
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Imidazol-2-ylidene-N′-phenylureate ligands in alkali and alkaline earth metal coordination spheres – heterocubane core to polymeric structural motif formation†
Kishor
Naktode
a,
Jayeeta
Bhattacharjee
a,
Hari Pada
Nayek
b and
Tarun K.
Panda
 *a
*a
aDepartment of Chemistry, Indian Institute of Technology Hyderabad, Ordnance Factory Estate, Yeddumailaram 502205, Telangana, India. E-mail: tpanda@iith.ac.in; Fax: +91 (40) 2301 6032; Tel: +91 (40) 2301 6036
bDepartment of Applied Chemistry, Indian School of Mines, Dhanbad, 826004, Jharkhand, India
First published on 9th March 2015
Abstract
The synthesis and isolation of two potassium, one lithium and two calcium complexes of imidazol-2-ylidene-N′-phenylureate ligands [ImRNCON(H)Ph] [(R = tBu (1a); Mes (1b) and Dipp (1c); Mes = mesityl, Dipp = 2,6-diisopropylphenyl] are described. Potassium complexes, [{κ2-(ImMesNCONPh)K}4] (2b) and [{κ3-(ImDippNCONPh)K}2{KN(SiMe3)2}2]n (2c), were prepared in good yields by the reactions of 1b and 1c, respectively, with potassium bis(trimethyl)silyl amide at ambient temperature in toluene. Lithium complex [{(2,6-tBu2-4-Me-C6H2O)Li(ImtBuNCON(H)Ph)}2{ImtBuNCON(H)Ph}] (3a) was isolated by a one-pot reaction between 1a and LiCH2SiMe3, followed by the addition of 2,6-tBu2-4-Me-C6H2OH in toluene. Calcium complex [{κ2-(ImtBuNCONPh)Ca{N(SiMe3)2}-{KN(SiMe3)2}]n (4a) was isolated by the one-pot reaction of 1a with [KN(SiMe3)2] and calcium diiodide in THF at ambient temperature. The solid-state structures of ligand 1a and complexes 2b, 2c, 3a and 4a were confirmed by single-crystal X-ray diffraction analysis. It was observed that potassium was coordinated to the oxygen atom of urea group and to the nitrogen atom of the imidazolin-2-imine group, in the solid-state structure of 2b. In complex 4a, the calcium ion was ligated to the monoanionic imidazol-2-ylidene-N′-phenylureate ligand in a bi-dentate (κ2) fashion through the oxygen and nitrogen atoms of the isocyanate building block leaving the imidazolin-2-imine fragment uncoordinated. In the solid state of the potassium complex 2c, tri-dentate (κ3) coordination from the imidazol-2-ylidene-N′-phenylureate ligand was observed through the oxygen and nitrogen atoms of the isocyanate building block and of the imidazolin-2-imine fragment. In contrast, in the dimeric lithium complex 3a, the neutral imidazol-2-ylidene-N′-phenylureate ligand was bound to the lithium centre in a mono-dentate fashion (κ1) through an oxygen atom of the isocyanate moiety. It is to be noted that in each complex thus observed, the elongated carbon–nitrogen bond distances indicate substantial electron delocalisation from the imidazole ring to the ureate group present in ligand 1.
Introduction
From an early stage, ligands with amine or imine functionality have played an important role in the field of coordination chemistry.1 After the remarkable discovery of the first stable and structurally characterised imidazolin-2-ylidene by Anthony Joseph Arduengo, III, N-heterocyclic carbenes (NHCs) immediately became indispensable to developments in many diverse research areas, such as homogeneous catalysis,2 materials science3 and medicinal chemistry.4The enhanced electron-donating capacity and high nucleophilicity observed in these carbenes are indicative of the capability of the imidazolium ring to effectively stabilise a positive charge. An analogous principle can be applied to organic imidazolin derivatives containing an exo-cyclic atom or an organic moiety X attached at the 2-position of the N-heterocycle such that, for species such as 2-methylene-, 2-imino-, 2-oxo- and 2-thioimidazolines (X = CH2, NH, O, S), a strong contribution from the ylidic mesomeric form 1B may be considered (Chart 1).5,6
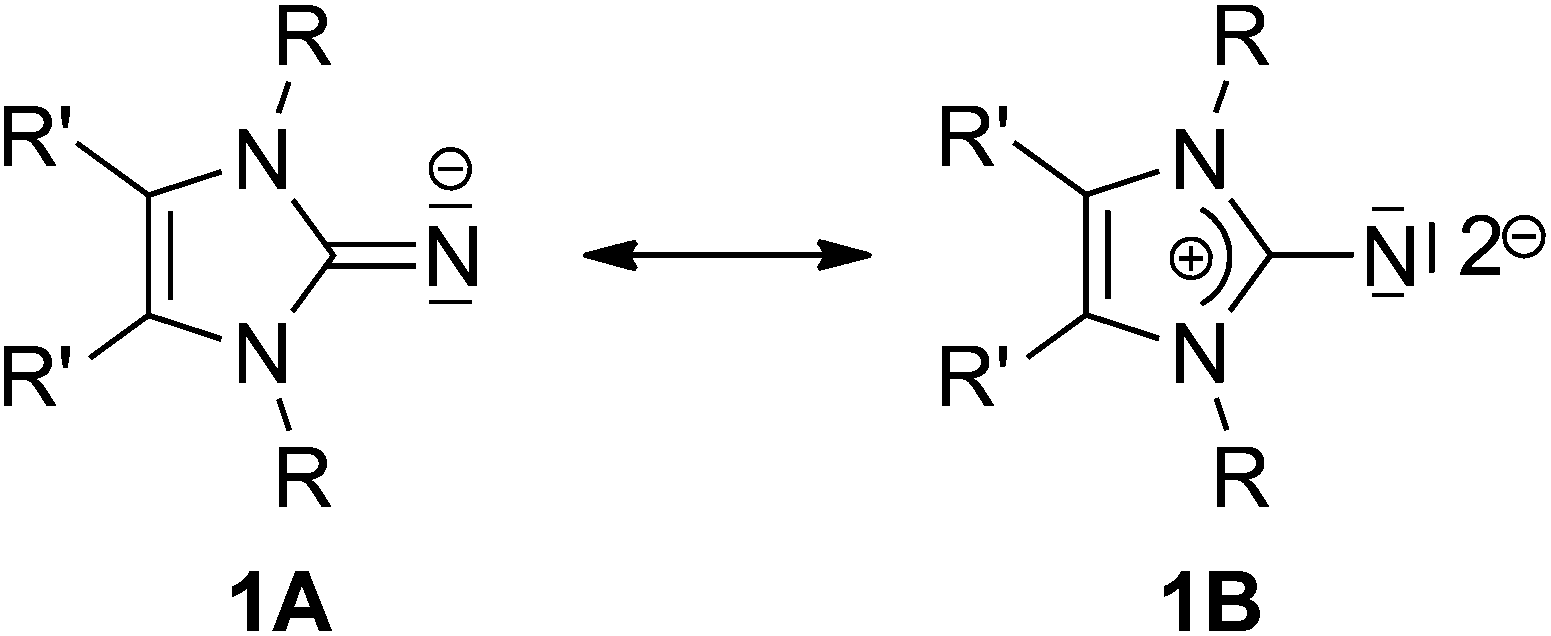 | ||
| Chart 1 Mesomeric forms of imidazolin-2-iminato ligand. | ||
It was established that, due to the resulting build-up of negative charge at the X atom due to the mesomeric form 1B, the atom X possesses enhanced basicity and nucleophilicity.7 In recent years, Tamm and co-workers exploited this concept by providing access to a large variety of novel imidazolin-2-imines (ImNH, X = NH) that can be used for the preparation of imidazolin-2-iminato complexes of transition metals and rare earth elements, and, more recently, of actinide metals, in order to achieve very short M–N bonds. This has led to the probability of a multiple bonding character between M–N bonds.8 The M–N bond possesses a multiple bonding character – its reactivity is described as being very similar to a M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N imido bond.9 It has been observed that various metal complexes supported by imidazolin-2-iminato ligands display high activity in ethylene (co)polymerisation and in alkyne metathesis.10
N imido bond.9 It has been observed that various metal complexes supported by imidazolin-2-iminato ligands display high activity in ethylene (co)polymerisation and in alkyne metathesis.10
Lavoie et al. recently reported the preparation of urea and the thio-urea derivatives imidazol-2-ylidene-N′-p-tolylureate and imidazol-2-ylidene-N′-p-tolylthioureate from imidazolin-2-imines and p-tolyl isocyanate and isothiocyanate, respectively.11 This family of neutral polydentate ligands incorporates the imidazolin-2-imine fragment in its fold. Lavoie et al. also introduced these urea- and thio-urea functionalised ligands into groups 4 and 10 in transition metal chemistry in order to explore their coordination behaviours. It was observed that these bi-dentate urea- and thio-urea functionalised ligands bound more electro positive titanium (Ti4+) ions through the nitrogen and oxygen atoms of the isocyanate building block, while leaving the imidazol-2-imine fragment uncoordinated. However, they found the lesser electropositive metal ions (Ni2+ and Pd2+) to be ligated through both the nitrogen atoms of the ligand. Nevertheless, their work was restricted to transition metal chemistry, and the structural aspects of alkali metal and alkaline earth metal complexes with these ligands have not been reported to date.
We earlier reported on phosphine-functionalised imidazolin-2-imines, imidazolin-2-ylidene-1,1-diphenylphosphine-amine and their chalcogenide derivatives (O, S, Se and Te).12 In our ongoing efforts to prepare functionalised imidazolin-2-imines, and with an interest to observe and record their coordination behaviour towards the main group organometallics, we prepared various alkali metal and alkaline earth metal complexes with imidazolin-2-imine urea derivatives.
It is in this context that herein we present the synthetic and structural details of two potassium complexes [{κ2-(ImMesNCONPh)K}4] (2b) and [{κ3-(ImDippNCONPh)K}2{KN(SiMe3)2}2]n (2c), one lithium complex (3a) and one calcium complex [{κ2-(ImtBuNCONPh)-Ca{N(SiMe3)2}{KN(SiMe3)2}]n (4a) of imidazol-2-ylidene-N′-phenylureate ligand [ImRNCON(H)Ph] [(R = tBu (1a); Mes (1b) and Dipp (1c); Mes = mesityl, Dipp = 2,6-diisopropylphenyl]. We also report the molecular structures of an imidazol-2-ylidene-N′-phenylthiourate ligand (1d) and a siloxane incorporated calcium complex [{κ3-(ImtBuNCONPh)}2Ca(OSiMe2OSiMe2O)2{κ3-(ImtBuNCONPh)CaK}2{KN(SiMe3)2}2{Ca(N(SiMe3)2)2}2]n (5a).
Results and discussion
Ligand synthesis
The imidazol-2-ylidene-N′-phenylureate and -thioureate ligands were prepared by an analogous method to that reported by Lavoie et al.11 Imidazolin-2-imines (ImRNH) with three different substituents over nitrogen atoms were charged with a slight excess of phenyl isocyanate (5% excess) at ambient temperature to give the corresponding imidazol-2-ylidene-N′-phenylureate [ImRNCON(H)Ph] (R = tBu) (1a); Mes (1b) and Dipp (1c) (Scheme 1). The analogous thioureate ligand [ImtBuNCSN(H)Ph] (1d) was isolated by the reaction of [ImtBuNH] with phenyl isothiocyanate in toluene (Scheme 1). All the ligands were characterised using standard analytical and spectroscopic techniques. The solid-state structures of 1a and 1d were established by single-crystal X-ray diffraction analysis.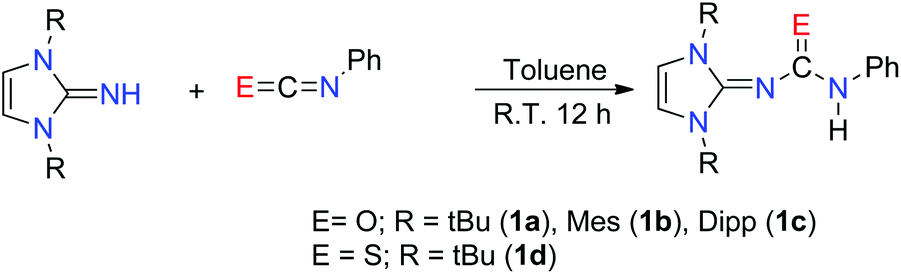 | ||
| Scheme 1 Synthesis of imidazol-2-ylidene-N′-phenylureate/thioureate ligands. | ||
The 1H NMR spectra measured in C6D6 for the compounds 1a–c and CDCl3 (1d) are similar and show a characteristic singlet resonance [δ 6.18 (1a), 5.80 (1b), 6.10 (1c) and 6.22 (1d) ppm] for imidazolium backbone olefinic protons. These can be observed as a downfield shift from that of the respective imidazol-2-imines [δ 5.96 (tBu); 5.71 (Mes); 5.87 (Dipp) ppm]. The resonances for 18 protons from the six methyl groups in 1a appeared as singlets at δ 1.43 ppm, whereas two singlets at δ 2.26 and 2.05 ppm could be assigned to the o- and p-methyl protons, respectively, in the mesityl groups. The distinct septet signal at δ 3.21 ppm, and the two doublet resonances with a coupling constant 6.8 Hz each, appeared at δ 1.45 and 1.16 ppm, respectively, due to the –CH hydrogen and isopropyl methyl hydrogen atoms of the ligand 1c. The CO stretching frequencies [ν 1627 (1a), 1647 (1b), 1650 (1c) cm−1] were considerably lower than that of the starting isocyanate (2170 cm−1), indicating a marked reduction in C–O strength upon formation of the urea. As the observed stretching frequencies were also slightly lower than those commonly observed in organic amides13 – a manifestation of the electron delocalisation from the imidazole ring to the acyl group – a further decrease of the C–O bond order can be realised. The resonance for the tert-butyl methyl protons in compound 1d could be detected as a sharp singlet at δ 1.65 ppm. The strong absorption band at 1400–1600 cm−1 in the FT-IR spectrum of 1d indicated the presence of a CS group. However, the exact band was difficult to identify due to the mixing of the other vibration modes.14
Single crystals of 1a and 1d were obtained from the concentrated toluene solution of the respective compounds at ambient temperature. Compound 1a crystallised in the monoclinic space group P21/c with four molecules in the unit cell. In contrast, the thiourea derivative 1d crystallised in the trigonal space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) with 18 molecules in the unit cell. The details of the structural parameters are given in Table TS1 in the ESI.† The solid-state structure of complex 1a is shown in Fig. 1, whereas Fig. S1 in the ESI† represents the solid-state structure of 1d. The C1–N1 bond distance of 1.346(3) Å in 1a is elongated, compared to ImtBuNH [1.295(2) Å];15 presumably due to enhanced electron donation to the imino nitrogen atom from the imidazolium ring. A slight elongation of the carbonyl C12–O1 bond [1.236(3) Å] was observed due to the delocalisation of electrons from imino nitrogen to the oxygen atom of the isocyanate building block. The slightly shorter N1–C12 bond [1.339 (3) Å], compared to the N4–C12 bond [1.389(3) Å], is also supportive of the above delocalisation over the N1–C12–O1 unit on ligand 1a. Short hydrogen bonding between the electronegative oxygen atom and hydrogen atoms from adjacent methyl groups, O1⋯H5a (2.706 Å) and O1⋯H9c (2.541 Å), is also observed (Fig. 1).
with 18 molecules in the unit cell. The details of the structural parameters are given in Table TS1 in the ESI.† The solid-state structure of complex 1a is shown in Fig. 1, whereas Fig. S1 in the ESI† represents the solid-state structure of 1d. The C1–N1 bond distance of 1.346(3) Å in 1a is elongated, compared to ImtBuNH [1.295(2) Å];15 presumably due to enhanced electron donation to the imino nitrogen atom from the imidazolium ring. A slight elongation of the carbonyl C12–O1 bond [1.236(3) Å] was observed due to the delocalisation of electrons from imino nitrogen to the oxygen atom of the isocyanate building block. The slightly shorter N1–C12 bond [1.339 (3) Å], compared to the N4–C12 bond [1.389(3) Å], is also supportive of the above delocalisation over the N1–C12–O1 unit on ligand 1a. Short hydrogen bonding between the electronegative oxygen atom and hydrogen atoms from adjacent methyl groups, O1⋯H5a (2.706 Å) and O1⋯H9c (2.541 Å), is also observed (Fig. 1).
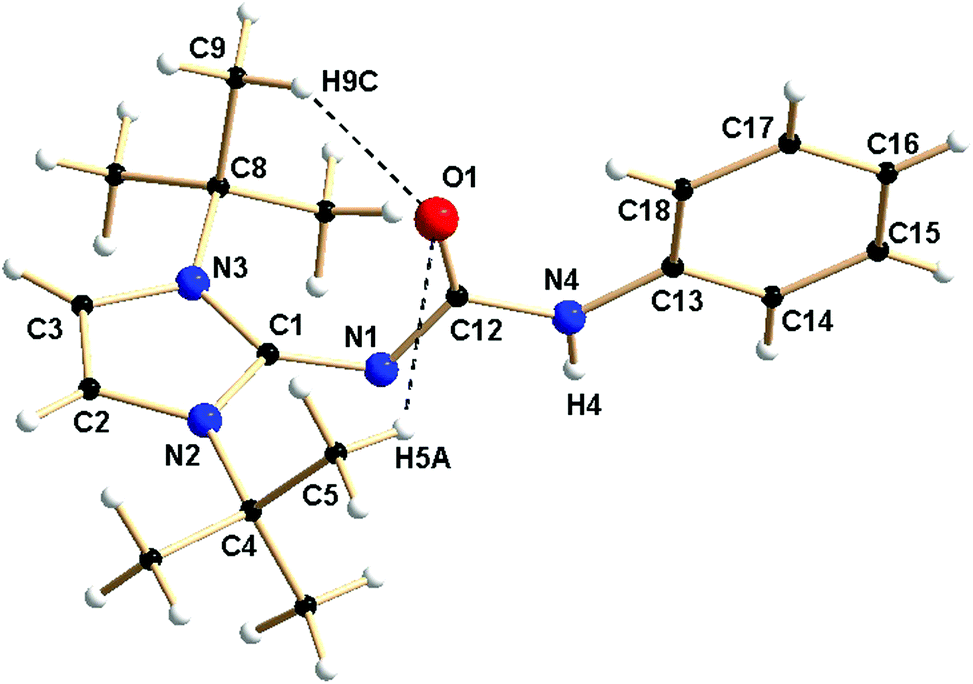 | ||
| Fig. 1 Solid-state structure of ligand 1a. Selected bond lengths [Å] and bond angles [°]: N1–C12 1.339(3), N4–C12 1.389(3), C12–O1 1.236(3), N1–C1 1.346(3), C1–N2 1.362(3), C1–N3 1.358(3), C2–C3 1.324(4), N1–C12–N4 111.1(2), N1–C12–O1 127.1(2), N4–C12–O1 121.8(2). | ||
Potassium complexes
In the recent past, we reported the syntheses and structural studies of highly reactive alkali metal complexes in order to apply them well-defined precursors for various salt metathesis reactions.16 To gain an additional insight into the structure–reactivity relationships of alkali metal complexes of imidazol-2-ylidene-N′-phenylureate ligands, we studied this chemistry further. The mesityl derivative of the ligand (1b) was reacted with potassium bis(trimethylsilyl)amide in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio and at ambient temperature in toluene to afford the tetra-nuclear structure 2b in a good yield (Scheme 2). The ligand 1d was reacted in a similar fashion with alkali and alkaline earth metal precursors; however, we could not isolate any of the corresponding alkali metal or alkaline earth metal complexes, probably due to the soft nature of the sulfur atom which did not have a preference for the hard metal ions. The polymeric potassium complex 2c was isolated in a good yield by the reaction of 1c and potassium bis(trimethylsilyl)amide in a 1:2 molar ratio in toluene at room temperature (Scheme 3). The air- and moisture-sensitive compounds 2b and 2c were characterised using 1H and 13C{1H} NMR spectroscopy, and the solid-state structures of complexes 2b and 2c were established by single-crystal X-ray diffraction analysis.
:1 molar ratio and at ambient temperature in toluene to afford the tetra-nuclear structure 2b in a good yield (Scheme 2). The ligand 1d was reacted in a similar fashion with alkali and alkaline earth metal precursors; however, we could not isolate any of the corresponding alkali metal or alkaline earth metal complexes, probably due to the soft nature of the sulfur atom which did not have a preference for the hard metal ions. The polymeric potassium complex 2c was isolated in a good yield by the reaction of 1c and potassium bis(trimethylsilyl)amide in a 1:2 molar ratio in toluene at room temperature (Scheme 3). The air- and moisture-sensitive compounds 2b and 2c were characterised using 1H and 13C{1H} NMR spectroscopy, and the solid-state structures of complexes 2b and 2c were established by single-crystal X-ray diffraction analysis.
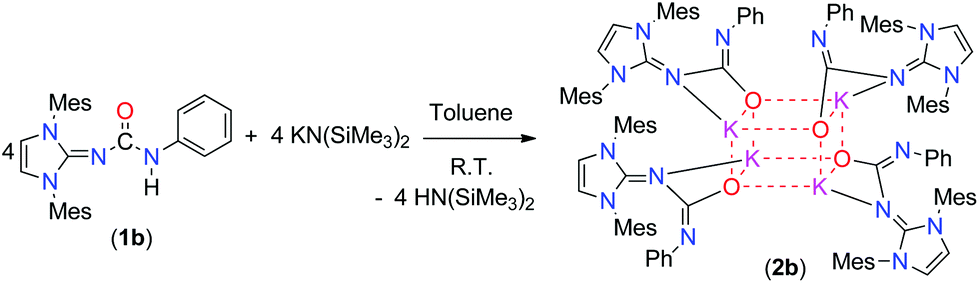 | ||
| Scheme 2 Synthesis of the heterocubane potassium complex 2b. | ||
 | ||
| Scheme 3 Synthesis of the polymeric potassium complex 2c. | ||
The 1H NMR spectra of complexes 2b and 2c, measured in C6D6, revealed one set of signals in each case. The two olefinic protons of the imidazole backbone resonated at δ 5.80 (2b) and 5.95 ppm (2c), respectively. These in a similar range to those of the starting urea ligands (5.80 ppm for 1b and 6.10 ppm for 1c). The methyl protons of the mesityl groups in 2b were observed to resonate at δ 2.26 and 2.06 ppm as two sharp singlets. Furthermore, in complex 2c, a broad signal at δ 3.16 ppm and two doublet resonances with a coupling constant of 8.4 Hz each appeared at δ 1.29 and 1.19 ppm, respectively, due to the –CH hydrogen and isopropyl methyl hydrogen atoms of the imidazolin-2-imine fragment of ligand 1c. A sharp singlet at δ 0.09 ppm was also observed for the trimethylsilyl group present in complex 2c. In the 13C{1H} NMR spectra of 2b and 2c, the chemical shift of the central imidazole carbon was observed at 149.6 ppm for 2b (versus 146.7 ppm for 1b) and at 147.5 ppm for 2c (versus 150.5 ppm for 1c), while the carbonyl carbon nucleus resonated at δ 158.2 ppm for 2b (versus 154.2 ppm 1b) and at 165.2 ppm for 2c (versus 157.2 ppm for 1c). The decrease in the CO stretching frequency (νCO = 1623 cm−1 for 2b and 1620 cm−1 for 2c) compared to that of the urea ligands 1b (νCO = 1647 cm−1) and 1c (νCO = 1647 cm−1) also supported coordination through the oxygen atom.
X-ray quality crystals of complex 2b were grown at −35 °C by the slow liquid diffusion of THF into a concentrated toluene solution, while single crystals of 2c were obtained at −35 °C from a concentrated solution of toluene. Complex 2b crystallised in the tetragonal space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 21c, with two molecules of complex 2b and two THF solvent molecules in the unit cell. In contrast, complex 2c crystallised in the monoclinic space group P21/c, with four molecules of 2c in the unit cell.
21c, with two molecules of complex 2b and two THF solvent molecules in the unit cell. In contrast, complex 2c crystallised in the monoclinic space group P21/c, with four molecules of 2c in the unit cell.
Complex 2b is tetra-nuclear monomeric, whereas complex 2c is polymeric in nature. Details of the structural parameters are given in Table TS1 in the ESI.† The solid-state structure of complex 2b is shown in Fig. 2. This solid-state structure of the tetra-nuclear potassium complex 2b confirmed the κ2-coordination mode of each ligand fragment forming four times four-membered metallacycles. Each potassium is ligated with the imidazolium-2-iminato nitrogen and the oxygen atom of the isocyanate building block. Four potassium and four oxygen atoms formed a heterocubane structure, with an average K–O distance of 2.659–2.709 Å and a K–O–K angle between 80.37(8) and 89.15(8)°, thus confirming the distorted nature of the heterocubane motif, which can be best described as a K4 tetrahedron consisting of four oxygen atoms capping the four triangular faces (Fig. 3). Significant electron donation from the imidazolium ring to the ureate group is evident from the unusual elongation of the N1–C27 (1.411(5) Å) and N1–C1 [1.285(5) Å] bonds. In addition, both the bond lengths C27–O1 [1.279(4) Å] and C27–N4 [1.309(5) Å] are between those of carbon–oxygen and carbon–nitrogen single bonds and double bonds, respectively, thus indicating extensive electron delocalisation over N1–C27–O1. A similar observation was reported in [CpTiCl2(ImMesNCONTol)](Tol = p-tolyl) by Lavoie et al.11 They, however, reported a bi-dentate mode of coordination of the ureate ligand through the oxygen and nitrogen atoms of the isocyanate building block, while leaving the imidazolin-2-imine fragment uncoordinated. To the best of our knowledge, complex 2b is the first reported ureate–potassium complex where the mono-anionic ligand is coordinated through the oxygen atom of the ureate group and the nitrogen atom from the imidazolin-2-imine fragment. An even similar pattern of bonding was discussed by Snaith et al., although it must be noted that such a heterocubane structural motif, derived using an ureate ligand in alkali metal chemistry, has not been reported so far.17
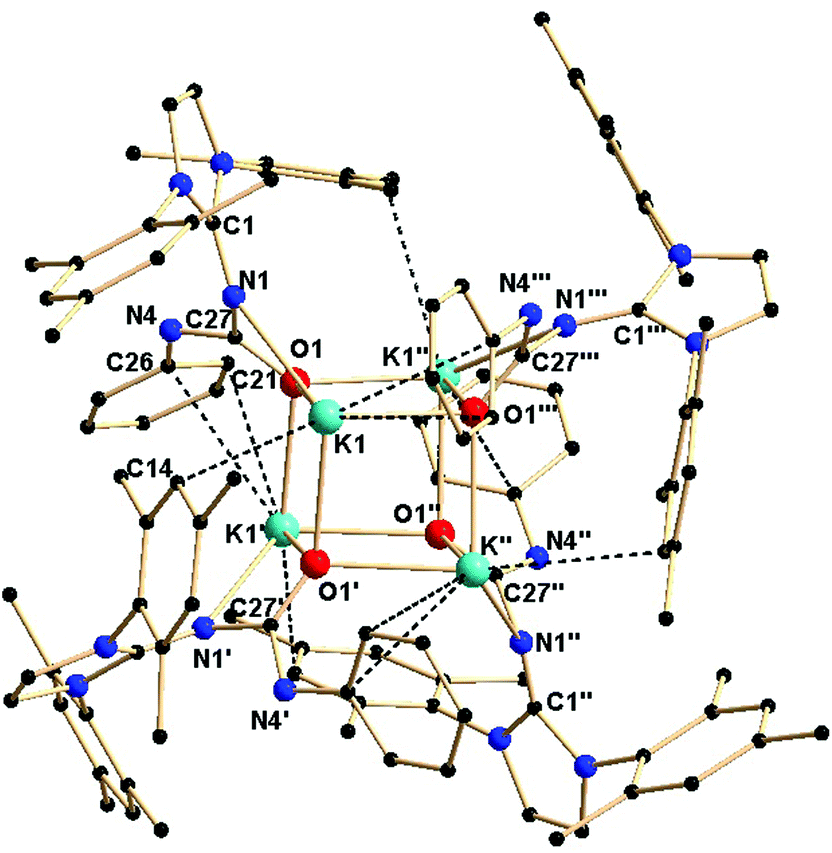 | ||
| Fig. 2 Solid-state structure of complex 2b. The hydrogen atoms are omitted for clarity. The secondary interactions of the potassium atoms and carbon atoms are shown. Selected bond lengths [Å] and bond angles [°]: K1–O1 2.709(2), K1–O1′ 2.659(3), K1–O1′′′ 2.662(3), K1–N1 2.803(3), N1–C27 1.411(5), N4–C27 1.309(5), C27–O1 1.279(4), N1–C1 1.285(5), K1–C14, 3.499(4), K1′–C21 3.423(7), O1–K1–N1 49.01(8), K1–N1–C27 90.0(2), N1–C27–O1 116.4(3), O1–K1–O1′ 80.42(8), O1–K1–O1′′′ 80.37(8), O1′–K1–O′′ 87.87(8), K1–O1–K1′ 99.56(8), K1–O1–K′′′ 99.64(9), K1′–O1–K1′′′ 89.15(8), N1–C27–N4 115.5(3). | ||
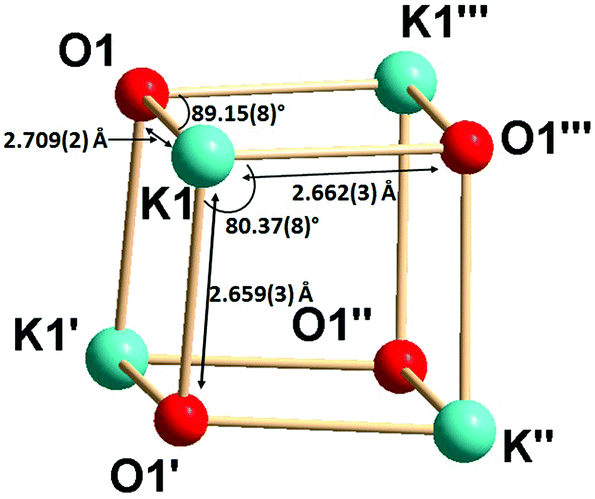 | ||
| Fig. 3 The heterocubane core in 2b formed by potassium and oxygen atoms. | ||
The effect of a substituent over the imidazol ring is very significant. The solid-state structure of complex 2c was found to be polymeric in nature, due to the use of two equivalents of potassium precursor in its preparation compared to one equivalent used for the synthesis of 2b. The asymmetric unit of complex 2c is shown in Fig. 4. The molecular structure of 2c confirms the κ3 coordination of the ureate ligand towards potassium. It further reveals that two molecules [KN(SiMe3)2] are also present in the asymmetric unit in order to stabilise the complex 2c. The nitrogen atoms from the imidazolin-2-imine and the ureate fragments bond with the potassium ion K1, whereas the third donor atom oxygen is also ligated to another adjacent potassium ion that is attached to the bis(trimethyl)silyl amide group. Two four-membered metallacycles, N1–K1–N4–C28 and N7–K4–N10–C71, are formed by the chelation of two nitrogen atoms of each ureate ligand with the potassium ion, while two six–membered metallacycles, N4–C28–O1–K3–C30–C29 and N10–C71–O2–K2–C37–C72, are observed by the ligation of an oxygen atom and a phenyl carbon of each ureate ligand with the potassium ion. The phenyl ring of the isocyanate building block plays a key role in holding the two [(ImDippNCONPh)K] units through the bridging of two phenyl–carbon atoms. Thus, an additional two six–membered metallacycles, K1–N4–C29–C34–K2–N5 and K3–N6–K4–N10–C72–C73, are observed as a result of the coordination of the phenyl carbon and nitrogen from the bis(trimethyl)silyl amido fragment. Further units of the molecule grow from the ends of the potassium ions K3 and K4 to lead to complex 2c as a polymer. The bond distances in complex 2c (C1–N1 1.289 Å, C44–N7 1.283(6) Å, N1–C28 1.418(5) Å, N7–C71 1.418(5) Å, N4–C28 1.3276) Å, C71–N10 1.324 (6) Å, C28–O1 1.248(5) Å, C71–O2 1.255(6) Å) are in a range similar to those in complex 2b and indicate a significant electron delocalisation over N1–C28–N4 and N7–C71–N10. To the best of our knowledge, complex 2c is the first example where the monoanionic ureate ligand acts as the κ3 coordination mode using its three donor atoms. A similar polymeric potassium complex {([K(μ4-oMp)(THF)][K(μ3-oMP)])5}∞ (oMP = o-methyl phenol) was reported by Boyle and his co-workers.18 Due to the fluxional nature of the complex 2c, only one set of NMR signals was observed.
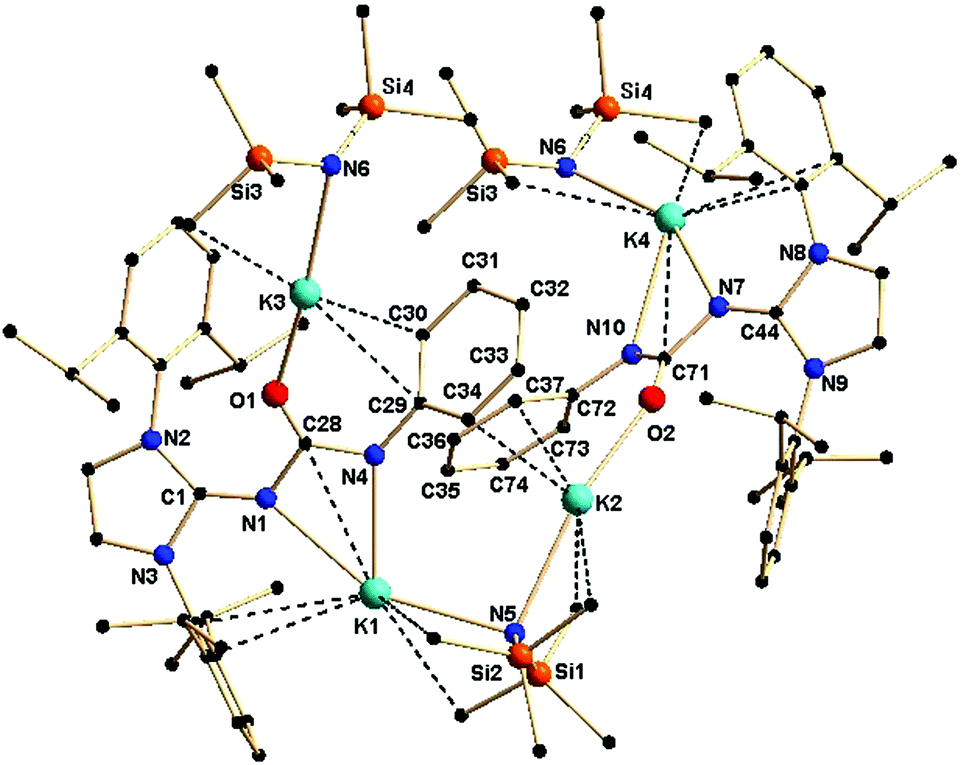 | ||
| Fig. 4 Solid-state structure of complex 2c. The hydrogen atoms are omitted for clarity. The secondary interactions of the potassium atoms and methyl carbon atoms are shown. Selected bond lengths [Å] and bond angles [°]: K1–N1 2.734(4), K1–N4 2.726(4), K1–N5 2.769(4), N1–C28 1.417(5), N4–C28 1.327(6), N4–C29 1.401(5), C28–O1 1.248(5), N1–C1 1.290(5), K2–O2 2.507(3), K2–N5 2.801(4), K2–C34 3.358(7), K3–N6 2.841(4), K3–O1 2.528(3), K3–C30 3.293(6), N5–Si1 1.670(4), N5–Si2 1.666(4), N1–C28–N4 111.2(4), N1–C28–O1 121.4(4), N4–C28–O1 127.2(4), C1–N1–C28 124.8(4), C28–N4–C29 117.9(4), N1–K1–N4 49.02(10), K1–N5–K2 40.61(8), O1–K3–N6 170.44(13), N5–K2–O2 160.99(14), Si1–N5–Si2 133.2(2). | ||
Lithium complex
As the ureate ligand has multiple donor atoms, as observed (κ2 and κ3) from the above potassium complexes 2b and 2c, multidentate coordination was expected from two nitrogen and oxygen atoms. We were interested in exploring the coordination behaviour of monoanionic ureate ligands towards lithium, a smaller alkali metal. As several attempts to crystallise the lithium complex did not succeed, we reacted the lithium complex prepared from 1a and LiCH2SiMe3 in a 1:1 molar ratio in toluene with one equivalent of 2,6-di-tert-butyl-4-methyl phenol to afford a lithium derivative, complex 3a (Scheme 4). Complex 3a was characterised using 1H, 13C{1H} NMR and combustion analysis, while its solid-state structure was established using single-crystal X-ray crystallography.
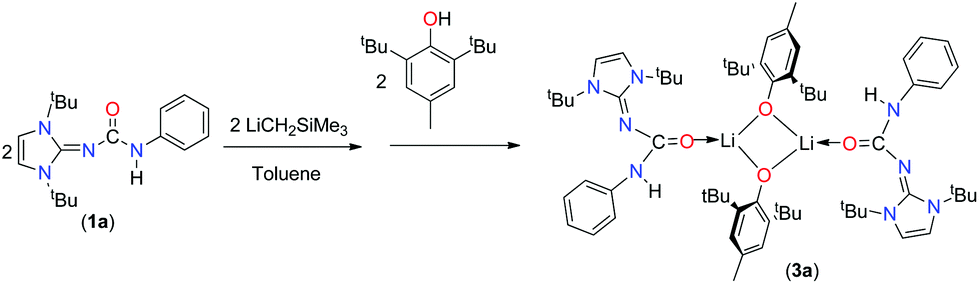 | ||
| Scheme 4 Synthesis of the lithium complex 3a. | ||
In the 1H NMR spectra of complex 3a measured in C6D6, the characteristic singlet at δ 6.28 ppm could be assigned to the olefinic protons of the imidazol backbone, which was in the same region (6.22 ppm) as that of ligand 1a. The tert-butyl methyl protons of the imidazol scaffold resonated at 1.46 ppm, whereas the tBu protons from the phenoxy ring were displayed as sharp singlets at 1.30 ppm. Additional singlet resonance at 2.10 ppm was obtained for the methyl protons located at position 4 of the phenoxy moiety. In the 13C spectra, resonances at 165.5 ppm for ipso carbon, 157.0 ppm for carbonyl carbon and 149.9 ppm for NCN group were observed for complex 3a.
Crystals of 3a were grown from a concentrated solution of toluene at −35 °C. Complex 3a crystallised in the asymmetric unit in the monoclinic space group P2/c along with a molecule of 1a. Details of the structural parameters are given in Table TS1 in the ESI.† The solid-state structure of the lithium fragment of complex 3a is shown in Fig. 5. The molecular structure clearly indicates that the neutral urea ligand 1a coordinated to the lithium ion in a κ1 mode through the oxygen atom of the isocyanate building block of the ligand. A four-membered Li2O2 diamond core was formed by the bridging coordination of two oxygen atoms from two phenoxy ligands. The terminal Li1–O1 bond distance [1.835(9) Å] where the oxygen atom is neutral is slightly shorter than the bridging Li–O2 bond (1.863(9) Å) where the oxygen atom is anionic. Dimeric lithium phenoxy complexes [(2,6-tBu2-4-Me-C6H2O)Li(THF)]2 and (2,6-tBu2-C6H2O)-Li(DMSO)]2, where one solvent (THF or DMSO) molecule is attached with each lithium centre to stabilise the respective complex, have been reported in the literature.19 Thus, the delocalisation of electrons over the N3–C12–O1 centres was observed in complex 3a. Furthermore, elongation of the C1–N3 bond [1.360(6) Å versus 1.346(3) Å for 1a] and the O1–C12 bond [1.270(6) Å versus 1.236(3) Å for 1a] also supports the above fact. It is assumed that the more nucleophilic lithium complex [ImtBuNCONPhLi] generated from 1a and LiCH2SiMe3 undergoes a protonolysis reaction with 2,6-di-tert-butyl-4-methyl phenol to form the neutral urea ligand and the lithium bis-phenoxo complex 3a. Thus, the neutral ligand 1a was trapped by and coordinated to the lithium ion, while another molecule of 1a remained uncoordinated in the asymmetric unit. Several hydrogen bonding interactions with the adjacent methyl protons from tBu groups with more electronegative oxygen atoms, important for the crystallisation of the compound (Fig. 5), were also observed in complex 3a.
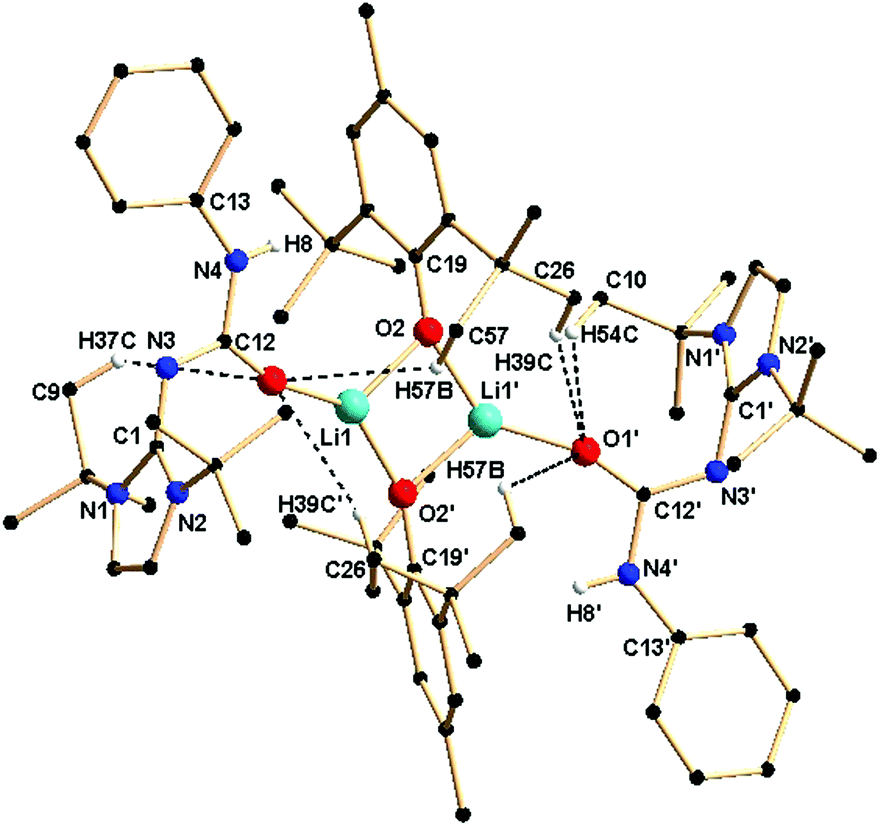 | ||
| Fig. 5 Solid-state structure of complex 3a. The hydrogen atoms are omitted for clarity except for those which have bonding interactions with oxygen atoms. Selected bond lengths [Å] and bond angles [°]: Li1–O1 1.835(9), Li1–O2 1.863(9), Li1–O2′ 1.911(8), O2–Li1′ 1.911(8), O1–C12 1.270(6), N3–C12 1.310(6), C12–N4 1.394(6), C1–N1 1.363(6), Li1–O2–Li1′ 49.0(3), O2–Li1–O2′ 95.9(4), O1–Li1–O2 140.9(5), O1–C12–N3 127.3(5), O1–C12–N4 115.8(5), N3–C12–N4 116.9(4). | ||
Calcium complex
Alkaline earth metal compounds have been recently employed in various catalytic applications in order to achieve the ring-opening polymerisation of various cyclic esters,20 the polymerisation of styrene and dienes,21 and hydroamination and hydrophosphination reactions of alkenes and alkynes.22 Determining the structure and reactivity of alkaline earth metal species is an important step towards the design and development of efficient catalysts. However, a full realisation of the catalytic potential of these elements still requires substantial advances to be made in order to understand their basic coordination and organometallic chemistry. We recently studied various group 2 metal complexes with amido-phosphine and related ligands in order to explore their structure, coordination behaviour and catalytic efficiency.23 In our ongoing study of alkaline earth metal chemistry, we aim to introduce the mono-anionic ureate ligand 1 into group 2 metal chemistry.The calcium potassium mixed metal complex 4a was isolated as a major product from a one-pot reaction with 1a and potassium precursor [KN(SiMe3)2] in a 1:3 molar ratio in THF, followed by the addition of one equivalent calcium diiodide at ambient temperature (Scheme 5). However, initial attempts to isolate the potassium free calcium iodo complex (1a)CaI(THF)n using the starting reagents in a 1:1:1 molar ratio did not meet success. The mixed Ca–K metal complex 4a was characterised using spectroscopic/analytic techniques and the molecular structure of 4a in its solid-state structure was established using single-crystal X-ray crystallography.
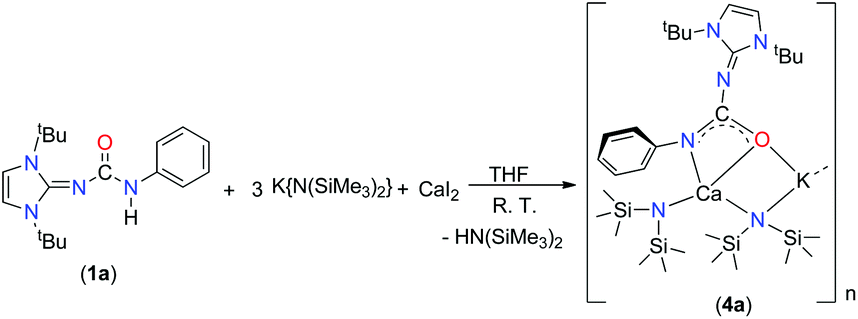 | ||
| Scheme 5 Synthesis of the calcium complex 4a. | ||
Complex 4a crystallised in the monoclinic space group P21/c, with four molecules in the unit cell. From the solid-state structure, it was evident that complex 4a was polymeric in nature. Details of the structural parameters are given in Table TS1 in the ESI;† the asymmetric unit of complex 4a is shown in Fig. 6, and the polymeric unit is given in Fig. 7. It is clearly seen in the asymmetric unit that two metal ions of calcium and potassium are, respectively, connected via μ2 bridging of the oxygen atom of the ureate ligand and the nitrogen atom from the bis(trimethylsilyl)amido group. This results in the four-fold coordinated central metal calcium ion adopting a distorted tetrahedral geometry due to the κ2 coordination of nitrogen and oxygen atoms from the isocyanate building block of ligand 1a while leaving the imidazolin-2-imine fragment uncoordinated. Two amido ligands bound the calcium ion to give two additional bonds. Thus, a four-membered metallacycle C1–O2–Ca1–N6 was formed to demonstrate the κ2 coordination of ligand 1a towards the calcium ion. The slight elongation of the C12–O1 bond [1.308(3) versus 1.236(3) Å for 1a] and the slight shortening of the C12–N4 bond [1.357(3) versus 1.389(3) Å] indicate an electron delocalisation over the O1–C12–N4 skeleton. The unperturbed bond distances of N1–C1 [1.338(3) versus 1.346(3) Å] and N1–C12 [1.342(3) versus 1.339(3) Å] are supportive of the non-interacting nature of the imidazolin-2-imine fragment towards ligation with both metal ions. In addition, a four-membered metallacycle O1–K1–N5–Ca1 was also formed. The bond distances Ca1–N4 [2.367(2) Å] and Ca1–O1 [2.355(2) Å] were similar to those [(2.4356(2) and 2.2805 (1) Å respectively] in the monomeric calcium ureate complex [(NacNac)Ca(η2-AdNC(NPh2)O](Ad = adamentyl) reported by Hill and co-workers.24 Similar mixed metal complexes with potassium–calcium, potassium–zinc, lithium–calcium and lithium–magnesium are reported in literature.25 The potassium ion K1 led the formation of the polymeric network by growing repetitive asymmetric units via η3 interactions with the phenyl ring of the adjacent ureate ligand fragment from another unit (Fig. 7). Such a polymeric network structure was realised due to the existence of multiple donor atoms in the ligand. In the NMR spectra of 4a, the presence of only one set of signals confirmed the fluxional nature of the complex.
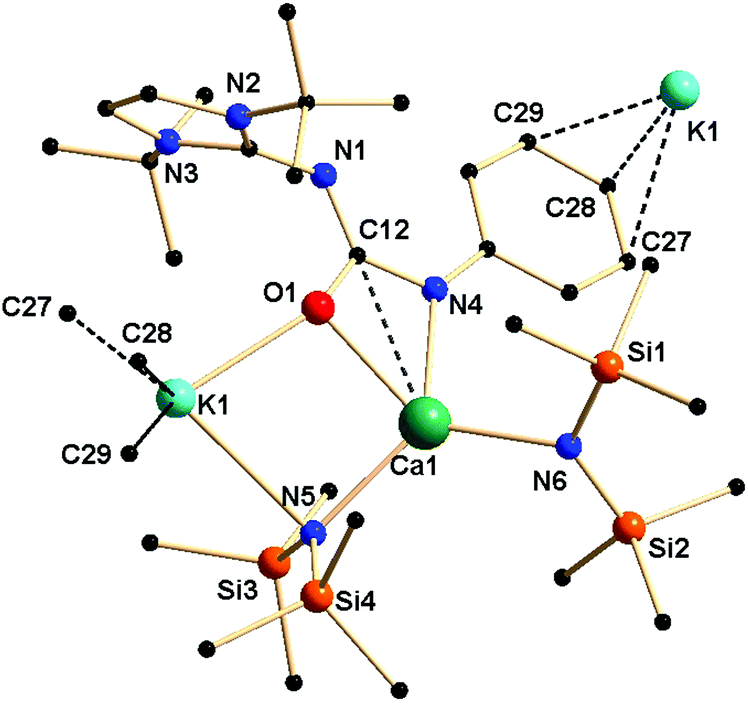 | ||
| Fig. 6 Solid-state structure of the asymmetric unit of complex 4a. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and bond angles [°]: Ca1–O1 2.3558(17), Ca1–N4 2.367(2), Ca1–N6 2.317(2), Ca1–N5 2.348(2), K1–O1 2.6199(18), K1–N5 2.989(2), O1–C12 1.308(3), N4–C12 1.357(3), C12–N1 1.342(3), N1–C1 1.338(3), N4–C13 1.389(3), O1–Ca1–N4 56.51(6), N4–Ca1–N6 104.47(8), N5–Ca1–N6 123.39(8), O1–K1–N5 73.68(5), K1–N5–Ca1 91.05(6), O1–C12–N4 114.0(2). | ||
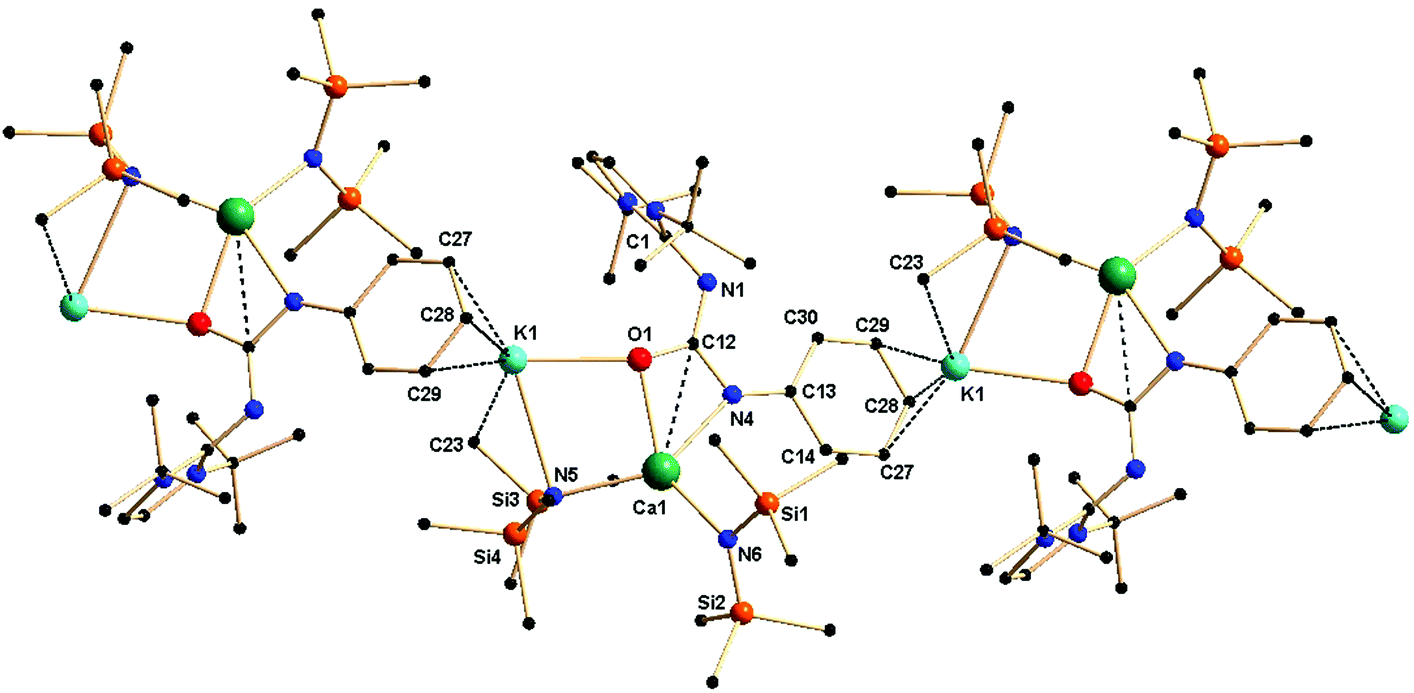 | ||
| Fig. 7 Polymeric network of complex 4a in the solid state. | ||
The synthesis of complex 4a was performed as a one-pot reaction, followed by a crystallisation process. An undesired minor product 5a was also isolated from the re-crystallisation of the second crop solution after complex 4a had been separated. Complex 5a was characterised using NMR spectroscopy, and the solid-state structure of 5a was established by single-crystal X-ray diffraction analysis.
Complex 5a showed one set of signals in the 1H NMR spectra measured in C6D6. The resonances for the olefinic protons were obtained at δ 6.12 ppm as a singlet, whereas the sharp singlet at δ 1.42 ppm could be assigned to the tert-butyl group in the imidazol fragment. In addition, a sharp singlet for the trimethylsilyl groups was also observed at δ 0.43 ppm, thus confirming the presence of SiMe3 groups in the complex. The 13C{1H} NMR spectra was also within the expected range and very similar to that of complex 4a.
Complex 5a crystallised in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , with one molecule in the unit cell. Details of the structural parameters are given in Table TS1 in the ESI,† and the asymmetric unit of complex 5a in its solid-state structure is shown in Fig. 8. The molecular structure is evidence of the incorporation of two fragments of dianionic disiloxane units [(Me2SiO)2O]2−, which bound three calcium ions in a bridging fashion. Three different chemical environments were observed for four calcium ions present in the molecule. The central calcium ion Ca1 was hexa-coordinated through the four oxygen atoms from two siloxane [(Me2SiO)2O]2− units and two oxygen atoms from two ureate ligands, resulting in the adoption of a distorted octahedral geometry around it. Each of the remaining two calcium ions had similar coordination spheres formed by the chelation of two μ3 oxygen atoms O1 and O2 of two siloxane [(Me2SiO)2O]2− units, and two nitrogen and two oxygen atoms of two monoanionic ureate ligands, resulting in the adoption of a distorted tetrahedral geometry around the calcium ion. Interestingly, for two ureate ligands, the nitrogen atoms from the isocyanate building block and the imidazolin-2-imine fragment alternatively bound either a calcium ion (Ca2) or a potassium ion (K2), thus making the ureate ligand a tridentate; the coordination mode can be best described as κ3 towards a metal ion. Apart from these calcium ions, the fourth calcium ion Ca3 exists as a [Ca{N(SiMe3)2}2] fragment in the molecule. Two kinds of potassium ions were also observed. The potassium ion K1 bound with two μ3 oxygen atoms (O1 and O2) from the two siloxane [(Me2SiO)2O]2− units and with two oxygen atoms (O3 and O4) from two isocyanate building blocks of the ureate ligand. Furthermore, a η6 attachment with an adjacent phenyl ring with a distance of (2.992–3.317 Å), which is quite common in potassium organometallic complexes, was also obtained.26 The second potassium atom K2, having one [N(SiMe3)2]− group attached, was coordinated by one imidazolin-2-imine nitrogen N3 along with a η6 arene interaction with the adjacent phenyl ring with a distance of (3.165–3.452 Å). Even the imidazol-2-iminato exocyclic C–N bond distances [N3–C24 1.342(6) Å and N6–C13 1.349(6) Å versus 1.346(3) Å for 1a] remained unchanged. The bond elongation of the ureate fragments [N3–C12 1.362(6) and N6–C11 1.391(6) Å versus 1.339(3) Å for 1a; C12–O4 1.299(6) and C11–O3 1.301(6) Å versus 1.236(3) Å for 1a] led to the conclusion that electron delocalisation occurs mainly on the N3–C12–O4 and N6–C11–O3 skeletons. The shortening of bonds C12–N7 1.341(6) and C11–N10 1.316(6) [versus 1.389(3) Å for 1a] indicated a localised carbon–nitrogen double bond rather than any involvement of electron delocalisation with the CO group. Thus, it can be assumed that compound 4a partly underwent decomposition with silicon grease under the reaction conditions so as to afford complex 5a. However, in a separate reaction, using a silicon-free grease, complex 4a was prepared without contamination from complex 5a. In complex 5a, the calcium ion Ca3 led to the repetition of the asymmetric unit, resulting in the growth of the polymeric chain.
, with one molecule in the unit cell. Details of the structural parameters are given in Table TS1 in the ESI,† and the asymmetric unit of complex 5a in its solid-state structure is shown in Fig. 8. The molecular structure is evidence of the incorporation of two fragments of dianionic disiloxane units [(Me2SiO)2O]2−, which bound three calcium ions in a bridging fashion. Three different chemical environments were observed for four calcium ions present in the molecule. The central calcium ion Ca1 was hexa-coordinated through the four oxygen atoms from two siloxane [(Me2SiO)2O]2− units and two oxygen atoms from two ureate ligands, resulting in the adoption of a distorted octahedral geometry around it. Each of the remaining two calcium ions had similar coordination spheres formed by the chelation of two μ3 oxygen atoms O1 and O2 of two siloxane [(Me2SiO)2O]2− units, and two nitrogen and two oxygen atoms of two monoanionic ureate ligands, resulting in the adoption of a distorted tetrahedral geometry around the calcium ion. Interestingly, for two ureate ligands, the nitrogen atoms from the isocyanate building block and the imidazolin-2-imine fragment alternatively bound either a calcium ion (Ca2) or a potassium ion (K2), thus making the ureate ligand a tridentate; the coordination mode can be best described as κ3 towards a metal ion. Apart from these calcium ions, the fourth calcium ion Ca3 exists as a [Ca{N(SiMe3)2}2] fragment in the molecule. Two kinds of potassium ions were also observed. The potassium ion K1 bound with two μ3 oxygen atoms (O1 and O2) from the two siloxane [(Me2SiO)2O]2− units and with two oxygen atoms (O3 and O4) from two isocyanate building blocks of the ureate ligand. Furthermore, a η6 attachment with an adjacent phenyl ring with a distance of (2.992–3.317 Å), which is quite common in potassium organometallic complexes, was also obtained.26 The second potassium atom K2, having one [N(SiMe3)2]− group attached, was coordinated by one imidazolin-2-imine nitrogen N3 along with a η6 arene interaction with the adjacent phenyl ring with a distance of (3.165–3.452 Å). Even the imidazol-2-iminato exocyclic C–N bond distances [N3–C24 1.342(6) Å and N6–C13 1.349(6) Å versus 1.346(3) Å for 1a] remained unchanged. The bond elongation of the ureate fragments [N3–C12 1.362(6) and N6–C11 1.391(6) Å versus 1.339(3) Å for 1a; C12–O4 1.299(6) and C11–O3 1.301(6) Å versus 1.236(3) Å for 1a] led to the conclusion that electron delocalisation occurs mainly on the N3–C12–O4 and N6–C11–O3 skeletons. The shortening of bonds C12–N7 1.341(6) and C11–N10 1.316(6) [versus 1.389(3) Å for 1a] indicated a localised carbon–nitrogen double bond rather than any involvement of electron delocalisation with the CO group. Thus, it can be assumed that compound 4a partly underwent decomposition with silicon grease under the reaction conditions so as to afford complex 5a. However, in a separate reaction, using a silicon-free grease, complex 4a was prepared without contamination from complex 5a. In complex 5a, the calcium ion Ca3 led to the repetition of the asymmetric unit, resulting in the growth of the polymeric chain.
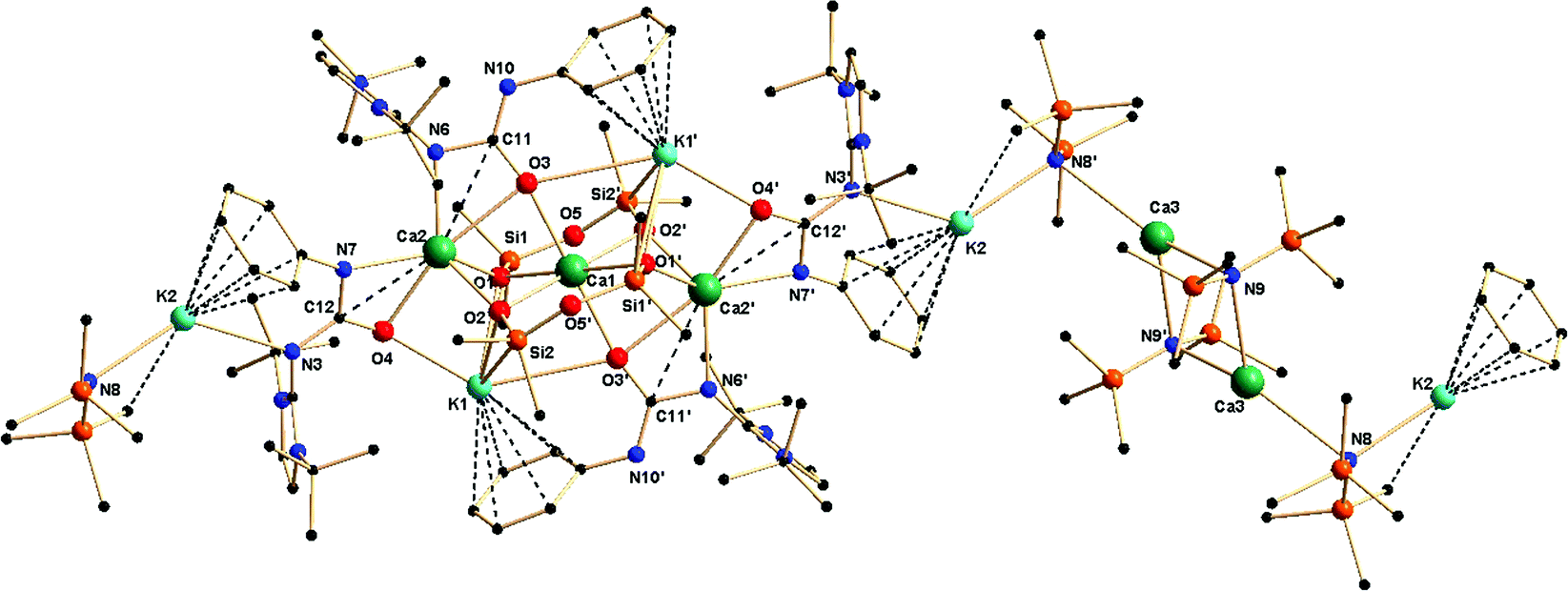 | ||
| Fig. 8 Solid-state structure of asymmetric unit of complex 5a. The hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and bond angles [°]: Ca1–O1 2.329(3), Ca1–O1′ 2.329(3), Ca1–O2 2.339(3), Ca1–O3 2.370(3), Ca2–O1 2.332(3), Ca2–O2 2.290(3), Ca2–O4 2.389(3), Ca2–N7 2.390(4), Ca2–N6 2.372(4), Ca2–C11 2.965(5), Ca2–C12 2.804(5), K1–O3′ 3.314(4), K1–O1 2.768(3), K1–O2 2.694(4), K2–N8 2.734(4), K2–N3 2.907(4), O3–C11 1.301(6), N6–C11 1.390(6), N10–C11 1.316(6), Si1–O5 1.634(4), Si1–O1 1.606(3), O1–Ca1–O2 79.75(11), O1–Ca1–O2 80.71(12), O1–Ca1–O2′ 100.25(11), O1–Ca1–O1′ 180.000(1), O1–Ca1–O3 83.22(12), O1–Ca2–O3 77.57(11), O1–Si1–O5 112.20(19), O5′–Si2–O2 110.86(19). | ||
Experimental
General consideration
All manipulations of air-sensitive materials were performed by rigorously excluding oxygen and moisture in flame-dried Schlenk-type glassware, either on a dual manifold Schlenk line interfaced to a high vacuum (10−4 torr) line, or in an argon-filled M-BRAUN glovebox. Toluene was distilled under nitrogen from LiAlH4 and stored in the glove box. 1H NMR (400 MHz) and 13C{1H} NMR spectra were recorded on a BRUKER AVANCE III-400 spectrometer. BRUKER ALPHA FT-IR was used for the FT-IR measurements. Elemental analyses were performed on a BRUKER EURO EA at the Indian Institute of Technology Hyderabad (IITH). Imidazolin-2-imines [(ImRNH); (R = tBu, Mes, Dipp)] and trimethylsilylmethyl lithium [LiCH2SiMe3] were prepared according to procedures specified in literature.15 Phenyl isocyanate, phenyl isothiocyanate, 2,6-di-tert-butyl-4-methyl phenol, [K{N(SiMe3)2}] and CaI2 were purchased from Alfa Aesar and used in their original forms. The NMR solvents CDCl3 and C6D6 were purchased from Sigma Aldrich and dried under Na/K alloy prior to use (for C6D6).Preparation of [ImRNCON(H)Ph] (R = tBu) (1a); Mes (1b) and Dipp (1c)
To a toluene solution (10 mL) of imidazole-2-imine (ImRNH) (1.56 mmol), 5% molar excess of phenyl-isocyanate (192 mg, 1.612 mmol) was added, and the reaction mixture was stirred for two hours. The solvent was evaporated in vacuo leaving behind a solid residue. This colourless solid was washed with n-pentane (5 mL) to afford a spectroscopically pure off-white solid. The title compounds were crystallised from concentrated toluene solution at 15 °C.
1a: Yield 420 mg, 86%. 1H NMR (400 MHz, C6D6): δ 7.9 (br, 1H, HN), 7.81 (d, 3JH–H = 7.75 Hz, 2H, Ph), 7.18 (m, 2H, Ph), 6.83 (t, 3JH–H = 7.32 Hz, 1H, Ph), 6.18 (s, 2H, HCCH), 1.43 (s, 18 H, C(CH3)3) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 158.8 (CO), 150.4 (NCN), 143.4 (Ar), 128.9 (Ar), 120.2 (Ar), 117.8 (Ar), 112.0 (HCCH), 58.4 (NC(CH3)3), 29.1 (C(CH)3) ppm. FTIR selected peaks (cm−1): 1627 (CO), 1574 (CN), 2979 (CH), 3155, 3287 (N–H). (C18H26N4O) (314.43) Calc. C 68.76, H 8.33, N 17.82; found C 68.23 H 8.11, N 17.59.
1b: Yield 515 mg, 75%. δ 7.09 (d, 3JHH = 7.88 Hz, 2H, Ph), 6.89 (m, 2H, Ph), 6.72 (s, 4H, m-CH(mesityl)), 6.62 (t, 3JHH = 7.48 Hz, 1H, Ph), 6.44 (br s, 1H, NH), 5.81 (s, 2H, HCCH), 2.26 (s, 12H, o-CH3(mesityl)), 2.05 (s, 6H, p-CH3(mesityl)) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 158.0 (CO), 149.8 (ipso-Ar), 141.7 (NCN), 138.4 (ipso-Ph), 135.8 (o-Ar), 133.8 (p-Ar), 129.4 (m-Ar), 128.3 (o-Ph), 120.8 (p-Ph), 118.2 (m-Ph), 115.5 (HCCH), 21.0 (CH3), 18.2 (CH3) ppm. FTIR selected peaks (cm−1): 1580 (CN), 1647 (CO), 2916, 3211 (N–H). (C28H30N4O) (438.56) Calc. C 76.68, H 6.89, N 12.78; found C 76.31 H 6.39, N 12.29.
1c: Yield 506 mg, 78%. 1H NMR (400 MHz, C6D6): δ 7.22–7.20 (m, 4H, Ar), 7.12–7.10 (m, 2H, Ar), 6.98 (d, 3JHH = 8.72 Hz, 2H, Ph), 6.92–6.88 (m, 2H, Ph), 6.63 (t, 3JHH = 7.20 Hz, 1H, Ph), 6.10 (s, 2H, HCCH), 3.12 (sept, 3JHH = 6.80 Hz, 4H, CH(CH3)2), 1.45 (d, 3JHH = 6.80 Hz, 12H, CH(CH3)2), 1.16 (d, 3JHH = 6.80 Hz, 12H, CH(CH3)2) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 157.1 (CO), 151.2 (NCN), 146.6 (ipso-Ar), 141.5 (ipso-Ph), 133.9 (o-Ar), 124.2 (o-Ph), 120.8 (p-Ph), 118.1 (m-Ph), 116.6 (HCCH), 29.2 (CH(CH3)2), 24.4 (CH(CH3)2), 23.7 (CH(CH3)2) ppm. FTIR selected peaks (cm−1): 1580 (CN), 1650 (CO), 2962, 3053, 3419 (N–H). (C34H42N4O) (522.72) Calc. C 78.12, H 8.10, N 10.72; found C 77.86, H 7.91, N 10.51.
Preparation of [ImtBuNCSN(H)Ph] (1d)
To a toluene solution (10 mL) of 1,3-di-tert-butylimidazole-2-imine a (300 mg, 1.536 mmol), 5% excess of phenyl-isothiocyanate (218 mg, 1.612 mmol) was added, and the reaction mixture was stirred for two hours. The solvent was evaporated in vacuo leaving behind a solid residue, which was washed with n-pentane (5 mL) to afford a spectroscopically pure off-white solid. The title compound 1d was re-crystallised from a concentrated solution of toluene.Yield: 436 mg 86%. 1H NMR (400 MHz, C6D6): δ 7.80 (s, 1H, HN), 7.62 (d, 3JH–H = 7.84 Hz, 2H, Ph), 7.20 (m, 2H, Ph), 6.89 (t, 3JH–H = 7.32 Hz, 1H, Ph), 6.77 (s, 2H, HCCH), 1.63 (s, 18 H, C(CH)3) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 178.2 (CO), 151.4 (ipso-Ph), 140.9 (NCN), 128.7 (Ph), 121.6 (Ph), 119.6 (Ph), 113.6 (HCCH), 58.5 (NC(CH3)3), 29.7 (C(CH3)3) ppm. FTIR selected peaks (cm−1): 1654 (CS), 1567 (CN), 2967, 3126, 3235 (N–H). (C18H26N4S) (330.49) Calc. C 65.42, H 7.93, N 16.95; found C 65.19 H 7.62, N 16.77.
Preparation of [{κ2-(ImMesNCONPh)K}4] (2b)
In an argon-filled glovebox, toluene (10 mL) solution of compound 1b (200 mg, 0.456 mmol) was added dropwise to a 25 ml Schlenk tube containing potassium bis(trimethylsilyl)amide (91 mg, 0.456 mmol) and 5 mL of toluene. The reaction mixture was kept for further stirring at room temperature for 12 hours. The solvent was evaporated in vacuo leaving behind a red-coloured residue, which was washed with n-hexane (10 × 3 mL) and dried under reduced pressure. The title compound 2b was crystallised from a mixture of THF and toluene (1:3) at −35 °C.
Yield 141 mg, 65%. 1H NMR (400 MHz, C6D6): δ 7.12–7.08 (m, 2H, Ph), 6.94–6.84 (m, 3H, Ph), 6.73 (s, 4H, m-CH(mesityl)), 5.80 (s, 2H, HCCH), 3.57 (thf), 2.26 (s, 12H, CH3), 2.06 (s, 6H, CH3), 1.40 (thf) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 158.2 (CO), 149.6 (NCN), 146.4 (ipso-Ar), 138.4 (ipso-Ph), 135.9 (o-Ar), 133.9 (Ph), 129.4 (Ph), 128.3 (Ar), 120.1 (Ph), 115.3 (HCCH), 20.9 (CH3), 18.2 (CH3) ppm. FTIR selected peaks (cm−1): 1623 (CO), 2956, 3078. (C128H148K N16O8) (2077.75) Calc. C 73.99, H 7.18, N 10.79; found C 73.54 H 6.94, N 10.32.
Preparation of [{κ3-(ImDippNCONPh)K}2{KN(SiMe3)2}2]n (2c)
Compound 1c (200 mg, 0.0.38 mmol) and potassium bis(trimethylsilyl)amide (153 mg, 0.765 mmol) were placed in a 25 ml Schlenk flask in an inert atmosphere. Toluene (8 ml) was added to the flask at room temperature. The resultant reaction mixture was stirred for another 12 hours. The solvent was evaporated under reduced pressure to obtain a red-coloured residue. The red solid was washed with n-hexane (3 × 5 ml) to obtain a red powder. The title compound 2c was re-crystallised from concentrated toluene at −35 °C.Yield 395 mg, 52%. 1H NMR (400 MHz, C6D6): δ 7.53 (m, 2H, Ar), 7.34 (m, 4H, Ar), 7.11 (m, 3H, Ph), 7.00 (m, 2H, Ph), 5.95 (s, 2H, HCCH), 3.16 (m, 4H, CH(CH3)2), 1.29 (d, 3JHH = 8.43 Hz, 12H, CH(CH3)2), 1.12 (d, 3JHH = 8.43 Hz 12H, CH(CH3)2), 0.09 (s, 18H, Si(CH3)3) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 165.2 (CO), 147.5 (NCN), 135.8 (ipso-Ar), 135.3 (ipso-Ph), 129.3 (Ar), 127.8 (Ph), 120.6 (Ph), 125.7 (Ar), 123.8 (Ar), 115.4 (HCCH), 29.0 (CH(CH3)2), 24.7 (CH(CH3)2), 23.5 (CH(CH3)2) 2.65 (Si(CH3)3) ppm. FTIR selected peaks (cm−1): 1620 (CO), 2962, 3053. (C80H118K4N10O2Si4) (1520.61) Calc. C 63.19, H 7.82, N 9.21; found C 62.72 H 7.41, N 9.01.
Preparation of [{(2,6-tBu2-4-Me-C6H2O)Li(ImtBuNCON(H)Ph)}2{ImtBuNCON(H)Ph}] (3a)
To a stirred solution of toluene (5 mL) and compound 1a (200 mg, 0.636 mmol), a toluene (3 mL) solution of trimethylsilyl methyllithium (60 mg, 0.636 mmol) was added dropwise at ambient temperature. The resulting reaction mixture was stirred for another 12 hours. 2,6-di-tert-butyl-4-methyl phenol (140 mg, 0.636 mmol) was added to this solution, and the reaction mixture was stirred for a further 2 hours. The solvent was evaporated and the solution mixture was reduced to 1/3 of the original volume. It was thereafter placed for crystallisation at −35° C. Colourless crystals were obtained two days later.Yield 335 mg, 62%. 1H NMR (400 MHz, C6D6): δ 7.57 (d, 3JH–H = 8.0 Hz, 2H, Ar), 7.22 (t, 3JH–H = 8.0 Hz, 2H, Ar), 7.02 (s, 2H, Ar), 6.85 (t, 3JH–H = 8.0 Hz, 1H, Ar), 6.28 (s, 2H, HCCH), 2.10 (6H, CH3), 1.46 (s, 18 H, C(CH3)3), 1.30 (18H, C(CH3)3) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 165.5 (ipso –Ar), 157.0 (CO), 149.9 (NCN), 141.1 (Ar), 133.1 (Ar), 129.3 (Ar), 128.5 (Ar), 128.3 (Ar), 125.8 (Ar), 120.4 (Ar), 119.3 (Ar), 112.1 (HCCH), 58.6 (NC(Me)2), 33.8 (C(CH3)3), 32.6 (C(CH3)3), 32.3 (CH)3), 29.1 (C(CH)3) ppm. FTIR selected peaks (cm−1): 1627 (CO), 2980, 3155, 3297 (N–H). (C102H150Li2N16O6) (1710.27) Calc. C 71.63, H 8.84, N 13.10; found C 71.33 H 8.37, N 12.87.
Preparation of [{κ2-(ImtBuNCONPh)Ca{N(SiMe3)2}{KN-(SiMe3)2}]n (4a)
In an argon-filled glovebox, to the toluene (5 mL) solution of calcium diiodide (186.9 mg, 0.636 mmol), a toluene (5 mL) solution of compound 1a (200 gm, 0.636) and potassium bis(trimethylsilyl)amide (380.6 mg, 1.90 mmol) was added dropwise at ambient temperature. The resulting reaction mixture was further stirred at room temperature for 12 hours. The white precipitate obtained from the reaction mixture was separated by filtration through a G4 frit. The solvent was evaporated under reduced pressure to obtain an off-white residue, which was washed with n-hexane (3 × 10 ml) and dried in vacuo. The title compound 4a was re-crystallised from a mixture of THF and toluene (1:3) at −35 °C.
Yield 426 mg, 57%. 1H NMR (400 MHz, C6D6): δ 7.80 (m, 2H, Ar), 7.29 (m, 3JH–H = 8.0 Hz, 2H, Ar)), 7.06 (m, 1H, Ar), 6.16 (s, 2H, HCCH), 3.42 (thf), 1.40 (thf), 1.49 (s, 18 H, C(CH3)3) 0.43 (s, 18H, Si(CH3)3), 0.21 (s, 18H, Si(CH3)3) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 164.1 (CO), 152.7 (NCN), 129.5 (Ar), 129.1 (Ar), 128.1 (Ar), 127.2 (Ar), 123.9 (Ar), 119.2 (Ar), 111.6 (HCCH), 68.5 (NC(CH3)3), 57.8 (thf), 29.4 (C(CH3)3) 25.2 (thf), 6.16 (Si(CH3)3), 2.90 (Si(CH3)3) ppm. FTIR selected peaks (cm−1): 1619 (CO), 2954. (C33H64CaKN6OSi4) (752.42) Calc. C 52.68, H 8.57, N 11.17; found C 52.22 H 8.13, N 10.88.
5a: Compound 5a was obtained as a minor product from the second crop solution after the crystals of 4a were isolated. Yield 195 mg, 8%. 1H NMR (400 MHz, C6D6): δ 7.81–7.68 (m, 2H, Ar), 7.31–7.27 (m, 2H, Ar)), 7.14–7.08 (m, 1H, Ar), 6.12 (s, 2H, HCCH), 3.57 (thf), 1.45 (thf), 1.42 (s, 18 H, C(CH3)3) 0.43 (s, 18H, Si(CH3)3), 0.21 (s, 18H, Si(CH3)3) 0.09 (s, 24H, OSi(CH3)2) ppm. 13C{1H} NMR (100 MHz, C6D6): δ 168.1 (CO), 156.3 (NCN), 129.8 (Ar), 129.3 (Ar), 128.0 (Ar), 127.6 (Ar), 125.1 (Ar), 120.2 (Ar), 115.4 (HCCH), 66.5 (NC(CH3)3), 57.8 (thf), 28.4 (C(CH3)3) 25.2 (thf), 7.15 (Si(CH3)3), 3.76 (Si(CH3)3), 1.02 (OSi(CH3)2) ppm. FTIR selected peaks (cm−1): 1672 (CO), 2956. (C118H212Ca5 K4N20O10Si12) (2764.96) Calc. C 51.26, H 7.73, N 10.13; found C 50.94 H 7.34, N 9.79.
X-Ray crystallographic studies of complexes 1a, 1d, 2b, 2c, 3a, 4a, 5a
Single crystals of compounds 1a and 1d were grown from concentrated toluene under an inert atmosphere at 15 °C. Crystals of 2c were grown from a concentrated solution of toluene at −35 °C, whereas single crystals of 2b, 4a and 5a were grown from a mixture of THF and toluene (1:3) at −35 °C. For all the complexes, a crystal of suitable dimensions was mounted on a CryoLoop (Hampton Research Corp.) with a layer of light mineral oil and placed either at room temperature (for 1a) or in a nitrogen stream at 150(2) K. All the measurements were made on an Agilent Supernova X-calibur Eos CCD detector with graphite-monochromatic Cu-Kα (1.54184 Å) radiation. The crystal data and structure refinement parameters are summarised in Table TS1 in the ESI.† The structures were solved by direct methods (SIR92)27 and refined on F2 by full-matrix least-squares methods using SHELXL-97.28 Non-hydrogen atoms were anisotropically refined. H atoms were included in the refinement in calculated positions riding on their carrier atoms. No restraints were made for any of the complexes. The function minimised was [∑w(Fo2 − Fc2)2] (w = 1/[s2(Fo2) + (aP)2 + bP]), where P = (max(Fo2, 0) + 2Fc2)/3 with s2(Fo2) from counting statistics. The function R1 and wR2 were (S||Fo| − |Fc||)/S|Fo| and [Sw(Fo2 − Fc2)2/S(wFo4)]1/2, respectively. The Diamond-3.0 program was used to draw the molecule. Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no CCDC 1046050–1046056.
Conclusion
We have demonstrated the synthetic and structural details of potassium, lithium and calcium complexes of the imidazol-2-ylidene-N′-phenylureate ligand, which display various coordination modes such as κ1, κ2 and κ3. From the solid-state structures it is evident that not only are the nitrogen and oxygen atoms of the isocyanate building block coordinating, but also that the imidazolin-2-imine nitrogen atom takes part in a coordination with the potassium and calcium ions. Due to the larger size of the potassium and calcium ions and the multi-dentate nature of the ureate ligand, polymeric structures were obtained in most cases. However, by controlling the substituent as mesityl groups over the imidazol ring, a heterocubane core K4O4 was isolated in potassium complex 2b.Acknowledgements
This work is supported by the Science and Engineering Research Board (SERB), Department of Science and Technology (DST), India, under project no. (SB/S1/IC/045/2013). The instrumental facilities were provided by the Indian Institute of Technology Hyderabad (IITH). K.N. and J.B. thank the University Grants Commission (UGC), India, for their PhD fellowships. We sincerely thank Prof. Kazushi Mashima and Dr Hayato Tsurugi, Osaka University for their generous support. We thank reviewers to improve the manuscript.Notes and references
- A. Werner, Z. Anorg. Chem., 1893, 3, 267–330 CrossRef.
- (a) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 36, 2162–2187 CrossRef CAS; (b) L. Jafarpour and S. P. Nolan, Adv. Organomet. Chem., 2000, 46, 181–222 CrossRef CAS; (c) T. Weskamp, V. P. W. Bohm and W. A. Herrmann, J. Organomet. Chem., 2000, 600, 12–22 CrossRef CAS; (d) W. A. Herrmann, T. Weskamp and V. P. W. Böhm, Adv. Organomet. Chem., 2001, 48, 1–69 CrossRef CAS; (e) L. Jafarpour and S. P. Nolan, J. Organomet. Chem., 2001, 617–618, 17–27 CrossRef CAS; (f) D. Enders and H. Gielen, J. Organomet. Chem., 2001, 617–618, 70–80 CrossRef CAS; (g) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS; (h) A. C. Hillier, G. A. Grasa, M. S. Viciu, H. M. Lee, C. Yang and S. P. Nolan, J. Organomet. Chem., 2002, 653, 69–82 CrossRef CAS; (i) D. Enders and T. Balensiefer, Acc. Chem. Res., 2004, 37, 534–541 CrossRef CAS PubMed; (j) V. Cesar, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619–636 RSC; (k) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239–2246 CrossRef CAS PubMed; (l) V. Nair, R. S. Menon and V. Sreekumar, Pure Appl. Chem., 2005, 77, 1191–1206 CrossRef CAS; (m) F. E. Hahn and M. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (n) S. D-Gonzalez, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (o) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed; (p) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (q) L. Benhamou, E. Chardon, G. Lavigne, S. B. Laponnaz and V. Cesar, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed; (r) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC; (s) M. N. Hopkinson, C. Richter1, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- (a) A. J. Boydston, K. A. Williams and C. W. Bielawski, J. Am. Chem. Soc., 2005, 127, 12496–12497 CrossRef CAS PubMed; (b) M. Bold, C. Lennartz, M. Prinz, H.-W. Schmidt, M. Thelakkat, M. Bate, C. Neuber, W. Kowalsky, C. Schildknecht and H.-H. Johannes, BASF AG, WO 2005019373, 2005 Search PubMed; (c) M. Egen, K. Kahle, M. Bold, T. Gessner, C. Lennartz, S. Nord, H.-W. Schmidt, M. Thelakkat, M. Bate, C. Neuber, W. Kowalsky, C. Schildknecht and H.-H. Johannes, BASF AG, WO2006056418, 2006 Search PubMed; (d) L. Mercsa and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC.
- (a) J. C. Garrison and W. C. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS PubMed; (b) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- (a) N. Kuhn, M. Gohner, G. Frenking and Y. Chen, in Unusual Structures and Physical Properties in Organometallic Chemistry, ed. M. Gielen, R. Willem and B. Wrack-meyer, John Wiley & Sons, The Atrium, West Sussex, UK, 2002, p. 337 Search PubMed; (b) N. Kuhn, M. Gohner, M. Grathwohl, J. Wiehoff, G. Frenking and Y. Chen, Z. Anorg. Allg. Chem., 2003, 629, 793–802 CrossRef CAS.
- (a) C. J. Carmalt and A. H. Cowley, Adv. Inorg. Chem., 2000, 50, 1–32 CrossRef CAS; (b) N. Kuhn and A. Al-Sheikh, Coord. Chem. Rev., 2005, 249, 829–857 CrossRef CAS PubMed.
- G. Frison and A. Sevin, J. Chem. Soc., Perkin Trans. 2, 2002, 1692–1697 RSC.
- (a) T. K. Panda, S. Randoll, C. G. Hrib, P. G. Jones, T. Bannenberg and M. Tamm, Chem. Commun., 2007, 5007–5009 RSC; (b) S. Beer, K. Brandhorst, J. Grunenberg, C. G. Hrib, P. G. Jones and M. Tamm, Org. Lett., 2008, 10, 981–984 CrossRef CAS PubMed; (c) D. Petrovic, L. M. R. Hill, P. G. Jones, W. B. Tolman and M. Tamm, Dalton Trans., 2008, 887–894 RSC; (d) D. Petrovic, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Organometallics, 2008, 27, 778–783 CrossRef CAS; (e) S. Beer, K. Brandhorst, C. G. Hrib, X. Wu, B. Haberlag, J. Grunenberg, P. G. Jones and M. Tamm, Organometallics, 2009, 28, 1534–1545 CrossRef CAS; (f) T. K. Panda, A. G. Trambitas, T. Bannenberg, C. G. Hrib, S. Randoll, P. G. Jones and M. Tamm, Inorg. Chem., 2009, 48, 5462–5472 CrossRef CAS PubMed; (g) A. Trambitas, T. K. Panda, J. Jenter, P. Roesky, C.-G. Daniliuc, C. Hrib, P. G. Jones and M. Tamm, Inorg. Chem., 2010, 49, 2435–2446 CrossRef CAS PubMed; (h) A. G. Trambitas, T. K. Panda and M. Tamm, Z. Anorg. Allg. Chem., 2010, 636, 2156–2171 CrossRef CAS; (i) T. K. Panda, C. G. Hrib, P. G. Jones and M. Tamm, J. Organomet. Chem., 2010, 695, 2768–2773 CrossRef CAS PubMed; (j) M. Tamm, A. G. Trambitas, C. Hrib and P. G. Jones, Terrae Rarae., 2010, 7, 1–4 Search PubMed; (k) A. G. Trambitas, D. Melcher, L. Hartenstein, P. W. Roesky, C. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 6753–6761 CrossRef CAS PubMed; (l) M. Eisen, I. Karmel, M. Botoshansky and M. Tamm, Inorg. Chem., 2014, 53, 694–696 CrossRef PubMed; (m) X. Wu and M. Tamm, Coord. Chem. Rev., 2014, 260, 116–138 CrossRef CAS PubMed.
- A. Glöckner, T. Bannenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Inorg. Chem., 2012, 51, 4368–4378 CrossRef PubMed.
- (a) M. Tamm, S. Randoll, E. Herdtweck, N. Kleigrewe, G. Kehr, G. Erker and B. Rieger, Dalton Trans., 2006, 459–467 RSC; (b) S. Beer, C. G. Hrib, P. G. Jones, K. Brandhorst, J. Grunenberg and M. Tamm, Angew. Chem., Int. Ed., 2007, 46, 8890–8894 CrossRef CAS PubMed; (c) S. H. Stelzig, M. Tamm and R. M. Waymouth, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 6064–6070 CrossRef CAS; (d) S. Beer, K. Brandhorst, J. Grunenberg, C. G. Hrib, P. G. Jones and M. Tamm, Org. Lett., 2008, 10, 981–984 CrossRef CAS PubMed; (e) T. Glöge, D. Petrovic, C. Hrib, P. G. Jones and M. Tamm, Eur. J. Inorg. Chem., 2009, 4538–4546 CrossRef; (f) S. Beer, K. Brandhorst, C. G. Hrib, X. Wu, B. Haberlag, J. Grunenberg, P. G. Jones and M. Tamm, Organometallics, 2009, 28, 1534–1545 CrossRef CAS; (g) B. Haberlag, X. Wu, K. Brandhorst, J. Grunenberg, C. G. Daniliuc, P. G. Jones and M. Tamm, Chem. – Eur. J., 2010, 16, 8868–8877 CrossRef CAS PubMed; (h) M. Tamm and X. Wu, Chemistry Today, 2010, 28, 60–63 CAS; (i) M. Tamm and X. Wu, Chim. Oggi., 2010, 28, 10–13 Search PubMed; (j) X. Wu and M. Tamm, Beilstein J. Org. Chem., 2011, 7, 82–93 CrossRef CAS PubMed; (k) A. G. Trambitas, J. Yang, D. Melcher, C. G. Daniliuc, P. G. Jones, Z. Xie and M. Tamm, Organometallics, 2011, 30, 1122–1129 CrossRef CAS; (l) X. Wu and M. Tamm, Beilstein J. Org. Chem., 2011, 7, 82–93 CrossRef CAS PubMed; (m) M. Sharma, H. Yameen, B. Tumanskii, S.-A. Filimon, M. Tamm and M. Eisen, J. Am. Chem. Soc., 2012, 134, 17234–17244 CrossRef CAS PubMed; (n) B. Haberlag, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2012, 51, 13019–13022 CrossRef CAS PubMed; (o) K. Nomura, H. Fukuda, W. Apisuk, A. Trambitas, B. Kitiyanan and M. Tamm, J. Mol. Catal. A: Chem., 2012, 363–364, 501–511 CrossRef CAS PubMed; (p) D. Shoken, M. Sharma, M. Botoshansky, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2013, 135, 12592–12595 CrossRef CAS PubMed; (q) S. Lysenko, C. G. Daniliuc, P. G. Jones and M. Tamm, J. Organomet. Chem., 2013, 744, 7–14 CrossRef CAS PubMed; (r) W. Apisuk, A. G. Trambitas, B. Kitiyanan, M. Tamm and K. Nomura, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2575–2580 CrossRef CAS; (s) J. Volbeda, P. G. Jones and M. Tamm, Inorg. Chim. Acta, 2014, 422, 158–166 CrossRef CAS PubMed; (t) K. Nomura, B. Bahuleyan, S. Zhang, P. V. Sharma, S. Katao, A. Igarashi, A. Inagaki and M. Tamm, Inorg. Chem., 2014, 53, 607–623 CrossRef CAS PubMed.
- (a) S. Dastgir and G. G. Lavoie, Dalton Trans., 2010, 39, 6943–6946 RSC; (b) S. Dastgir and G. G. Lavoie, Dalton Trans., 2012, 41, 9651–9658 RSC; (c) M. B. Harkness, E. Alvarado, A. C. Badaj, B. C. Skrela, L. Fan and G. G. Lavoie, Organometallics, 2013, 32, 3309–3321 CrossRef CAS.
- K. Naktode, S. D. Gupta, A. Kundu, S. K. Jana, H. P. Nayek, B. S. Mallik and T. K. Panda, Aust. J. Chem., 2015, 68, 127–136 CrossRef CAS.
- (a) R. M. Silverstein, G. C. Bassler and T. C. Morrill, Spectrometric Identification of Organic Compounds, John Wiley and Sons, New York, 1981 Search PubMed; (b) J. Workman and L. Weyer, Practical Guide to Interpretive near-Infrared Spectroscopy, CRC Press, Boca Raton, FL, 2008 Search PubMed.
- (a) A. Yamaguchi, R. B. Penland, S. Mizushima, T. J. Lane, C. Curran and J. V. Quagliano, J. Am. Chem. Soc., 1958, 80, 527–529 CrossRef CAS; (b) C. N. R. Rao and R. Venkataraghavan, Spectrochim. Acta, 1962, 18, 541–547 CrossRef CAS; (c) R. K. Gosavi, U. Agarwala and C. N. R. Rao, J. Am. Chem. Soc., 1967, 89, 235–239 CrossRef CAS.
- M. Tamm, D. Petrovic, S. Randoll, S. Beer, T. Bannenberg, P. G. Jonesa and J. Grunenberg, Org. Biomol. Chem., 2007, 5, 523–530 CAS.
- (a) R. K. Kottalanka, S. Anga, K. Naktode, P. Laskar, H. P. Nayek and T. K. Panda, Organometallics, 2013, 32, 4473–4482 CrossRef CAS; (b) R. K. Kottalanka, K. Naktode and T. K. Panda, J. Mol. Struct., 2013, 1036, 189–195 CrossRef PubMed; (c) R. K. Kottalanka, S. Anga, S. K. Jana and T. K. Panda, J. Organomet. Chem., 2013, 740, 104–109 CrossRef CAS PubMed.
- (a) K. Gregory, P. Von, R. Schleyer and R. Snaith, Adv. Inorg. Chem., 1991, 37, 47–142 CrossRef CAS; (b) X. He, B. C. Noll, A. Beatly, R. E. Mulvey and K. W. Henderson, J. Am. Chem. Soc., 2004, 126, 7444–7445 CrossRef CAS PubMed; (c) J. J. Morris, B. C. Noll, A. J. Schultz, P. M. B. Piccoli and K. W. Henderson, Inorg. Chem., 2007, 46, 10473–10475 CrossRef CAS PubMed; (d) S. A. Cantalupo, J. S. Lum, M. C. Buzzeo, C. Moore, A. G. Dipasquale, A. L. Rheingold and L. H. Doerrer, Dalton Trans., 2010, 39, 374–383 RSC.
- T. J. Boyle, N. L. Andrews, M. A. Rodriguez, C. Campana and T. Yiu, Inorg. Chem., 2003, 42, 5357–5366 CrossRef CAS PubMed.
- (a) L. Matilainen, M. Klinga and M. Leskelä, Polyhedron, 1995, 14, 635–638 CrossRef CAS; (b) W. Clegg, E. Lamb, S. T. Liddle, R. Snaith and A. E. H. Wheatley, J. Organomet. Chem., 1999, 573, 305–312 CrossRef CAS; (c) I. Fernández, R. D. Price, P. D. Bolton, M. F. Mahon, M. G. Davidson and F. López-Ortiz, J. Organomet. Chem., 2004, 689, 1890–1896 CrossRef PubMed.
- (a) S. M. Li, I. Rashkov, J. L. Espartero, N. Manolova and M. Vert, Macromolecules, 1996, 29, 57–62 CrossRef CAS; (b) P. Dobrzyński, J. Kasperczyk and M. Bero, Macromolecules, 1999, 32, 4735–4737 CrossRef; (c) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC; (d) Z. Zhong, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Macromolecules, 2001, 34, 3863–3868 CrossRef CAS; (e) M. Westerhausen, S. Schneiderbauer, A. N. Kneifel, Y. Söltl, P. Mayer, H. Nöth, Z. Zhong, P. J. Dijkstra and J. Feijen, Eur. J. Inorg. Chem., 2003, 3432–3439 CrossRef CAS; (f) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48–49 RSC; (g) M. S. Hill and P. B. Hitchcock, Chem. Commun., 2003, 1758–1759 RSC; (h) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS PubMed; (i) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (j) Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC; (k) D. J. Darensbourg, W. Choi, P. Ganguly and C. P. Richers, Macromolecules, 2006, 39, 4374–4379 CrossRef CAS; (l) M. G. Davidson, C. T. O'Hara, M. D. Jones, C. G. Keir, M. F. Mahon and G. Kociok-Köhn, Inorg. Chem., 2007, 46, 7686–7688 CrossRef CAS PubMed; (m) D. J. Darensbourg, W. Choi and C. P. Richers, Macromolecules, 2007, 40, 3521–3523 CrossRef CAS; (n) D. J. Darensbourg, W. Choi, O. Karroonnirun and N. Bhuvanesh, Macromolecules, 2008, 41, 3493–3502 CrossRef CAS; (o) V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820–9827 RSC; (p) C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832–4861 RSC; (q) C. M. Thomas, Chem. Soc. Rev., 2010, 39, 165–173 RSC; (r) X. Xu, Y. Chen, G. Zou, Z. Ma and G. Li, J. Organomet. Chem., 2010, 695, 1155–1162 CrossRef CAS PubMed; (s) Y. Sarazin, D. Rosca, V. Poirier, T. Roisnel, A. Silvestru, L. Maron and J.-F. Carpentier, Organometallics, 2010, 29, 6569–6577 CrossRef CAS; (t) Y. Sarazin, B. Liu, T. Roisnel, L. Maron and J.-F. Carpentier, J. Am. Chem. Soc., 2011, 133, 9069–9087 CrossRef CAS PubMed.
- (a) S. Harder, F. Feil and K. Knoll, Angew. Chem., Int. Ed., 2001, 40, 4261–4264 CrossRef CAS; (b) S. Harder and F. Feil, Organometallics, 2002, 21, 2268–2274 CrossRef CAS; (c) P. Jochmann, T. S. Dols, T. P. Spaniol, L. Perrin, L. Maron and J. Okuda, Angew. Chem., Int. Ed., 2009, 48, 5715–5719 CrossRef CAS PubMed.
- (a) A. G. M. Barrett, M. R. Crimmin, M. S. Hill and P. A. Procopiou, Proc. R. Soc. London, Ser. A, 2010, 466, 927 CrossRef CAS; (b) S. Harder, Chem. Rev., 2010, 110, 3852–3876 CrossRef CAS PubMed.
- (a) R. K. Kottalanka, K. Naktode, S. Anga, H. P. Nayek and T. K. Panda, Dalton Trans., 2013, 42, 4947–4956 RSC; (b) K. Naktode, R. K. Kottalanka, S. K. Jana and T. K. Panda, Z. Anorg. Allg. Chem., 2013, 639, 999–1003 CrossRef CAS; (c) K. Naktode, J. Bhattacharjee, S. D. Gupta, H. P. Nayek, B. S. Mallik and T. K. Panda, Z. Anorg. Allg. Chem., 2014, 640, 994–999 CrossRef CAS; (d) R. K. Kottalanka, A. Harinath, J. Bhattacharjee, H. V. Babu and T. K. Panda, Dalton Trans., 2014, 43, 8757–8766 RSC.
- A. G. M. Barrett, T. C. Boorman, M. R. Crimmin, M. S. Hill, G. Kociok-Köhna and P. A. Procopiouc, Chem. Commun., 2008, 5206–5208 RSC.
- (a) R. P. Davies, Inorg. Chem. Commun., 2000, 3, 13–15 CrossRef CAS; (b) A. R. Kennedy, R. E. Mulvey and R. B. Rowlings, J. Organomet. Chem., 2002, 648, 288–292 CrossRef CAS; (c) W. Clegg, G. C. Forbes, A. R. Kennedy, R. E. Mulvey and S. T. Liddle, Chem. Commun., 2003, 406–407 RSC; (d) X. He, B. C. Noll, A. Beatty, R. E. Mulvey and K. W. Henderson, J. Am. Chem. Soc., 2004, 126, 7444–7445 CrossRef CAS PubMed.
- (a) G. W. Gokel, L. J. Barbour, R. Ferdani and J. Hu, Acc. Chem. Res., 2002, 35, 878–886 CrossRef CAS PubMed; (b) R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 52, 11470–11487 CrossRef CAS PubMed.
- A. Altomare, M. C. Burla, G. Camalli, G. Cascarano, C. Giacovazzo, A. Gualiardi and G. Polidori, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: For crystallographic details in CIF. CCDC 1046050–1046056. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00490j |
| This journal is © The Royal Society of Chemistry 2015 |