
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic evidence for selenium(IV) dimerization in aqueous solution†
J.
Kretzschmar
a,
N.
Jordan
*a,
E.
Brendler
b,
S.
Tsushima
 a,
C.
Franzen
a,
H.
Foerstendorf
a,
M.
Stockmann
a,
K.
Heim
a and
V.
Brendler
a
a,
C.
Franzen
a,
H.
Foerstendorf
a,
M.
Stockmann
a,
K.
Heim
a and
V.
Brendler
a
aHelmholtz-Zentrum Dresden-Rossendorf, Institute of Resource Ecology, Bautzner Landstraße 400, 01328 Dresden, Germany. E-mail: n.jordan@hzdr.de; Fax: +49 351 260 13233; Tel: +49 351 260 2148
bInstitute of Analytical Chemistry, Technical University Bergakademie Freiberg, Leipziger Straße 29, 09599 Freiberg, Germany
First published on 1st May 2015
Abstract
The aqueous speciation of selenium(IV) was elucidated by a combined approach applying quantum chemical calculations, infrared (IR), Raman, and 77Se NMR spectroscopy. The dimerization of hydrogen selenite (HSeO3−) was confirmed at concentrations above 10 mmol L−1 by both IR and NMR spectroscopy. Quantum chemical calculations provided the assignment of vibrational bands observed to specific molecular modes of the (HSeO3)22− ion. The results presented will provide a better understanding of the chemistry of aqueous Se(IV) which is of particular interest for processes occurring at mineral/water interfaces.
Introduction
Depending on its chemical form and concentration, selenium is either essential or hazardous for animals and human beings, with a very narrow difference between its deficiency and toxic level.1 The natural background is strongly varying (extremes are 0.03 to 1200 mg kg−1),1 and there are various anthropogenic influxes (coal combustion, processing of copper, use of phosphate fertilizers, etc.).1 In addition, the isotope 79Se is a long-lived (τ1/2 = 3.27 × 105 a)2 fission product contributing significantly to the potential radiation dose from nuclear waste repositories.3–5 Consequently, a detailed knowledge of the mobility and bioavailability of selenium in its different oxidation states is of great importance for constraining environmental hazards and developing remediation strategies where required.Retardation of water-soluble selenium oxyanions, selenate (SeO42−) and selenite (SeO32−), is governed by the respective thermodynamics of both the aqueous phase and the interactions with mineral surfaces. The still most comprehensive overview about selenium chemistry is presented in the respective volume of the OECD/NEA Thermochemical Database (NEA TDB) by Olin et al.6 However, although many papers have been published since 2005, some gaps still need to be closed. One of the open questions is a detailed acquisition of the dimerization of aqueous SeO32− ions, which is expected to start at concentrations around 1 mmol L−1.7 The NEA TDB reported a broad variety of conductometry and cryometry,8–11 potentiometric,12–14 calorimetric,15 and kinetic16–19 studies supporting this phenomenon. Olin et al.6 despite considering the existence of Se(IV) binuclear species, could not recommend stability constants for these species. Torres et al.7,20 presented a set of thermodynamic constants for this system recently. However, their list of species is not backed-up by any independent spectroscopic evidence and solely derived from best-fits to potentiometric titrations.
Fortunately, several pathways were opened during the last few decades that can shed light on this issue. Here, in situ Attenuated Total Reflection Fourier Transform Infrared Spectroscopy (ATR FT-IR) and Raman spectroscopy are to be mentioned, though a reliable interpretation of the spectra and, thus, the identification of the dissolved and sorbed species can only be given when the assignment of bands is unequivocal. As highlighted by Su and Suarez,21 the assignment of IR bands to individual species for a 1.0 mol L−1 Na2SeO3 solution in 1.0 mol L−1 NaCl at pH 5.0 or 8.0 was not possible due to the presence of several binuclear species. Here, new approaches are required. Results from IR and Raman spectroscopy can be checked and further refined by quantum chemical calculations using the second-order Møller–Plesset perturbation theory (MP2). MP2 calculations provide vibrational data including the identification of the vibrational modes for different molecular geometries.
A complementary technique is provided by Nuclear Magnetic Resonance (NMR) spectroscopy. The spin 1/2 nucleus of the stable isotope 77Se is well suited to be directly observed by NMR spectroscopy. This method is in particular a valuable tool for probing the electronic environment of the Se nucleus, thereby providing information on the Se oxidation or protonation states. A drawback is its natural abundance of only 7.63%, which can be overcome by isotopic enrichment.
A combination of these methods is exploited here. This approach enables a more consistent and trustworthy description of selenium chemistry in natural and anthropogenic waters, eventually improving respective prognostic modelling.
Experimental
Reagents and solutions
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cH+).22,23 To keep the total ionic strength constant, samples were adjusted to I = 3 mol L−1 by addition of respective amounts of NaCl if necessary, considering pH corrections due to ionic strength adjustment.
cH+).22,23 To keep the total ionic strength constant, samples were adjusted to I = 3 mol L−1 by addition of respective amounts of NaCl if necessary, considering pH corrections due to ionic strength adjustment.
Methods & instrumentation
Results and discussion
Speciation calculations
Speciation calculations were done using the geochemical computer program PHREEQC.30 They were performed at 0.3 mol L−1 total ionic strength (between pH 4 and 7) with the equilibrium constants of the NEA TDB (Olin et al.6), who did not recommend values for the Se(IV) dimeric species (Fig. 1A). Further calculations using the equilibrium constants (including Se dimeric species) proposed by Torres et al.7 (derived from potentiometric titration) are presented (Fig. 1B–D). All equilibrium constants are listed in Table S1 in the ESI.† At pHc 13, the selenite anion is the only species present in solution and will serve as a reference system. For Se concentrations higher than 0.01 mol L−1, the (HSeO3)22− dimer is predicted to become predominant between pH 4 and 7.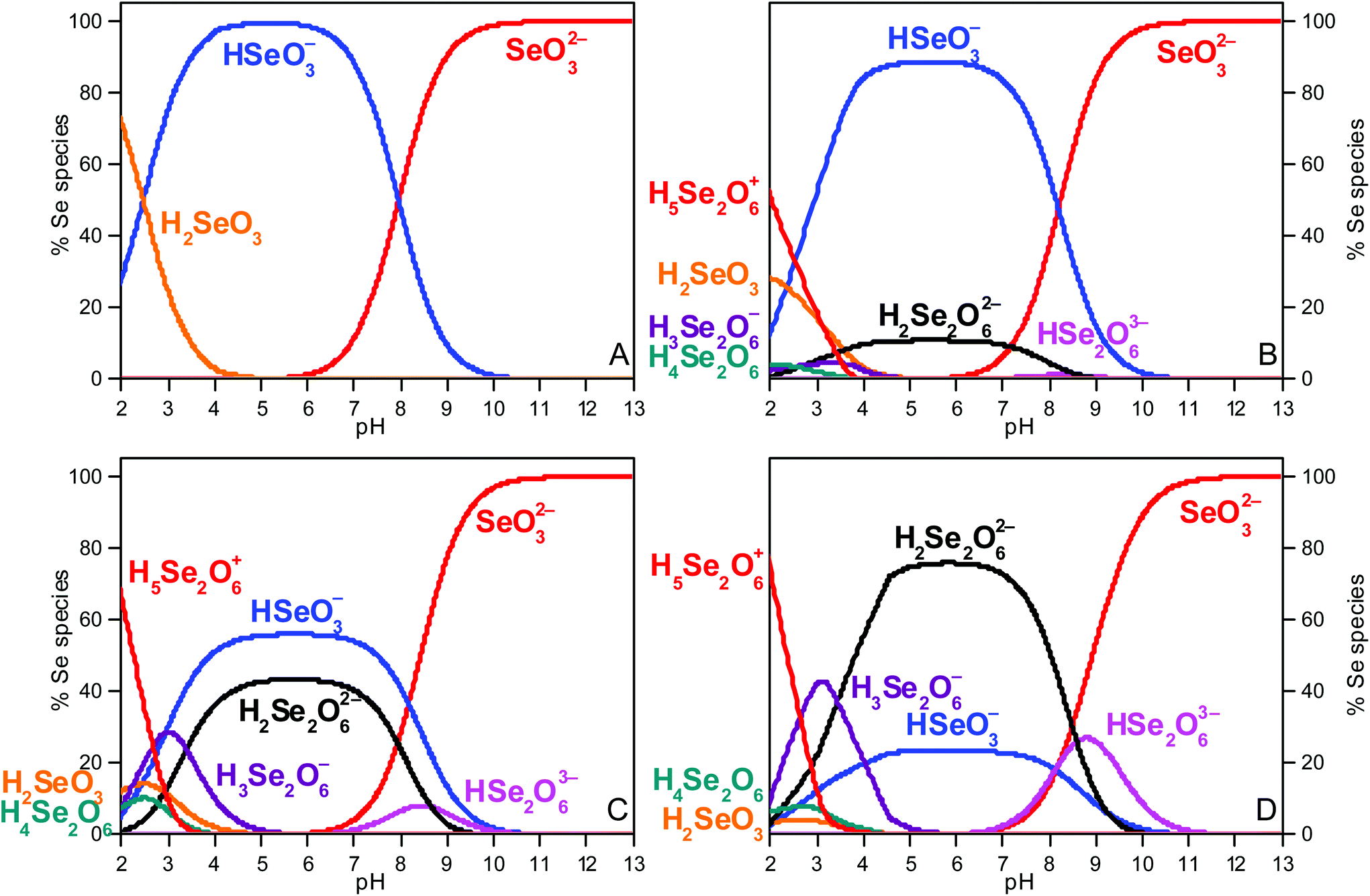 | ||
| Fig. 1 Se(IV) speciation diagrams (I = 0.3 mol L−1 between pH 4 and 7) (A) at total Se concentration of 0.1 mol L−1 (0.145 mol L−1 NaCl); (B) at total Se concentrations of 1 mmol L−1 (0.3 mol L−1 NaCl), (C) of 10 mmol L−1 (0.285 mol L−1 NaCl) and (D) 0.1 mol L−1 (0.113 mol L−1 NaCl). Note that an increase of ionic strength from 0.34–0.62 mol L−1 for pH 7.5–13 (A), from 0.32–0.44 mol L−1 for pH 12–13 (B), from 0.31–0.46 mol L−1 for pH 8–13 (C) and from 0.32–0.59 mol L−1 for pH 7.5–13 (D) is predicted. | ||
Quantum chemical calculations
For MP2 calculations of small dissolved molecules, the question arises as to how many water molecules should be considered to obtain an accurate hydration shell. An estimate can be derived from the hydrogen bonding of hydrated selenite which was recently studied by Large Angle X-ray Scattering (LAXS).31 For the selenite ion, the mean Se–O distance between selenium and the oxygen of H-bonded water molecules (Se⋯H–Ow) was found to be 3.87 Å. The O–O distance between oxygens of selenite anion and oxygens of H-bonded water molecules (O2Se–O⋯H–Ow) was ranging between 2.83 and 2.86 Å. The likelihood for equilibrium between two and three water molecules hydrogen binding to the selenite oxygens was revealed. For the three water molecules clustered outside the selenite lone pair, a distance of 4.36 Å was reported.31Consequently, we used nine water molecules surrounding the selenite ion (6 close to the Se–O bonds and three above the lone pair). However, MP2 calculations (data not shown) considering nine water molecules in the first hydration shell did not reduce the shift between theoretical and experimental frequencies in the IR and Raman spectra. This is presumably due to the oxygens of the nine water molecules remaining undersaturated in terms of hydrogen bonding. Unfortunately, results from solutions containing HSeO3− and (HSeO3)22− are not available since LAXS measurements were only performed for selenite.31 Therefore, no water was added for the structure optimisation of the systems investigated here.
The geometry of the Se(IV) monomers and dimers and their vibration modes predicted by MP2 are shown in Fig. 2. The pyramidal selenite ion has a C3v symmetry (Schoenflies notation, further used to describe symmetry groups in this study)21,32,33 showing six fundamental vibrations, two pairs of which are degenerate, leading to four normal vibrational modes.32,33 All these modes are both IR and Raman active: the ν1(A1) (symmetric Se–O stretching), ν2(A1) (symmetric O–Se–O bending), ν3(E) (asymmetric Se–O stretching) and ν4(E) (asymmetric O–Se–O bending). Both ν3(E) and ν4(E) asymmetric modes are doubly degenerate.
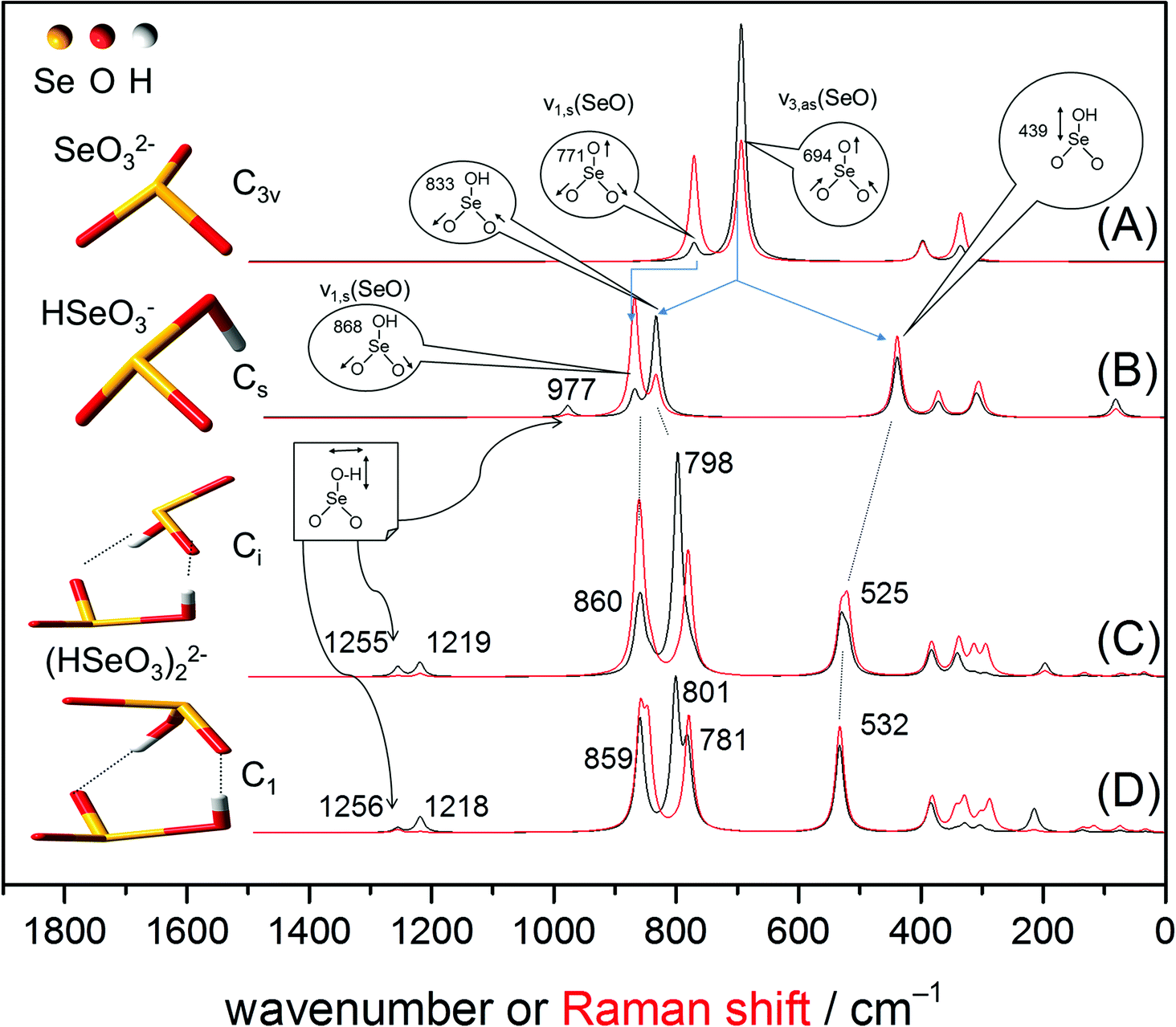 | ||
| Fig. 2 Optimized structures of the anions: SeO32− (A), HSeO3− (B) and two configurations of the Se(IV) dimer, (HSeO3)22− (C, D). In the dimeric structures the monomers are linked by hydrogen bonds. Theoretical (MP2) IR (black line) and Raman (red line) spectra. For clarity, only frequencies of the predicted IR spectra are shown (values of the Raman spectra are given in Table 1). | ||
In the following discussion only the stretching modes, i.e. ν1(A1) and ν3(E), will be considered in IR spectroscopy because the bending vibrations usually appear outside the range of the used MCT detector (<600 cm−1). The calculated IR and Raman spectra of the aqueous species are given in Fig. 2. Frequencies of MP2 predicted IR and Raman spectra are summarized in Table 1.
a
| Species | SeO32− | HSeO3− | (HSeO3)22− | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Symmetry | C 3v | C s | C 1 | C i | C 1/Ci | |||||
| Spectroscopy | MP2 | IR | Raman | MP2 | MP2 (IR) | MP2 (Raman) | MP2 (IR) | MP2 (Raman) | IR | Raman |
| a ν: stretching vibration; δ: bending vibration. | ||||||||||
| δ-OH | 1256 | 1256 | 1255 | 1255 | 1200 | |||||
| 1218 | 1218 | 1219 | 1219 | |||||||
| δ-OH | 977 | |||||||||
| ν 1,s(Se–O) | 771 | 807 | 807 | 868 | 859 | 859–846 | 860 | 860 | 849 | 857 |
| ν 3,as(Se–O) | 694 | 734 | 728 | 833 | 801–781 | 780 | 798 | 781 | 820 | 810 |
| ν(Se–OH) | 439 | 532 | 532 | 525 | 525 | 614 | ||||
The calculated IR (black traces) and Raman (red traces) spectra of selenite exhibits two bands at 771 cm−1 and 694 cm−1 representing the symmetric Se–O stretching ν1(A1) and asymmetric Se–O stretching ν3(E) modes, respectively (Fig. 2A). Although MP2 calculations underestimate the absolute frequencies of these two modes by around 40 cm−1, their band gap is well predicted: 77 cm−1 in MP2 vs. 73 cm−1 in experimental IR (Fig. 3A) and vs. 79 cm−1 in experimental Raman (Fig. 3C). This gives confidence on the accuracy of our MP2 calculations in the mid-IR region.
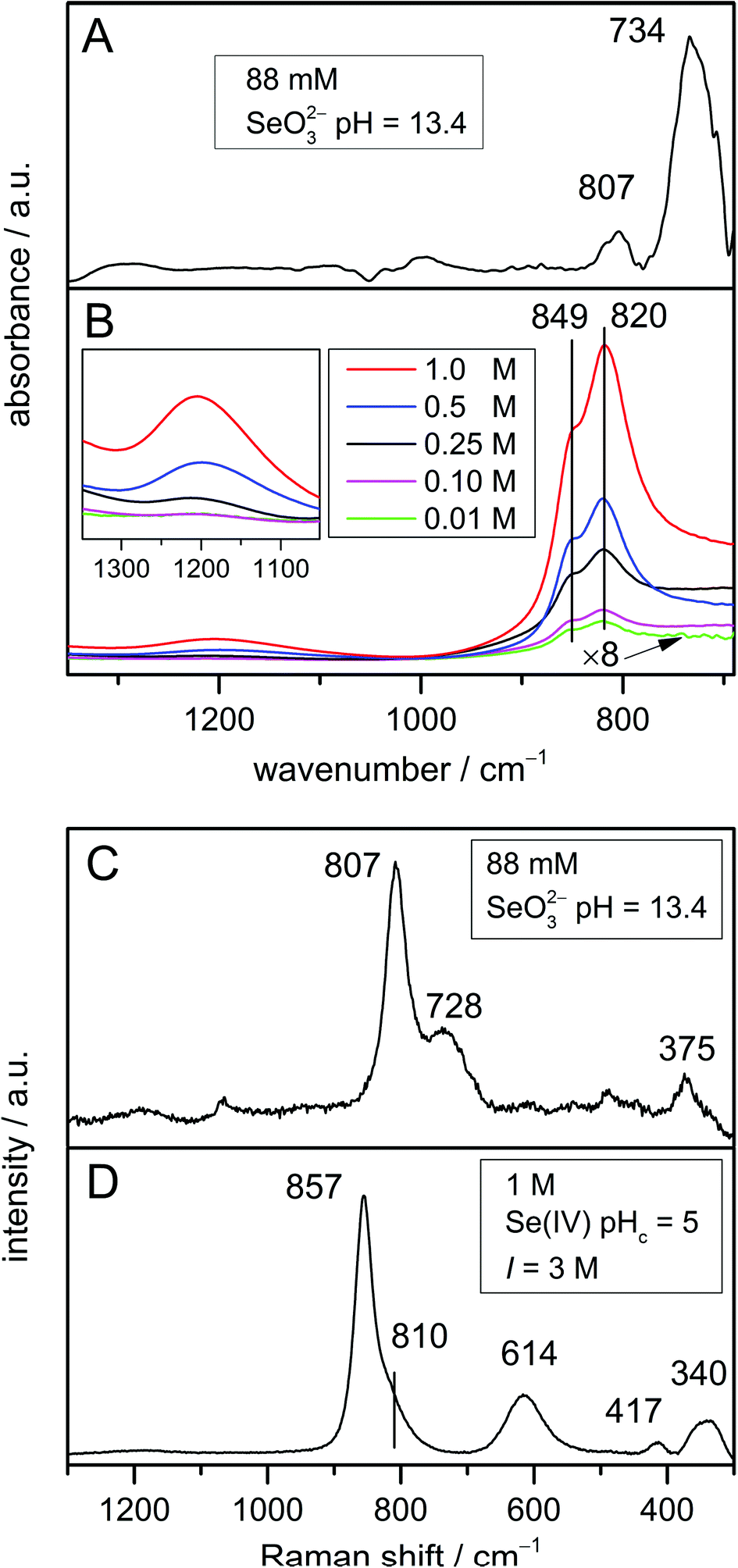 | ||
| Fig. 3 (A) IR spectrum of 88 mmol L−1 Se(IV) in H2O at pH 13.4; (B) IR spectra (as obtained) of solutions of different Se(IV) concentrations, pHc = 5, I = 3 mol L−1. The spectrum of the lowest concentration (0.01 mol L−1) is presented to an enlarged scale (×8). The insert shows an expansion of the dimer characteristic features; (C) Raman spectrum of 88 mmol L−1 Se(IV) in H2O at pH 13.4; (D) Raman spectrum of 1 mol L−1 Se(IV) in H2O at pHc = 5, I = 3 mol L−1 M denoting mol L−1. | ||
The HSeO3− ion shows Cs symmetry. In this symmetry group, there are two symmetrical operations: the identity E and a plane of reflection σ (principle axis included in the plane, but not the outer atoms). The nonlinear HSeO3− molecule exhibits two types of vibrational modes, A′ (in-plane) and A′′ (out-of-plane), which are distributed into nine fundamental vibrations as follows: Γvib = 6A′ + 3A′′.34 All these modes are both IR and Raman active. As expected, the frequencies and assignment of the vibrational modes predicted in Raman spectroscopy for the HSeO3− ion are identical to those predicted in the IR spectrum (except for the predominating intensities of the Se–O stretching modes calculated to appear at 868 and 833 cm−1). For the HSeO3− monomer, MP2 predicts a hypsochromic shift of about 100 cm−1 of the symmetric stretching Se–O vibration, calculated to be observed at 868 cm−1 (Fig. 2B). Regarding the doubly degenerate asymmetric Se–O stretching ν3(E) in SeO32−, a split into two modes, the antisymmetric Se–O stretching (at 833 cm−1) and the Se–OH stretching mode (at 439 cm−1), is estimated (Fig. 2B). The frequency of this stretching vibration is increased due to the addition of a proton reducing the resonance energy.35 In addition, a Se–OH bending mode of weak intensity, where the proton lies in the symmetry plane, is predicted at 977 cm−1.
The Gibbs energies of two selenite dimers (Fig. 2C and D) are virtually identical with an energy difference of only 0.6 kJ mol−1 (slightly favouring structure C) which is smaller than the hydrogen bond energy and comparable to the kinetic energy at room temperature. Therefore, two configurations are likely to coexist and equilibrate.
In both structures shown in Fig. 2(C and D), the orientation of the two HSeO3− molecules disrupt the plane of reflection σ present in the single HSeO3− molecule. In structure C, the symmetrical group is consequently reduced to Ci and should have two types of vibrational modes, Ag and Au, distributed into 24 fundamental vibrations as follows: Γvib = 12Ag + 12Au. This is in agreement with the recently proposed structure of cyclic dimers of HSeO3− ions intercalated in LDH phases36 and present in solid NaHSeO3.37,38 These dimers (Ci symmetry) are joined by hydrogen bonds around their centre of symmetry. Note that in the solid state of NaHSeO3 disordered H-bonds were noticed by differential scanning calorimetry, NMR, and neutron diffraction.37,38 However, in structure D, the dimer can be assigned to the C1 symmetry group. This structure exhibits vibrational modes distributed into 24 A fundamental vibrations. Thus, besides the assumed coexistence of the dimers in solution, they potentially exhibit the same number of vibrational modes.
No significant spectral differences among the two proposed dimer structures are envisioned by MP2 (Fig. 2C and D). The results of MP2 calculations predict that the symmetric and antisymmetric Se–O stretching vibrations of the SeO2 group appear in the IR spectrum (Fig. 2C) at 860 and 798 cm−1, respectively. In Raman spectroscopy, the symmetric and antisymmetric Se–O stretching vibrations of the SeO2 group are predicted at similar frequencies as that for IR, with a slightly higher gap of 80 cm−1 (Fig. 2C and D). The Se–OH stretching mode is estimated to be observable in both vibrational spectroscopies at about 530 cm−1. In addition, at higher frequencies, two in-plane deformation δ-OH vibrations of weak intensity are calculated at 1255 and 1219 cm−1 (Fig. 2C and D).
In summary, MP2 calculations revealed no significant spectral differences in the Se–O stretching region between HSeO3− and the (HSeO3)22− dimer in the IR and Raman spectra. However, two in-plane deformation δ-OH vibrations of weak intensity (very weak for Raman) at 1255 and 1219 cm−1 are envisioned to occur specifically with the dimer.
IR and Raman spectroscopy
The IR spectrum of the selenite ion (Fig. 3A) exhibits the symmetric Se–O stretching ν1(A1) at 807 cm−1 and the asymmetric Se–O stretching ν3(E) mode at 734 cm−1 as observed earlier.32,33 In addition, the higher intensity of the asymmetric Se–O stretching ν3(E) mode in comparison with the symmetric Se–O stretching ν1(A1) mode in the IR spectrum is also nicely predicted by MP2 (Fig. 2A).The respective Raman spectrum (Fig. 3C) exhibits in accordance to Siebert39 the symmetric Se–O stretching ν1(A1) at 807 cm−1 and the asymmetric Se–O stretching ν3(E) mode at 728 cm−1. It was found that the frequencies of the ν1(A1) and ν3(E) modes are not impacted by the excitation wavelength (spectra not shown). This means that Raman diffusion is not resonant at three different excitation wavelengths applied (633, 532, and 473 nm). The respective bands are observed at slightly higher frequencies as predicted by MP2. However, the band gap of 79 cm−1 fits very well with the MP2 calculations (77 cm−1). Only the relative intensities of these bands are different in the experimental spectrum.
Moreover, the modes ν2(A1) (symmetric O–Se–O bending) and ν4(E) (asymmetric O–Se–O bending), which are only Raman active, should be present at 432 and 374 cm−1, respectively.33,39 In fact, the ν4(E) mode is obviously observed at 375 cm−1 (Fig. 3C), whereas the ν2(A1) is hardly detectable due to low intensity.
Since the exclusive existence of the monomeric HSeO3− is predicted only at concentrations below 1 mmol L−1, the acquisition of a vibrational spectrum of the pure species is not achievable with a sufficient signal to noise ratio (and this applies even more to Raman spectroscopy which intrinsically exhibits significantly lower detection limit). Therefore, the verification of the MP2-predicted spectra of the HSeO3− monomer by IR and Raman spectra is not feasible.
The dimerization of the hydrogen selenite ions was investigated at five different Se(IV) concentrations ranging from 0.01 mol L−1 to 1 mol L−1 at pHc 5. The respective experimental IR spectra are shown in Fig. 3B. The spectra match perfectly with the data published by Su and Suarez.21
The IR spectra are characterized by strong bands at 849 and 820 cm−1 which can be assigned to the symmetric and antisymmetric Se–O stretching vibrations of the SeO2 group, respectively. The spectral splitting of these two modes is predicted (MP2) to be 80 cm−1, whereas only 30 cm−1 are found in the spectrum. In addition, the two in-plane deformation δ-OH vibrational modes are observed as one weak broad band centred around 1200 cm−1, obviously strongly overlapping in this spectral region to appear as a single feature. It has to be noted that not only the position of the characteristic bands of the dimer around 1200 cm−1, but also the intensity ratio between the deformation and the stretching modes predicted by MP2 are in very good agreement with IR experimental data.
In the lower frequency range of the vibrational spectra below 600 cm−1, the Se–OH stretching mode of HSeO3− is predicted to occur at 439 cm−1 (Fig. 2B). Upon dimerization, this mode is estimated to be shifted by about 100 cm−1 to higher frequencies and was calculated around 530 cm−1 for the (HSeO3)22− dimer (Fig. 2C and D). This band is not observed in the IR spectra due to the strong water absorption in this spectral region and the cut-off of the MCT detector. However, IR data obtained with the far infrared setup of our IR instrument suggest the presence of this band at this frequency (data not shown).
Varying Se(IV) concentrations ranging from 0.1 mol L−1 to 1 mol L−1, at pHc 5 and I = 3 mol L−1 were studied by Raman spectroscopy. At lower concentrations, no Raman spectra were obtained. The Raman spectrum of Se(IV) solution at pHc 5 was found to be independent of the concentration and the excitation wavelength (data not shown). The symmetric and antisymmetric Se–O stretching vibrations of the SeO2 group appear at 857 and 810 cm−1 (Fig. 3D), with a gap of 47 cm−1 smaller than MP2 predictions. The in-plane deformation δ-OH vibration modes around 1200 cm−1 are, however, not observed in the Raman spectrum (Fig. 3D) since they are generally of very low intensity (see also Fig. 2C and D). As the lower frequency range can be easily detected by Raman spectroscopy, the identification of the Se–OH stretching mode at a frequency of 614 cm−1 becomes feasible. This frequency is 100 cm−1 higher than that predicted by MP2.
Interestingly, no significant spectral differences for the Se–O stretching vibration are observed in the IR experimental spectra (Fig. 3B) with increasing concentration. Therefore, these bands are not suitable to differentiate between the monomer and the dimer, as expected from MP2 calculations (Fig. 2B–D). Though being intrinsically weak, the dimer specific feature around 1200 cm−1 evidences the continuously increasing content of the dimer with increasing total Se(IV) concentration (Fig. 3B).
NMR spectroscopy
NMR spectra were recorded at pHc 5 and 13 at different Se(IV) concentrations (Fig. 4). The selenium chemical shift δ is slightly concentration dependent (Fig. 4D), showing increasing values for pHc 5 and decreasing values for pHc 13, with overall changes of approximately 1 and 0.5 ppm, respectively. These changes in δ are small considering a chemical shift range for aqueous Se(IV) species of about 50 ppm, but nevertheless significant. The formation of lower or higher protonated species can be excluded from speciation calculation and pH dependent measurements. Our investigations and literature data40 show that this would cause a decrease of the chemical shift. | ||
| Fig. 4 77Se NMR of Se(IV) at pHc 5 (A) and 13 (B) with concentrations from 1 mmol L−1 through 1 mol L−1 and constant total ionic strength (3 mol L−1). Dependence of line width (C) and chemical shift (D) at pHc 5 (▲) and 13 (○) on selenium concentration. | ||
As the selenium's electronic environment changes only slightly upon dimerization, the monomer and the dimer are expected to possess only minor differences in chemical shifts. However, there is only one signal observed. According to the species calculation, this signal represents a molar fraction weighted averaged signal due to selenium site exchange between the monomer and the dimer. With increasing selenium concentration, the centre of the apparent signal is shifted downfield towards the chemical shift of the dimer, indicating an increasing content of the latter. Measurements carried out at different magnetic field strengths did not result in changes of the spectral behaviour (cf. Fig. S1, ESI†).
The analysis of the line width Δν1/2 (i.e. the signal's width at half amplitude) clearly shows that the pHc 5 solution exhibits a strong line width dependence on concentration ranging over two orders of magnitude, whereas the line width of the pHc 13 solution and of the external standard is virtually constant. Since other concentration-dependent effects such as changes in susceptibility or viscosity would influence δ at both pHc and have been excluded by keeping the ionic strength constant, these cannot be reasons for the broadening of the signal at pHc 5 (see also Fig. S2, ESI†). We therefore attribute the line broadening to exchange processes between different species in the solution – the HSeO3− monomer and dimer. These results are in agreement with the line broadening observed in 2 M Se(IV) solutions in the pH range 4–7 by Kolshorn and Meier.40 They suggested that additional species, e.g. (HSeO3)22−, are involved in the equilibrium.
Speciation calculations performed at total ionic strength of 0.3 mol L−1 predicted the (HSeO3)22− dimer to increase rapidly in concentration in the range of 0.001–0.1 mol L−1 and to be predominant at concentrations of 0.1 mol L−1 at pHc 5 (Fig. 1 and Table S1 (ESI†)). Significant NMR spectral changes (i.e. both chemical shift and line width) as well as the dimer specific IR feature were revealed at Se concentrations above 0.1 mol L−1. This threshold does not match exactly the one predicted by our speciation calculations (based on complexation constants derived from potentiometric titrations). Indeed, spectroscopic experiments were performed at I = 3 mol L−1, at which the speciation (and hence the dimer/monomer ratio) might be different in comparison with lower ionic strength used in calculation. To perform speciation calculations at high ionic strength using the SIT or Pitzer model, one would need a consistent set of interaction coefficients, which are to our knowledge not available.
To provide further evidence for the presence of Se(IV) dimers, we performed EXAFS spectroscopy of a 1 mol L−1 Se(IV) solution in H2O, pHc 5, I = 3 mol L−1. EXAFS did not reveal the expected Se–Se interaction. This does not necessarily contradict the existence of the dimers, but is rather due to the vibrational or static disorder causing destructive interference of the Se–Se paths (data not shown).
Conclusions
The combination of vibrational (IR and Raman) and 77Se NMR spectroscopy with a theoretical approach (MP2) was shown to be a helpful strategy in determining the aqueous speciation of selenium(IV). Based on predicted characteristic vibrational modes, their experimental verification in the IR spectra and further 77Se NMR spectroscopic support, the existence of the hydrogen selenite dimer was unambiguously proven for the first time. Its formation definitely occurs in aqueous solution above a critical concentration of ∼10 mmol L−1. These findings contribute to a deeper understanding of aqueous selenium chemistry in general and may be useful for further investigations addressing the mobility of selenium oxyanions in the environment. These results might serve as references for future spectroscopic investigations of the Se(IV) sorption processes on mineral phases. Indeed, the evaluation of the surface reactions requires a detailed knowledge of the spectral properties of the dominating aqueous species present at the interfaces under investigation.Acknowledgements
This work was supported by the BMWi (Germany) as a part of the VESPA project (contract number 02E10790). The authors would like to thank Aline Ritter for ICP-MS measurements. We are also grateful to the ROBL team at ESRF, Grenoble (France). We would like to acknowledge Sophia Kostudis for her support with dispersive Raman spectroscopy. All of the quantum chemical calculations were performed using PC-Farm Atlas at the Zentrum für Informationdienste und Hochleistungsrechnen at the Dresden University of Technology, Dresden, Germany, using the library program Gaussian 09.Notes and references
- A. Fernandez-Martinez and L. Charlet, Rev. Environ. Sci. Biotechnol., 2009, 8, 81–110 CrossRef CAS.
- G. Jörg, R. Buhnemann, S. Hollas, N. Kivel, K. Kossert, S. Van Winckel and C. L. V. Gostomski, Appl. Radiat. Isot., 2010, 68, 2339–2351 CrossRef PubMed.
- ANDRA, Synthèse: Evaluation de la faisabilité du stockage géologique en formation argileuse, Agence Nationale pour la gestion des Déchets RAdioactifs, Châtenay-Malabry, 2005 Search PubMed.
- T. Brasser, J. Droste, I. Müller-Lyda, J. Neles, M. Sailer, G. Schmidt and M. Steinhoff, Endlagerung wärmeentwickelnder radioaktiver Abfälle in Deutschland. GRS-247, Report ISBN 978-3-939355-22-9, Gesellschaft für Anlagen und Reaktorsicherheit (GRS) mbH and Öko-Institut e.V., Braunschweig/Darmstadt, 2008 Search PubMed.
- ONDRAF/NIRAS, Technical overview of the SAFIR 2 report – Safety assessment and feasibility interim report 2 (NIROND 2001–05 E), Belgian Agency for Radioactive Waste and Enriched Fissile Materials, Brussels, 2001.
- A. Olin, B. Noläng, E. G. Osadchii, L.-O. Öhman and E. Rosén, Chemical thermodynamics of selenium, Elsevier Science Publishers B. V., Amsterdam, 2005 Search PubMed.
- J. Torres, V. Pintos, S. Dominguez, C. Kremer and E. Kremer, J. Solution Chem., 2010, 39, 1–10 CrossRef CAS.
- J. Janickis and H. Gutmanaite, Z. Anorg. Allg. Chem., 1936, 227, 1–16 CrossRef CAS PubMed.
- H. Ley and E. König, Z. Phys. Chem., Abt. B, 1938, 41, 365–387 Search PubMed.
- A. Rosenheim and L. Krause, Z. Anorg. Allg. Chem., 1921, 118, 177–192 CrossRef CAS PubMed.
- A. Miolati and E. Mascetti, Gazz. Chim. Ital., 1901, 31, 93–139 Search PubMed.
- L. Barcza and L. G. Sillen, Acta Chem. Scand., 1971, 25, 1250–1260 CrossRef CAS PubMed.
- R. Sabbah and G. Carpeni, J. Chim. Phys., 1966, 63, 1549–1554 CAS.
- E. S. Ganelina, V. P. Kuz'micheva and M. B. Krasnopol'skaya, Russ. J. Inorg. Chem., 1973, 18, 698–700 Search PubMed.
- R. Arnek and L. Barcza, Acta Chem. Scand., 1972, 26, 213–217 CrossRef CAS PubMed.
- J. N. Cooper, M. Woods, J. C. Sullivan and E. Deutsch, Inorg. Chem., 1976, 15, 2862–2864 CrossRef CAS.
- L. S. A. Dikshitulu, P. Vani and B. V. Kumar, J. Indian Chem. Soc., 1984, 61, 385–388 CAS.
- L. S. A. Dikshitulu, P. Vani and V. H. Rao, Indian J. Chem., Sect. A, 1981, 20, 36–39 Search PubMed.
- S. Nadimpalli, R. Rallabandi and D. S. A. Lanka, Transition Met. Chem., 1990, 15, 191–196 CrossRef CAS.
- J. Torres, V. Pintos, L. Gonzatto, S. Dominguez, C. Krerner and E. Kremer, Chem. Geol., 2011, 288, 32–38 CAS.
- C. M. Su and D. L. Suarez, Soil Sci. Soc. Am. J., 2000, 64, 101–111 CrossRef CAS.
- M. Altmaier, V. Metz, V. Neck, R. Muller and T. Fanghanel, Geochim. Cosmochim. Acta, 2003, 67, 3595–3601 CrossRef CAS.
- M. Altmaier, V. Neck and T. Fanghanel, Radiochim. Acta, 2008, 96, 541–550 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Revision A.02 edn, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. Head-Gordon and T. Head-Gordon, Chem. Phys. Lett., 1994, 220, 122–128 CrossRef CAS.
- M. Head-Gordon, J. A. Pople and M. J. Frisch, Chem. Phys. Lett., 1988, 153, 503–506 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- W. Koch, O. Lutz and A. Nolle, Z. Naturforsch., A J. Phys. Sci., 1978, 33, 1025–1028 Search PubMed.
- D. L. Parkhurst and C. A. J. Apello, U.S. Geological Survey, 1999 Search PubMed.
- L. Eklund and I. Persson, Dalton Trans., 2014, 43, 6315–6321 RSC.
- K. Nakamoto, Infrared and Raman spectra of Inorganic and Coordination Compounds. Part A: Theory and Applications in Inorganic Chemistry, Wiley-Interscience. John Wiley & Sons, Inc., New York, 5th edn, 1997 Search PubMed.
- K. Nakamoto, in Handbook of Vibrational Spectroscopy, ed. J. M. Chalmers and P. R. Griffiths, vol. 3: Sample Characterization and Spectral Data Processing, Wiley, 2002, pp. 1872–1892 Search PubMed.
- J. Weidlein, U. Müller and K. Dehnicke, Schwingungsspektroskopie: Eine Einführung. 2., überarbeitete Auflage, Georg Thieme Verlag Stuttgart, New York, 1988 Search PubMed.
- A. Simon and R. Paetzold, Z. Elektrochem., 1960, 64, 209–212 CAS.
- J. H. Lee, Y. S. Lee, H. Kim and D.-Y. Jung, Eur. J. Inorg. Chem., 2011, 3334–3339 CrossRef CAS PubMed.
- A. R. Lim, S. W. Jang and J. H. Chang, Solid State Nucl. Magn. Reson., 2007, 31, 124–130 CrossRef CAS PubMed.
- E. É. Rider, V. A. Sarin, N. N. Bydanov and I. S. Vinogradova, Sov. Phys. – Crystallogr., 1986, 31, 155–159 Search PubMed.
- H. Siebert, Z. Anorg. Allg. Chem., 1954, 275, 225–240 CrossRef CAS PubMed.
- H. Kolshorn and H. Meier, J. Chem. Res., 1977, 338–339 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Brief literature review on Se(IV) dimerization studies, stability constants used for speciation calculations, 77Se NMR spectra of Se(IV) at pHc 5, and varying I, both at 9.4 T. See DOI: 10.1039/c5dt00730e |
| This journal is © The Royal Society of Chemistry 2015 |