
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective B–B bond activation in an unsymmetrical diborane(4) by [(Me3P)4Rh–X] (X = Me, OtBu): a switch of mechanism?†
C.
Borner
,
K.
Brandhorst
 and
C.
Kleeberg
*
and
C.
Kleeberg
*
Technische Universität Carolo-Wilhelmina zu Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: ch.kleeberg@tu-braunschweig.de
First published on 14th April 2015
Abstract
The unsymmetrical diborane(4) pinB–B((RN)2(C6H4)) (R = Me, Bn) reacts with [(Me3P)4Rh–X] (X = Me, OtBu) giving predominantly either [(Me3P)4Rh–Bpin] or [(Me3P)3Rh–B((RN)2(C6H4))] depending on X. At low temperatures in the presence of excess PMe3 the unprecedented equatorial boryl complex [(Me3P)4Rh–B((MeN)2(C6H4))] is formed.
Rhodium boryl complexes have been studied extensively as crucial intermediates in various catalytic transformations such as C–H functionalization, hydroboration and diboration. More fundamentally rhodium boryl complexes have also been studied regarding their coordination chemistry, the oxidative addition chemistry of RhI boryl complexes to form RhIII tris-boryl complexes or, more recently, their use as subunits in boryl pincer complexes.1–4 In particular, rhodium boryl complexes of the type [(R3P)nRh–B(OR′)2] (R = Me, Et; n = 3, 4; (OR′)2 = OCMe2CMe2O (pin), 1,2-O2C6H4 (cat), OCH2CMe2CH2O (neop)) have been explored (Scheme 1).2,3 These complexes are accessible by the reaction of a precursor complex [(Me3P)nRh–X] and a symmetrical diborane(4) derivative (B2(OR)2) (Scheme 1).2
 | ||
| Scheme 1 General synthesis and two examples of [(R3P)nRh–B(OR)2] complexes.2 | ||
Marder and co-workers have studied in some detail the chemistry of [(Me3P)4Rh–B(OR)2] complexes and especially their formation from [(Me3P)4Rh–Me] (1a) and B2(OR)2, presumably proceeding via an oxidative addition/reductive elimination pathway.2a–c,3a More recently Braun and co-workers demonstrated that also the rhodium phenolato (X = OPh) and fluorido (X = F) complexes [(Et3P)3Rh–X] are effective in such B–B bond activation reactions (Scheme 1) and have studied their reactivity to some extent.2d,e,3b–d The unsymmetrical diborane(4) derivatives pinB–Bdan (dan = 1,8-(NH)2C10H6) have recently gained considerable attention as reagents in transition metal as well as Lewis base catalysed diboration reactions.5 Moreover, we have recently reported a class of well accessible unsymmetrical diborane(4) derivatives of the type pinB–B((NR)2C6H4) (R = Me (2a), Bn (2b), TMS) and demonstrated their utility as precursors for diaminoboryl complexes of CuI and PtII.6 Herein we extend our study to RhI boryl complexes, in particular addressing the influence of the ligand X on the B–B bond activation process with complexes [(Me3P)4Rh–X] (X = Me (1a), OtBu (1b)). Whilst 1a is an established reagent in related reactions, 1b has not been described earlier. However, related tris-phosphine complexes have been used in B–B bond activation reactions (vide supra).2d Complex 1b is formed virtually quantitatively from equimolar amounts of [(Me3P)4Rh]Cl and KOtBu but appears to be only stable in solution and all experiments were conducted with in situ prepared 1b. Several attempts to isolate 1b led to the isolation of the dimer [(Me3P)2Rh–CH2–PMe2]2.7,8 Nevertheless, in solution 1b was found to be sufficiently stable (no significant decomposition was observed within 30 h).8 Complex 1b was characterised by in situ NMR spectroscopy. Most characteristically the 31P NMR spectrum at low temperatures shows a doublet of quartets and a doublet of doublets as the AM3 part of an AM3X spin system indicative of a five-coordinate, approximately trigonal-bipyramidal rhodium complex with the alkoxido ligand occupying an apical position.8
To study the B–B bond activation of 2a by 1a and 1b the respective reactions were monitored by in situ31P{1H} and 11B{1H} NMR spectroscopy (Fig. 1). For both reactions, two 11B NMR signals with shift >40 ppm (45 and 52 ppm), characteristic of boryl complexes, were observed. This suggests the formation of the same two boryl complexes in both reactions, albeit in different relative amounts. Furthermore, the 11B NMR data give evidence for the complementary formation of pinB–Me, dmabB–Me and pinB–OtBu, respectively (δ11B: 34, 31 and 22 ppm; these compounds, as well as dmabB–OtBu, were also detected by GC/MS analysis).8,9 In addition, unreacted 2a, and for the reaction of 1b, so far unidentified tetracoordinated boron species are observed.10 The corresponding 31P{1H} NMR spectra indicate overall the formation of two different major rhodium phosphine complexes. For the reaction of 1a with 2a, the only major signal observed was a doublet at −22 ppm that is assigned to the complex [(Me3P)4Rh–Bpin] (3) by comparison with an authentic sample.2b,8 For the reaction of 1b with 2a, this compound is only present in minor amounts and the major very broad signal at −27 ppm was eventually assigned to the novel tris-trimethylphosphine rhodium diaminoboryl complex [(Me3P)3Rh–Bdmab] (4a) in rapid exchange with free PMe3 (vide infra).8 In addition, for both reactions, small amounts of by-products/impurities (especially [(Me3P)4Rh–H]) and, in the case of 1b, unreacted starting material were detected.8,11 The selectivity of the B–B bond activation of 2a with 1a/b (hence the [3]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[4a] ratio) is cautiously estimated using combined NMR and GC/MS data to be in the order of 10:1 for 1a and 1:10 for 1b, respectively.8,12
:[4a] ratio) is cautiously estimated using combined NMR and GC/MS data to be in the order of 10:1 for 1a and 1:10 for 1b, respectively.8,12
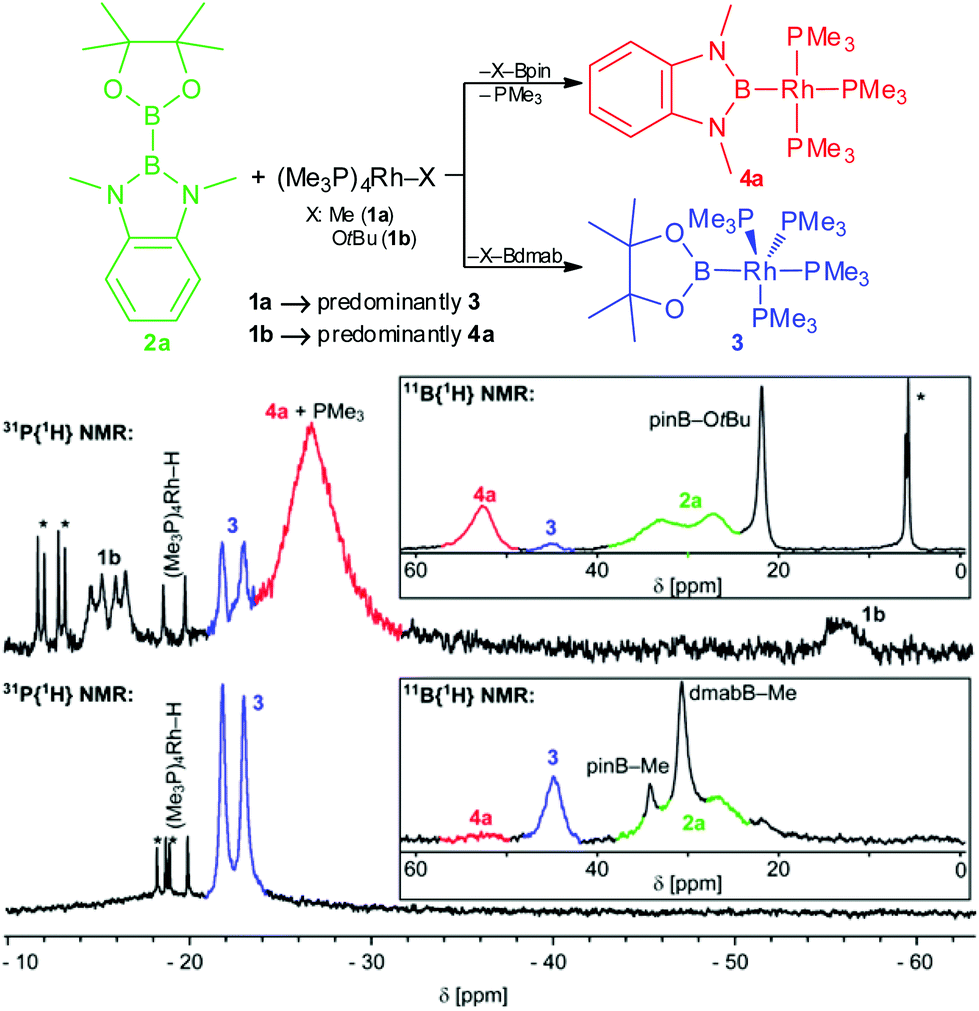 | ||
| Fig. 1 Reaction pathways and in situ31P{1H} and 11B{1H} NMR spectra of the reaction of 2a with 1a (bottom, in C6D6 after 20 h at rt) and 1b (top, in THF-d8 after 20 h at rt) (rt, 122 MHz/96 MHz, *unidentified impurities).10,11 | ||
In order to elucidate further the reaction of 1b with 2a a reaction altering the order of addition (first 2a + KOtBu then addition of [(Me3P)4Rh]Cl) as well as the reaction of an isolated alkoxide adduct of 2a, [(18-C-6)K][dmabB–B(OtBu)pin], with [(Me3P)4Rh]Cl were studied by in situ NMR spectroscopy.6,8 In both cases, corroborating the above results, the predominant formation of 4a is observed.
It may be considered that the formation of 4a from 1b proceeds via its decomposition product [(Me3P)2Rh–CH2–PMe2]2. Indeed, the latter complex reacts with 2a giving both 3 and 4a, however, this reaction is appreciably slower than the B–B activation with in situ formed 1b, furthermore, no indications were found that the decomposition product is present to a substantial amount under the reaction conditions.8 Moreover, the detection of pinB–OtBu and dmabB–OtBu as concomitantly formed by-products is also suggestive for a reaction pathway via the alkoxido complex 1b.
It may be proposed that an oxidative addition/reductive elimination pathway appears likely in the case of 1a, in agreement with the literature,2a whereas a different (predominant) reaction pathway may be operative in the reaction of 1b, e.g. σ-bond metathesis or abstraction of OtBu− to form the anionic sp2–sp3 diboron species of the type [dmabB–B(OtBu)pin]−. Furthermore, it should be emphasised that the loss of a phosphine ligand concomitant with a B–B bond activation of a diborane(4) derivative is unprecedented amongst the related dialkoxy-/aryloxyboryl systems. This may be rationalised by the increased steric demand of the NR groups compared to an oxygen atom.2a,b Nevertheless, tris-phosphine rhodium boryl complexes are readily obtained from tris-phosphine precursors [(R3P)3Rh–X] (Scheme 1).2c–e
On a preparative scale, complex 4a was isolated from reactions of 1b and 2a in 43% yield.8 Analogous reactions of 1b with 2b and with B2pin2, respectively, gave [(Me3P)3Rh–Bdbab] (4b) and 3 in 15% and 24% isolated yield, respectively.2b
The 31P NMR data (in THF-d8) of 4a suggest a square-planar solution structure: at lower temperatures, a doublet of triplets for the phosphine ligands trans and a doublet of doublets for the phosphine ligand cis to the boryl ligand are observed (−80 °C; −23.6 ppm, 1 P, dt, 1JRh–P = 107 Hz, 2JP–P = 33 Hz, Δw1/2 = 11 Hz; −12.8 ppm, 2 P, dd, 1JRh–P = 148 Hz, 2JP–P = 33 Hz, Δw1/2 = 3 Hz). Towards ambient temperature both signals broaden, the latter (Δw1/2 = 7 Hz) much less than the first one which loses any fine-structure (Δw1/2 = 194 Hz). The more pronounced broadening of the signal of the trans-phosphine ligand may suggest dynamic processes specific to that ligand (e.g. exchange with solvent) in accordance with it being trans to the strongly trans-influencing boryl ligand. Noteworthily, in the less coordinating solvent C6D6 comparable differences in linewidth are observed.8
The analogous benzyl-substituted complex 4b exhibits similar NMR spectroscopic data.8
For the complexes 4a,b, single crystals were obtained and tris-phosphine RhI boryl complexes were thus structurally characterised for the first time (Fig. 2). Complex 4a crystallises in the space group P2/n and is situated on the two-fold axis exhibiting C2 symmetry and consequently disorder of the trans-boryl PMe3 ligand.8 Complex 4b, in contrast, exhibits no molecular symmetry and crystallises in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) as a PhMe/n-pentane solvate.8 Both complexes adopt distorted square-planar structures (sum of angles at Rh = 360° and 360.6°, respectively). The Rh–P distances trans to the boryl ligand in 4a,b are significantly longer (0.05 and 0.08 Å, respectively) than the respective distances cis to the boryl ligand indicating the very strong trans influence of the boryl ligand.2a,6,13 These P–Rh distances may be compared to the respective distances in [(Me3P)4Rh–Bcat]; despite the trigonal-bipyramidal structure of this complex a similar trend is observed (B–Rh 2.047(2) Å, Peq–Rh 2.29–2.31 Å, Ptrans–Rh 2.3404(6) Å).2a As an effect of the steric crowding in the coordination sphere of these complexes the boryl ligand plane (defined by the BN2C6H4 moiety) and the coordination plane (defined by the RhP3B subunit) are virtually perpendicular to one another with angles of 84.41(3)° (4a) and 89.47(4)° (4b), respectively.
as a PhMe/n-pentane solvate.8 Both complexes adopt distorted square-planar structures (sum of angles at Rh = 360° and 360.6°, respectively). The Rh–P distances trans to the boryl ligand in 4a,b are significantly longer (0.05 and 0.08 Å, respectively) than the respective distances cis to the boryl ligand indicating the very strong trans influence of the boryl ligand.2a,6,13 These P–Rh distances may be compared to the respective distances in [(Me3P)4Rh–Bcat]; despite the trigonal-bipyramidal structure of this complex a similar trend is observed (B–Rh 2.047(2) Å, Peq–Rh 2.29–2.31 Å, Ptrans–Rh 2.3404(6) Å).2a As an effect of the steric crowding in the coordination sphere of these complexes the boryl ligand plane (defined by the BN2C6H4 moiety) and the coordination plane (defined by the RhP3B subunit) are virtually perpendicular to one another with angles of 84.41(3)° (4a) and 89.47(4)° (4b), respectively.
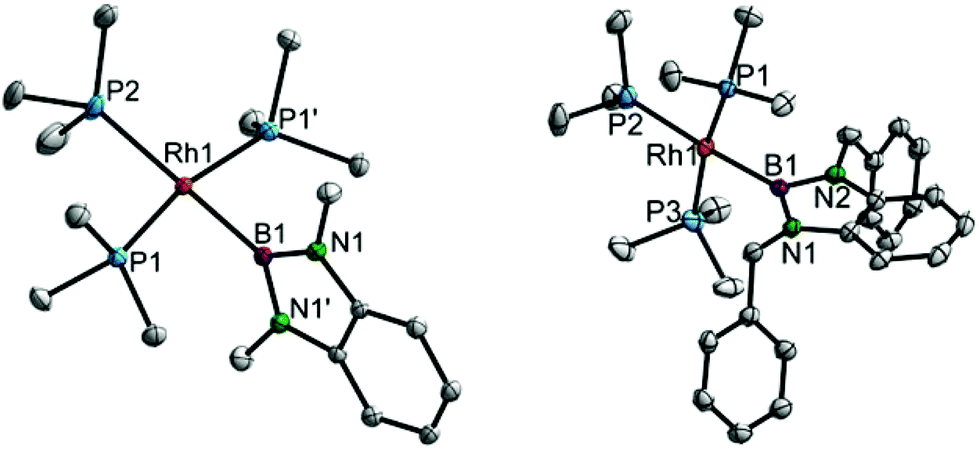 | ||
| Fig. 2 Molecular structures of 4a (left) and 4b (right) at 100 K. Selected bond lengths and angles for 4a: B1–Rh1 2.074(3) Å, P1–Rh1 2.2697(5) Å, P2–Rh1 2.3194(7) Å, B1–Rh1–P2 180°, B1–Rh1–P1 83.73(1)°, P1–Rh1–P1′ 167.46(2)°, P1–Rh1–P2 96.27(1)° (symmetry equivalent atom denoted by a prime (2-fold axis)); for 4b, B1–Rh1 2.090(2) Å, P1–Rh1 2.2659(6) Å, P2–Rh1 2.3455(6) Å, P3–Rh1 2.2651(7) Å, B1–Rh1–P2 174.95(7)°, B1–Rh1–P1 83.58(6)°, B1–Rh1–P3 85.37(6)°, P1–Rh1–P3 167.10(2)°, P1–Rh1–P2 96.73(2)°, P2–Rh1–P3 94.87(2)°. Hydrogen atoms and disorder (at P2 in 4a) are omitted for clarity; thermal ellipsoids are drawn at the 50% probability level.8 | ||
The ability of the tris-triethylphosphine boryl complex [(Et3P)3Rh–Bpin] to activate C–H bonds of aromatics to give a rhodium hydrido complex and an aryl boronate has been studied in some detail and employed in catalytic reactions.2d,3c Hence, it is not surprising that the related diaminoboryl complex 4a reacts with benzene under C–H activation. An initial in situ study of this reaction suggests – according to NMR and GC/MS analysis – the formation of the hydrido complex [(Me3P)4Rh–H] as well as dmabB–Ph along with B2dmab2 and further unidentified species.8 It can be stated that the C–H bond activation of C6H6 (and C6D6) by 4a is a comparably slow process and that NMR measurements of 4a may be conducted in C6D6 within a few hours.8
To gain insight into the fundamental coordination chemistry of 4a its reaction with PMe3 was investigated. Upon addition of PMe3 to a solution of 4a the colour changes from red-orange to yellow and the resulting 31P NMR spectrum exhibits only one broad signal (Fig. 3, bottom).8 The chemical shift of this signal changes with the relative amount of PMe3 added and is well described by the weighted average of the 31P NMR chemical shifts of 4a and PMe3, whilst the 11B NMR shift does not change noticeably.8 This indicates that, at ambient temperature, the PMe3 ligands in 4a are in rapid exchange with free PMe3, whilst other species, in particular [(Me3P)4Rh–Bdmab] 5 (vide infra), are only present in minor amounts (<10 mol%).8 However, at lower temperatures a more complicated behaviour is observed (Fig. 3). The single broad signal (−52 ppm) observed at ambient temperature splits, upon cooling, into a relatively sharp signal indicative of free PMe3 (−61 ppm) and a very broad signal at −22 ppm (−22 °C). Upon further cooling, the latter first sharpens and then broadens again (−22 to −54 °C), indicating the presence of more than one dynamic process. Finally, at even lower temperatures, this signal splits into two signals of equal intensity (−95 °C: −5.9 ppm, Δw1/2 = 400 Hz, and −38.4 ppm, Δw1/2 = 430 Hz; though not fully resolved, the peak shape suggests a pair of broad doublets). These data are consistent with rapid intermolecular exchange of free PMe3 with coordinated PMe3 in 4a at and slightly below ambient temperature. At lower temperatures, however, a single new rhodium species becomes predominant exhibiting intramolecular Pax–Peq exchange, but slower intermolecular exchange with free PMe3.
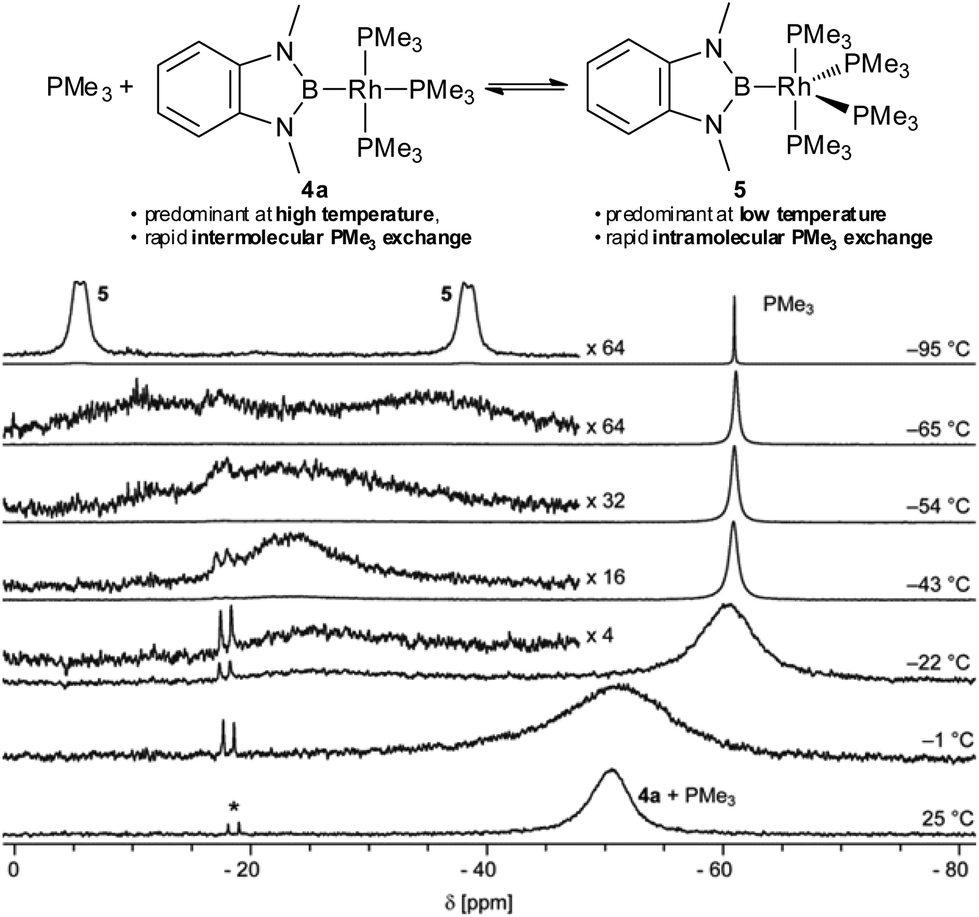 | ||
| Fig. 3 Selected 31P{1H} VT-NMR spectra of 4a in the presence of 10.2 eq. of PMe3 (THF-d8, 162 MHz, *denotes an impurity of [(Me3P)4Rh–H]).8,11 | ||
A likely candidate for the novel species observed at low temperature is the tetrakis-trimethylphosphine complex [(Me3P)4Rh–Bdmab] 5, though, the intensity of the 31P NMR signals imply a trigonal-bipyramidal structure with an equatorial boryl ligand that is unprecedented amongst related complexes such as [(Me3P)4Rh–Bcat] or 3.2a,3a,8 Attempts to crystallise 5 from solutions of 4a in THF and/or n-pentane in the presence of excess PMe3 at low temperatures lead repeatedly to a yellow crystalline material. However, these crystals decompose upon removal from the mother liquor and the residue was identified by NMR analysis as 4a. Eventually, single crystals of 5 suitable for X-ray structure determination were obtained by in situ crystallisation from a saturated solution in neat PMe3 on the diffractometer.8
Complex 5 indeed adopts a trigonal-bipyramidal structure with an equatorial boryl ligand (Fig. 4). As this coordination mode is unprecedented amongst related Rh boryl complexes, direct comparison is difficult; however, increased Peq–Rh distances compared to the cis boryl Pax–Rh distance are observed.2a,8 Noteworthy are the small (∼80°) P1–Rh1–B1 and P3–Rh1–B1 angles indicating significant steric encumbrance of this complex.
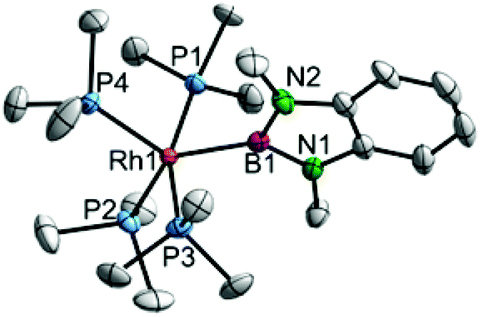 | ||
| Fig. 4 Molecular structures of 5 at 175 K. Selected bond lengths and angles B1–Rh1 2.109(3) Å, P1–Rh1 2.2765(7) Å, P2–Rh1 2.3396(7) Å, P3–Rh1 2.2660(7) Å, P4–Rh1 2.3616(7) Å, B1–Rh1–P1 79.20(8)°, B1–Rh1–P2 130.45(8)°, B1–Rh1–P3 80.02(8)°, B1–Rh1–P4 121.15(8)°, P1–Rh1–P2 96.04(3)°, P1–Rh1–P3 158.92(3)°, P1–Rh1–P4 96.08(3)°, P2–Rh1–P3 94.69(3)°, P2–Rh1–P4 108.40(3)°, P3–Rh1–P4 97.67(3)°. Hydrogen atoms are omitted for clarity; thermal ellipsoids are drawn at the 50% probability level.8 | ||
DFT computations were performed to elucidate further the reaction of 4a with PMe3 and the formation of 5 (Fig. 5).8,14 The geometries of 4a and 5 were optimised starting from the X-ray structures of 4a and for 5 from a putative isomeric apical boryl complex based on the X-ray structure of 3. It should be mentioned that the fully optimised structure of 5 represents the equatorial isomer and all attempts to locate a minimum structure for the respective (expected) apical isomer have failed. This is in contrast to the related dialkoxyboryl complexes where the axial boryl complex is preferred for electronic reasons.3a The geometrical parameters of the fully optimised structures correspond well with the crystallographically determined data, albeit, in agreement with literature data, the Rh–P bonds are systematically too long.3a
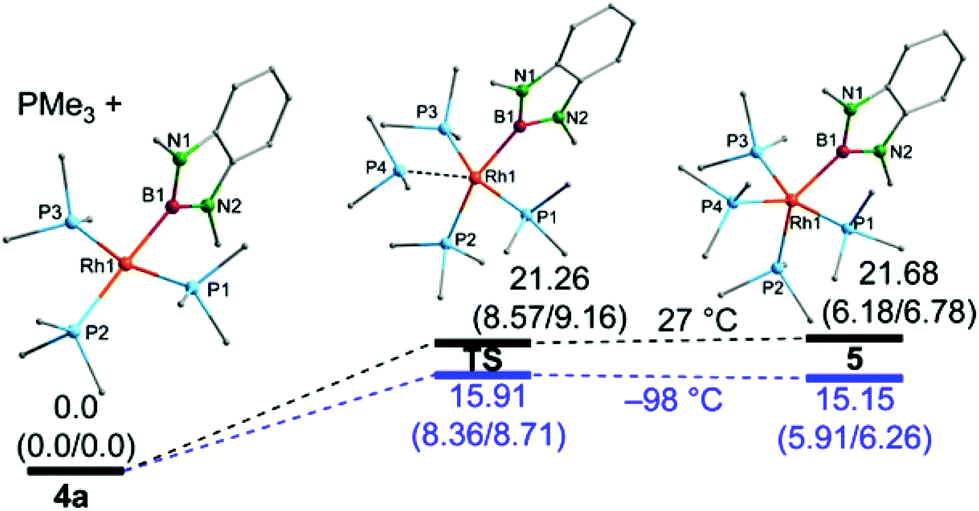 | ||
| Fig. 5 Calculated structures and relative energies ΔG(ΔH/ΔE) in kcal mol−1 for 4a, 5 and the respective transition states for the two temperatures 27 °C and −98 °C at the B3LYP/6-311G(d,p) level of theory; for further details see ESI.†8 | ||
In terms of free energy, the dissociation of a PMe3 ligand from 5 to give 4a is found to have a marginal or even no barrier at all and, hence, the isolation of 4a, in agreement with the experimental data, is expected. Moreover, the data are also fundamentally in agreement with a rapid intermolecular exchange of PMe3 ligands in 4a with free PMe3, possibly involving 5 as an intermediate. The 5-coordinate complex 5 is calculated to be higher in energy than 4 at all temperatures examined, but much less so at lower temperatures, consistent with the NMR spectra recorded in the presence of excess PMe3 at low temperature. As the computed enthalpies/energies were determined in the gas-phase, they cannot be expected to match the experimental findings exactly, which is especially true for the Gibbs free energies which contain entropic contributions, that are expected to be smaller in the condensed phase.
Conclusions
The regioselectivity of B–B bond activation of the unsymmetrical diborane(4) derivative pinB–B((MeN)2(C6H4)) by [(Me3P)4Rh–X] (X = Me, OtBu) (1a,b) is controlled by the nature of X. Either the dialkoxido boryl complex [(Me3P)4Rh–Bpin] (3) for X = Me or, accompanied by dissociation of a PMe3 ligand, the diaminoboryl complex [(Me3P)3Rh–B((MeN)2(C6H4))] (4a) for X = OtBu is obtained predominantly. This strongly suggests that two fundamentally different mechanisms are operating for B–B activation. The expected penta-coordinated product [(Me3P)4Rh–B((MeN)2(C6H4))] (5), however, is only stable at low temperatures in the presence of excess PMe3 and exhibits an unprecedented equatorial boryl ligand. In agreement with DFT computations, this is rationalised considering the steric demand of the NR groups that causes the expected five-coordinate apical boryl complex to be disfavoured over its equatorial isomer 5 and, moreover, favours dissociation of a PMe3 ligand to give the tris-phosphine complex 4a. Further studies to elucidate the mechanisms of the B–B bond activation processes with 1a,b, as well as a detailed study on the reactivity of 4a are in progress.Acknowledgements
C. K. and C. B. thank the Fonds der Chemischen Industrie for the generous support by a Liebig- and a Doktoranden-Stipendium. We also gratefully acknowledge support by the Deutsche Forschungsgemeinschaft (DFG). The authors wish to thank T. B. Marder (Würzburg) for very helpful discussions and sharing data prior to publication as well as for a sample of 1a. The authors thank BASF SE for a generous supply of B2(NMe2)4.Notes and references
- For selected reviews, see: (a) J. F. Hartwig, Acc. Chem. Res., 2012, 45, 864–873 CrossRef CAS PubMed; (b) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (c) T. Ishiyama and N. Miyaura, Chem. Rec., 2004, 3, 271–280 CrossRef CAS PubMed; (d) K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS; (e) G. J. Irvine, M. J. G. Lesley, T. B. Marder, N. C. Norman, C. R. Rice, E. G. Robins, W. R. Roper, G. R. Whittell and L. J. Wright, Chem. Rev., 1998, 98, 2685–2722 CrossRef CAS PubMed; (f) T. B. Marder and N. C. Norman, Top. Catal., 1998, 5, 63–73 CrossRef CAS; (g) D. L. Kays and S. Aldridge, Struct. Bonding, 2008, 130, 29–122 CrossRef CAS.
- (a) C. Dai, G. Stringer, T. B. Marder, A. J. Scott, W. Clegg and N. C. Norman, Inorg. Chem., 1997, 36, 272–273 CrossRef CAS; (b) C. Dai, Ph. D. Thesis, University of Waterloo, Canada, 1998 Search PubMed; (c) T. B. Marder , personal communication; (d) M. Teltewskoi, J. A. Panetier, S. A. Macgregor and T. Braun, Angew. Chem., Int. Ed., 2010, 49, 3947–3951 CrossRef CAS PubMed; (e) M. Teltewskoi, S. I. Kalläne, T. Braun and R. Herrmann, Eur. J. Inorg. Chem., 2013, 5762–5768 CrossRef CAS.
- (a) K. C. Lam, W. H. Lam, Z. Lin, T. B. Marder and N. C. Norman, Inorg. Chem., 2004, 43, 2541–2547 CrossRef CAS PubMed; (b) S. I. Kalläne, T. Braun, B. Braun and S. Mebs, Dalton Trans., 2014, 43, 6786–6801 RSC; (c) S. I. Kalläne and T. Braun, Angew. Chem., Int. Ed., 2014, 53, 9311–9315 CrossRef PubMed; (d) S. I. Kalläne, M. Teltewskoi, T. Braun and B. Braun, Organometallics, 2015 DOI:10.1021/om500952x.
- For further examples of Rh boryl complexes see: (a) W. Clegg, F. J. Lawlor, T. B. Marder, P. Nguyen, N. C. Norman, A. G. Orpen, M. J. Quayle, C. R. Rice, E. G. Robins, A. J. Scott, F. E. S. Souza, G. Stringer and G. R. Whittell, J. Chem. Soc., Dalton Trans., 1998, 301–309 RSC; (b) P. Nguyen, G. Lesley, N. J. Taylor, T. B. Marder, N. L. Pickett, W. Clegg, M. R. J. Elsegood and N. C. Norman, Inorg. Chem., 1994, 33, 4623–4624 CrossRef CAS; (c) M. Hasegawa, Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2012, 51, 6956–6960 CrossRef CAS PubMed; (d) M. V. Câmpian, J. L. Harris, N. Jasim, R. N. Perutz, T. B. Marder and A. C. Whitwood, Organometallics, 2006, 25, 5093–5104 CrossRef; (e) R. J. Lindup, T. B. Marder, R. N. Perutz and A. C. Whitwood, Chem. Commun., 2007, 3664–3666 RSC; (f) B. Procacci, Y. Jiao, M. E. Evans, W. D. Jones, R. N. Perutz and A. C. Whitwood, J. Am. Chem. Soc., 2015, 137, 1258–1272 CrossRef CAS PubMed.
- (a) N. Iwadate and M. Suginome, J. Am. Chem. Soc., 2010, 132, 2548–2549 CrossRef CAS PubMed; (b) J. Cid, J. J. Carbó and E. Fernández, Chem. – Eur. J., 2014, 20, 3616–3620 CrossRef CAS PubMed.
- C. Borner and C. Kleeberg, Eur. J. Inorg. Chem., 2014, 2486–2489 CrossRef CAS.
- [(Me3P)2Rh–CH2–PMe2]2 was previously reported: V. V. Mainz and R. A. Andersen, Organometallics, 1984, 3, 675–678 CrossRef CAS.
- See ESI† for details.
- (a) R. Goetze, H. Nöth, H. Pommerening, D. Sedlak and B. Wrackmeyer, Chem. Ber., 1981, 114, 1884–1893 CrossRef CAS; (b) H. C. Brown, W. S. Park, J. S. Cha, B. T. Cho and C. A. Brown, J. Org. Chem., 1986, 51, 337–342 CrossRef CAS; (c) B. L. Tran, D. Adhikari, H. Fan, M. Pink and D. J. Mindiola, Dalton Trans., 2010, 358–360 RSC; (d) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350–5354 CrossRef CAS PubMed; (e) C. Kleeberg and C. Borner, Eur. J. Inorg. Chem., 2013, 2799–2806 CrossRef CAS.
- The 11B{1H} NMR spectrum of the reaction of 1b with 2a exhibits additionally two sharp signals at 6.0 and 5.8 ppm (Δw1/2 = 3.4, 9.0 Hz) indicating the formation of tetra-coordinated boron species, possibly Lewis acid/base adducts of tBuO− and pinB–OtBu, dmabB–OtBu or 2a.
- The formation of small amounts of [(Me3P)4Rh–H] in related reactions was reported earlier and may be associated with the presence of adventitious moisture.2a,e Alternatively it is formed slowly by C–H activation of aromatics.2d,3c,d,8 For spectroscopic data see: (a) D. Zargarian, P. Chow, N. J. Taylor and T. B. Marder, J. Chem. Soc., Chem. Commun., 1989, 540–544 RSC; (b) M. Paneque and P. M. Maitlis, J. Chem. Soc., Chem. Commun., 1989, 105–106 RSC.
- Performing the reaction of 1a with 2a in THF-d8 led to a slight preference for 4a, accompanied by substantial formation of unidentified species.8.
- (a) J. Zhu, Z. Lin and T. B. Marder, Inorg. Chem., 2005, 44, 9384–9390 CrossRef CAS PubMed; (b) H. Braunschweig, P. Brenner, A. Müller, K. Radacki, D. Rais and K. Uttinger, Chem. – Eur. J., 2007, 13, 7171–7176 CrossRef CAS PubMed.
- M. J. Frisch et al. , Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental and analytical data as well as additional crystallographic and NMR spectroscopic material. CCDC 1037662–1037666. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00618j |
| This journal is © The Royal Society of Chemistry 2015 |