
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and characterization of mixed-valence manganese fluorophosphate and analogues with clathrate-like structures: MnIII6F12(PO3(OH))8[Na8(Kx(H3O)4−x(H2O)2)MIV(OH)6] (MIV = Mn, Ti, Ge)†
Wei-Jian
Ren
a,
Jing-Quan
Wang
a,
Ya-Xi
Huang
a,
Zhi-Mei
Sun
b,
Yuanming
Pan
c and
Jin-Xiao
Mi
 *a
*a
aFujian Provincial Key Laboratory of Advanced Materials, Department of Materials Science and Engineering, College of Materials, Xiamen University, Xiamen 361005, Fujian Province, People's Republic of China. E-mail: jxmi@xmu.edu.cn
bSchool of Materials Science and Engineering, Beihang University, Beijing 100191, People's Republic of China
cDepartment of Geological Sciences, University of Saskatchewan, 114 Science Place, Saskatoon, SK S7N 5E2, Canada
First published on 20th March 2015
Abstract
A novel, mixed- and high-valence manganese (Mn3+/Mn4+) fluorophosphate, MnIII6F12(PO3(OH))8[Na8(Kx(H3O)4−x(H2O)2)MnIV(OH)6] (denoted as MN), has been prepared via a water-deficient hydrothermal route with phosphoric acid as the sole solvent. This compound features a cubic three-dimensional open-framework structure built from corner-sharing [MnIIIO4F2] octahedra and [HPO4] groups, which encapsulates a clathrate-like “guest cluster” of Na8(Kx(H3O)4−x(H2O)2)MnIV(OH)6. The guest cluster is architecturally composed of a [MnIV(OH)6] octahedron in a cubic cage of Na+ cations, which in turn is surrounded by an octahedral arrangement of K+/H2O ions, resulting in an unprecedented octahedral @ cubic @ octahedral @ cubic arrangement (OCOC). The +4 oxidation state of Mn in the guest cluster has been confirmed by the synthesis of isotypic Ti- and Ge- analogues (denoted as TI and GE) using TiO2 and GeO2 as the replacement for MnO2 in the starting materials. The compounds MN, TI and GE are not stable in aqueous solution and are peeled off layer-by-layer after the absorption of water. This report provides a new route for the synthesis of mixed- and high-valence manganese phosphates that cannot be produced by conventional hydrothermal methods.
1. Introduction
With the development of novel molecular materials and in-depth studies on the origin and process of life, it is now recognized that many optical,1 electrical2 and magnetic properties,3 as well as superconductivity4 and electronic transmissions within organisms,5 are closely related to the mixed-valence phenomenon. Although mixed-valence compounds were produced by chemists a century ago for their unusual color and non-stoichiometric valence, there are only four mixed-valence manganese phosphates among hundreds of inorganic manganese phosphates: BiMnII/III6PO12,6 bermanite MnII(H2O)4[MnIII2(OH)2(PO4)2],7 αββα-K3Na11[MnIII2MnII2(H2O)2(P2W15O56)2]·40H2O8 and K14Na17[(MnIII13MnIIO12(PO4)4(PW9O34)4]·∼56H2O.9 It is also noteworthy that all of these four compounds are the mixtures of MnII and MnIII, although manganese has diverse valence chemistry: MnII, MnIII, MnIV, MnV, MnVI, MnVII, etc. High-valence manganese compounds have important applications in oxidation, catalysis and magnetism. However, the instability of high-valence manganese ions in aqueous solution makes the synthesis of these compounds difficult.To the best of our knowledge, only a few dozen inorganic trivalent manganese phosphates10 have been reported and there are no tetravalent or higher-valence manganese phosphates, with one exception being polyoxometalate.11 Thus, in an attempt to enrich the structural chemistry of mixed-valence compounds and obtain high valence manganese compounds, we initiated a comprehensive synthesis program to explore manganese phosphates. The results reported herein demonstrate that we have succeeded in the synthesis of the first-ever, inorganic, mixed- and high-valence manganese phosphate with both MnIII and MnIV. Mixed MnIII and MnIV organic compounds are not uncommon.12 This mixed- and high-valence manganese phosphate MnIII6F12(PO3(OH))8[Na8(K3.74(H3O)0.26(H2O)2)MnIV(OH)6] (denoted as MN hereafter) has been synthesized by using a water-deficient hydrothermal method and exhibits a three-dimensional (3D) open-framework structure containing clathrate-like Na8(K3.74(H3O)0.26(H2O)2)MnIV(OH)6 guest clusters.
Of particular interest, the structure of the new mixed- and high-valence manganese phosphate resembles that of the clathrate compounds. Clathrates have been known for over two hundred years and are of growing interests for diverse applications from hydrogen storage13 to superconductivity14 and semiconductivity,15etc. These materials possess open framework structures with large cages in the crystal lattice, which can incorporate guest molecules. Previous studies showed that clathrate structures are commonly cubic.16 The structure of the title compound MN is also cubic and is assembled by guest clusters residing in a 3D open framework. The +4 oxidation state of Mn in the guest cluster has been confirmed by the formation of isotypic Ti- and Ge-analogues, MnIII6F12(PO3(OH))8[Na8(K2.97(H3O)1.03(H2O)2)Ti(OH)6] and MnIII6F12(PO3(OH))8[Na8(K2.79(H3O)1.21(H2O)2)Ge(OH)6] (denoted as TI and GE), in synthesis experiments using TiO2 or GeO2 as starting materials as the replacement for MnO2. All three isotypic fluorophosphates have been characterized by single X-ray diffraction (XRD) analysis, Fourier-transform infrared spectroscopy (FTIR), X-ray photoelectron spectroscopy (XPS) and magnetic studies.
2. Experimental section
2.1. Synthesis
Reactions of a transition metal fluoride with a phosphate source in a low-water containing, high-fluoride system have been proven to be a powerful route for synthesizing new fluorophosphate compounds.10,17–19 Here we have synthesized the title compounds via a similar water-deficient hydrothermal route with phosphoric acid as the sole solvent. For MN, in a typical synthesis procedure, the starting materials of NaF (0.4 g, 9.5 mmol), KPF6 (0.8 g, 4.3 mmol) and KMnO4 (0.5 g, 3.2 mmol) were dissolved in H3PO4 (85%, 1 mL, 14.6 mmol) without adding any water. The resulting mixture with the molar ratio of NaF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :KPF6:KMnO4:H3PO4 = 3:1.3:1:4.6 was transferred into a 25 mL Teflon-lined stainless-steel autoclave and heated at 240 °C for 3 days under static conditions. The resultant product is composed of crystals in highly viscous gel-like materials. Rosy-red truncated-octahedral crystals in high yields (above 90%, based on Mn, but poor quality) were isolated by filtering after the viscous materials were dissolved in deionized water for a few minutes.
:KPF6:KMnO4:H3PO4 = 3:1.3:1:4.6 was transferred into a 25 mL Teflon-lined stainless-steel autoclave and heated at 240 °C for 3 days under static conditions. The resultant product is composed of crystals in highly viscous gel-like materials. Rosy-red truncated-octahedral crystals in high yields (above 90%, based on Mn, but poor quality) were isolated by filtering after the viscous materials were dissolved in deionized water for a few minutes.
Manganese ions of high oxidation states are well known to be unstable in acidic hydrothermal conditions, e.g. KMnO4 spontaneously decomposes to K2MnO4 + MnO2 + O2. Consequently, the high valence Mn7+ ions are readily reduced to low valence ions such as Mn4+, Mn3+ and Mn2+. In contrast, low-valence manganese ions (i.e., Mn2+ or Mn3+) are relatively stable in hydrothermal conditions, and cannot be spontaneously oxidized to high valence ions. It is not possible to synthesize high valence manganese compounds using low valence manganese reactants as starting materials without the addition of high oxidant reagents. In other words, manganese in the starting materials must be tetravalent or higher in valence state in order to synthesize MN that contains both trivalent and tetravalent manganese.
As an alternative route for the synthesis of MN, we adopted a similar experimental method as above but used MnO2 (0.278 g, 3.2 mmol) instead of KMnO4 and with the addition of KCl (0.239 g, 3.2 mmol). This method did not produce a single phase but a mixture of high-quality single crystals of MN with minor amounts of KMnF2(PO3F).10 The high-quality crystals of MN produced via this route are suitable for single crystal XRD analysis (see below). Further synthesis investigation shows that Mn2O3 or MnCl2 instead of MnO2 resulted in water-soluble, highly viscous gel-like materials without MN. Runs using Mn-metal powder as a starting material produced a new compound (K0.59(H3O)0.41MnII(P(O0.5F0.5)3OH)2Cl2, to be reported elsewhere), supporting the above discussion that the synthesis of the title compound requires high-valence Mn in the starting materials. Hand-picked crystals used for further characterization were first confirmed by a powder X-ray diffraction (PXRD) analysis, which also indicated MN to be a new phase and is consistent with the calculated pattern from the single-crystal structure analysis below (Fig. S1†).
Additional synthesis experiments with Ti and Ge have been carried out to confirm that the title compound contains MnIV as suggested by the single-crystal structure analysis. It is interesting to note that synthesis experiments using the same ingredients and conditions as described above, plus additional TiO2 or GeO2 in the stoichiometric proportion, resulted in only the compound MN owing to the existence of MnIV. The Ti and Ge analogues of the title compound (denoted as TI and GE hereafter) were synthesized by using Mn2O3 instead of MnO2. In a typical synthesis experiment for the compound TI, NaF (0.4 g, 9.5 mmol), KPF6 (0.8 g, 4.3 mmol), KCl (0.239 g, 3.2 mmol), Mn2O3 (0.216 g, 1.37 mmol) and TiO2 (0.036 g, 0.46 mmol) were dissolved in H3PO4 (85%, 1 mL, 14.6 mmol). The molar ratio of NaF/KPF6/KCl/Mn2O3/TiO2/H3PO4 was about 20.7:9.3:7.0:3.0:1.0:31.7. Finally, rosy-red truncated-octahedral crystals in high yields (about 90%, based on Ti) were obtained as a pure phase. For the compound GE, a similar recipe was used, except that GeO2 (0.048 g, 0.46 mmol) was added. Rosy-red truncated-octahedral crystals in high yields (about 90%, based on Ge) were also obtained. The PXRD patterns of both TI and GE are consistent with the calculated ones from the single-crystal X-ray diffraction analyses shown below, and confirm that they are isostructural with MN (Fig. S2 and 3†).
2.2. Characterization methodologies
The existence of the Na, K, Mn/Ti/Ge, P, O and F atoms in the title compounds was confirmed by the use of an Energy Dispersive X-ray Spectrometer (Oxford Instruments) (Fig. S4–6†). The powder samples checked by PXRD were used for thermal investigations, FTIR and XPS analyses and magnetic measurements. Thermal investigations were performed on a TG-209F1 thermogravimetric/differential thermal analyzer (TG-DTA) in a N2 atmosphere with a heating rate of 10 K min−1. The FTIR spectra recorded were of powder samples mixed with KBr in pressed pellets on a Nicolet 330 FTIR spectrometer in the range of 400–4000 cm−1. XPS analyses were performed using a Physical Electronics Quantum 2000 scanning ESCA microprobe equipped with a standard focused monochromatic Al Kα (1486.7 eV) X-ray source. Magnetic susceptibility was measured in the temperature range from 2 to 300 K, using a Quantum Design MPMS XL-7 SQUID magnetometer.2.3. Crystal structure determination
Single crystals of the compounds MN, TI and GE were carefully selected on the basis of clarity and uniformity under a petrographic microscope and were glued onto a thin glass capillary for single-crystal X-ray diffraction analysis. Data collection was performed at 173(2) K, using a Bruker Apex CCD diffractometer or an Oxford Diffraction Xcalibur Sapphire 3 CCD diffractometer, equipped with a graphite-monochromatic MoKα radiation (λ = 0.71073 Å). A total of 6146 observed reflections were collected from 2.21° < θ < 27.93°, yielding 462 unique reflections (Rint = 0.025) with 394 I > 2σ(I) for the compound MN. 2364 observed reflections were collected from 3.60° < θ < 29.75°, yielding 495 unique reflections (Rint = 0.042) with 409 I > 2σ(I) for the compound TI. 6452 observed reflections were collected from 2.21° < θ < 28.75°, yielding 493 unique reflections (Rint = 0.048) with 424 I > 2σ(I) for the compound GE.The crystal structures of the compounds MN, TI and GE were solved in a cubic space group of Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) by direct methods and refined by the full-matrix least-squares method using the SHELXS-2014 and SHELXL-2014 software packages.20 The final refinements converged at R1 [I > 2σ(I)] = 0.033, 0.036 and 0.042, as well as wR2 (for all) = 0.106, 0.112 and 0.121 for the compounds MN, TI and GE, respectively. Experimental details for the structure determinations are summarized in Table 1. Detailed information may be found in the ICSD (no. 429030–429032).
by direct methods and refined by the full-matrix least-squares method using the SHELXS-2014 and SHELXL-2014 software packages.20 The final refinements converged at R1 [I > 2σ(I)] = 0.033, 0.036 and 0.042, as well as wR2 (for all) = 0.106, 0.112 and 0.121 for the compounds MN, TI and GE, respectively. Experimental details for the structure determinations are summarized in Table 1. Detailed information may be found in the ICSD (no. 429030–429032).
| MN | TI | GE | |
|---|---|---|---|
| a Weight parameters (w = 1/[σ2(Fo2) + (W1P)2 + W2P] where P = (Fo2 + 2Fc2)/3) were used. | |||
| Formula sum, weight (g mol−1) | F12H18.78K3.74Mn7Na8O40.26P8, 1853.58 | F12H21.10K2.97Mn6Na8O41.03P8Ti, 1831.03 | F12GeH21.62K2.79Mn6Na8O41.21P8, 1852.21 |
| Crystal size (mm3), color | 0.27 × 0.27 × 0.27, rosy-red | 0.32 × 0.32 × 0.32, rosy-red | 0.30 × 0.30 × 0.30, rosy-red |
| Crystal system, space-group | Cubic, Fm (no. 202) |
Cubic, Fm (no. 202) |
Cubic, Fm (no. 202) |
| Cell parameters (Å) | a = 15.968(2) | a = 16.0001(2) | a = 15.981(7) |
| Cell volume (Å3), Z, Calc. density (g cm−3) | 4071.5, 4, 3.024 | 4096.05, 4, 2.969 | 4082, 4, 3.014 |
| Rad., wavelength (Å), temp. (K) | MoKα, 0.71073, 173(2) | MoKα, 0.71073, 173(2) | MoKα, 0.71073, 173(2) |
| μ (mm−1), F(000) | 3.067, 3612 | 2.858, 3575 | 3.392, 3609 |
| 2θmax (°), Npara | 55.86, 43 | 59.49, 55 | 57.51, 55 |
| Miller-index | −20 ≤ h ≤ 13, −21 ≤ k ≤ 20, −17 ≤ l ≤ 20 | −21 ≤ h ≤ 22, −20 ≤ k ≤ 15, −21 ≤ l ≤ 8 | −21 ≤ h ≤ 21, −21 ≤ k ≤ 9, −20 ≤ l ≤ 20 |
| R int, R1, wR2 | 0.025, 0.033, 0.106 | 0.042, 0.036, 0.112 | 0.048, 0.042, 0.121 |
| S, N, N (I > 2σ(I)) | 1.184, 462, 394 | 1.144, 495, 409 | 1.100, 493, 424 |
| Weight parameters (W1, W2)a | 0.0597, 19.4949 | 0.0535, 8.1128 | 0.0648, 45.4995 |
| Δρmax, Δρmin (e Å−3) | 0.780, −0.742 | 0.955, −0.495 | 1.317, −0.949 |
3. Results and discussion
3.1. Crystal structure description
The crystal structures of the three compounds, MN, TI and GE, are isotypic and are characterized by [MnIIIO4F2] octahedra sharing O-corners with [HPO4] tetrahedra to form a three-dimensional open-framework of {MnIII6F12(PO3(OH))8}10− with an eight-membered ring channel running along the direction of <100>, in which the {Na8(Kx(H3O)4−x(H2O)2)MIV(OH)6}10+ (M = Mn, Ti, Ge) clusters are encapsulated.In the crystal structure of MN, Mn ions of two different valences (BVS: 3.05 and 4.02 vu.)21 occur separately at two crystallographically distinct positions: Mn(1)3+ and Mn(2)4+ at Wyckoff 24d and 4b sites, respectively. Each Mn(1)3+ ion is coordinated to four oxygen atoms at 2.066(2) Å and two fluorine atoms at 1.818(2) Å to form a compressed [Mn(1)3+O4F2] octahedron, whereas the Mn(2)4+ ion is surrounded by six hydroxyl groups (OH) at 1.901(5) Å in a regular octahedron. The compressed [Mn(1)IIIO4F2] octahedron may suggest that the Mn3+ ion adopts the high-spin electronic configuration of four unpaired 3d electrons with one electron in one eg (i.e., dx2 − y2) orbital. Each [Mn(1)IIIO4F2] octahedron links to four [HPO4] groups via the sharing of equatorial O-corners, and four bidentate Na+ ions in the axial sides. The [HPO4] tetrahedron shares three O-corners, each with a different [MnIIIO4F2] octahedron, and has a terminal OH group. Each bidentate Na+ ion also links to three [Mn3+O4F2] octahedra. Six neighboring [Mn(1)IIIO4F2] octahedra in octahedral distribution enclose a mini-cube cage, where four involved [HPO4] groups and four bidentate Na+ alternately locate at the mini-cube corners in tetrahedral patterns. The mini-cube cage has a diameter of 5.0 Å (Fig. 1). By viewing along the direction of <100>, three-dimensional (3D) interconnected channels contain a narrow 8-membered ring neck consisting of alternating four [Mn(1)IIIO4F2] octahedra and four [HPO4] groups. The octahedrally distributed K/H3O ions and water molecules (denoted as [K6]) and the [MnIV(OH)6]@Na8 clusters locate within the channels and are arranged in the positions of the NaCl-type structure (Fig. 1). Interestingly, the [MnIV(OH)6]@Na8 cluster is octahedrally surrounded by six potassium or water molecules to form a new [MnO6]@Na8@K/H2O cluster in the channels. The [MnIV(OH)6] octahedra are not involved in the construction of the 3D open framework structure, and are separated from the framework by Na+, K+ and H2O. As such, the [MnIV(OH)6] octahedra and surrounding Na+, K+ and H2O form a clathrate-like “guest cluster” of Na8(Kx(H3O)4−x(H2O)2)MnIV(OH)6 (Fig. 2).
 | ||
| Fig. 1 The crystal structure of MN. (a) The framework composed of corner-sharing [MnIIIO4F2] and [PO4], with encapsulated cages that are located at the centers of the 8-minicubes of the cubic cell, as F in the fluorite (CaF2) structure; (b) illustration of the relationship of the framework, cages and guest clusters (the green spheres represent the encapsulated cages in the framework; and the cyan spheres denote the spaces surrounded by six K(H3O)/H2O ions); (c) guest clusters and potassium ions in channels arranged in a NaCl-type pattern. | ||
 | ||
| Fig. 2 Illustration of the relationship of the framework and guest clusters. Heptavalent MnVII(d0) ions of the starting materials were reduced to low-valence ions: with tetravalent MnIV(d3) ions in the guest clusters and trivalent MnIII(d4) ions forming the framework. | ||
3.2. Thermal properties
Thermal analysis of the compound MN shows that the first-step weight-loss of 2.32% at the temperature range of 250–450 °C in the TG curve (Fig. S7†) can be ascribed to the removal of 2.39 × H2O per formula unit (calcd, 2.40%). The powder X-ray diffraction analysis shows that the solid residue of MN after annealing at 477 °C for 2 hours mainly consists of Na2MnPO4F with a minor amount of KMnF3 and an unidentified phase22,23 (Fig. S8†).3.3. Infrared spectroscopy
The FTIR spectra of the compounds MN and TI show similar absorption bands (see Fig. S9†). The peaks at 3439/3437 cm−1 (medium, broad) can be attributed to the asymmetric O–H stretching, while the peaks at 1620/1640 cm−1 (weak, broad) and 3599/3597 cm−1 (sharp, weak) are assigned to the H–O–H bending mode. The free PO43− ion with the perfect Td symmetry has four normal modes: ν1(A1), the symmetric stretching mode, ν2(E), the OPO symmetric bending mode, and ν3(F2) and ν4(F2), the asymmetric stretching and bending modes, respectively (all Raman allowed, but only ν3 and ν4 IR allowed). The bands at 1151/1157 and 1023/1030 cm−1 are attributable to the antisymmetric stretching mode (ν3) of [HPO4], and those at 585/592 and 517/522 cm−1 are ascribed to the antisymmetric bending mode (ν4). Two inactive IR modes (i.e., ν1 & ν2) are not observed, suggesting that the IR forbidden modes are not lifted, and this consistent with the high symmetry of the lattice. The band at 793/826 cm−1, which is beyond the ranges of occurrences for the four normal modes of [HPO4], may be attributed to the water librations.243.4. XPS analysis
X-ray photoelectron spectroscopic analysis (Fig. S10†) was performed to identify the oxidation states of manganese and confirm the existence of other elements (i.e., Na, K, F, O, and P) in the compound MN. Fig. S10 shows that the XPS spectrum of MN exhibits two strong peaks at 642.5 and 653.9 eV in the energy region of Mn 2p3/2 and Mn 2p1/2, respectively. The distance between the two main peaks is about 11.4 eV, which may suggest that the manganese mainly has an oxidation state at +3.25,26 It is noteworthy that the BVS calculations suggest only one Mn4+ ion but six Mn3+ ions per formula unit in the compound MN, and thus the signal of the former in the XPS spectrum is probably overshadowed by the much more abundant Mn3+.3.5. Magnetic properties
Compounds MN and TI show linear Curie–Weiss behavior in the χ−1vs. T data in the entire 2–300 K temperature range, yielding C1 = 20.33 emu K mol−1, θC1 = −2.2 K and C2 = 18.35 emu K mol−1, θC2 = −2.7 K, respectively (Fig. 3). The observed effective moment μeff1 = 12.75μB for the compound MN is in excellent agreement with the predicted value of 12.61μB, assuming a spin-only J value for one Mn4+ and six high-spin Mn3+ ions. Considering the d0 state of Ti4+, the observed effective moment μeff2 = 12.11μB for the compound TI is also in good agreement with the predicted value of 12.00μB, assuming a spin-only J value for six high-spin Mn3+ ions. Weak antiferromagnetic (AF) Mn–Mn interactions are expected to predominate in both compounds, which explains the small negative Weiss temperatures, as observed. The gradual increase in the χT product versus temperature also conforms to the AF couplings.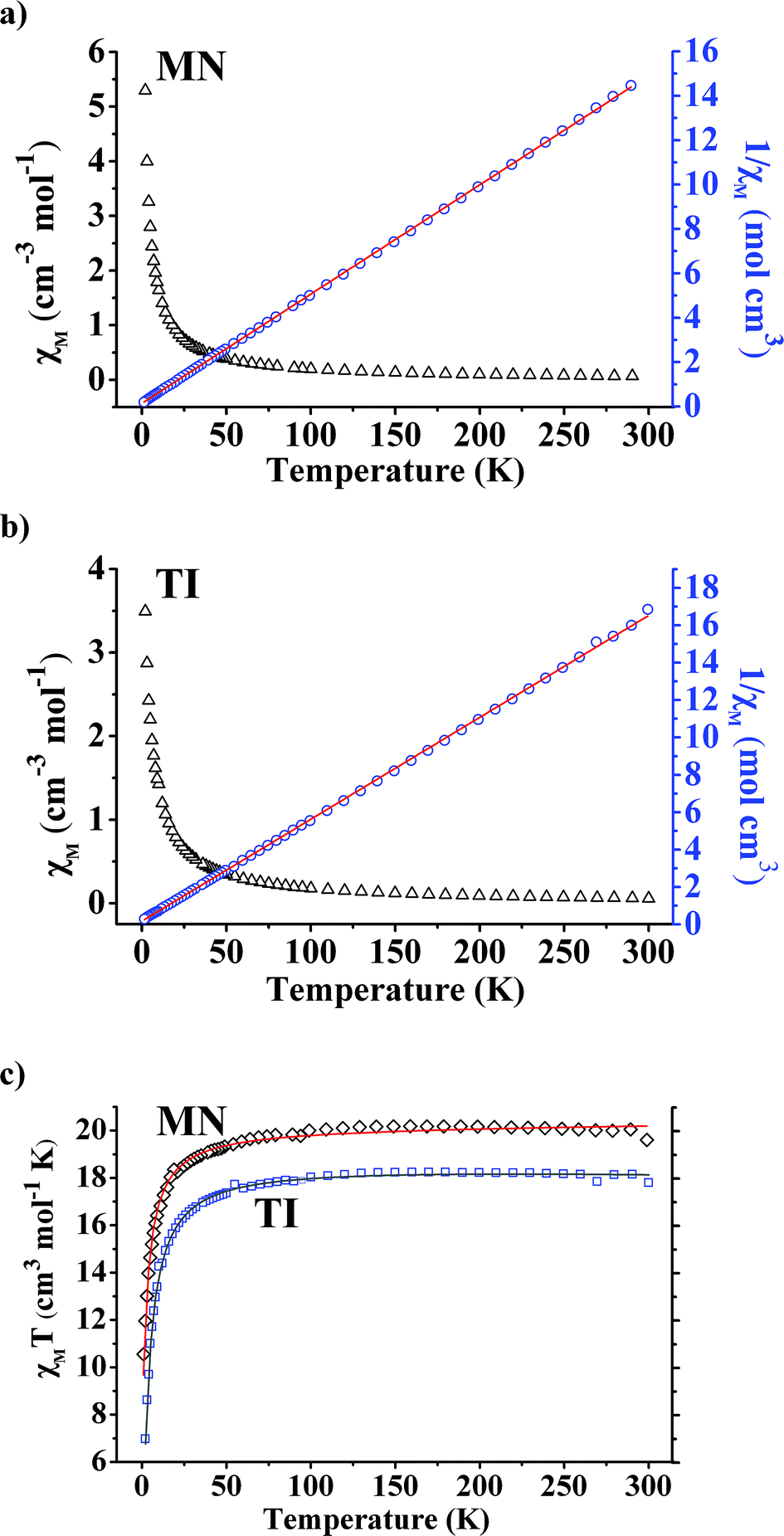 | ||
| Fig. 3 Magnetic properties of the compounds MN and TI. Magnetic susceptibility (χM) and reciprocal susceptibility (χ−1) vs. temperature at 1000 Oe for MN (a) and TI (b), respectively; their χMT product vs. temperature (c). | ||
3.6. Structural flexibilities and stabilities
The ideal phosphate ion, PO43−, has four O2− ions arranged at the corners of a regular tetrahedron. In the compound MN, however, each phosphorus atom is split into two positions, and correspondingly, one of the oxygen atoms is also distributed in two sites. This disordered distribution of the [HPO4] tetrahedron is supposedly caused by self-adjusting of the fundamental building units for fitting the whole structure. As described above, the “guest cluster” of [MnIV(OH)6] in the channel can be substituted by [Ge(OH)6] or [Ti(OH)6] to form the isotypic compounds of TI and GE. In both compounds TI and GE, there is a small residue peak observed around the P-positions, suggesting that a slight disorder of the [HPO4] tetrahedra also occurs in TI and GE. However, the occupancies of the disordered P atoms are less than 5% in those compounds; they can be considered to be in an almost fully ordered arrangement. In contrast to MN, the P atoms in TI and GE are nearly fully ordered in the framework, but the “guest clusters” in the channel are disordered. Every oxygen atom (24e) in the [TiIV(OH)6] or [GeIV(OH)6] octahedron is split into three positions: at Wyckoff 24e, 48h and 48h sites, respectively. So the crystal structures of the title compounds have high structural flexibilities, which are tuned by order and disorder alteration.It is interesting to note that the crystals of MN, TI and GE are highly unstable in aqueous solution. When the crystals were soaked in water for one hour, they were peeled off layer-by-layer along {100} and {111} after being pressed slightly (Fig. 4). This pattern of dissolution is observed in all three title compounds, MN, TI and GE, and suggests a preferential removal of the channel constituent oriented along {100}. This suggestion is further supported by the presence of minor crystalline Na(OH) and K2SiF6 in the dissolved glassware, although the majority of the solid dissolution products from the title compounds are amorphous (Fig. S11†). Such instability of the title compounds in aqueous solution is not unexpected and is consistent with the fact that high-valence manganese phosphates cannot be synthesized under normal hydrothermal conditions. The successful syntheses of the title compounds in the present study are attributable to our usage of phosphoric acid as the sole solvent under water-deficient hydrothermal conditions. Our previous works have shown that the water content is a crucial factor in the synthesis of other chloride,27 phosphate28 and borate29 compounds.
 | ||
| Fig. 4 Illustration of the decomposition of the title compounds in water. All crystals of MN, TI and GE peeled off layer-by-layer after soaking in aqueous solution for 1 hour. | ||
4. Conclusions
In this paper, we have demonstrated three isotypic new compounds: MnIII6F12(PO3(OH))8[Na8(Kx(H3O)4−x(H2O)2)MIV(OH)6] (MIV = Mn, Ti, Ge). The first-ever, mixed- and high-valence manganese fluorophosphate with both Mn3+ and Mn4+ has been synthesized by a water-deficient hydrothermal method with phosphoric acid as the sole solvent. This compound features a clathrate-like structure with a cubic three-dimensional open-framework structure encapsulating the unprecedented Na8(K3.74(H3O)0.26(H2O)2)MnIV(OH)6 guest clusters in the channels. Successful syntheses of the isotypic compounds TI and GE confirm the presence of Mn4+ in the guest clusters. This report provides a new hydrothermal route for the synthesis of mixed- and high-valence manganese compounds.Acknowledgements
This work is supported by the National Science Foundation for Distinguished Young Scientists of China (Grant no. 51225205), the National Natural Science Foundation of China (no. 21233004, 61274005 and 21201144), the Fundamental Research Funds for the Central Universities (no. 2013121020) and financial support from the Natural Science and Engineering Research Council of Canada.References
- B. S. Brunschwig, C. Creutz and N. Sutin, Chem. Soc. Rev., 2002, 31, 168–184 RSC.
- E. G. Walton, P. J. Corvan, D. B. Brown and P. Day, Inorg. Chem., 1976, 15, 1737–1739 CrossRef CAS.
- M. Viret, L. Ranno and J. M. D. Coey, Phys. Rev. B: Condens. Matter, 1997, 55, 8067–8070 CrossRef CAS.
- S. P. Pai, J. Jasudasan, P. R. Apte, R. Pinto, J. Kurian, P. K. Sajith, J. James and J. Koshy, Europhys. Lett., 1997, 39, 669–673 CrossRef CAS.
- M. Di Vaira, F. Mani and P. Stoppioni, J. Chem. Soc., Dalton Trans., 1997, 1375–1379 RSC.
- O. Cousin, O. Mentre, M. Huve and F. Abraham, J. Solid State Chem., 2001, 157, 123–133 CrossRef CAS.
- A. R. Kampf and P. B. Moore, Am. Mineral., 1976, 61, 1241–1248 CAS.
- S. Yao, J. H. Yan, Y. C. Yu and E. B. Wang, J. Coord. Chem., 2012, 65, 1451–1458 CrossRef CAS.
- Q. Wu, Y. G. Li, Y. H. Wang, E. B. Wang, Z. M. Zhang and R. Clerac, Inorg. Chem., 2009, 48, 1606–1612 CrossRef CAS PubMed.
- J. A. Armstrong, E. R. Williams and M. T. Weller, Dalton Trans., 2013, 42, 2302–2308 RSC.
- X. K. Fang and P. Koegerler, Angew. Chem., Int. Ed., 2008, 47, 8123–8126 CrossRef CAS PubMed.
- C. Lampropoulos, A. E. Thuijs, K. J. Mitchell, K. A. Abboud and G. Christou, Inorg. Chem., 2014, 53, 6805–6816 CrossRef CAS PubMed.
- H. Lee, J. W. Lee, D. Y. Kim, J. Park, Y. T. Seo, H. Zeng, I. L. Moudrakovski, C. I. Ratcliffe and J. A. Ripmeester, Nature, 2005, 434, 743–746 CrossRef CAS PubMed.
- H. Kawaji, H. Horie, S. Yamanaka and M. Ishikawa, Phys. Rev. Lett., 1995, 74, 1427–1429 CrossRef CAS.
- G. S. Nolas, T. J. R. Weakley and J. L. Cohn, Chem. Mater., 1999, 11, 2470–2473 CrossRef CAS.
- J. A. Ripmeester, J. S. Tse, C. I. Ratcliffe and B. M. Powell, Nature, 1987, 325, 135–136 CrossRef CAS.
- J. A. Armstrong, E. R. Williams and M. T. Weller, J. Am. Chem. Soc., 2011, 133, 8252–8263 CrossRef CAS PubMed.
- A. C. Keates, J. A. Armstrong and M. T. Weller, Dalton Trans., 2013, 42, 10715–10724 RSC.
- J. A. Armstrong, E. R. Williams and M. T. Weller, Dalton Trans., 2012, 41, 14180–14187 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- O. V. Yakubovich, O. V. Karimova and O. K. Melnikov, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1997, 53, 395–397 Search PubMed.
- K. Knox, Acta Crystallogr., 1961, 14, 583–585 CrossRef CAS.
- V. G. Koleva, Spectrochim. Acta, Part A, 2007, 66, 413–418 CrossRef PubMed.
- Y. F. Han, F. X. Chen, Z. Y. Zhong, K. Ramesh, L. W. Chen and E. Widjaja, J. Phys. Chem. B, 2006, 110, 24450–24456 CrossRef CAS PubMed.
- F. Moro, V. Corradini, M. Evangelisti, V. De Renzi, R. Biagi, U. del Pennino, C. J. Milios, L. F. Jones and E. K. Brechin, J. Phys. Chem. B, 2008, 112, 9729–9735 CrossRef CAS PubMed.
- T. T. Zhu, W. Sun, Y. X. Huang, Z. M. Sun, Y. M. Pan, L. Balents and J. X. Mi, J. Mater. Chem. C, 2014, 2, 8170–8178 RSC.
- L. Z. Sun, W. Sun, W. J. Ren, J. Y. Zhang, Y. X. Huang, Z. M. Sun, Y. M. Pan and J. X. Mi, J. Solid State Chem., 2014, 212, 48–57 CrossRef CAS PubMed.
- B. C. Zhao, W. Sun, W. J. Ren, Y. X. Huang, Z. C. Li, Y. M. Pan and J. X. Mi, J. Solid State Chem., 2013, 206, 91–98 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The figures of experimental and simulated powder X-ray diffraction patterns, EDS/SEM images, TG-DTA, FT-IR and XPS. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00646e |
| This journal is © The Royal Society of Chemistry 2015 |