
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An iron(II) spin-crossover metallacycle from a back-to-back bis-[dipyrazolylpyridine]†
Laurence J.
Kershaw Cook
a,
Julie
Fisher
a,
Lindsay P.
Harding
b and
Malcolm A.
Halcrow
*a
aSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, UK LS2 9JT. E-mail: m.a.halcrow@leeds.ac.uk; Fax: +44 (0)113 343 6565; Tel: +44 (0)113 343 6506
bDepartment of Chemical and Biological Sciences, University of Huddersfield, Huddersfield, UK HD1 3DH
First published on 16th April 2015
Abstract
The syntheses of 4-mercapto-2,6-di(pyrazol-1-yl)pyridine (bppSH) and bis[2,6-di(pyrazol-1-yl)pyrid-4-yl]disulfide (bppSSbpp) are reported. In contrast to previously published “back-to-back” bis-[2,6-di(pyrazol-1-yl)pyridine] derivatives, which form coordination polymers with transition ions that are usually insoluble, bppSSbpp yields soluble oligomeric complexes with iron(II) and zinc(II). Mass spectrometry and DOSY data show that [{Fe(μ-bppSSbpp)}n]2n+ and [{Zn(μ-bppSSbpp)}n]2n+ form tetranuclear metallacycles in nitromethane solution (n = 4), although 1H NMR and conductivity measurements imply the iron compound may undergo more fragmentation than its zinc congener. Both [{Fe(bppSH)2]2+ and [{Fe(μ-bppSSbpp)}n]2n+ exhibit thermal spin-crossover in CD3NO2 solution, with midpoint temperatures near 245 K. The similarity of these equilibria implies there is little cooperativity between the iron centres in the metallacyclic structures.
Introduction
One of the most useful series of compounds in the field of spin-crossover research1–5 has proven to be [Fe(bpp)2]2+, where bpp is 2,6-di(pyrazol-1-yl)pyridine or a derivative of it (Scheme 1).6–8 Two particular properties of [Fe(bpp)2]2+ centres account for their ubiquity. First, is that procedures are available to functionalise every position of the bpp ligand skeleton with a range of substituents.1,3 Second, is that they often exhibit spin-state transitions at or near room temperature.1,2 Thus, this has afforded molecular switches based on [Fe(bpp)2]2+ spin-transition centres bearing peripheral fluorescent,9 conducting10 or photo-isomerisable substituents,11 yielding complexes combining spin-crossover with those functionalities. Alternatively, [Fe(bpp)2]2+ complexes with appropriate tether substituents have been used to form switchable surface nanostructures12–14 and single-molecule junctions.15 [Fe(bpp)2]2+ derivatives bearing protic or basic pyridyl substituents have proven useful in studies of the crystal engineering of supramolecular switching assemblies.13,16,17 Finally, ‘back-to-back’ ligands with two bpp metal-binding domains linked through their pyridyl groups, have yielded 1D coordination polymers with spin-crossover functionality.14,18,19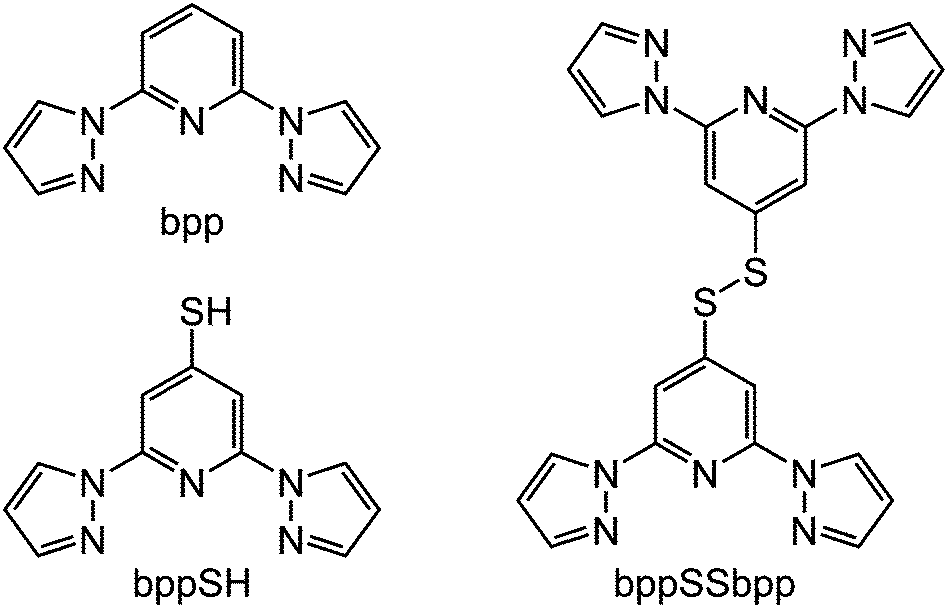 | ||
| Scheme 1 The ligands discussed in this work. | ||
A group of compounds from the bpp family that have not been investigated until now are derivatives bearing sulfur substituents at their pyridyl ring, of which 4-mercapto-2,6-di(pyrazol-1-yl)pyridine (bppSH) is the parent (Scheme 1). While such compounds could be used in self-assembled monolayers‡, bppSH is also of interest as a precursor to larger assembly structures through disulfide formation, or alkylation or ligation of the peripheral thiyl group. Such assemblies have been produced using closely related [Fe(terpySH)2]2+ as a component (terpySH = 4′-mercapto-2,2′:6′,2′′-terpyridine).20–23 For example, the disulfide bis[2,2′:6′,2′′-terpyridine-4′-yl]disulfide (terpySSterpy) produces a cyclic oligomer [{Fe(μ-terpySSterpy)}4]8+ when complexed to iron(II), which was crystallographically characterised (Fig. 1).20,24 We reasoned that a comparable metallacycle based on bis[2,6-di(pyrazol-1-yl)pyrid-4-yl]disulfide (bppSSbpp) could be a new type of multimetallic spin-crossover assembly.25–28 We therefore now report the synthesis of bppSH and bppSSbpp, and a study of their coordination chemistry.
 | ||
| Fig. 1 Crystallographic structure of the [{Fe(μ-terpySSterpy)}4]8+ metallacycle (data taken from ref. 20). The [{M(μ-bppSSbpp)}4]8+ (M = Fe or Zn) assemblies in this work are proposed to have the same connectivity as this structure. Colour code: C, white; Fe, green; N, blue; S, pink. | ||
Results and discussion
Following a literature method for terpySH,20 reaction of 4-iodo-2,6-di(pyrazolyl)pyridine16 with NaSH in refluxing dmf affords bppSH in almost quantitative yield. Interestingly, its characterisation data imply that bppSH exists as its thiol tautomer (form A, Scheme 2), rather than as the alternative thione tautomer adopted by 4-mercaptopyridine (form B).29 The evidence includes a strong ν(S–H) vibration at 2526 cm−1, and the absence of a comparable ν(N–H) stretch, in the solid state IR spectrum of bppSH; and, the appearance of its pyridyl C4 NMR resonance at 152.1 ppm in CDCl3, essentially identical to that shown by the disulfide bppSSbpp described below (a thione tautomer would exhibit this peak near 195 ppm29). In contrast, terpySH exists as the thione tautomer in the single crystal,20 but has been reported to be either pure thione or a mixture of both forms in solution.20,21 The preference of bppSH for the thiol form may reflect the electron withdrawing nature of its pyrazolyl substituents, which could make its central pyridyl group insufficiently basic to deprotonate the thiol group (pKa ≈ 530). | ||
| Scheme 2 The tautomeric equilibrium exhibited by 4-mercaptopyridines, between thiol (A) and thione (B) forms. bppSH (R = pyrazolyl) exhibits form A, but terpySH (R = pyridyl) predominantly adopts form B. | ||
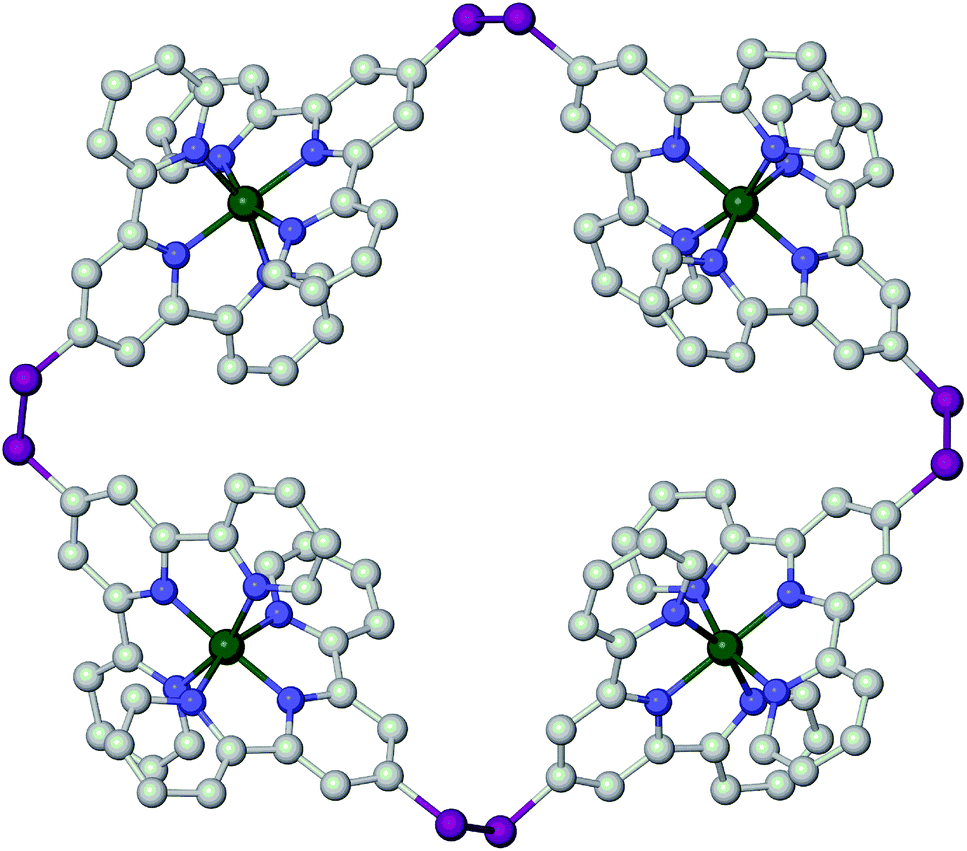
Solutions of bppSH in air oxidise slowly to bis[2,6-di(pyrazol-1-yl)pyrid-4-yl]disulfide (bppSSbpp) over a period of days. The same reaction was achieved almost quantitatively by oxidation of bppSH with I2 in the presence of base. The identity of bppSSbpp was confirmed by mass spectrometry (m/z = 507.1, corresponding to [Na(bppSSbpp)]+) and by a crystal structure determination (Fig. 2). The coplanar, transoid disposition of the heterocyclic rings in each dipyrazolylpyridyl (bpp) fragment is typical for this class of compound.31 The torsion angle about the disulfide group is 82.45(7)° and the two bpp metal-binding domains in the molecule are almost perfectly perpendicular to each other, the dihedral angle between them being 88.86(2)°. The molecules in the crystal associate into 1D stacks via π⋯π interactions between bpp fragment N(19)–C(36) and its symmetry equivalents N(19i)–C(36i) and N(19ii)–C(36ii) (symmetry codes (ii) −x, 2 − y, −z; (ii) 1 − x, 2 − y, −z), which are related by crystallographic inversion centres translating along a. These stacks are in turn linked by another pairwise π⋯π interaction, between bpp the moieties N(1)–C(16) and N(1iii)–C(16iii) (symmetry code (iii) 1 − x, 2 − y, 1 − z). In each case the interacting bpp fragments are coplanar by symmetry, and separated by 3.508(4)–3.510(4) Å (ESI†).
 | ||
| Fig. 2 View of the unique molecule in the crystal structure of bppSSbpp, emphasising the right-angled disposition of the metal-binding bpp moieties. Atomic displacement ellipsoids are drawn at the 50% probability level. Colour code: C, white; H, grey; N, blue; S, pink. | ||
Two salts [Fe(bppSH)2]X2 (X− = BF4−, 1[BF4]2; X− = BF4−, 1[ClO4]2) were prepared by treatment of the appropriate metal salt with 2 equiv. bppSH. Both salts precipitate from organic solvents as mixtures of polycrystalline and amorphous material by X-ray powder diffraction, which contain lattice water according to microanalysis (ESI†). Neither salt was obtained in single crystal form. Magnetic susceptibility measurements showed that both salts exhibit temperature-dependent spin-state behaviour in the solid state. However, only 1[ClO4]2 exhibits a well-defined spin-transition, which involves ca. 60% of the sample and is superimposed on a much more gradual spin-equilibrium involving the remainder of the material. The transition occurs with midpoint temperature T1/2 = 240 K and, unusually, exhibits a 15 K thermal hysteresis loop (ESI†). The first derivative of the χMT vs. T curve implies that both sides of the hysteretic transition are split into two components separated by 3–4 K, which was confirmed by a DSC measurement. Since 1[ClO4]2 has the greatest degree of crystallinity of the two salts, its hysteretic spin-transition can be attributed to the crystalline fraction of the material. However, the structural origin of this unusually cooperative transition could not be investigated further in the absence of a single crystal structure.
Treatment of hydrated Fe[BF4]2 or Fe[ClO4]2 with 1 equiv. bppSSbpp in nitromethane yields homogeneous solutions, whose orange colour is characteristic of [Fe(bpp)2]2+-type centres. Attempted crystallisation of the products from these solutions by slow solvent diffusion methods yielded intractable oils, and the complexes could only be isolated as solids by evaporation of the solutions to dryness. The solid residues were washed with diethyl ether, dried in vacuo and analysed without further purification. This yielded [{Fe(μ-bppSSbpp)}n]X2n (X− = BF4−, 2[BF4]2; X− = ClO4−, 2[ClO4]2). Since the iron compounds are paramagnetic (see below), the diamagnetic zinc(II) analogue [{Zn(μ-bppSSbpp)}n][BF4]2n (3[BF4]2) was also prepared by the same method, for the purposes of NMR characterisation. All three of these compounds retained 1/4–1/2 equiv. of diethyl ether per metal ion in the solid state by microanalysis, which was also visible in their 1H NMR spectra. The solubility of 2[BF4]2, 2[ClO4]2 and 3[BF4]2 in organic solvents is striking, since other back-to-back bis-bpp ligands without solubilising substituents afford insoluble coordination polymers when complexed to iron(II).18,19
These materials are completely amorphous powders by X-ray powder diffraction (ESI†). At 298 K, solid 2[BF4]2 and 2[ClO4]2 both exhibit χMT (per Fe atom) = 2.9 cm3 mol−1 K, showing they are predominantly high-spin at room temperature; a small low-spin population at room temperature is consistent with their red-orange colouration. Both samples undergo gradual and incomplete thermal spin-crossover equilibria, which are almost monotonic between 100–350 K but exhibit a 40–50% residual high-spin fraction frozen in below that temperature (Fig. 3). Such gradual and poorly defined spin-crossover is typical for amorphous samples, with a distribution of molecular environments in a heterogeneous solid matrix.
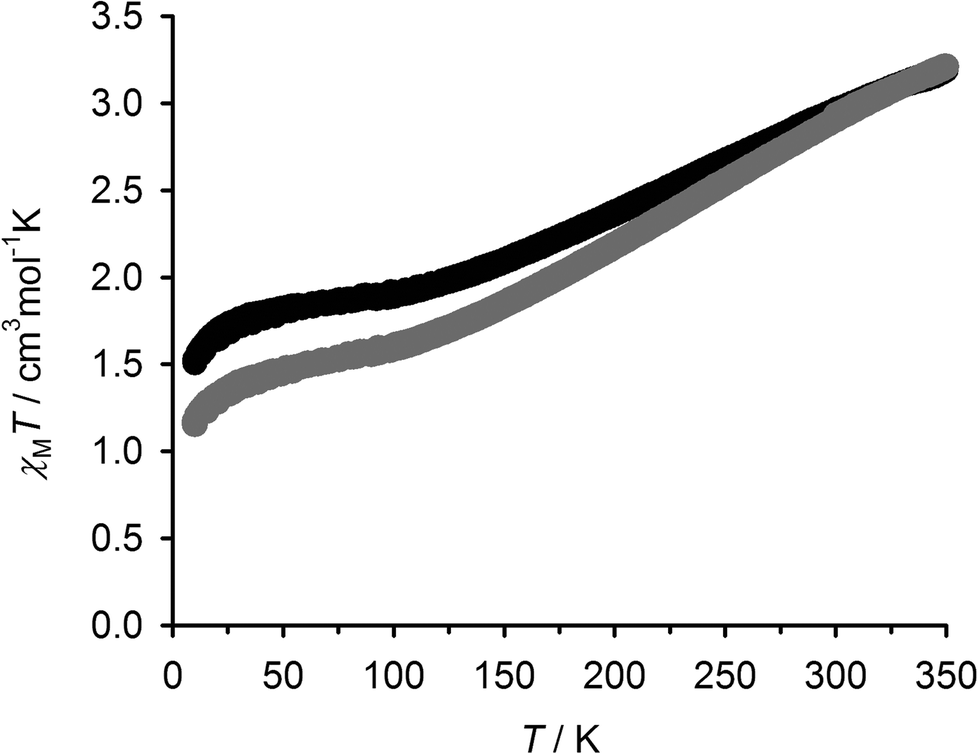 | ||
Fig. 3 Variable temperature magnetic susceptibility data for solid 2[BF4]2 (●) and 2[ClO4]2 ( ). The χMT data are quoted per mole equivalent of Fe. ). The χMT data are quoted per mole equivalent of Fe. | ||
The room temperature 1H NMR spectrum of 3[BF4]2 in the weakly interacting solvent CD3NO2 is broadened, but contains peaks attributable to one metal-bound bpp domain, and a second minor component that may be an uncoordinated bpp fragment based on its chemical shifts. The integral ratio of coordinated![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :uncoordinated bpp residues in the spectrum is ca. 9:1. In contrast, the 1H NMR spectra of paramagnetic 2[BF4]2 and 2[ClO4]2 contain contact-shifted peaks attributable to two metal-bound bpp environments with comparable intensities, and one uncoordinated bpp environment in the diamagnetic region. The ratio of coordinated:uncoordinated bpp residues cannot be quantified in these spectra, because the integrals of diamagnetic and contact-shifted NMR peaks are not directly comparable.32 None-the-less, the spectra confirm that 2[BF4]2 and 2[ClO4]2 are high-spin at room temperature in solution, as well as in the solid state. The observation of different numbers of coordinated bpp environments for 2X2 (X− = BF4− and ClO4−) and 3[BF4]2 was unexpected, and could have two explanations. The distribution of assembly structures in all the spectra could be similar, but in more rapid chemical exchange in 3[BF4]2 than in 2X2. That seems unlikely, since zinc(II) and high-spin iron(II) exhibit comparable rates of ligand exchange in solution.33 Alternatively, the CD3NO2 solutions of 2X2 and 3[BF4]2 may contain different molecular assemblies.
:uncoordinated bpp residues in the spectrum is ca. 9:1. In contrast, the 1H NMR spectra of paramagnetic 2[BF4]2 and 2[ClO4]2 contain contact-shifted peaks attributable to two metal-bound bpp environments with comparable intensities, and one uncoordinated bpp environment in the diamagnetic region. The ratio of coordinated:uncoordinated bpp residues cannot be quantified in these spectra, because the integrals of diamagnetic and contact-shifted NMR peaks are not directly comparable.32 None-the-less, the spectra confirm that 2[BF4]2 and 2[ClO4]2 are high-spin at room temperature in solution, as well as in the solid state. The observation of different numbers of coordinated bpp environments for 2X2 (X− = BF4− and ClO4−) and 3[BF4]2 was unexpected, and could have two explanations. The distribution of assembly structures in all the spectra could be similar, but in more rapid chemical exchange in 3[BF4]2 than in 2X2. That seems unlikely, since zinc(II) and high-spin iron(II) exhibit comparable rates of ligand exchange in solution.33 Alternatively, the CD3NO2 solutions of 2X2 and 3[BF4]2 may contain different molecular assemblies.
These data were supported by variable temperature conductivity measurements in MeNO2. A plot of (Λ0 − Λe) vs. c1/2 (Λ0 = conductivity at infinite dilution, Λe = measured conductivity and c = concentration) should yield a straight line whose slope is characteristic of the electrolyte composition of the solution.34 Since such linear behaviour was exhibited by all the compounds, the speciation of the [{M(μ-bppSSbpp)}n]2n+ (M = Fe and Zn) metallacycles does not change across the measured concentration range (Fig. 4).35 The slope of the plot for 1[ClO4]2 is 509, which is within the range expected for a 2:1 metal complex electrolyte in MeNO2.36 The slopes for 2[BF4]2 (1312) and 2[ClO4]2 and (1347) are identical within experimental error, implying that their anions do not contribute to their aggregation structures.37 Unexpectedly, however, a higher slope was obtained for 3[BF4]2 (1738). All these values are greater than expected for a 3:1 electrolyte in this solvent,36 although the electrolyte type for these compounds cannot be determined more accurately because of the lack of appropriate reference data.34
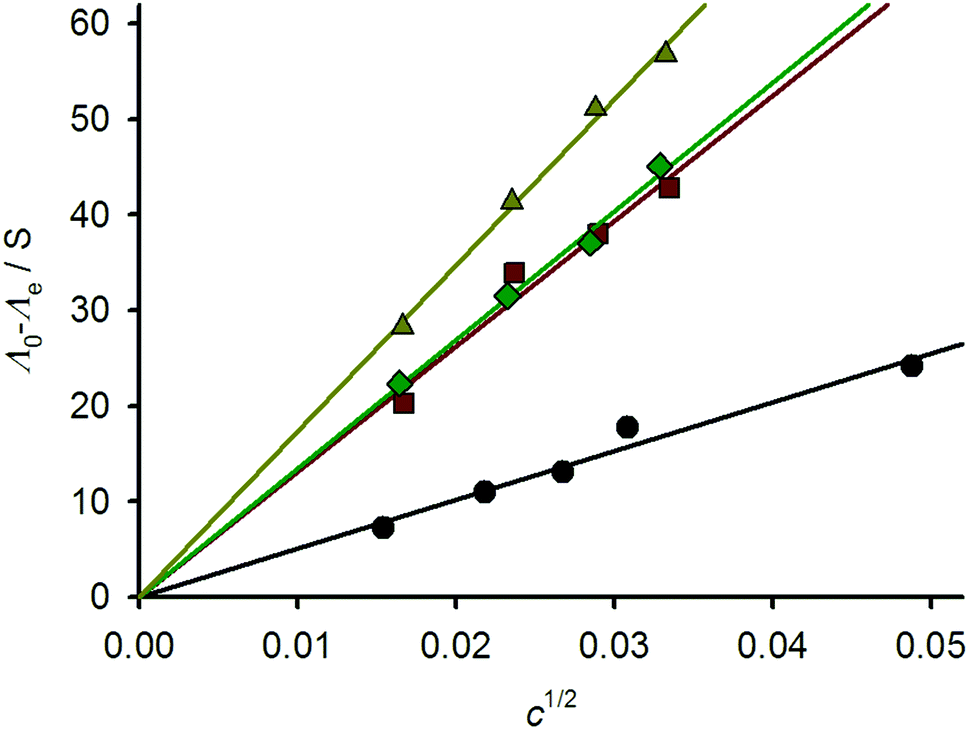 | ||
| Fig. 4 Variable concentration conductivity plots for 1[ClO4]2 (black circles), 2[BF4]2 (red squares), 2[ClO4]2 (green diamonds) and 3[BF4]2 (yellow triangles). | ||
The solution structure of 3[BF4]2 in CD3NO2 at 293 K was probed further by a diffusion oriented spectroscopy (DOSY) experiment,38 which has been shown previously to be useful for metallacyclic assemblies.39–41 The diffusion coefficient for the free bppSSbpp ligand under these conditions was 7.69 ± 0.56 × 10−10 m2 s−1. The DOSY analysis of 3[BF4]2 yielded just one aggregated species, with a diffusion coefficient of 3.58 ± 0.14 × 10−10 m2 s−1 (ESI†). A smaller fraction of free bppSSbpp was also detected, supporting the presence of trace free ligand in the complex's 1H NMR spectrum (see above). The ratio of the two diffusion coefficients is 0.46, which is lower than might be expected for a dimeric [{Zn(μ-bppSSbpp)}n]2n+ assembly (n = 2; theoretical monomer:dimer ratio ≈ 0.75) or an equilateral trimer (n = 3; monomer:trimer ratio ≈ 0.6),42 but is significantly higher than has been measured for a cylindrical 16-mer assembly.43
Assuming a stick solvent/solute relationship,44 the isotropic hydrodynamic radius of the [{M(μ-bppSSbpp)}n]2n+ assembly in 3[BF4]2 is 9.66 Å. That agrees excellently with the DOSY hydrodynamic radii for another series of tetranuclear metallacycles containing [M(terpy)2]2+ centres (rH = 9.7 Å).39 In contrast, larger hydrodynamic radii have been measured for hexameric (rH = 11.3 Å) and nonameric (rH = 11.8 Å) macrocycles of [M(terpy)2]2+ units.41 The crystallographic dimensions of the [{Fe(μ-terpySSterpy)}4]8+ tetramer are ca. 11 × 25.5 × 25.5 Å,20 yielding an averaged diameter of 20.7 Å, or radius of 10.3 Å. That is in reasonable agreement with the rH value for 3[BF4]2, particularly since the bpp metal-binding residues in bppSSbpp are slightly smaller than the terpy moieties in the literature compound.
Electrospray mass spectra of 2[BF4]2, 2[ClO4]2 and 3[BF4]2 all show strong molecular ions, corresponding to the same three species: [M(bppSSbpp)2X]+, [M2(bppSSbpp)2X3]+ and [M4(bppSSbpp)4X6]2+ (M = Fe or Zn, X− = anion; Fig. 5 and ESI†). Each spectrum also contains a number of weaker ions with relative intensities 3–10%, most of which can be derived from the cyclic tetramer species by loss or gain of bppSSbpp, loss or gain of metal cations and anions, or fragmentation. Only 2[ClO4]2 shows clear evidence of other cyclic oligomers through weak ions assignable to [Fe3(bppSSbpp)3(ClO4)4]2+ and [Fe5(bppSSbpp)5(ClO4)8]2+, each of 2–3% relative abundance (ESI†). Hence, the mass spectrometry data also imply that tetrameric [{M(μ-bppSSbpp)}4]8+ (M = Fe or Zn) is the predominant oligomeric species in solutions of these compounds.
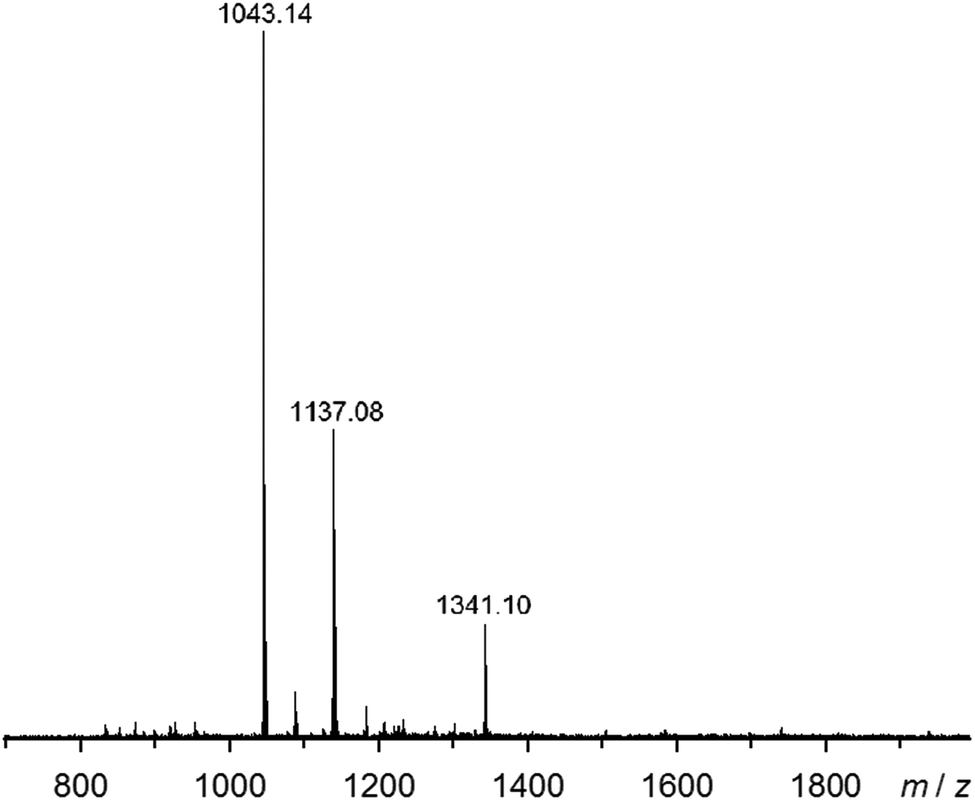 | ||
| Fig. 5 Electrospray mass spectrum of 2[BF4]2 from MeNO2 solution. The highlighted ions are [Fe(bppSSbpp)2F]+ (m/z = 1043.1); [Fe2(bppSSbpp)2(F)3]+ (m/z = 1137.1) and [Fe4(bppSSbpp)4(BF4)6]2+ (m/z = 1341.1). The F− ions arise from metal-promoted hydrolysis of BF4− under the conditions of the experiment. Other mass spectra and peak assignments are in the ESI.† | ||
Variable temperature Evans method magnetic susceptibility measurements in CD3NO2 solution from 1[BF4]2 and 2[BF4]2 gave very similar results. Both complexes are almost completely high-spin at room temperature under these conditions, but exhibit the onset of a thermal spin equilibrium on cooling (Fig. 6). The midpoint temperatures of the equilibria (T1/2) are both near the freezing point of the solvent, and can be estimated at 246 ± 1 K for 1[BF4]2 and 243 ± 1 K for 2[BF4]2. These values are almost identical to the parent complex [Fe(bpp)2]2+ (Scheme 1), which exhibits T1/2 = 248 K in (CD3)2CO45 (the use of a different organic solvent will have a minimal effect on these data46). The thermodynamic parameters for the equilibria are likewise similar, with 1[BF4]2 exhibiting ΔH = 29(2) kJ mol−1 and ΔS = 120(8) J mol−1 K−1; and for 2[BF4]2, ΔH = 25(2) kJ mol−1 and ΔS = 104(8) J mol−1 K−1 (per mole of Fe). The larger-than-normal errors on those values arise because the equilibria could not be fully measured within the liquid range of the CD3NO2 solvent; use of a solvent with a lower freezing point in this work was precluded on solubility grounds. The slightly higher ΔH value for 1[BF4]2 might be evidence for ligand dissociation in the high-spin state of that compound, which could be promoted by the nucleophilic thiol substituents. Be that as it may, such a rapid pre-equilibrium would have no effect on the spin-crossover T1/2.47 Conversely, the ΔH value for 2[BF4]2 is at the high end of the usual range for [Fe(bpp)2]2+ derivatives in inorganic solvents.45,46,48 That implies ligand displacement reactions play, at most, a minor role in the solution chemistry of 2[BF4]2.47 Hence, incorporation of the isolated [Fe(bpp)2]2+ centres in 1[BF4]2 into a metallacyclic structure in 2[BF4]2 has little effect on their spin state properties.
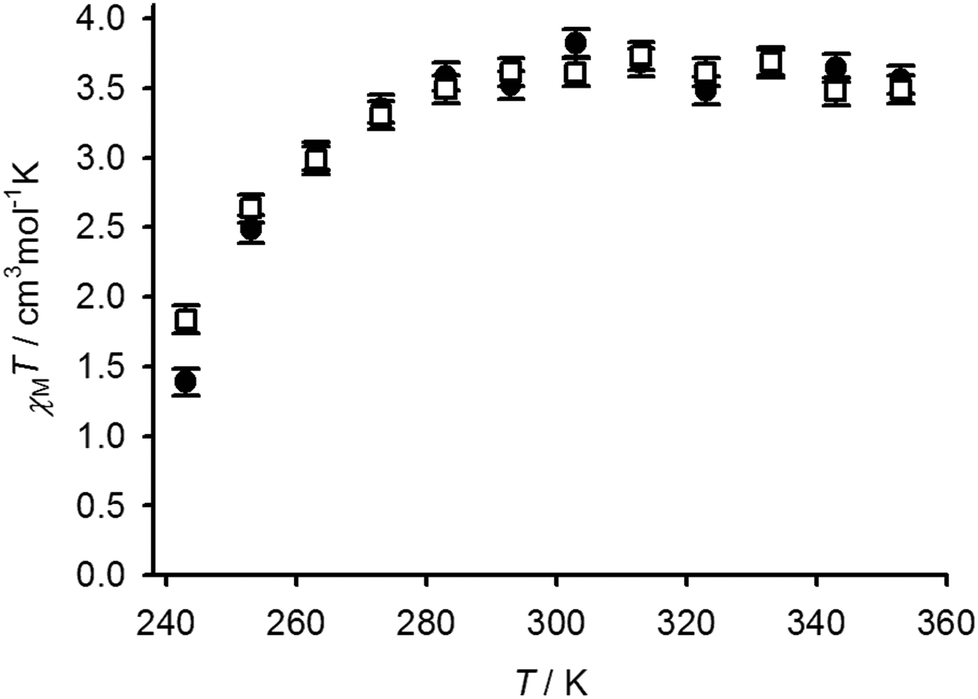 | ||
| Fig. 6 Variable temperature magnetic susceptibility data in CD3NO2 solution for 1[BF4]2 (●) and 2[BF4]2 (□). | ||
Conclusions
The new thiolated ligand 4-mercapto-2,6-di(pyrazol-1-yl)pyridine (bppSH) has been synthesised,‡ and converted to the dinucleating disulfide ligand bppSSbpp (Scheme 1). While iron(II) or zinc(II) complexes of other back-to-back bis-[dipyrazolylpyridine] derivatives have been made before, bppSSbpp is unique in forming soluble oligomer assemblies upon complexation with metal ions, rather than insoluble 1D coordination polymers. Mass spectrometry and DOSY data are both consistent with tetrameric metallacycles [{M(μ-bppSSbpp)}4]8+ (M = Fe or Zn) being the predominant species in solutions of these compounds. However, the conductivity data (Fig. 4) and 1H NMR (ESI†) imply that the [{M(μ-bppSSbpp)}4]8+ assembly may undergo a greater degree of fragmentation or ring-opening when M = Fe than when M = Zn, under the conditions in this work. Since high-spin Fe2+ and Zn2+ are both labile metal ions that is an unexpected observation, but it might reflect the pronounced Jahn–Teller distortion that is often exhibited by high-spin [Fe(bpp)2]2+ centres.6 The symmetry of the metallacycle would be disrupted if a fraction of the iron centres in 2[BF4]2 and 2[ClO4]2 adopt this distorted geometry at room temperature, potentially leading to this increased fragmentation as observed. That would not be a factor for the zinc centres in 3[BF4]2 or the low-spin iron sites in [{Fe(μ-terpySSterpy)}4]8+ (Fig. 1), which are not Jahn–Teller active.This study compliments Constable et al.'s earlier report of [{Fe(μ-terpySSterpy)}4]8+. That was characterised as a tetrameric metallacycle on the basis of a crystal structure (Fig. 1), but no solution phase data were reported. While we have been unable to crystallise 2[BF4]2, 2[ClO4]2 or 3[BF4]2, they form cyclic tetranuclear assemblies with minimal evidence for higher or lower cyclic oligomers, in the weakly associating solvent MeNO2.20 The apparent preference of these back-to-back bis-tridentate disulfides to form soluble, square metallacyclic complexes reflects the acute R–S–S–R torsions favoured by the disulfido group, which place the metal-binding domains approximately at right angles to each other (Fig. 2).20 This contrasts with the transoid R–CH2–CH2–R torsion adopted by 1,2-bis[2,6-di(pyrazol-1-yl)pyrid-4-yl]ethane (bppC2H4bpp), for example, which instead forms insoluble coordination polymers upon reaction with iron(II) salts.18
Solid 2[BF4]2 and 2[ClO4]2 exhibit thermal spin-crossover, as predicted at the outset of this work, making this a new type of supramolecular spin-crossover assembly.25–28 However, their spin-crossover transitions are gradual and incomplete at low temperatures, reflecting their amorphous nature in the solid state. In solution, 2[BF4]2 undergoes a thermal spin-state equilibrium at an almost identical temperature to the mononuclear precursor 1[BF4]2, implying that the four iron centres in [{Fe(μ-bppSSbpp)}4]8+ undergo spin-crossover independently of each other under these conditions.
Experimental
Unless otherwise stated, all reactions were carried out in air using non-pre-dried AR-grade solvents. 4-Iodo-2,6-di(pyrazol-1-yl)pyridine was synthesised by the literature method,16 while all other reagents were used as commercially supplied. Mass spectra for the metallacyclic complexes are tabulated and assigned in the ESI.†Synthesis of 4-mercapto-2,6-di(pyrazol-1-yl)pyridine (bppSH)
Addition of NaSH·H2O (2.70 g, 36.4 mmol) to 4-iodo-2,6-di(pyrazol-1-yl)pyridine (0.49 g, 1.45 mmol) in dimethylformamide (100 cm3) caused a darkening of the solution to a deep green color. The solution was refluxed for 4 h, then cooled to room temperature. The solvent was removed in vacuo leaving a pale yellow residue which was taken up in water (50 cm3) and neutralised with concentrated HCl which precipitated a cream-coloured solid. This was collected by filtration and dried in vacuo. Yield 0.35 g, 96%. Found: C 54.4, H 3.55, N 28.5%. Calcd for C11H9N5S C 54.3, H 3.73, N 28.8%. M.p. 133–135 °C. ES mass spectrum: m/z 266.0 [Na(bppSH)]+. 1H NMR (300 MHz, {CD3}2SO); δ, ppm 6.60 (dd, J = 1.7 and 2.5 Hz, 2H, pz H4), 7.82 (s, 2H, py H3/5), 7.84 (d, 1.7 Hz, 2H, pz H3), 8.87 (d, 2.5 Hz, 2H, pz H5). 13C NMR (75.5 MHz, {CD3}2SO); δ, ppm 106.6 (2C, py C3/5), 108.5 (2C, pz C4), 128.2 (2C, pz C5), 142.7 (2C, pz C3), 149.5 (2C, pz C2/6), 152.1 (1C, py C4).Synthesis of bis(2,6-di[pyrazol-1-yl]pyridin-4-yl)disulfide (bppSSbpp)
NaOH (29 mg, 0.0.73 mmol) and I2 (84 mg, 0.33 mmol) were added to a stirring suspension of bppSH (0.15 g, 0.62 mmol) in H2O (15 cm3) at room temperature. Stirring was continued for 20 h, yielding a cream-coloured precipitate. The off-white solid was collected, washed with further H2O, EtOH and Et2O and dried over P2O5. Yield 0.14 g, 91%. Found C 54.3, H 3.25, N 29.1%. Calcd for C22H16N10S2 C 54.5, H 3.33, N 28.9%. M.p. 240–242 °C. ES mass spectrum: m/z 507.1 [Na(bppSSbpp)]+. 1H NMR (300 MHz, CDCl3); δ, ppm 6.47 (t, 2.1 Hz, 4H, pz H4), 7.74 (br s, 4H, pz H3), 8.02 (s, 4H, py H3/5), 8.51 (d, 2.1 Hz, 4H, pz H5); 13C NMR (75.5 MHz, CDCl3); δ, ppm 105.4 and 108.2 (both 2C, py C3/5 and pz C4), 127.3 (2C, pz C5), 142.7 (2C, pz C3), 150.4 (2C, Py C2/6), 152.4 (1C, Py C4).Synthesis of [Fe(bppSH)2][BF4]2 (1[BF4]2)
A mixture of Fe[BF4]2·6H2O (0.11 g, 0.32 mmol) and bppSH (0.17 g, 0.64 mmol) in MeNO2 (40 cm3) was stirred under N2 for 2 h, until all the solid had dissolved. The opaque yellow-orange solution was filtered, and excess Et2O (100 cm3) was added to the filtrate, precipitating the yellow product which was collected and desiccated. Yield 0.18 g, 76%. Found: C, 35.1; H, 2.70; N, 18.6%. Calcd for C22H18B2FeN10F8S2·2H2O: C, 35.1; H, 2.95; N, 18.6%. 1H NMR (CD3NO2) δ 36.9 and 38.1 (both 4H, Py H3/5 and Pz H5), 55.4 (4H, Pz H4), 64.8 (4H, Pz H3).Synthesis of [Fe(bppSH)2][ClO4]2 (1[ClO4]2)
Method as for 1[BF4]2, using Fe[ClO4]2·6H2O (0.12 g, 0.32 mmol). Slow addition of degassed iPr2O (25 cm3) to the filtered solution caused the formation of a yellow solid. This was collected by filtration, washed with a further few drops of iPr2O and dried in vacuo. Yield 0.18 g, 73%. Found: C, 35.1; H, 2.40; N, 18.2%. Calcd for C22H18Cl2FeN10O8S2·H2O: C, 34.8; H, 2.65; N, 18.4%.Synthesis of [Fe(bppSSbpp)][BF4]2 (2[BF4]2)
Solid Fe[BF4]2·6H2O (49 mg, 0.15 mmol) was added to a suspension of bppSSbpp (70 mg, 0.14 mmol) in MeNO2 (30 cm3). The solution was stirred at room temperature for 30 min, and then filtered. The solvent was removed in vacuo to give a brick-red microcrystalline solid, which was washed with diethyl ether and dried in vacuo. Yield 77.0 mg, 77%. Found: C, 38.2; H, 3.05; N, 18.6%. Calcd for C22H16B2F8N10S2Fe·1/2Et2O C, 38.4; H, 2.82; N, 18.6%. 1H NMR (CD3NO2) species 1: δ 37.5 and 38.1 (both 4H, Py H3 + Pz H5), 56.4 (4H, Pz H4), 65.7 (4H, Pz H3); species 2: δ 37.3 and 38.7 (both 4H, Py H3 + Pz H5), 56.9 (4H, Pz H4), 66.1 (4H, Pz H3); species 3: δ 6.55 (br s, 2H, pz H4), 7.69 and 7.80 (both br s, 2H, pz H3 and py H3/5), 8.66 (br s, 2H, pz H5).Synthesis of [Fe(bppSSbpp)][ClO4]2 (2[ClO4]2)
Method as for 1[BF4]2, using Fe[ClO4]2·6H2O (54 mg, 0.15 mmol). The product was an orange-brown solid. Yield 52 mg, 44%. Found: C, 36.1; H, 2.60; N, 18.2%. Calcd for C22H16Cl2FeN10O8S2·1/4Et2O: C, 36.4; H, 2.72; N, 18.4%. 1H NMR (CD3NO2) δ 37.4 and 38.1 (both 4H, Py H3 + Pz H5), 55.5 (4H, Pz H4), 64.9 (4H, Pz H3).Synthesis of [Zn(bppSSbpp)][BF4]2 (3[BF4]2)
Solid Zn[BF4]2·6H2O (51 mg, 0.15 mmol) and bppSSbpp (70 mg, 0.14 mmol) were stirred together in a 1:2 MeNO2–MeCN solvent mixture (20 cm3) for 1 h. The solvents were removed from the filtered solution and the pale yellow oily residue was triturated in Et2O (80 cm3), yielding a colourless polycrystalline solid that was collected by filtration. Yield 65.0 mg, 64%. Found: C, 38.1; H, 2.90; N, 18.5%. Calcd for C22H16B2F8N10S2Zn·1/2Et2O: C, 37.9; H, 2.78; N, 18.4%. 1H NMR (CD3NO2) major species: δ 6.67 (br s, 4H, Pz H4), 7.65 (br s, 4H, Pz H3), 8.18 (br s, 4H, Py H3/5), 8.68 (br s, 4H, Pz H5): minor species: δ 6.38, 7.58, 7.77; 13C NMR (CD3NO2) major species: δ 106.9 (Py C3/5), 113.1 (Pz C4), 131.5 (Pz C5), 144.5 (Pz C3), 147.0 (Py C2/6), 155.6 (Py C4).
Single crystal X-ray structure determination
Diffraction data were measured using a Bruker X8 Apex II diffractometer, with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å) generated by a rotating anode. The diffractometer was fitted with Oxford Cryostream low-temperature device. Experimental details of the structure determination are given in Table 1. The structure was solved by direct methods (SHELXS9749), and developed by full least-squares refinement on F2 (SHELXL9749). Crystallographic figures were prepared using XSEED,50 which incorporates POVRAY.51| a R = ∑[∣Fo∣ − ∣Fc∣]/∑∣Fo∣. b wR = [∑w(Fo2 − Fc2)/∑wFo4]1/2. | |||
|---|---|---|---|
| Molecular formula | C22H16N10S2 | V (Å3) | 1144.5(3) |
| M r | 484.57 | Z | 2 |
| Crystal class | Triclinic | μ (mm–1) | 0.266 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
T (K) | 150(2) |
| a (Å) | 8.9223(13) | Measured reflections | 36263 |
| b (Å) | 10.2903(14) | Independent reflections | 7730 |
| c (Å) | 13.341(2) | R int | 0.052 |
| α (°) | 90.512(6) | R 1, I > 2σ(I)a | 0.041 |
| β (°) | 107.862(7) | wR2, all datab | 0.141 |
| γ (°) | 100.271(6) | Goodness of fit | 1.148 |
No disorder is present in the model, and no restraints were applied. All non-H atoms were refined anisotropically, and all H atoms were located in the Fourier map and allowed to refine freely. Refined C–H distances range from 0.93(2)–1.01(2) Å.
Other measurements
Elemental microanalyses were performed by the University of Leeds School of Chemistry microanalytical service. Electrospray mass spectra were recorded from 10−3 M solutions on a Bruker MicroTOF-q instrument, from CHCl3 solution (organic compounds) or CH3NO2 solution (metal complexes). X-ray powder diffraction patterns were measured using a Bruker D2 Phaser diffractometer.Variable temperature magnetic susceptibility measurements were performed on a Quantum Design SQUID/VSM magnetometer, in an applied field of 5000 G. A diamagnetic correction for the sample was estimated from Pascal's constants;52 a diamagnetic correction for the sample holder was also measured separately, and applied to the data. Magnetic susceptibility measurements in solution were obtained by Evans method using a Bruker DRX500 spectrometer operating at 500.13 MHz.53 A diamagnetic correction for the sample,52 and a correction for the variation of the density of the CD3NO2 solvent with temperature,54 were applied to these data. All magnetochemical data manipulation was carried out using SIGMAPLOT.55
DOSY NMR measurements in CD3NO2 were obtained using a Varian Inova 500 MHz spectrometer, with a 5 mm ID probe. A bipolar pulse pair stimulated echo (BBPSTE) pulse sequence56 was employed, operating in the ONESHOT experiment.57 Additional parameters: number of different gradient levels, 15; gradient stabilization delay, 0.003 s; gradient length, 0.02–0.03 s; relaxation delay, 12 s; acquisition time, 1.3 s; number of data points, 8192 pairs; spectral width, 6000 Hz. Data were processed with a 1 Hz line broadening prior to Fourier transformation and baseline correction. The concentration of the sample was 0.8 mM.
Acknowledgements
This work was funded by the EPSRC grants EP/H015639/1 (studentship to LJKC) and EP/K00512X/1 (SQUID/VSM magnetometer). The authors thank Dr Oscar Cespedes (University of Leeds) and Dr Floriana Tuna (University of Manchester) for help with the magnetic susceptibility measurements.Notes and references
- Spin Crossover in Transition Metal Compounds I-III, ed. P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, vol. 233–235 Search PubMed.
- Spin-crossover materials - properties and applications, ed. M. A. Halcrow, John Wiley & Sons, Chichester, 2013, p. 568 Search PubMed.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Chem. Soc. Rev., 2011, 40, 3313 RSC.
- M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119 RSC.
- For other recent reviews see: M. C. Muñoz and J. A. Real, Coord. Chem. Rev., 2011, 255, 2068 CrossRef CAS PubMed; J. Tao, R.-J. Wei, R.-B. Huang and L.-S. Zheng, Chem. Soc. Rev., 2012, 41, 703 RSC; P. Gütlich, Eur. J. Inorg. Chem., 2013, 581 CrossRef PubMed; P. Gütlich, A. B. Gaspar and Y. Garcia, Beilstein J. Org. Chem., 2013, 9, 342 CrossRef PubMed; P. Guionneau, Dalton Trans., 2014, 43, 382 RSC.
- M. A. Halcrow, Coord. Chem. Rev., 2009, 253, 2493 CrossRef CAS PubMed; L. J. Kershaw Cook, R. Mohammed, G. Sherborne, T. D. Roberts, S. Alvarez and M. A. Halcrow, Coord. Chem. Rev., 2015, 289–290, 2 CrossRef PubMed.
- J. Olguín and S. Brooker, Coord. Chem. Rev., 2011, 255, 203 CrossRef PubMed.
- M. A. Halcrow, New J. Chem., 2014, 38, 1868 RSC.
- R. González-Prieto, B. Fleury, F. Schramm, G. Zoppellaro, R. Chandrasekar, O. Fuhr, S. Lebedkin, M. Kappes and M. Ruben, Dalton Trans., 2011, 40, 7564 RSC.
- M. Nihei, N. Takahashi, H. Nishikawa and H. Oshio, Dalton Trans., 2011, 40, 2154 RSC.
- Y. Hasegawa, K. Takahashi, S. Kume and H. Nishihara, Chem. Commun., 2011, 47, 6846 RSC; K. Takahashi, Y. Hasegawa, R. Sakamoto, M. Nishikawa, S. Kume, E. Nishibori and H. Nishihara, Inorg. Chem., 2012, 51, 5188 CrossRef CAS PubMed.
- M. S. Alam, M. Stocker, K. Gieb, P. Müller, M. Haryono, K. Student and A. Grohmann, Angew. Chem., Int. Ed., 2010, 49, 1159 CrossRef CAS PubMed.
- M. Cavallini, I. Bergenti, S. Milita, J. C. Kengne, D. Gentili, G. Ruani, I. Salitros, V. Meded and M. Ruben, Langmuir, 2011, 27, 4076 CrossRef CAS PubMed.
- K. Botcha, S. Basak and R. Chandrasekar, RSC Adv., 2014, 4, 34760 RSC.
- D. Secker, S. Wagner, S. Ballmann, R. Härtle, M. Thoss and H. B. Weber, Phys. Rev. Lett., 2011, 106, 136807 CrossRef; V. Meded, A. Bagrets, K. Fink, R. Chandrasekar, M. Ruben, F. Evers, A. Bernand-Mantel, J. S. Seldenthuis, A. Beukman and H. S. J. van der Zant, Phys. Rev. B: Condens. Matter, 2011, 83, 245415 CrossRef.
- C. Rajadurai, F. Schramm, S. Brink, O. Fuhr, M. Ghafari, R. Kruk and M. Ruben, Inorg. Chem., 2006, 45, 10019 CrossRef CAS PubMed.
- I. Šalitroš, J. Pavlik, R. Boča, O. Fuhr, C. Rajadurai and M. Ruben, CrystEngComm, 2010, 12, 2361 RSC; A. Abhervé, M. Clemente-León, E. Coronado, C. J. Gómez-García and M. López-Jordà, Dalton Trans., 2014, 43, 9406 RSC.
- J. Elhaïk, C. M. Pask, C. A. Kilner and M. A. Halcrow, Tetrahedron, 2007, 63, 291 CrossRef PubMed.
- C. Rajadurai, O. Fuhr, R. Kruk, M. Ghafari, H. Hahn and M. Ruben, Chem. Commun., 2007, 2636 RSC.
- E. C. Constable, B. A. Hermann, C. E. Housecroft, M. Neuburger, S. Schaffner and L. J. Scherer, New J. Chem., 2005, 29, 1475 RSC.
- M. J. J. Pereira Silva, P. Bertoncello, N. N. Daskalakis, N. Spencer, B. M. Kariuki, P. R. Unwin and Z. Pikramenou, Supramol. Chem., 2007, 19, 115 CrossRef.
- E. P. L. van der Geer, G. van Koten, R. J. M. Klein Gebbink and B. Hessen, Inorg. Chem., 2008, 47, 2849 CrossRef CAS PubMed.
- Q. Wang, P. Day, J.-P. Griffiths, H. Nie and J. D. Wallis, New J. Chem., 2006, 30, 1790 RSC; C. W. Machan, M. Adelhardt, A. A. Sarjeant, C. L. Stern, J. Sutter, K. Meyer and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 16921 CrossRef CAS PubMed.
- For other metallamacrocycles based on linked [M(terpy)2]n+ building blocks see: S.-S. Sun and A. J. Lees, Inorg. Chem., 2001, 40, 3154 Search PubMed; G. R. Newkome, T. J. Cho, C. N. Moorefield, R. Cush, P. S. Russo, L. A. Godinez, M. J. Saunders and P. Mohapatra, Chem. – Eur. J., 2002, 8, 2946 Search PubMed; E. C. Constable, C. E. Housecroft, M. Neuburger, S. Schaffner and E. J. Shardlow, Dalton Trans., 2005, 234 Search PubMed; E. C. Constable, E. L. Dunphy, C. E. Housecroft, W. Kylberg, M. Neuburger, S. Schaffner, E. R. Schofield and C. B. Smith, Chem. – Eur. J., 2006, 12, 4600 Search PubMed; A. Schultz, X. Li, B. Barkakaty, C. N. Moorefield, C. Wesdemiotis and G. R. Newkome, J. Am. Chem. Soc., 2012, 134, 7672 Search PubMed; A. Schultz, Y. Cao, M. Huang, S. Z. D. Cheng, X. Li, C. N. Moorefield, C. Wesdemiotis and G. R. Newkome, Dalton Trans., 2012, 41, 11573 Search PubMed; T.-Z. Xie, K. Guo, M. Huang, X. Lu, S.-Y. Liao, R. Sarkar, C. N. Moorefield, S. Z. D. Cheng, C. Wesdemiotis and G. R. Newkome, Chem. – Eur. J., 2014, 20, 11291 Search PubMed.
- [Fe4(μ-CN)4]8+ molecular squares exhibiting spin-crossover: M. Nihei, M. Ui, M. Yokota, L. Han, A. Maeda, H. Kishida, H. Okamoto and H. Oshio, Angew. Chem., Int. Ed., 2005, 44, 6484 Search PubMed; T. Nishihara, M. Nihei, H. Oshio and K. Tanaka, J. Phys.: Conf. Ser., 2009, 148, 012033 Search PubMed; M. Nihei, M. Ui and H. Oshio, Polyhedron, 2009, 28, 1718 Search PubMed; I. Boldog, F. J. Muñoz-Lara, A. B. Gaspar, M. C. Muñoz, M. Seredyuk and J. A. Real, Inorg. Chem., 2009, 48, 3710 Search PubMed; R.-J. Wei, Q. Huo, J. Tao, R.-B. Huang and L.-S. Zheng, Angew. Chem., Int. Ed., 2011, 50, 8940 Search PubMed; R.-J. Wei, J. Tao, R.-B. Huang and L.-S. Zheng, Eur. J. Inorg. Chem., 2013, 916 Search PubMed; O. Hietsoi, P. W. Dunk, H. D. Stout, A. Arroyave, K. Kovnir, R. E. Irons, N. Kassenova, R. Erkasov, C. Achim and M. Shatruk, Inorg. Chem., 2014, 53, 13070 Search PubMed.
- Tetrametallic grid complexes exhibiting spin-crossover: M. Ruben, E. Breuning, J.-M. Lehn, V. Ksenofontov, F. Renz, P. Gütlich and G. B. M. Vaughan, Chem. – Eur. J., 2003, 9, 4422 Search PubMed; D.-Y. Wu, O. Sato, Y. Einaga and C.-Y. Duan, Angew. Chem., Int. Ed., 2009, 48, 1475 Search PubMed; B. Schneider, S. Demeshko, S. Dechert and F. Meyer, Angew. Chem., Int. Ed., 2010, 49, 9274 Search PubMed; A. R. Stefankiewicz, G. Rogez, J. Harrowfield, A. N. Sobolev, A. Madalan, J. Huuskonen, K. Rissanen and J.-M. Lehn, Dalton Trans., 2012, 41, 13848 Search PubMed; Y.-T. Wang, S.-T. Li, S.-Q. Wu, A.-L. Cui, D.-Z. Shen and H.-Z. Kou, J. Am. Chem. Soc., 2013, 135, 5942 Search PubMed; T. Matsumoto, G. N. Newton, T. Shiga, S. Hayami, Y. Matsui, H. Okamoto, R. Kumai, Y. Murakami and H. Oshio, Nat. Commun., 2014, 5, 3865 Search PubMed.
- Tetrahedral coordination cages exhibiting spin-crossover: A. Ferguson, M. A. Squire, D. Siretanu, D. Mitcov, C. Mathonière, R. Clérac and P. E. Kruger, Chem. Commun., 2013, 49, 1597 Search PubMed; R. A. Bilbeisi, S. Zarra, H. L. C. Feltham, G. N. L. Jameson, J. K. Clegg, S. Brooker and J. R. Nitschke, Chem. – Eur. J., 2013, 19, 8058 Search PubMed; D.-H. Ren, D. Qiu, C.-Y. Pang, Z. Li and Z.-G. Gu, Chem. Commun., 2015, 51, 788 Search PubMed.
- Other spin-crossover compounds with high nuclearity: M. Shatruk, A. Dragulescu-Andrasi, K. E. Chambers, S. A. Stoian, E. L. Bominaar, C. Achim and K. R. Dunbar, J. Am. Chem. Soc., 2007, 129, 6104 Search PubMed; M. B. Duriska, S. M. Neville, B. Moubaraki, J. D. Cashion, G. J. Halder, K. W. Chapman, C. Balde, J.-F. Létard, K. S. Murray, C. J. Kepert and S. R. Batten, Angew. Chem., Int. Ed., 2009, 48, 2549 Search PubMed; R. Boča, I. Šalitroš, J. Kožíšek, J. Linares, J. Moncol and F. Renz, Dalton Trans., 2010, 39, 2198 Search PubMed; M. B. Duriska, S. M. Neville, B. Moubaraki, K. S. Murray, C. Baldé, J.-F. Létard, C. J. Kepert and S. R. Batten, ChemPlusChem, 2012, 77, 616 Search PubMed.
- G. B. Barlin, D. J. Brown and M. D. Fenn, Aust. J. Chem., 1984, 37, 2391 Search PubMed.
- H.-Z. Yu, Y.-M. Yang, L. Zhang, Z.-M. Dang and G.-H. Hu, J. Phys. Chem. A, 2014, 118, 606 Search PubMed.
- C. A. Bessel, R. F. See, D. L. Jameson, M. R. Churchill and K. J. Takeuchi, J. Chem. Soc., Dalton Trans., 1992, 3223 Search PubMed.
- I. Bertini and C. Luchinat, NMR of paramagnetic substances , Coord. Chem. Rev., 1996, 150, 292 Search PubMed.
- S. F. Lincoln, Helv. Chim. Acta, 2005, 88, 523 Search PubMed.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81 Search PubMed; R. K. Boggess and D. A. Zatko, J. Chem. Educ., 1975, 52, 649 Search PubMed.
- See e.g.: A. Davison, D. V. Howe and E. T. Shawl, Inorg. Chem., 1967, 6, 458 Search PubMed; R. L. Dutta, D. W. Meek and D. H. Busch, Inorg. Chem., 1970, 9, 1215 Search PubMed; S. Ruttimann, C. Piguet, G. Bernardinelli, B. Bocquet and A. F. Williams, J. Am. Chem. Soc., 1992, 114, 4230 Search PubMed; N. C. Habermehl, P. M. Angus, N. L. Kilah, L. Norén, A. D. Rae, A. C. Willis and S. B. Wild, Inorg. Chem., 2006, 45, 1445 Search PubMed.
- R. D. Feltham and R. G. Hayter, J. Chem. Soc., 1964, 4587 Search PubMed.
- P. H. Smith, Z. E. Reyes, C.-W. Lee and K. N. Raymond, Inorg. Chem., 1988, 27, 4154 Search PubMed.
- S. A. Willis, T. Stait-Gardner, A. S. Virk, R. Masuda, M. Zubkov, G. Zheng and W. S. Price, in Modern NMR Techniques for Synthetic Chemists, ed. J. Fisher, CRC Press, Baton Rouge, FL, USA, 2013, ch. 4, p. 125–176 Search PubMed.
- J. E. Beves, E. C. Constable, C. E. Housecroft, M. Neuburger, S. Schaffner and E. J. Shardlow, Dalton Trans., 2007, 1593 Search PubMed.
- E. C. Constable, K. Harris, C. E. Housecroft and M. Neuburger, Dalton Trans., 2011, 40, 1524 Search PubMed.
- Y.-T. Chan, X. Li, J. Yu, G. A. Carri, C. N. Moorefield, G. R. Newkome and C. Wesdemiotis, J. Am. Chem. Soc., 2011, 133, 11967 Search PubMed.
- V. V. Krishnan, J. Magn. Reson., 1997, 124, 468 Search PubMed.
- M. S. Kaucher, Y.-F. Lam, S. Pieraccini, G. Gottarelli and J. T. Davis, Chem. – Eur. J., 2005, 11, 164 Search PubMed.
- R. Davi and D. Ben-Amotz, Chem. Phys., 1994, 183, 385 Search PubMed.
- J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman and M. A. Halcrow, J. Chem. Soc., Dalton Trans., 2002, 548 Search PubMed.
- S. A. Barrett, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2011, 40, 12021 Search PubMed.
- N. Hassan, A. B. Koudriavtsev and W. Linert, Pure Appl. Chem., 2008, 80, 1281 Search PubMed.
- R. Pritchard, C. A. Kilner, S. A. Barrett and M. A. Halcrow, Inorg. Chim. Acta, 2009, 362, 4365 Search PubMed; R. Mohammed, G. Chastanet, F. Tuna, T. L. Malkin, S. A. Barrett, C. A. Kilner, J.-F. Létard and M. A. Halcrow, Eur. J. Inorg. Chem., 2013, 819 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 Search PubMed.
- POVRAY, v. 3.5, Persistence of Vision Raytracer Pty. Ltd, Williamstown, Victoria, Australia, 2002. http://www.povray.org Search PubMed.
- C. J. O'Connor, Prog. Inorg. Chem., 1982, 29, 203 Search PubMed.
- D. F. Evans, J. Chem. Soc., 1959, 2003 Search PubMed; E. M. Schubert, J. Chem. Educ., 1992, 69, 62 Search PubMed.
- J. C. Philip and H. B. Oakley, J. Chem. Soc., Dalton Trans., 1924, 1189 Search PubMed.
- SIGMAPLOT, v. 8.02, SPSS Scientific Inc., Chicago, IL, 2002 Search PubMed.
- M. D. Pelta, H. Barjat, G. A. Morris, A. L. Davis and S. J. Hammond, Magn. Reson. Chem., 1998, 36, 706 Search PubMed.
- M. D. Pelta, G. A. Morris, M. J. Stchedroff and S. J. Hammond, Magn. Reson. Chem., 2002, 40, S147 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional crystallographic figures and tables, magnetic susceptibility, X-ray powder diffraction, 1H and DOSY NMR data. CCDC 1006369. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00732a |
| ‡ The chemistry of bppSH and its iron complexes on a gold surface will be described in a future publication. |
| This journal is © The Royal Society of Chemistry 2015 |