followed by hydrolysis with traces of water present in air.
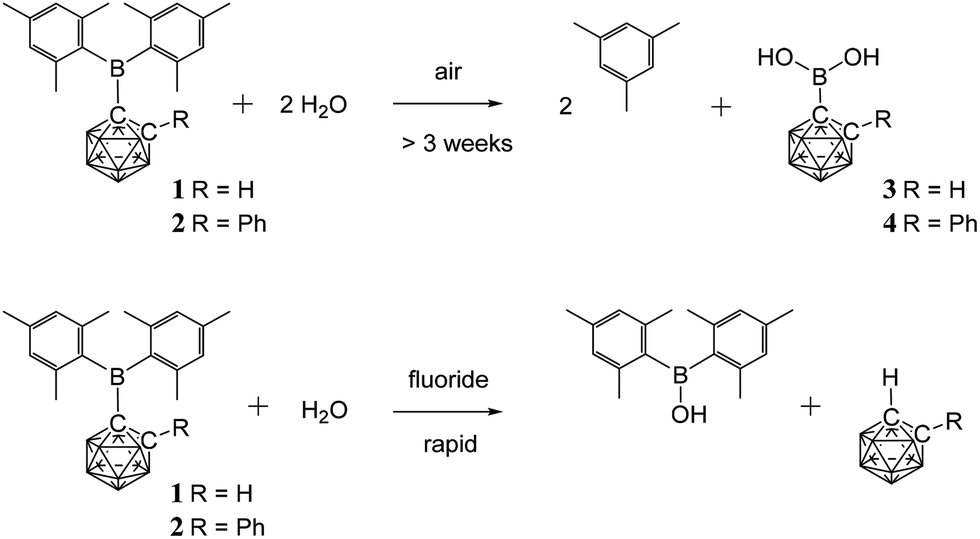 | ||
| Fig. 2 Hydrolysed products from reactions of 1 and 2 with air and fluoride ions. | ||
As many organic dimesitylboranes have been explored as fluoride sensors,7,29 the reactivity of 1 and 2 towards fluoride ions was of interest. Chloroform solutions of 1 and 2 were treated with an excess of tetra-n-butylammonium fluoride hydrate (TBAFH) while acetonitrile solutions of 1 and 2 were added with potassium fluoride (KF) and 18-crown-6 to obtain the desired fluoride adducts [1·F]− and [2·F]−, respectively, where the fluoride ion is bound to the boryl boron atom. Hydrolysis took place instead in all cases to give Mes2BOH, and the corresponding unsubstituted carborane, 1,2-C2B10H12 or 1-Ph-1,2-C2B10H11, as detected by 1H, 11B, 13C and 19F NMR spectroscopies on the reaction mixtures (Fig. 2). These reactions were complicated by fluoride-ion deboronation processes on the unsubstituted carboranes to give 11B and 19F NMR peaks corresponding to fluoroborates of the boron atom initially removed from the cluster.40
It is possible that fluorodimesitylborane, Mes2BF, is initially formed in the reaction as Mes2BF is known to be easily hydrolysed to Mes2BOH.39,41 However, careful 19F NMR monitoring of the reaction mixtures from 1 and 2 with TBAFH in the first few minutes did not reveal any evidence of an intermediate such as the fluoride adducts [1·F]−, [2·F]− or Mes2BF. The reactions of potassium hydroxide (KOH) and 18-crown-6 with 1 and 2 in acetonitrile gave Mes2BOH and the corresponding unsubstituted carborane. Deboronation products were also present in the latter reactions as the combination of KOH and 18-crown-6 is a strong deboronating agent.42
X-ray crystallography
Single crystals of 1 and 2 were grown from n-hexane/dichloromethane mixtures and their molecular structures were determined by X-ray diffraction (Fig. 3 and 4, Table 1). The BMes2 groups adopt orientations with C2–C1–B1–C3 torsion angles at 36.0(2)° and 22.5(2)° and C2–C1–B1–C12 at −144.5(2)° and −157.8(2)° in 1 and 2 respectively (Table 1). Thus the empty pz-orbitals are approximately in plane with the C1–B3 bonds in the clusters and these C1–B3 bonds are shorter than the C1–B6 bonds by 0.02–0.03 Å in both compounds. However, all B–B and B–C bonds lengths in the clusters are within the usual range.24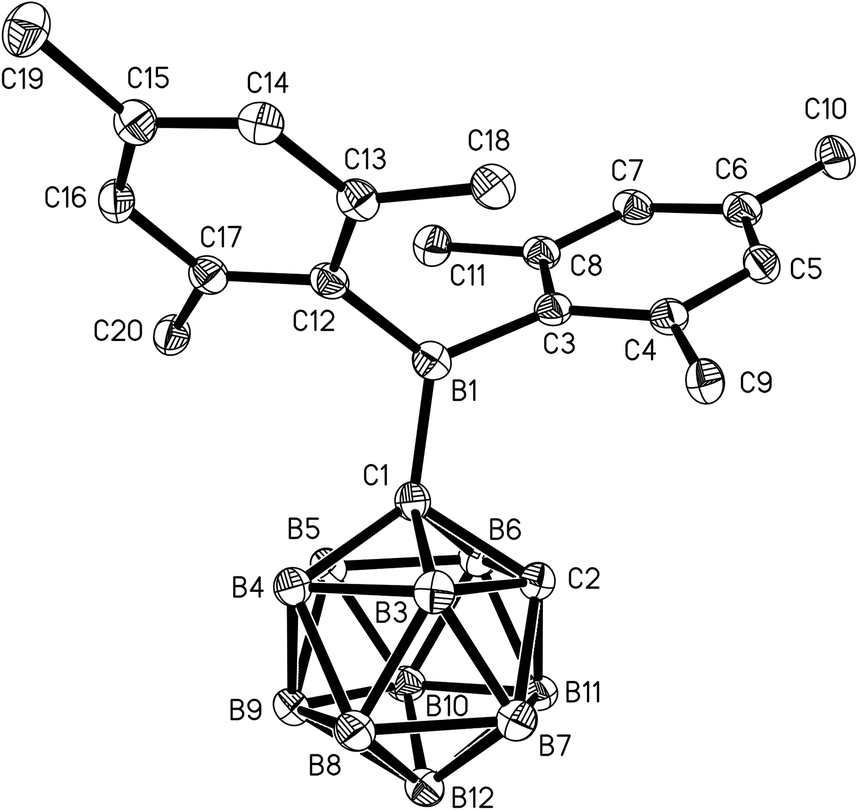 | ||
| Fig. 3 Molecular structure of 1 with hydrogen atoms omitted for clarity. Thermal ellipsoids are drawn at 50% probability. | ||
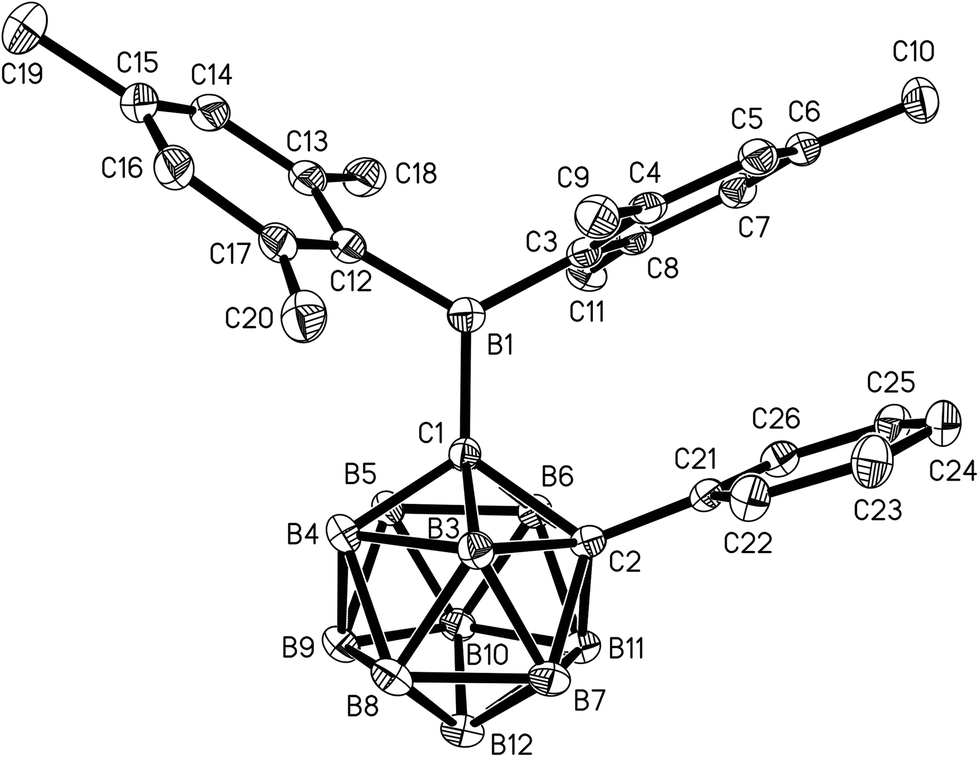 | ||
| Fig. 4 Molecular structure of 2 with hydrogen atoms omitted. Thermal ellipsoids are plotted at 50% probability. | ||
| 1 | 2 | |||
|---|---|---|---|---|
| Exp. | Calcd.a | Exp. | Calcd.a | |
| a Calculated values from optimised geometries of 1 and 2 at B3LYP/6-31G*. | ||||
| Bond lengths [Å] | ||||
| C1–C2 | 1.677(3) | 1.671 | 1.761(2) | 1.791 |
| C1–B1 | 1.635(3) | 1.641 | 1.629(2) | 1.648 |
| C1–B3 | 1.728(3) | 1.723 | 1.727(2) | 1.724 |
| C1–B6 | 1.756(3) | 1.757 | 1.748(2) | 1.751 |
| B1–C3 | 1.595(3) | 1.601 | 1.590(2) | 1.597 |
| B1–C12 | 1.581(3) | 1.593 | 1.602(2) | 1.602 |
| Bond angles [°] | ||||
| C1–B1–C3 | 115.7(2) | 116.9 | 118.2(1) | 120.7 |
| C1–B1–C12 | 121.0(2) | 121.0 | 119.4(1) | 118.3 |
| C3–B1–C12 | 123.3(2) | 122.2 | 122.4(1) | 120.9 |
| Torsion angles [°] | ||||
| C2–C1–B1–C3 | 36.0(2) | 34.9 | 22.5(2) | 37.1 |
| C2–C1–B1–C12 | −144.5(2) | −145.1 | −157.8(2) | −145.8 |
| B3–C1–B1–C3 | 102.7(2) | 102.1 | 95.8(2) | 110.6 |
| B3–C1–B1–C12 | −77.8(2) | −77.8 | −84.5(2) | −72.3 |
| Interplanar angles [°] | ||||
| (C1,B1,C3,C12)(C3–C8) | 77.9(1) | 74.2 | 78.6(1) | 69.8 |
| (C1,B1,C3,C12)(C12–C17) | 54.1(1) | 58.3 | 65.5(1) | 64.0 |
The C1–C2 distance of 1.677(3) Å in 1 agrees within 3 esd with C1–C2 bond lengths of 1.667(1)–1.673(1) Å found in other C-monoboryl-ortho-carboranes.24,33,36 By contrast, the C1–C2 bond of 1.761(2) Å in 2 is significantly longer than in its 1,3-diethyl-1,3,2-benzodiazaborol-2-yl analogue (1.701(2)–1.730(2) Å),24 in 1,2-diphenyl-ortho-carborane (1.720(4)–1.733(4) Å)43 and 1,2-diboryl-ortho-carboranes (1.695(1)–1.725(2) Å).26,36 The longer C1–C2 bond in 2 is explained by the different steric interactions between the BMes2 group at C1 and the phenyl ring at C2 in 2.
The BMes2 groups are linked to the cage carbon atoms by B–C single bonds (C1–B1) with lengths of 1.635(3) Å in 1 and 1.629(2) Å in 2, which is at the upper edge of the range determined for other C-boryl-ortho-carboranes (1.607(4)–1.649(12) Å).24–26,33,36 The B–C bond lengths between the mesityl rings and the boryl-boron atoms (B1–C3/C12 1.581(3)–1.602(2) Å) are typical for dimesitylboranes. As a consequence of the three-dimensional shape of the cluster in both structures, the interplanar angle enclosed by the mesityl ring pointing towards the second cage carbon atom and the plane defined by the boryl-boron atoms and the three neighbouring carbon atoms (77.9(1)° (1), 78.6(1)° (2)) is larger than in most reported structures of BMes2 compounds.44 A virtually perpendicular orientation of the phenyl substituent in 2 with respect to the C1–C2 axis (torsion angles = C1–C2–C21–C22 94.3(2)°, C1–C2–C21–C26 −91.5(2)°) corresponds to the situation in other disubstituted phenyl-ortho-carboranes and is preferred due to sterics.43,45
Photophysics
Photophysical data for 1 and 2 are listed in Table 2. The absorption maxima of both C-dimesitylboryl-ortho-carboranes (Fig. S1‡) in solvents of different polarity do not display any significant solvatochromism. The lack of solvatochromism points to very similar dipole moments in the electronic ground state and the initial excited state indicating that local transitions within the dimesitylboryl unit give rise to the absorption bands observed. The presence of the phenyl ring at C2 in 2 appears to lower the HOMO–LUMO energy gap as the absorption maxima of 2 (330 nm–333 nm) are bathochromically shifted by approximately 12–14 nm compared to 1 (318–319 nm) in all solvents used. The energy difference in the absorption maxima between 1 (329 nm) and 2 (332 nm) in the solid state is smaller.| Solid | CyH | CHCl3 | THF | DCM | ||
|---|---|---|---|---|---|---|
| a In L mol−1 cm−1. | ||||||
| Absorption λmax [nm] (ε)a | 1 | 329 | 318 (8300) | 319 (7350) | 319 (7060) | 318 (7920) |
| 2 | 332 | 332 (6750) | 331 (7330) | 333 (5710) | 330 (7760) | |
| Emission λmax [nm] (relative height) | 1 | 567 | 400, 541 (0.15:1) |
396, 641 (0.14:1) |
408, 653 (0.91:1) |
406, 664 (0.48:1) |
| 2 | 550 | 544 | 620 | 640 | 643 | |
| Stokes shift [cm−1] | 1 | 12310 |
5500, 12300 |
5780, 15610 |
6240, 15600 |
6600, 16170 |
| 2 | 9210 | 11920 |
14310 |
14660 |
14800 |
|
Emission maxima of both compounds in cyclohexane are virtually identical at 541 nm for 1 and 544 nm for 2 with large Stokes shifts of 12300 cm−1 for 1 and 11920 cm−1 for 2. The luminescence spectra (Fig. 5) reveal positive solvatochromism with emission maxima in the more polar solvent dichloromethane in the red emission region at 664 nm for 1 and 643 nm for 2. By using the Lippert–Mataga method46,47 with an Onsager radius of 4.00 Å estimated from the molecular structures, the calculated transition dipole moments are 10.4 D (1) and 9.2 D (2) – similar to transition dipole moments of C-benzodiazaborolyl-ortho-carboranes (6.9–10.9 D).24–26 The results show that the carborane cluster plays a part in the emission process acting as the electron acceptor of the charge transfer process after excitation. The solid-state emissions occur at longer wavelengths than in cyclohexane (Fig. 6). This bathochromic shift is more pronounced for 1 (567 nm) than for 2 (550 nm) and is presumably caused by fluorophore–fluorophore interactions. The quantum yields (Φ) are very low in all solvents (<1%) with higher values of 2% (1) and 7% (2) found in the solid state.
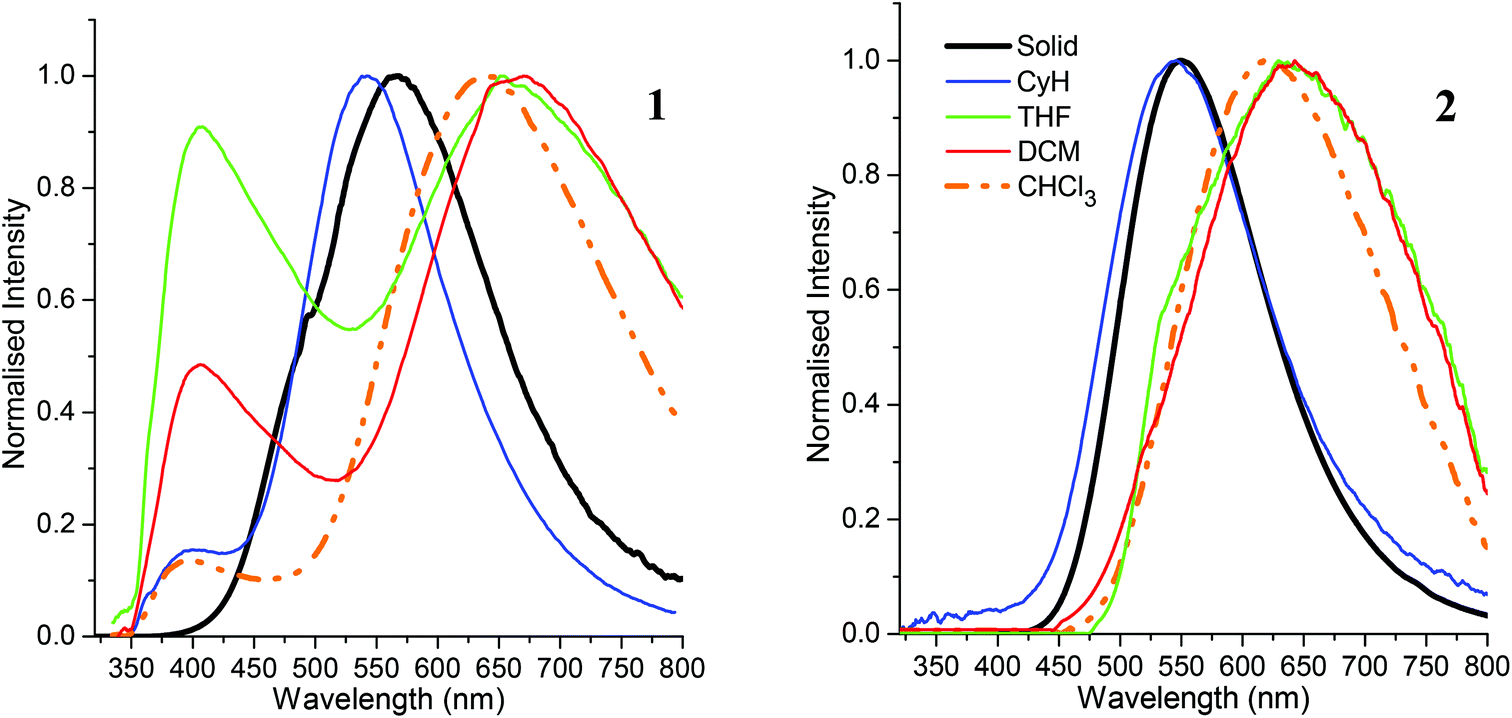 | ||
| Fig. 5 Emission spectra of 1 and 2 in the solid state and in various solvents. | ||
 | ||
| Fig. 6 Crystals of 1 and 2. Left column: Without UV irradiation. Right column: Under UV irradiation at 350 nm. | ||
In addition to these low-energy emissions, compound 1 displays a weaker emission band at the violet edge of the visible spectrum in polar solvents. The high-energy emissions with smaller Stokes shifts of 5500–6600 cm−1 probably originate from local transitions at the BMes2 group.48 Similar dual emissions originating from both local and CT transitions have been reported for some ortho-carboranes with substituents at one or both cluster carbon atoms.20,24–27
Electrochemistry
The electrochemical properties of both C-dimesitylboryl-ortho-carboranes 1 and 2 were investigated by cyclic voltammetry (CV, Fig. 7). The peak potentials measured in acetonitrile and dichloromethane solutions, with platinum and glassy carbon working electrodes are listed in Table S1.‡ The traces resemble reported CV data on reductions of carboranes elsewhere20,21,25–27,49–54 and, by inference, reductions take place at the carborane clusters in 1 and 2. CV traces for reductions of the BMes2 group would involve a simple one-electron reversible wave. In contrast, the one-electron reduction wave associated with PhBMes2 occurs at −2.30 V (vs. the ferrocenium/ferrocene couple at 0.0 V)2 and hence the BMes2 localised reduction in 1 and 2 likely falls outside the electrochemical window examined here.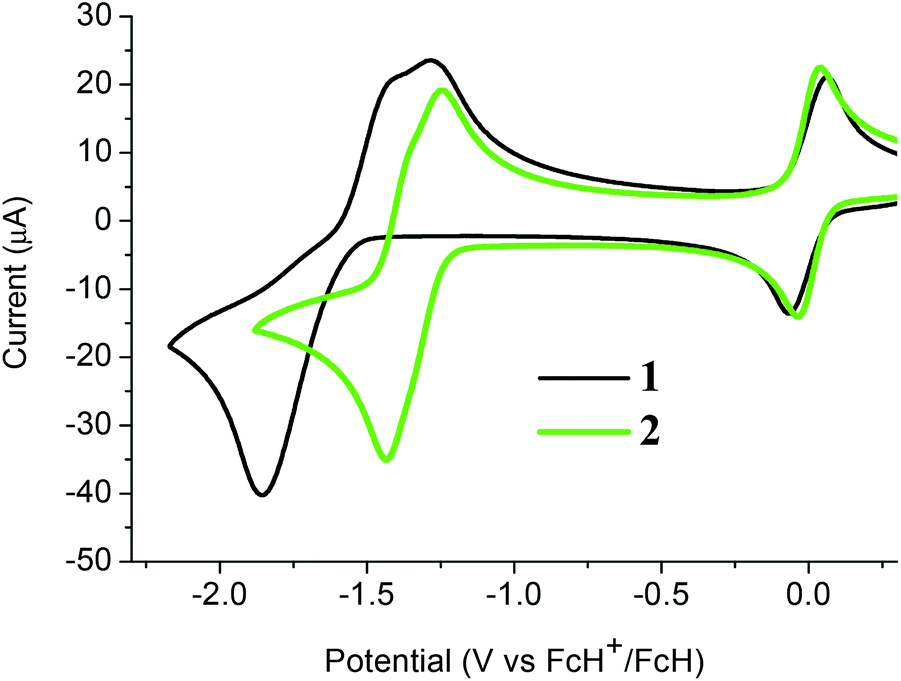 | ||
| Fig. 7 Cyclic voltammograms of 1 and 2 with a glassy carbon working electrode in acetonitrile and internal ferrocenium/ferrocene couples at 0.0 V. | ||
A CV trace for ortho-carborane or a C-monosubstituted-ortho-carborane usually shows a two-electron cathodic wave and an anodic wave that is not of the same current intensity as the cathodic wave.25,49,50 Often the peak–peak separation between the cathodic and anodic peaks of the wave can be several hundred millivolts due to the structural rearrangement of the dianion on the CV timescale. Decomposition and proton-coupled electron transfer (PCET) processes can also complicate the electrochemical response.49,55
The CV of 1 in acetonitrile with a glassy carbon working electrode shows a two-electron cathodic wave at −1.86 V and two anodic waves at −1.43 V and −1.29 V with values referenced to the ferrocenium/ferrocene redox couple at 0 V (Fig. 7). The cathodic wave value of −1.80 V for 1 means that 1 is more easily reduced than C-monophenyl-ortho-carborane at −2.25 V50 reflecting the substantial electron-withdrawing effect of the BMes2 group. Similar CV traces are observed for 1 with a platinum working electrode and with DCM as solvent (Fig. S2 and Table S1‡). The non-equivalent current intensities between the forward and reverse waves for 1 suggest that the dianion [1]2− is not stable and would be difficult to isolate.
A CV trace for ortho-carborane with aryl substituents at both cluster carbon atoms generally shows a reversible wave (or two) on reduction.20,25–27,49,51–54 In several cases, a stepwise reduction involving two separated one-electron reduction steps has been found, with the initial reduction process giving rise to a radical anion with an unusual 2n + 3 skeletal electron (SE) count. One example is diphenyl-ortho-carborane 5 where the radical anion has been shown to be stable enough to be observed spectroscopically at ambient temperature in solution (Fig. 8).51 The first one-electron reduction process on the CV timescale is usually slow due to the rearrangement of the cluster and is often immediately followed by a second one-electron process giving an apparently two-electron cathodic wave. The latter wave is usually evident in DCM for these carboranes.26 However, on back oxidation two separate anodic waves (with the combined current intensities similar to the current intensity of the cathodic wave) are evident corresponding to two one-electron processes.25,26
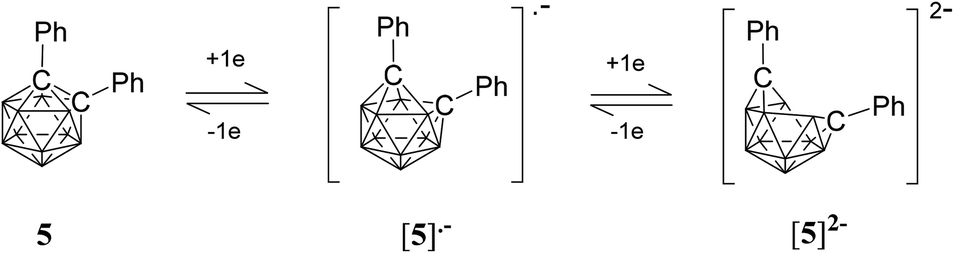 | ||
| Fig. 8 Two one-electron reductions for 1,2-diphenyl-ortho-carborane 5. | ||
The CV of 2 in acetonitrile here shows two very closely spaced and barely resolved one-electron waves (Fig. 7). These waves are separated by less than 80 mV based on gaussian analyses of square-wave potential traces (Fig. S3‡). The two half-wave potentials are estimated to be −1.31 and −1.39 V which indicate that 2 is reduced more readily than diphenyl-ortho-carborane 3 at −1.57 and −1.72 V under the same CV conditions. Thus, compound 2 does form a radical anion with a 2n + 3 skeletal electron count. However, the very low comproportionation constant (Kc) associated with the intermediate [2]˙− means that spectroscopic observation is challenging, and the monoanion could not be isolated in any appreciable concentration in the comproportionated mixture obtained following one-electron reduction (Fig. S4‡). The CV of 2 in DCM shows the expected CV pattern with a one two-electron cathodic wave and two one-electron anodic waves (Fig. S2 and Table S1‡).
Computations
Calculated bond lengths and angles from geometries of 1 and 2 optimised at B3LYP/6-31G* are in good agreement with the experimental values (Table 1). The lengthening of the C1–B6 bonds compared to the C1–B3 bonds is attributed to steric repulsion between an ortho-methyl group of one of the mesityl rings and the B6–H unit. Computed energy barriers for the rotation around the C1–B1 bonds are 6.1 kcal mol−1 in 1 and 18.6 kcal mol−1 in 2. Comparison between computed GIAO-NMR and experimental 11B NMR chemical shifts for 1–4 are in very good agreement (Table S2‡).The HOMO is a combination of π-orbitals at the mesityl groups (π(Mes)) and the LUMO consists mainly of the empty p-orbital of the boryl boron atom (p(B)) (Fig. 9). Antibonding orbitals with significant cluster contributions have much higher energies (>−0.16 eV) and thus the influence of the clusters on the absorption process is merely inductive in both cases. According to TD-DFT calculations π(Mes)–p(B) transitions with oscillator strengths (f) of 0.0065 to 0.0698 can be assigned to the absorption bands of both compounds (Tables S3 and S4‡). Therefore, the electron density in the initially formed excited state is shifted within the BMes2-unit only which is not expected to entail strong changes in the overall dipole moment. This is in agreement with the lack of solvatochromism in the absorption spectra. Weak transitions between the π-orbitals of the phenyl group of 2 and the p(B) orbital as well as π–π* transitions within the phenyl ring occur at considerably higher energy far in the UV region. The HOMO–LUMO gap energy of 2 is 0.10 eV smaller than that in 1 which agrees well with the observed bathochromic shift of the absorption maximum of 2 compared to 1.
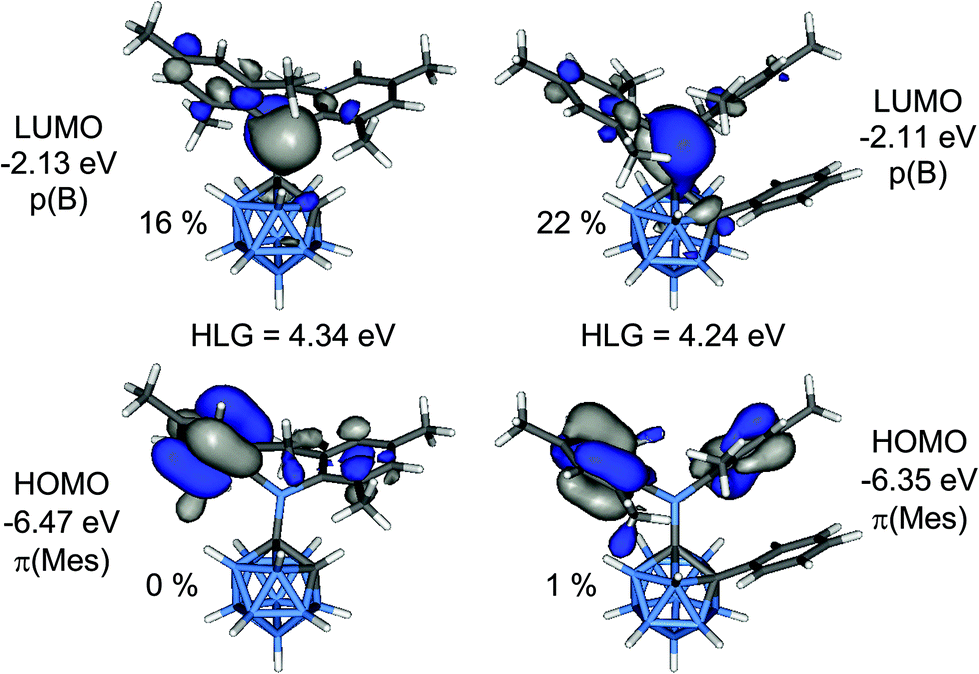 | ||
| Fig. 9 Frontier molecular orbitals of 1 (left) and 2 (right). The percentage values are the cluster contributions to the molecular orbitals. | ||
In order to elucidate the origin of the visible CT emission of 1 and 2, their geometries were optimised at the first excited singlet state (S1). In both cases “open” cluster geometries were found with C1–C2 distances expanded to 2.384 Å (1) and 2.440 Å (2), respectively. The frontier orbitals of the S1 geometries are depicted in Fig. 10. These orbitals were calculated at the ground state, S0 thus HOMO and LUMO correspond to the highest and second highest singly occupied orbitals in the S1 state. The HOMO is a π(Mes) orbital but the LUMO, in contrast, is an antibonding cluster orbital (cage*) with small contributions from the exopolyhedral boron atom (1: 15%, 2: 8%). Thus, the HOMO–LUMO transition corresponds to a charge transfer between the cluster and the mesityl groups from the excited state and the two compounds can be regarded as donor–acceptor systems. TD-DFT calculations predicted low-energy emissions with low oscillator strengths at 859 nm (1; f = 0.0016) and 783 nm (2; f = 0.0066) for these transitions and gave the same trend as experimentally observed with 1 emitting at longer wavelength than 2.
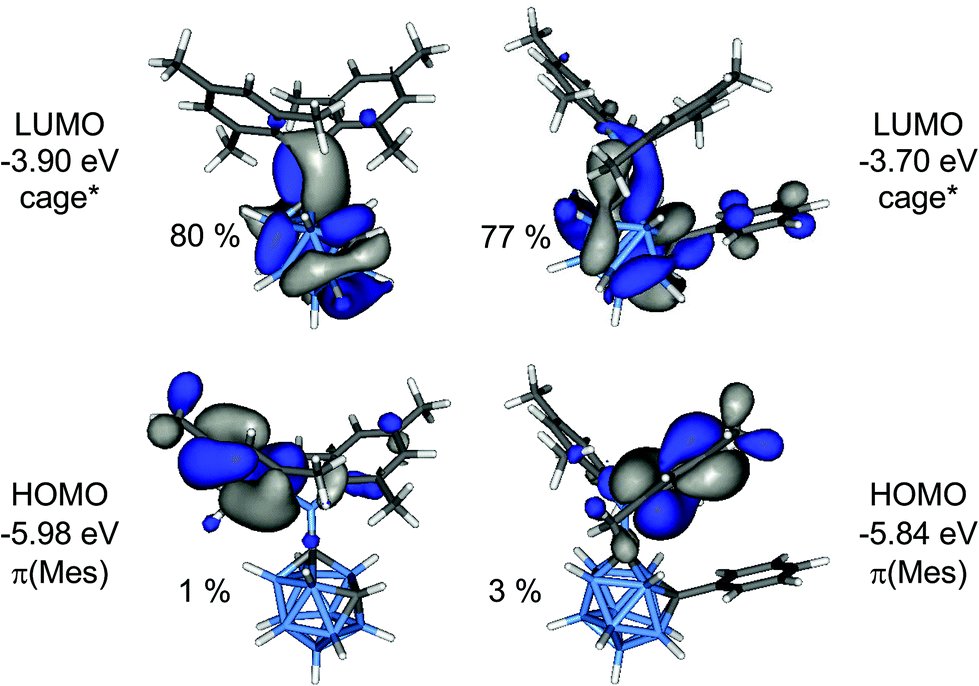 | ||
| Fig. 10 Frontier orbitals of the S1 geometries of 1 (left) and 2 (right). The percentage values are the cluster contributions to the molecular orbitals. | ||
The geometries of the fluoride adducts [1·F]− and [2·F]− were optimised in order to examine why fluoride adducts were not observed experimentally. The fluoride ion affinities of 1 and 2 are 132.9 and 127.3 kcal mol−1, respectively, which are higher than the fluoride ion affinities of many aryldimesitylboranes (Table 3).8 Although the strong carborane electron-withdrawing character increases the affinities for the fluoride ion in 1 and 2 with respect to the affinities of the aryldimesitylborane analogues, the optimised geometries [1·F]− and [2·F]− contain unusually long B–C(carboranyl) bonds of 1.786 and 1.823 Å respectively. These bond lengths suggest that they would be easily cleaved either before the B–F bond is formed or by hydrolysis as observed experimentally.
Geometries of the dianions [2]2−and [5]2−
While closo-dicarbadodecaboranes all adopt the pseudo-icosahedral geometry, several different geometries of nido-dicarbadodecaborane dianions have been determined crystallographically (Fig. 11).56 Dianions with almost planar C2B4 open faces like [6]2− and [7]2− are observed in Group 1 metallacarboranes.57–60 Bowl-shaped geometries have been observed in carborane dianions like [8]2− and [9]2− with tethers at both cage carbons.59,61 The bowl geometry in [9]2− differs from that in [8]2− where [9]2− has a notably smaller open face.61 These bowl-shaped geometries are similar to geometries determined for neutral 12-vertex tetracarbadodecaboranes by X-ray crystallography.62 The neutral compound 10 may also be regarded as a genuine 12-vertex dicarbadodecaborane dianion [R2C2B10H10]2− with two positively charged phosphonium groups.63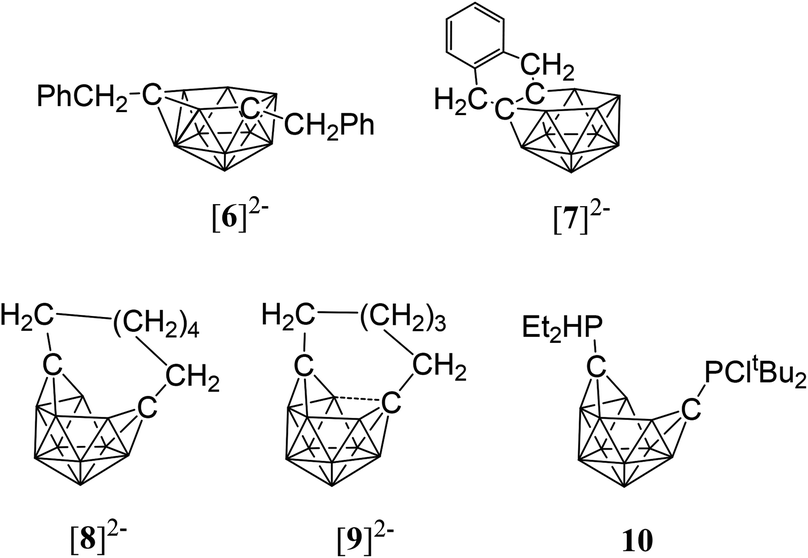 | ||
| Fig. 11 Geometries of nido-dicarbadodecaboranes determined by X-ray crystallography. | ||
While there are several structural studies published on nido-dicarbadodecaborane dianions, the geometries of nido-dicarbadodecaborane dianions in solutions have not been determined. The successful method64 of comparing observed and computed 11B NMR data to determine carborane cluster geometries is applied here for dianion [2]2−. Before discussing the dianion [2]2− made by chemical reductions on 2, the dianion [5]2− generated from diphenyl-ortho-carborane 5 is described here to demonstrate the use of the combined experimental and calculated 11B NMR method in determining the geometry of its dianion. Dianion [5]2− has been proposed to have a geometry65 with a C2B4 open face A or a geometry54,66 with a C2B2 open face B (Fig. 12). Thus, the geometry of the dianion [5]2− has not been established even though this dianion has been known for many decades.67
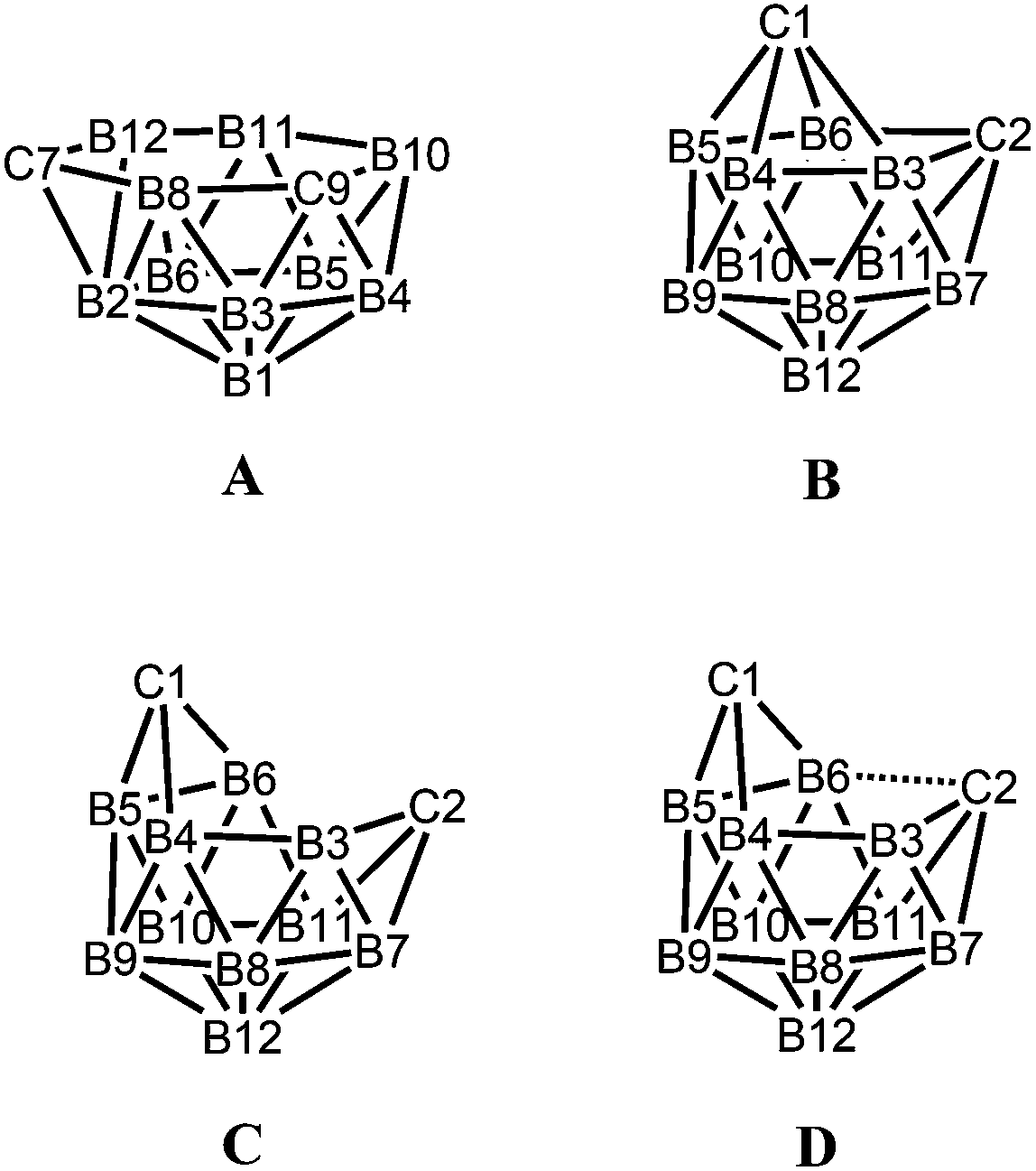 | ||
| Fig. 12 Geometries A–D of nido-dicarbadodecaborane dianions with atom numbering. | ||
Chemical reductions of 5 were carried out with alkali metals (Li, Na, K) in THF solutions. The reactions were monitored by 11B NMR spectroscopy until all the peaks corresponding to the starting carborane and the red colours of the solutions due to the radical monoanions disappeared. The 11B{1H} spectra of the dianions [5]2− in clear yellow solutions show 2:2:4:2 patterns which are not significantly influenced by the different alkali metal cations (Fig. 13). This pattern suggests a geometry of high symmetry or two mirror-image geometries that are fluctional in solution for [5]2− with the metal cations not strongly interacting in solution.
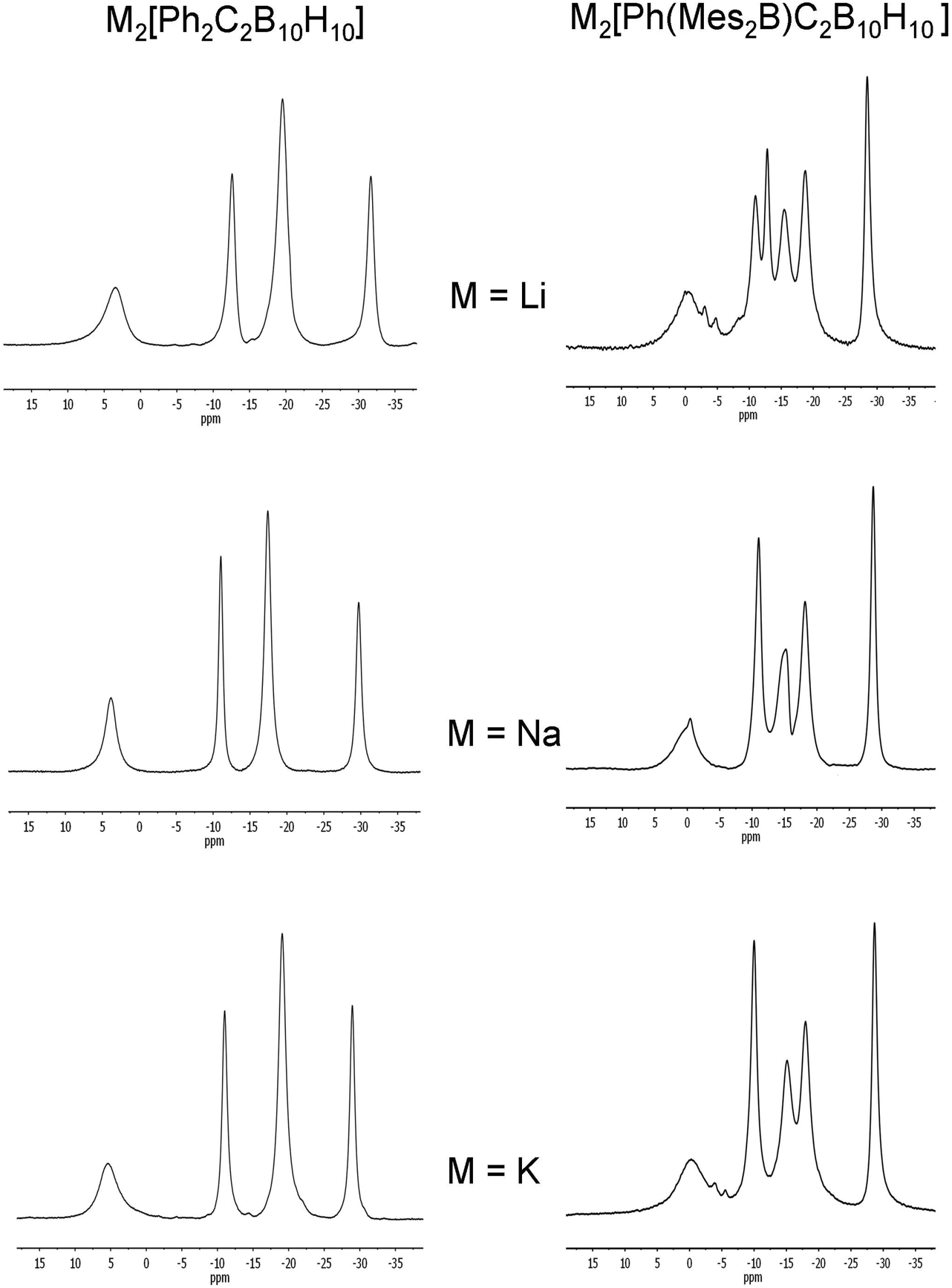 | ||
| Fig. 13 11B{1H} NMR spectra of M2[5]2− and M2[2]2− (M = Li, Na or K) in THF. The broad peaks at 65–70 ppm corresponding to the boryl boron in [2]2− are not shown here. | ||
Geometry optimisations on [5]2− reveal that the bowl-shaped geometry C is more stable than A and B by 5.5 and 11.0 kcal mol−1 respectively. More importantly, the computed GIAO 11B NMR chemical shifts of the bowl-shaped geometry fit well with observed shifts when fluctionality between the two mirror-image geometries of C takes place in solution (Table 4). The geometry D found in [9]2− could not be located for [5]2− where the initial geometry D rearranged to C on optimisation.
| Geometry | B3,6 | B12 | B9 | B4,5 | B7,11 | B8,10 | BMes2 | Rel. E | |
|---|---|---|---|---|---|---|---|---|---|
| a All borons are non-equivalent in geometry A of [2]2−, values are calculated assuming same fluctionality process as in geometry A of [5]2−; 64.6 (BMes2), 14.1 (B12), 12.1 (B10), 5.7 (B11), −0.7 (B4), −1.7 (B6), −7.3 (B8), −18.2 (B5), −20.3 (B1), −23.9 (B3), −24.8 (B2). b Experimental data for sodium salt in CD3CN, Fig. 14. c Calculated values are averaged assuming fluctionality between two mirror-image geometries; 10.0 (B10,12), −0.8 (B11), −6.0 (B8), −9.3 (B5,6), −18.8 (B3), −20.6 (B2,4), −25.6 (B1). d Experimental data for sodium salt in d8-THF. | |||||||||
| [2]2− | A | 14.0 | |||||||
| C | 13.3 | −1.2 | −8.4 | −17.4 | −21.3 | −23.7 | 60.2 | 0.0 | |
| D | −4.1 | −10.5 | −11.8 | −15.4 | −19.5 | −29.2 | 56.0 | 0.8 | |
| Exp.b | 0.1 | −9.2 | −10.1 | −14.0 | −17.8 | −27.2 | 67.5 | ||
| [5]2− | A | 11.0 | |||||||
| B | −23.5 | −20.0 | −20.0 | −18.6 | −18.6 | −55.7 | 5.5 | ||
| C | 12.2 | −7.6 | −7.6 | −20.7 | −20.7 | −25.5 | 0.0 | ||
| Exp.d | 7.0 | −9.3 | −9.3 | −17.3 | −17.3 | −27.5 | |||
Reduction of 2 with sodium metal in 1,2-dimethoxyethane (DME) yielded a dark red solid identified as [Na(DME)n]2[2] by 1H, 11B and 13C NMR spectroscopy. Purification of this extremely air-sensitive salt by crystallization was not successful. Salts of dicarbadodecaborane dianions are known to be extremely air- and moisture-sensitive.57 The 11B{1H} NMR spectrum recorded in CD3CN revealed a 2:1:1:2:2:2 pattern for the cluster atoms and a very broad signal at 67.5 ppm corresponding to the boryl boron atom (Fig. 14). The latter peak is considerably shifted to higher field by about 13 ppm compared to the neutral starting material.
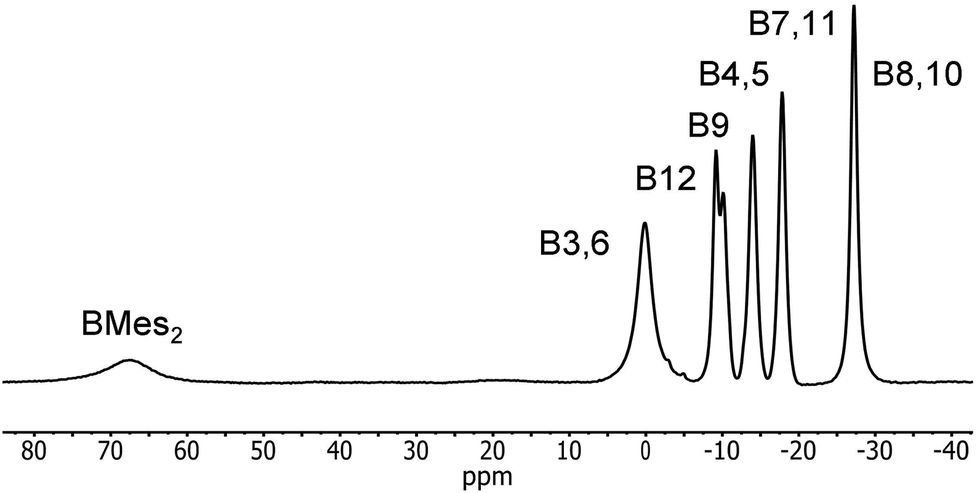 | ||
| Fig. 14 11B{1H} NMR spectrum of [2]2− in CD3CN with peak assignments based on GIAO-NMR data. | ||
Chemical reductions of the Mes2B compound 2 in THF with alkali metals were carried out as for 5. After observation of the purple colours corresponding to the radical species and the disappearance of the peaks corresponding to the starting material, the clear solutions containing the dianions [2]2− were dark red. 11B NMR spectra of the dianions [2]2− show either a 2:1:1:2:2:2 (Li salt) or a 2:2:2:2:2 (Na, K) peak pattern (Fig. 13) and are similar to peaks found for the diphenylcarborane dianions [5]2− when taking into account the lower symmetry in [2]2−.
The similarities in the 11B NMR peaks observed for [2]2− and [5]2− suggest that both have bowl-shaped geometries. Geometry optimisations on the BMes2 species [2]2− reveal that the starting geometry B was rearranged to the bowl-shaped geometry D. The most stable geometry for [2]2− is C with D only 0.8 kcal mol−1 higher in energy. However, computed 11B NMR shifts from geometry D fit better with observed 11B NMR shifts than geometry C for [2]2− assuming fluctionalities between mirror-image geometries occur in solution (Fig. 15).
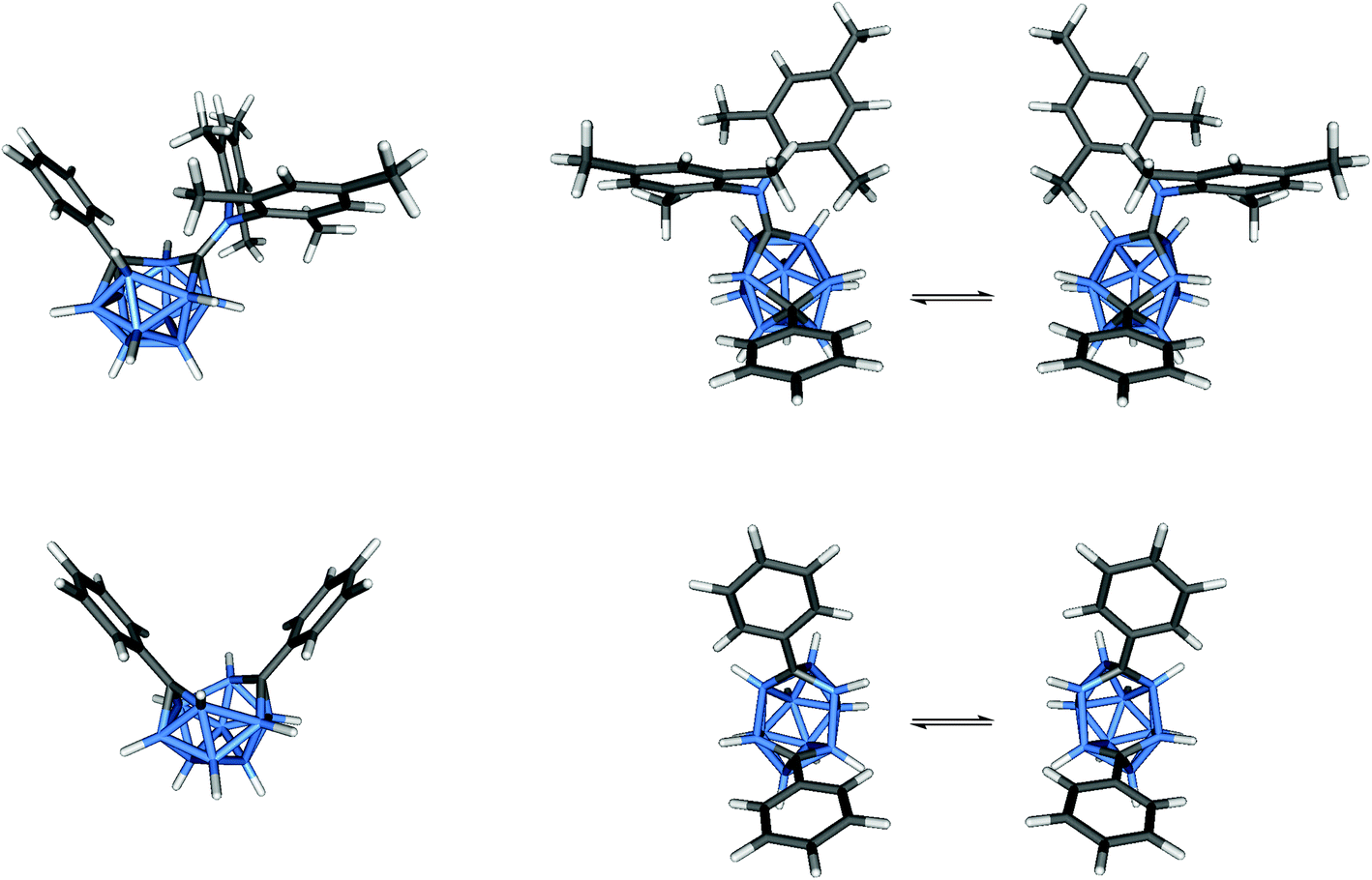 | ||
| Fig. 15 Optimised geometries of [2]2− (top) and [5]2− (bottom) and the fluctional processes. | ||
It is concluded here that bowl-shaped geometries are present in solutions of 12-vertex nido-dicarbadodecaborane dianions with fluctional cluster geometries of C and D in dianions of C,C′-diphenyl-carborane [5]2− and C-dimesitylboryl-C′-phenyl-carborane [2]2− respectively. The calculated geometries for [2]2− and [5]2− are similar to the experimental cluster geometries61 of [9]2− and [8]2− respectively as shown from comparison of distances involving the cluster carbon atoms, C1 and C2, in Table 5. The combined experimental and calculated 11B NMR method is shown to be useful in determining nido-12-vertex geometries in solutions and will aid further progress on the intriguing range of nido-12-vertex geometries in the future.
| [2]2− calc. | [9]2− obs. | [5]2− calc. | [8]2− obs. | |
|---|---|---|---|---|
| C1⋯C2 | 2.666 | 2.687(6) | 2.915 | 2.87(1) |
| C1⋯B3 | 2.528 | 2.302(9) | 2.606 | 2.59(1) |
| C1⋯B6 | 1.624 | 1.617(9) | 1.546 | 1.51(1) |
| C2⋯B3 | 1.586 | 1.534(8) | 1.546 | 1.51(1) |
| C2⋯B6 | 1.878 | 2.083(8) | 2.606 | 2.55(1) |
Conclusions
Two C-dimesitylboryl-1,2-dicarba-closo-dodecaboranes were synthesised from fluorodimesitylborane and the corresponding lithio-carboranes and structurally characterised by X-ray crystallography. Photophysical studies and TD-DFT calculations showed that the absorptions correspond to local transitions within the BMes2 groups whereas visible emissions with Stokes shifts up to 16170 and 14800 cm−1 in dichloromethane originate from intramolecular CT transitions between the mesityl rings and the cluster. Compound 2 with a phenyl substituent at the second cage carbon atom can be easily reduced to a stable dianion [2]2− by cyclic voltammetry and chemical reductions with alkali metals. Based on experimental and calculated 11B NMR data, a dynamic bowl-shaped nido-cage geometry is determined for the dianion. These findings indicate that the ortho-carboranyl group is a stronger electron acceptor than the BMes2 group and C-dimesitylboryl-1,2-dicarba-closo-dodecaboranes are dyads with the mesityl group as the donor and the carborane as the acceptor.
Experimental section
General
The reactions were performed under an atmosphere of dry oxygen-free argon using Schlenk techniques unless otherwise stated. All solvents were dried by standard methods and freshly distilled prior to use. Fluorodimesitylborane68 and 1-phenyl-1,2-dicarba-closo-dodecaborane69 were prepared as described in the literature. 1,2-Dicarba-closo-dodecaborane was purchased commercially (KatChem). NMR spectra were recorded from solutions at room temperature on a Bruker AM Avance DRX500 (1H, 11B, 13C), a Bruker Avance III 500 and a Bruker Avance 400 Spectrometer (1H{11B}, 19F) with SiMe4 (1H, 13C), BF3·OEt2 (11B) and CFCl3 (19F) as external standards. 1H- and 13C{1H} NMR spectra were calibrated on the solvent signal [CDCl3: 7.24 (1H), 77.16 (13C); CD3CN: 1.94 (1H), 118.25, 1.32 (13C); d8-THF: 3.58, 1.73 (1H), 67.57, 25.46 (13C)]. The 13C NMR peaks were assigned with the aid of observed 13C DEPT spectra and computed 13C NMR shifts. Electron Ionisation (EI) and Atmospheric pressure Solids Analysis Probe (ASAP) mass spectra were recorded with a VG Autospec sector field (Micromass) and Xevo QTOF (Waters) mass spectrometers respectively.Hydrolyses of 1 and 2
A drop of water (excess) was mixed with a solution of 1 or 2 in deuterated chloroform in an NMR tube and the mixture was analysed by NMR spectroscopy. No change in the spectra was observed after a week.Mesitylene: 1H-NMR (CDCl3): δ [ppm] = 6.81, 2.25; 13C{1H}-NMR (CDCl3): δ [ppm] = 137.9, 127.0, 21.3.
(1,2-Dicarba-closo-dodecaboranyl)-1-borinic acid (3): 1H{11B}-NMR (CDCl3): δ [ppm] = 2.05 (2H, BH), 2.17 (2H, BH), 2.26 (2H, BH), 2.33 (4H, BH), 3.67 (1H, B10H10C2H), 4.91 (2H, OH); 13C{1H}-NMR (CDCl3): δ [ppm] = 57.4 (s, CB10H10CH), the 13C peak corresponding to CB(OH)2 is not observed; 11B{1H}-NMR (CDCl3): δ [ppm] = −12.6 (s), −7.8 (s), −2.1 (s), −1.0 (s) (skeletal boron atoms), 26.5 (s, exopolyhedral boron atom) see Fig. S11‡ for 11B NMR spectra of 3; MS (ASAP, M = C2H13B11O2): m/z = 188.0 (M+, 21%), 171.2 (M+ − OH, 100%).
(2-Phenyl-1,2-dicarba-closo-dodecaboranyl)-1-borinic acid (4): 1H NMR (CDCl3): δ [ppm] = 2.32 (2H, BH), 2.42 (3H, BH), 2.48 (2H, BH), 2.61 (1H, BH), 2.92 (2H, BH), 4.46 (2H, OH), 7.34 (dd, 3JHH = 7.4 Hz, 3JHH = 7.6 Hz, 2 H, Hmeta), 7.41 (t, 3JHH = 7.4 Hz, 1 H, Hpara), 7.64 (d, 3JHH = 7.6 Hz, 2 H, Hortho); 13C{1H} NMR (CDCl3): δ [ppm] = 20.8 (s, para-CH3), 26.8 (s, ortho-CH3), 86.5 (s, CB10H10CPh), 87.3 (s, CB10H10CBMes2), 127.7 (s, Cmeta, Ph), 129.3 (s, Cpara, Ph), 129.4 (s, CHMes), 130.3 (s, Cortho, Ph), 131.7 (s, Cipso, Ph), 138.8 (s, Cipso, Mes), 139.1 (s, Cpara, Mes), 139.4 (s, Cortho, Mes); 11B{1H} NMR (CDCl3): δ [ppm] = −11.7 (s), −10.6 (s), −8.3 (s), −3.1 (s), 0.9 (s) (skeletal boron atoms), 26.5 (s, exopolyhedral boron atom) see Fig. S12‡ for 11B NMR spectra of 4; MS (ASAP, M = C8H17B11O2): m/z = 263.2 (M − 1+, 44%), 247.2 (M+ − OH, 100%).
Reductions of 2 and 5
:1 ratio. Method 2 was also used in the reductions of 5 with alkali metals (Li, Na, K) but with deuterated THF in all cases and NMR data of M2[5] (M = Li, Na, K) are listed in Table S6.‡
Photophysical measurements
For all solution state measurements, samples were contained in quartz cuvettes of 10 × 10 mm (Hellma type 111-QS, suprasil, optical precision). Cyclohexane was used as received from commercial sources (p.a. quality), the other solvents were dried by standard methods prior to use. Concentrations varied from 20 to 100 μM in order to get analysable emission spectra due to the low quantum yields. Effects of the concentration on the shape of the observed emission spectra were excluded in this concentration range. Solid samples were prepared by vacuum sublimation on quartz plates (35 × 10 × 1 mm) using standard Schlenk equipment and conditions. Each plate was laid in a 100 mL round bottom flask and a crystal of the sample substance placed below it was sublimed. Absorption was measured with a UV/VIS double-beam spectrometer (Shimadzu UV-2550), using the solvent as a reference.The output of a continuous Xe-lamp (75 W, LOT Oriel) was wavelength-separated by a first monochromator (Spectra Pro ARC-175, 1800 l mm−1 grating, Blaze 250 nm) and then used to irradiate a sample. The fluorescence was collected by mirror optics at right angles and imaged on the entrance slit of a second spectrometer while compensating astigmatism at the same time. The signal was detected by a back-thinned CCD camera (RoperScientific, 1024\256 pixels) in the exit plane of the spectrometer. The resulting images were spatially and spectrally resolved. As the next step, one averaged fluorescence spectrum was calculated from the raw images and stored in the computer. This process was repeated for different excitation wavelengths. The result is a two-dimensional fluorescence pattern with the y-axis corresponding to the excitation, and the x-axis to the emission wavelength. The wavelength range is λex = 230–430 nm (in 1 nm increments) for the UV light and λem = 305–894 nm for the detector. The time to acquire a complete EES is typically less than 15 min. Post-processing of the EES includes subtraction of the dark current background, conversion of pixel to wavelength scales, and multiplication with a reference file to take the varying lamp intensity as well as grating and detection efficiency into account. Stokes shifts were calculated from excitation and emission maxima, which were extracted from spectra that were converted from wavelength to wavenumbers beforehand. The quantum yields in solution were determined against POPOP (p-bis-5-phenyl-oxazolyl(2)-benzene) (ΦF = 0.93) as the standard.
The solid-state fluorescence was measured by addition of an integrating sphere (Labsphere, coated with Spectralon, Ø 12.5 cm) to the existing experimental setup. At the exit slit of the first monochromator the exciting light was transferred into a quartz fibre (LOT Oriel, LLB592). It passed a condenser lens and illuminated a 1 cm2 area on the sample in the centre of the sphere. The emission and exciting light was imaged by a second quartz fibre on the entrance slit of the detection monochromator. Post-processing of the spectra was done as described above. The measurement and calculation of quantum yields was performed according to the method described by Mello.71
Electrochemistry
Cyclic voltammetric measurements were carried out using an EcoChemie Autolab PG-STAT 30 potentiostat at 298 K with a platinum or glassy carbon working electrode and platinum wires as counter and reference electrodes in a nitrogen-containing glove box with 0.1 M tetra-n-butylammonium hexafluorophosphate [Bu4N][PF6] in DCM or acetonitrile. Scan rates of 100 mV s−1 and analyte concentrations of 10−3 M were used. The ferrocene/ferrocenium FcH/FcH+ couple served as internal reference at 0.0 V for potential measurements and peak–peak separations of this couple were generally in the region of 90–110 mV.The spectroelectrochemical (SEC) experiment on 2 was performed at room temperature in an airtight optically transparent thin-layer electrochemical (OTTLE) cell72 equipped with Pt minigrid working and counter electrodes (32 wires cm−1), Ag wire pseudo-reference electrode and CaF2 windows for a 200 μm path-length solvent compartment. Acetonitrile containing 0.1 M [Bu4N][PF6] electrolyte was used in the cell which was fitted into the sample compartment of a Cary 5000 (UV-Vis-NIR) spectrophotometer. Bulk electrolysis was carried out using an Autolab PG-STAT 30 potentiostat.
Crystallographic studies
Single crystals were coated with a layer of hydrocarbon oil and attached to a glass fiber. Crystallographic data were collected with a Bruker AXS X8 Prospector Ultra APEX II diffractometer with Cu Kα radiation (graphite monochromator, λ = 1.54178 Å) at 100 K. Crystallographic programs used for structure solution and refinement were from SHELX-97.73 The structures were solved by direct methods and were refined by using full-matrix least squares on F2 of all unique reflections with anisotropic thermal parameters for all non-hydrogen atoms. The hydrogen atoms bonded to the carborane units were refined isotropically, all other hydrogen atoms were refined using a riding model with U(H) = 1.5Ueq for CH3 groups and U(H) = 1.2Ueq for all others. Crystallographic data for the compounds are listed in Table S7.‡ CCDC 1048027 (1) and CCDC 1048028 (2) contain the supplementary crystallographic data for this paper.Computational details
All computations were carried out with the Gaussian 09 package.74 The model geometries were fully optimised with the B3LYP functional75 with no symmetry constraints using the 6-31G* basis set76 for all atoms. Frequency calculations on all optimised geometries revealed no imaginary frequencies. Computed absorption data were obtained from TD-DFT77 calculations on S0 geometries whereas computed emission data were from the S1 geometries. The MO diagrams and MO compositions were generated with the Molekel78 and GaussSum79 packages, respectively. Calculated 11B and 13C NMR chemical shifts obtained at the GIAO80-B3LYP/6-31G*//B3LYP/6-31G* level on the optimised geometries were referenced to BF3·OEt2 for 11B: δ(11B) = 111.7–σ(11B) and referenced to TMS for 13C: δ(13C) = 189.4–σ(13C). Computed NMR values reported here were averaged where possible.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) and the Engineering and Physical Sciences Research Council (EPSRC) for financial support. P.J.L. gratefully acknowledges an EPSRC Leadership Fellowship, and currently holds an ARC Future Fellowship (FT120100073).References
- (a) C. D. Entwistle and T. B. Marder, Angew. Chem., 2002, 114, 3051–3056 ( Angew. Chem., Int. Ed. , 2002 , 41 , 2927–2931 ) CrossRef; (b) C. D. Entwistle and T. B. Marder, Chem. Mater., 2004, 16, 4574–4585 CrossRef CAS; (c) S. Yamaguchi and A. Wakamiya, Pure Appl. Chem., 2006, 78, 1413–1424 CAS; (d) F. Jäkle, Coord. Chem. Rev., 2006, 250, 1107–1121 CrossRef.
- A. Schulz and W. Kaim, Chem. Ber., 1989, 122, 1863–1868 CrossRef CAS.
- M. E. Glogowski and J. L. R. Williams, J. Organomet. Chem., 1981, 218, 137–146 CrossRef CAS.
- (a) T. Noda and Y. Shirota, J. Am. Chem. Soc., 1998, 120, 9714–9715 CrossRef CAS; (b) M. Kinoshita, N. Fujii, T. Tsukaki and Y. Shirota, Synth. Met., 2001, 121, 1571–1572 CrossRef CAS.
- (a) W.-L. Jia, D.-R. Bai, T. Mc Cormick, Q.-D. Liu, M. Motala, R.-Y. Wang, C. Seward, Y. Tao and S. Wang, Chem. – Eur. J., 2004, 10, 994–1006 CrossRef CAS PubMed; (b) W.-L. Jia, D. Feng, D.-R. Bai, Z. H. Lu, S. Wang and G. Vamvounis, Chem. Mater., 2005, 17, 164–170 CrossRef CAS; (c) M. Mazzeo, V. Vitale, F. Della Sala, M. Anni, G. Barbarella, L. Favaretto, G. Sotgui, R. Cingolani and G. Gigli, Adv. Mater., 2005, 17, 34–39 CrossRef CAS.
- (a) T. Noda, H. Ogawa and Y. Shirota, Adv. Mater., 1999, 11, 283–285 CrossRef CAS; (b) T. Noda and Y. Shirota, J. Lumin., 2000, 87–89, 1168–1170 CrossRef CAS; (c) Y. Shirota, M. Kinoshita, T. Noda, K. Okumuto and T. Ohara, J. Am. Chem. Soc., 2000, 122, 11021–11022 CrossRef CAS; (d) W.-Y. Wong, S.-Y. Poon, M.-F. Lin and W.-K. Wong, Aust. J. Chem., 2007, 60, 915–922 CrossRef CAS; (e) G.-J. Zhou, G.-L. Ho, W.-Y. Wong, Q. Wang, D.-G. Ma, L.-X. Wang, Z.-Y. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499–511 CrossRef CAS; (f) C.-L. Ho;, B. Yao, B. Zhang, K.-L. Wong, W.-Y. Wong, Z. Xie, L. Wang and Z. Lin, J. Organomet. Chem., 2013, 730, 144–155 CrossRef.
- (a) E. Sakuda, A. Funahashi and N. Kitamura, Inorg. Chem., 2006, 45, 10670–10677 CrossRef CAS PubMed; (b) M. Melaimi and F. P. Gabbaï, J. Am. Chem. Soc., 2005, 127, 9680–9681 CrossRef CAS PubMed; (c) Y. Kim and F. P. Gabbaï, J. Am. Chem. Soc., 2009, 131, 3363–3369 CrossRef CAS PubMed; (d) S.-B. Zhao, T. McCormick and S. Wang, Inorg. Chem., 2007, 46, 10965–10967 CrossRef CAS PubMed; (e) M.-S. Yuan, Z.-Q. Liu and Q. Fang, J. Org. Chem., 2007, 72, 7915–7922 CrossRef CAS PubMed; (f) X. Y. Liu, D. R. Bai and S. Wang, Angew. Chem., 2006, 118, 5601–5604 ( Angew. Chem., Int. Ed. , 2006 , 45 , 5475–5478 ) CrossRef; (g) D.-R. Bai, X.-Y. Liu and S. Wang, Chem. – Eur. J., 2007, 13, 5713–5723 CrossRef CAS PubMed; (h) M. H. Lee, T. Agou, J. Kobayashi, T. Kawashima and F. P. Gabbaï, Chem. Commun., 2007, 1133–1135 RSC; (i) T. W. Hudnall, M. Melaïmi and F. P. Gabbaï, Org. Lett., 2006, 8, 2747–2749 CrossRef CAS PubMed; (j) H.-P. Shi, J.-X. Dai, L. Xu, L.-W. Shi, L. Fang, S.-M. Shuang and C. Dong, Org. Biomol. Chem., 2012, 10, 3852–3858 RSC; (k) C.-W. Chiu and F. P. Gabbaï, J. Am. Chem. Soc., 2006, 128, 14248–14249 CrossRef CAS PubMed; (l) S. Yamaguchi, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2001, 123, 11372–11375 CrossRef CAS PubMed; (m) S. Yamaguchi, T. Shirasaka, S. Akiyama and K. Tamao, J. Am. Chem. Soc., 2002, 124, 8816–8817 CrossRef CAS PubMed; (n) Y. Kubo, M. Yamamoto, M. Ikeda, M. Takeuchi, S. Shinkai, S. Yamaguchi and K. Tamao, Angew. Chem., 2003, 115, 2082–2086 ( Angew. Chem., Int. Ed. , 2003 , 42 , 2036–2040 ) CrossRef; (o) S. Solé and F. P. Gabbaï, Chem. Commun., 2004, 1284–1285 RSC; (p) T. W. Hudnall and F. P. Gabbaï, J. Am. Chem. Soc., 2007, 129, 11978–11986 CrossRef CAS PubMed; (q) A. Sundararaman, M. Victor, R. Varughese and F. Jäkle, J. Am. Chem. Soc., 2005, 127, 13748–13749 CrossRef CAS PubMed; (r) K. Parab, K. Venkatasubbaiah and F. Jäkle, J. Am. Chem. Soc., 2006, 128, 12879–12885 CrossRef CAS PubMed; (s) W.-J. Xu, S.-H. Liu, X. Zhao, N. Zhao, X.-Q. Yu and W. Huang, Chem. – Eur. J., 2013, 19, 621–629 CrossRef CAS PubMed; (t) Z. Zhang, R. M. Edkins, J. Nitsch, K. Fucke, A. Eichhorn, A. Steffen, Y. Wang and T. B. Marder, Chem. – Eur. J., 2015, 21, 177–190 CrossRef CAS PubMed.
- L. Weber, D. Eickhoff, J. Kahlert, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann and M. A. Fox, Dalton Trans., 2012, 41, 10328–10346 RSC.
- S.-B. Zhao, P. Wucher, Z. M. Hudson, T. M. McCormick, X.-Y. Liu, S. Wang, X.-D. Feng and Z.-H. Lu, Organometallics, 2008, 27, 6446–6456 CrossRef CAS.
- (a) Y. Kim, H.-S. Huh, M. H. Lee, I. L. Lenov, H. Zhao and F. P. Gabbaï, Chem. – Eur. J., 2011, 17, 2057–2062 CrossRef CAS PubMed; (b) C. Wang, J. Jia, W.-N. Zhang, H.-Y. Zhang and C. H. Zhao, Chem. – Eur. J., 2014, 17, 16590–16601 CrossRef PubMed; (c) J. Jia, P. Xue, Y. Zhang, Q. Xu, G. Zhang, T. Huang, H. Zhang and R. Lu, Tetrahedron, 2014, 70, 5499–5504 CrossRef CAS.
- C. Bresner, C. J. E. Haynes, D. A. Addy, A. E. J. Broomsgrove, P. Fitzpatrick, D. Vidovic, A. L. Thompson, I. A. Fallis and S. Aldridge, New J. Chem., 2010, 34, 1652–1659 RSC.
- R. N. Grimes, Carboranes, Academic Press (Elsevier), New York, 2nd edn, 2011 Search PubMed.
- For other reviews on carboranes see: (a) B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, New J. Chem., 2011, 35, 1955–1972 RSC; (b) I. B. Sivaev and V. V. Bregadze, Eur. J. Inorg. Chem., 2009, 1433–1450 CrossRef CAS; (c) F. Issa, M. Kassiou and L. M. Rendina, Chem. Rev., 2011, 111, 5701–5722 CrossRef CAS PubMed; (d) M. Scholz and E. Hey-Hawkins, Chem. Rev., 2011, 111, 7035–7062 CrossRef CAS PubMed; (e) J. F. Valliant, K. J. Guenther, A. S. King, P. Morel, P. Schaffer, O. O. Sogbein and K. A. Stephenson, Coord. Chem. Rev., 2002, 232, 173–230 CrossRef CAS; (f) V. N. Kalinin and V. A. Ol'shevskaya, Russ. Chem. Bull., 2008, 57, 815–836 CrossRef CAS; (g) V. I. Bregadze, Chem. Rev., 1992, 92, 209–223 CrossRef CAS; (h) A. F. Armstrong and J. F. Valliant, Dalton Trans., 2007, 4240–4251 RSC; (i) L. A. Leites, Chem. Rev., 1992, 92, 279–323 CrossRef CAS; (j) T. J. Wedge and M. F. Hawthorne, Coord. Chem. Rev., 2003, 240, 111–128 CrossRef CAS; (k) D. Olid, C. Viñas and F. Teixidor, Chem. Soc. Rev., 2013, 42, 3318–3336 RSC; (l) C. Viñas, R. Núñez and F. Teixidor, Large molecules containing icosahedral boron clusters designed for potential applications, in Boron Science, ed. N. S. Hosmane, CRC Press, New York, 2012, ch. 27 Search PubMed.
- (a) B. P. Dash, R. Satapathy, J. A. Maguire and N. S. Hosmane, Chem. Commun., 2009, 3267–3269 RSC; (b) Y. A. Kabachii and P. M. Valetskii, Int. J. Polym. Mater., 1990, 14, 9–19 CrossRef CAS; (c) K. Hideaki, O. Koichi, I. Motokuni, S. Toshiya, K. Shigeki and A. Isao, Chem. Mater., 2003, 15, 355–362 CrossRef; (d) E. Hao, B. Fabre, F. R. Fronczek and M. G. H. Vicente, Chem. Commun., 2007, 4387–4389 RSC; (e) E. Hao, B. Fabre, F. R. Fronczek and M. G. H. Vicente, Chem. Mater., 2007, 19, 6195–6205 CrossRef CAS; (f) M. A. Fox and K. Wade, J. Mater. Chem., 2002, 12, 1301–1306 RSC; (g) O. K. Farha, A. M. Spokoyny, K. L. Mulfort, M. F. Hawthorne, C. A. Mirkin and J. T. Hupp, J. Am. Chem. Soc., 2007, 129, 12680–12681 CrossRef CAS PubMed.
- (a) J. Llop, C. Viñas, J. M. Oliva, F. Teixidor, M. A. Flores, R. Kivekäs and R. Sillanpää, J. Organomet. Chem., 2002, 657, 232–238 CrossRef CAS; (b) J. M. Oliva, N. L. Allan, P. v. R. Schleyer, C. Viñas and F. Teixidor, J. Am. Chem. Soc., 2005, 127, 13538–13547 CrossRef CAS PubMed; (c) D. A. Brown, W. Clegg, H. M. Colquhoun, J. A. Daniels, I. R. Stephenson and K. Wade, J. Chem. Soc., Chem. Commun., 1987, 889–891 RSC; (d) L. A. Boyd, W. Clegg, R. C. B. Copley, M. G. Davidson, M. A. Fox, T. G. Hibbert, J. A. K. Howard, A. Mackinnon, R. J. Peace and K. Wade, Dalton Trans., 2004, 2786–2799 RSC.
- (a) T. D. Getman, C. B. Knobler and M. F. Hawthorne, J. Am. Chem. Soc., 1990, 112, 4593–4594 CrossRef CAS; (b) T. D. Getman, C. B. Knobler and M. F. Hawthorne, Inorg. Chem., 1992, 31, 101–105 CrossRef CAS.
- For recent articles and seminal reviews on ortho-carborane derivatives, see: (a) S. V. Svidlov, O. A. Varzatskii, T. V. Potapova, A. V. Vologzhanina, S. S. Bukalov, L. A. Leites, Y. Z. Voloshin and Y. N. Bubnov, Inorg. Chem. Commun., 2014, 43, 142–145 CrossRef CAS; (b) J. Marshall, J. Hooton, Y. Han, A. Creamer, R. S. Ashraf, Y. Porte, T. D. Anthopoulos, P. N. Stavrinou, M. A. McLachlan, H. Bronstein, P. Beavis and M. Heeney, Polym. Chem., 2014, 5, 6190–6199 RSC; (c) J. Marshall, Z. Fei, C. P. Yau, N. Yaacobi-Gross, S. Rossbauer, T. D. Anthopoulos, S. E. Watkins, P. Beavis and M. Heeney, J. Mater. Chem. C, 2014, 2, 232–239 RSC; (d) J. Fajardo, A. L. Chan, F. S. Tham and V. Lavallo, Inorg. Chim. Acta, 2014, 422, 206–208 CrossRef CAS; (e) B. Wrackmeyer, E. V. Klimkina and W. Milius, Eur. J. Inorg. Chem., 2014, 233–246 CrossRef CAS; (f) Z. Qiu, Tetrahedron Lett., 2015, 56, 963–971 CrossRef CAS; (g) J. U. Kahlert, A. Rawal, J. M. Hook, L. M. Rendina and M. Choucair, Chem. Commun., 2014, 50, 11332–11334 RSC; (h) M. E. El-Zaria, K. Keskar, A. R. Genady, J. A. Ioppolo, J. McNulty and J. F. Valliant, Angew. Chem., Int. Ed., 2014, 53, 5156–5160 CAS; (i) K. Junold, J. A. Baus, C. Burschka, M. Finze and R. Tacke, Eur. J. Inorg. Chem., 2014, 5099–5102 CrossRef CAS; (j) R. N. Grimes, Dalton Trans., 2015, 44, 5939–5956 RSC; (k) J. M. Ludlow III, M. Tominaga, Y. Chujo, A. Schultz, X. Lu, T. Xie, K. Guo, C. N. Moorefield, C. Wesdemiotis and G. R. Newkome, Dalton Trans., 2014, 43, 9604–9611 RSC; (l) A. Kreienbrink, M. B. Sárosi, R. Kuhnert, P. Wonneberger, A. I. Arkhypchuk, P. Lönnecke, S. Ott and E. Hey-Hawkins, Chem. Commun., 2015, 51, 836–838 RSC; (m) Y. Quan and Z. Xie, J. Am. Chem. Soc., 2015, 137, 3502–3505 CrossRef CAS PubMed; (n) Y. Nie, Y. Wang, J. Miao, D. Bian, Z. Zhang, Y. Cui and G. Sun, Dalton Trans., 2014, 43, 5083–5094 RSC; (o) V. I. Bregadze, Russ. Chem. Bull., 2014, 63, 1021–1026 CrossRef CAS; (p) K. Cao, Y. Huang, J. Yang and J. Wu, Chem. Commun., 2015, 51, 7257–7260 RSC.
- (a) V. Z. Paschenko, R. P. Evstigneeva, V. V. Gorokhov, V. N. Luzgina, V. B. Tusov and A. B. Rubin, J. Photochem. Photobiol., B, 2000, 54, 162–167 CrossRef CAS; (b) V. N. Luzgina, V. A. Ol'shevskaya, A. V. Sekridova, A. F. Mironov, V. N. Kalinin, V. Z. Pashchenko, V. V. Gorokhov, V. B. Tusov and A. A. Shtil’, Russ. J. Org. Chem., 2007, 43, 1243–1251 CrossRef CAS; (c) B. P. Dash, R. Satapathy, E. R. Galliard, K. M. Norton, J. A. Maguire, N. Chug and N. S. Hosmane, Inorg. Chem., 2011, 50, 5485–5493 CrossRef CAS PubMed; (d) A. Ferrer-Ugalde, E. J. Juárez-Pérez, F. Teixidor, C. Viñas, R. Sillanpää, E. Pérez-Inestrosa and R. Núñez, Chem. – Eur. J., 2012, 18, 544–553 CrossRef CAS PubMed; (e) L. Zhu, W. Lv, S. Liu, H. Yan, Q. Zhao and W. Huang, Chem. Commun., 2013, 49, 10638–10640 RSC; (f) A. Ferrer-Ugalde, A. González-Campo, C. Viñas, J. Rodriguez-Romero, R. Santillan, N. Farfán, R. Sillanpää, A. Sousa-Pedrares, R. Núñez and F. Teixidor, Chem. – Eur. J., 2014, 20, 9940–9951 CrossRef CAS PubMed; (g) G. F. Jin, Y.-J. Cho, K.-R. Wee, S. A. Hong, I.-H. Suh, H.-J. Son, J.-D. Lee, W.-S. Han, D. W. Cho and S. O. Kang, Dalton Trans., 2015, 44, 2780–2787 RSC.
- (a) K. Kokado and Y. Chujo, Macromolecules, 2009, 42, 1418–1420 CrossRef CAS; (b) K. Kokado, Y. Tokoro and Y. Chujo, Macromolecules, 2009, 42, 9238–9242 CrossRef CAS; (c) K. Kokado, A. Nagai and Y. Chujo, Macromolecules, 2010, 43, 6463–6468 CrossRef CAS; (d) K. Kokado and Y. Chujo, Polym. J., 2010, 42, 363–367 CrossRef CAS; (e) K. Kokado, A. Nagai and Y. Chujo, Tetrahedron Lett., 2011, 52, 293–296 CrossRef CAS; (f) K. Kokado and Y. Chujo, Dalton Trans., 2011, 40, 1919–1923 RSC; (g) K. Kokado and Y. Chujo, J. Org. Chem., 2011, 76, 316–319 CrossRef CAS PubMed; (h) M. Tominaga, H. Naito, Y. Morisaki and Y. Chujo, Asian J. Org. Chem., 2014, 3, 624–631 CrossRef CAS; (i) M. Tominaga, H. Naito, Y. Morisaki and Y. Chujo, New J. Chem., 2014, 38, 5686–5690 RSC.
- (a) K.-R. Wee, W.-S. Han, D. W. Cho, S. Kwon, C. Pac and S. O. Kang, Angew. Chem., 2012, 124, 2731–2734 ( Angew. Chem., Int. Ed. , 2012 , 51 , 2677–2680 ) CrossRef; (b) S. Kwon, K.-R. Wee, Y.-J. Cho and S. O. Kang, Chem. – Eur. J., 2014, 20, 5953–5960 CrossRef CAS PubMed.
- S. Inagi, K. Hosoi, T. Kubo, N. Shida and T. Fuchigami, Electrochemistry, 2013, 81, 368–370 CrossRef CAS.
- (a) M. Eo, M. H. Park, T. Kim, Y. Do and M. H. Lee, Polymer, 2013, 54, 6321–6328 CrossRef CAS; (b) H. J. Bae, H. Kim, K. M. Lee, T. Kim, Y. S. Lee, Y. Do and M. H. Lee, Dalton Trans., 2014, 43, 4978–4985 RSC; (c) H. Naito, Y. Morisaki and Y. Chujo, Angew. Chem., 2015, 127, 5173–5176 ( Angew. Chem., Int. Ed. , 2015 , 54 , 5084–5087 ) CrossRef.
- (a) A. R. Davis, J. J. Peterson and K. R. Carter, ACS Macro Lett., 2012, 1, 469–472 CrossRef CAS; (b) J. J. Peterson, A. R. Davis, M. Werre, E. B. Coughlin and K. R. Carter, ACS Appl. Mater. Interfaces, 2011, 3, 1796–1799 CrossRef CAS PubMed; (c) J. J. Peterson, M. Werre, Y. C. Simon, E. B. Coughlin and K. R. Carter, Macromolecules, 2009, 42, 8594–8598 CrossRef CAS.
- L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, R. A. Harder and M. A. Fox, Chem. – Eur. J., 2012, 18, 8347–8357 CrossRef CAS PubMed.
- L. Weber, J. Kahlert, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann, R. A. Harder, P. J. Low and M. A. Fox, Dalton Trans., 2013, 42, 2266–2281 RSC.
- L. Weber, J. Kahlert, R. Brockhinke, L. Böhling, J. Halama, A. Brockhinke, H.-G. Stammler, B. Neumann, C. Nervi, R. A. Harder and M. A. Fox, Dalton Trans., 2013, 42, 10982–10996 RSC.
- K.-R. Wee, Y.-J. Cho, J. K. Song and S. O. Kang, Angew. Chem., 2013, 125, 9864–9867 ( Angew. Chem., Int. Ed. , 2013 , 52 , 9682–9685 ) CrossRef.
- K.-R. Wee, Y.-J. Cho, S. Jeong, S. Kwon, J.-D. Lee, I.-H. Suh and S. O. Kang, J. Am. Chem. Soc., 2012, 134, 17982–17990 CrossRef CAS PubMed.
- J. O. Huh, H. Kim, K. M. Lee, Y. S. Lee, Y. Do and M. H. Lee, Chem. Commun., 2010, 46, 1138–1140 RSC.
- K. C. Song, H. Kim, K. M. Lee, Y. S. Lee, Y. Do and M. H. Lee, Dalton Trans., 2013, 42, 2351–2354 RSC.
- Z. Yuan, C. D. Entwistle, J. C. Collings, D. Albesa-Jove, A. S. Batsanov, J. A. K. Howard, H. M. Kaiser, D. E. Kaufmann, S.-Y. Poon, W.-Y. Wong, C. Jardin, S. Fatallah, A. Boucekkine, J.-F. Halet and T. B. Marder, Chem. – Eur. J., 2006, 12, 2758–2771 CrossRef CAS PubMed.
- (a) J. L. Boone, R. J. Brotherton and L. L. Petterson, Inorg. Chem., 1965, 4, 910–912 CrossRef CAS; (b) Y. Z. Voloshin, S. Y. Erdyakov, I. G. Makarenko, E. G. Lebed’, T. V. Potapova, S. V. Svidlov, Z. A. Starikova, E. V. Pol'shin, M. E. Gurskii and Y. N. Bubnov, Russ. Chem. Bull., 2007, 56, 1787–1794 CrossRef CAS; (c) S. Y. Erdyakov, Y. Z. Voloshin, I. G. Makarenko, E. G. Lebed, T. V. Potapova, A. V. Ignatenko, A. V. Vologzhanina, M. E. Gurskii and Y. N. Bubnov, Inorg. Chem. Commun., 2009, 12, 135–139 CrossRef CAS; (d) K. Ohta, T. Goto, H. Yamazaki, F. Pichierri and Y. Endo, Inorg. Chem., 2007, 46, 3966–3970 CrossRef CAS PubMed.
- D. A. Brown, H. M. Colquhoun, J. A. Daniels, J. A. H. MacBride, I. R. Stephenson and K. Wade, J. Mater. Chem., 1992, 2, 793–804 RSC.
- G. Zi, H.-W. Li and Z. Xie, Organometallics, 2002, 21, 3850–3855 CrossRef CAS.
- (a) G. Zi, H.-W. Li and Z. Xie, Organometallics, 2002, 21, 1136–1145 CrossRef CAS; (b) Y. Nie, J. Miao, H. Wadepohl, H. Pritzkow, T. Oeser and W. Siebert, Z. Anorg. Allg. Chem., 2013, 639, 1188–1193 CrossRef CAS.
- Y. Nie, J. Miao, H. Pritzkow, H. Wadepohl and W. Siebert, J. Organomet. Chem., 2013, 747, 174–177 CrossRef CAS.
- N. M. D. Brown, F. Davidson, R. McMullan and J. W. Wilson, J. Organomet. Chem., 1980, 193, 271–282 CrossRef CAS.
- R. Anulewicz-Ostrowska, S. Luliński, J. Serwatowski and K. Suwińska, Inorg. Chem., 2000, 39, 5763–5767 CrossRef CAS PubMed.
- H. C. Brown and V. H. Dodson, J. Am. Chem. Soc., 1957, 79, 2302–2306 CrossRef CAS.
- (a) H. Tomita, H. Luu and T. Onak, Inorg. Chem., 1991, 30, 812–815 CrossRef CAS; (b) M. A. Fox, J. A. H. MacBride and K. Wade, Polyhedron, 1997, 16, 2449–2507 Search PubMed; (c) J. Yoo, J.-W. Hwang and Y. Do, Inorg. Chem., 2001, 40, 568–570 CrossRef CAS PubMed.
- (a) J. J. Eisch, B. Shafii, J. D. Odom and A. L. Rheingold, J. Am. Chem. Soc., 1990, 112, 1847–1853 CrossRef CAS; (b) M. H. Chisholm, K. Folting, S. T. Haubrich and J. D. Martin, Inorg. Chim. Acta, 1993, 213, 17–24 CrossRef CAS.
- D. C. Busby and M. F. Hawthorne, Inorg. Chem., 1982, 21, 4101–4103 CrossRef CAS.
- Z. G. Lewis and A. J. Welch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, 49, 705–710 CrossRef.
- A search in the CCDC database in May 2012 revealed that 95% of the B–CMes bond lengths in 37 structures of p-substituted dimesitylborylbenzene derivatives are in the range of 1.55 Å–1.61 Å. The interplanar angles between the mesityl rings and the planes defined by the boryl-boron atom and the three neighbouring carbon atoms were found between 42.9° and 67.5° in these structures.
- (a) T. D. McGrath and A. J. Welch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1995, 51, 646–649 CrossRef; (b) E. S. Alekseyeva, M. A. Fox, J. A. K. Howard, J. A. H. MacBride and K. Wade, Appl. Organomet. Chem., 2003, 17, 499–508 CrossRef CAS.
- S. Schwedler, D. Eickhoff, R. Brockhinke, D. Cherian, L. Weber and A. Brockhinke, Phys. Chem. Chem. Phys., 2011, 13, 9301–9310 RSC.
- (a) E. Lippert, Z. Naturforsch., A: Phys. Sci., 1955, 10, 541–545 Search PubMed; (b) E. Lippert, Z. Elektrochem., 1957, 61, 962–975 CAS; (c) N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1955, 28, 690–691 CrossRef CAS; (d) N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS; (e) G. Weber and F. J. Farris, Biochemistry, 1979, 18, 3075–3078 CrossRef CAS PubMed; (f) A. C. Benniston, A. Harriman and J. P. Rostron, Phys. Chem. Chem. Phys., 2005, 7, 3041–3047 RSC.
- L. Weber, D. Eickhoff, T. B. Marder, M. A. Fox, P. J. Low, A. D. Dwyer, D. J. Tozer, S. Schwedler, A. Brockhinke, H.-G. Stammler and B. Neumann, Chem. – Eur. J., 2012, 18, 1369–1382 CrossRef CAS PubMed.
- (a) M. V. Yarosh, T. V. Baranova, V. L. Shirokii, A. A. Érdman and N. A. Maier, Elektrokhimiya, 1993, 29, 921–922 CAS. (Russian; English version Russ. J. Electrochem., 1993, 29, 789–790); (b) M. V. Yarosh, T. V. Baranova, V. L. Shirokii, A. A. Érdman and N. A. Maier, Elektrokhimiya, 1994, 30, 406–408 CAS . (Russian; English version Russ. J. Electrochem., 1994, 30, 366–368).
- R. A. Harder, J. A. H. MacBride, G. P. Rivers, D. S. Yufit, A. E. Goeta, J. A. K. Howard, K. Wade and M. A. Fox, Tetrahedron, 2014, 70, 5182–5189 CrossRef CAS.
- M. A. Fox, C. Nervi, A. Crivello and P. J. Low, Chem. Commun., 2007, 2372–2374 RSC.
- H. Tricas, M. Colon, D. Ellis, S. A. Macgregor, D. McKay, G. M. Rosair, A. J. Welch, I. V. Glukhov, F. Rossi, F. Laschi and P. Zanello, Dalton Trans., 2011, 40, 4200–4211 RSC.
- (a) M. A. Fox, C. Nervi, A. Crivello, A. S. Batsanov, J. A. K. Howard, K. Wade and P. J. Low, J. Solid State Electrochem., 2009, 13, 1483–1495 CrossRef CAS; (b) G. F. Jin, J.-H. Hwang, J.-D. Lee, K.-R. Wee, I.-H. Suh and S. O. Kang, Chem. Commun., 2013, 49, 9398–9400 RSC; (c) J. Kahlert, H.-G. Stammler, B. Neumann, R. A. Harder, L. Weber and M. A. Fox, Angew. Chem., 2014, 126, 3776–3779 ( Angew. Chem., Int. Ed. , 2014 , 53 , 3702–3705 ) CrossRef.
- K. Hosoi, S. Inagi, T. Kubo and T. Fuchigami, Chem. Commun., 2011, 47, 8632–8634 RSC.
- (a) A. V. Lebedev, A. V. Buchtiarov, N. N. Golyshin, Y. G. Kudryatsev, I. Y. Lovchinovsky and L. N. Rozhkov, Organomet. Chem. USSR, 1991, 4, 205–208 Search PubMed. (English Transl.); (b) A. V. Lebedev, A. V. Bukhtiarov, Y. G. Kudryavtev and I. N. Rozhkov, Organomet. Chem. USSR, 1991, 4, 208–212 Search PubMed. (English Transl.); (c) A. V. Bukhtiarov, V. N. Golyshin, A. V. Lebedev, Y. G. Kudryavtsev, I. A. Rodnikov, L. I. Zakharkin and O. V. Kuz'min, Dokl. Acad. Sci. USSR, 1989, 45–48 Search PubMed . (English Transl.).
- M. A. Fox, Polyhedral Carboranes, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mingos, Elsevier, Oxford, 2007, ch. 3.02 Search PubMed.
- K. Chui, H.-W. Li and Z. Xie, Organometallics, 2000, 19, 5447–5453 CrossRef CAS.
- (a) G. Zi, H.-W. Li and Z. Xie, Organometallics, 2001, 20, 3836–3838 CrossRef CAS; (b) G. Zi, H.-W. Li and Z. Xie, Chem. Commun., 2001, 1110–1111 RSC.
- G. Zi, H.-W. Li and Z. Xie, Organometallics, 2002, 21, 5415–5427 CrossRef CAS.
- (a) M.-S. Cheung, H.-S. Chan and Z. Xie, Organometallics, 2004, 23, 517–526 CrossRef CAS; (b) H. Shen, H.-S. Chan and Z. Xie, Organometallics, 2006, 25, 2617–2625 CrossRef CAS; (c) L. Deng, H.-S. Chan and Z. Xie, Inorg. Chem., 2007, 46, 2716–2724 CrossRef CAS PubMed.
- L. Deng, M.-S. Cheung, H.-S. Chen and Z. Xie, Organometallics, 2005, 24, 6244–6249 CrossRef CAS.
- (a) T. L. Venable, R. B. Maynard and R. N. Grimes, J. Am. Chem. Soc., 1984, 106, 6187–6193 CrossRef CAS; (b) J. T. Spencer, M. R. Pourian, R. J. Butcher, E. Sinn and R. N. Grimes, Organometallics, 1987, 6, 335–343 CrossRef CAS; (c) N. S. Hosmane, T. J. Colacot, H. Zhang, J. Yang, J. A. Maguire, Y. Wang, M. B. Ezhova, A. Franken, T. Demissie, K.-J. Lu, D. Zhu, J. L. C. Thomas, J. D. Collins, T. G. Gray, S. N. Hosmane and W. N. Lipscomb, Organometallics, 1998, 17, 5294–5309 CrossRef CAS.
- J. P. H. Charmant, M. F. Haddow, R. Mistry, N. C. Norman, A. G. Orpen and P. G. Pringle, Dalton Trans., 2008, 1409–1411 RSC.
- (a) M. Bühl and P. v. R. Schleyer, J. Am. Chem. Soc., 1992, 114, 477–491 CrossRef; (b) P. v. R. Schleyer, J. Gauss, M. Bühl, R. Greatrex and M. A. Fox, J. Chem. Soc., Chem. Commun., 1993, 1766–1768 RSC; (c) C. E. Willans, C. A. Kilner and M. A. Fox, Chem. – Eur. J., 2010, 16, 10644–10648 CrossRef CAS PubMed.
- S. Zlatogorsky, D. Ellis, G. M. Rosair and A. J. Welch, Chem. Commun., 2007, 2178–2180 RSC.
- J. Zhang, X. Fu, Z. Lin and Z. Xie, Inorg. Chem., 2015, 54, 1965–1973 CrossRef CAS PubMed.
- (a) L. I. Zakharkin, V. N. Kalinin and L. S. Podvisotskaya, Bull. Acad. Sci. USSR Div. Chem. Sci., 1966, 1444 CrossRef. (English Transl.); (b) L. I. Zakharkin, V. N. Kalinin and L. S. Podvisotskaya, Bull. Acad. Sci. USSR Div. Chem. Sci., 1967, 2212–2217 CrossRef. (English Transl.); (c) L. I. Zakharkin, Pure Appl. Chem., 1972, 29, 513–526 CrossRef CAS; (d) L. I. Zakharkin, V. N. Kalinin, V. A. Antonovich and E. G. Rhys, Bull. Acad. Sci. USSR Div. Chem. Sci., 1976, 1009–1014 CrossRef . (English Transl.).
- A. Pelter, K. Smith and H. C. Brown, Borane Reagents, Academic Press, London, 1988, p. 428 Search PubMed.
- L. I. Zakharkin, V. I. Bregadze and O. Y. Okhlobystin, J. Organomet. Chem., 1966, 6, 228–234 CrossRef CAS.
- E. S. Alekseyeva, A. S. Batsanov, L. A. Boyd, M. A. Fox, T. G. Hibbert, J. A. K. Howard, J. A. H. MacBride, A. Mackinnon and K. Wade, Dalton Trans., 2003, 475–482 RSC.
- J. C. de Mello, H. F. Wittmann and R. H. Friend, Adv. Mater., 1997, 9, 230–232 CrossRef CAS.
- M. Krejcik, M. Danek and F. Hartl, J. Electroanal. Chem., 1991, 317, 179–187 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09 (Revision A.02), Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- (a) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS; (b) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- U. Varetto, MOLEKEL Version, Swiss National Supercomputing Centre, Mann, Switzerland Search PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- (a) R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS; (b) C. M. Rohling, L. C. Allen and R. Ditchfield, Chem. Phys., 1984, 87, 9–15 CrossRef; (c) K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
Footnotes |
| † In memory of Ken Wade, a brilliant chemist and mentor. |
| ‡ Electronic supplementary information (ESI) available: Absorption spectra for 1 and 2, detailed CV data for 1 and 2, TD-DFT data for 1 and 2, spectroelectrochemical data for 2, computed GIAO-NMR data for 1–4, NMR spectra for 1–4 and dianions [2]2− and [5]2−, crystallographic data for 1 and 2 and Cartesian coordinates for eleven optimised geometries. CCDC 1048027–1048028. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00758e |
| This journal is © The Royal Society of Chemistry 2015 |