
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHomoleptic borates and aluminates containing the difluorophosphato ligand – [M(O2PF2)x]y− – synthesis and characterization†
Christoph
Schulz
a,
Philipp
Eiden
a,
Petra
Klose
a,
Andreas
Ermantraut
a,
Michael
Schmidt
b,
Arnd
Garsuch
b and
Ingo
Krossing
*a
aInstitut für Anorganische und Analytische Chemie and Freiburger Materialforschungszentrum (FMF), Universität Freiburg, Albertstr. 21, 79104 Freiburg, Germany. E-mail: krossing@uni-freiburg.de
bBASF SE, Ludwigshafen, Germany
First published on 18th March 2015
Abstract
Weakly coordinating anions (WCAs) with the difluorophosphato ligand (O2PF2) were the target of this study. Initial experiments were conducted towards the preparation of homoleptic aluminates of the well-studied [Al(OR)4]−-type. The preparation of the initial target structure Li[Al(O2PF2)4] failed due to the remaining Lewis acidic character of the central aluminum atom. Instead, the formation of Li3[Al(O2PF2)6] and Al(O2PF2)3 was observed with hexacoordinate aluminum atoms and verified by NMR, IR and X-ray crystallography. A possible mechanism towards these compounds was postulated in the solvent induced dismutation of the tetracoordinate Li[Al(O2PF2)4]. A singly charged WCA was realized by the exchange of the central aluminum atom for boron. The [B(O2PF2)4]− anion was prepared starting from BH3·S(CH3)2 and boron tribromide leading to the protic room temperature Ionic Liquid (IL) [H(S(CH3)2)][B(O2PF2)4] and the neat liquid Brønsted acid H[B(O2PF2)4], respectively, representing a significantly improved synthesis with regard to the first experiments of Dove et al. The basicity of the [B(O2PF2)4]− anion and its WCA quality were investigated on the basis of the IR-spectroscopic NH-scale and the salt [H(N(Oct)3)][B(O2PF2)4] that places it better than all oxyanions and close to the carboranate based WCAs. A pathway to the solvent free pure Li[B(O2PF2)4] salt was established on a multi-gram scale with excellent purities enabling electrochemical applications (verified by NMR, IR, X-ray crystallography and cyclovoltammetry).
Introduction
The preparation of stable salts of reactive cations imposes special requirements upon the respective anions, such as low nucleophilicity and basicity. A decrease in the electrostatic interactions of the respective ions is achieved in the design of complex anions with a large surface area leading to a delocalization of the negative charge. Thereby, the otherwise stronger cation–anion interactions are transformed into multiple weak interactions,2,3 determining the nomenclature of this substance class as “weakly coordinating anions”, or WCAs. Other desirable properties of any type of WCA include stability against electrophilic attack, oxidation and/or reduction. Ongoing research on WCAs is devoted to the development of novel WCAs by using sterically demanding, often partially fluorinated ligands. Other approaches towards new classes of anions have been developed in the use of polyhedral boranates [BnHn]2− (n = 10, 12)4 or carboranates [HCBnHn]− (n = 9, 11)5 and their halogenated derivatives.6Working with weakly coordinating anions (WCAs)2,3,7 of the type [MIII(ORF)4]− (MIII = B, Al, ORF = fluorinated alkoxide)8 and [MV(ORF)6]− (ref. 9) (MV = Nb, Ta) led to an expertise in our group with respect to the stabilization of weakly bound complexes,10 highly reactive cations,11 as well as the induction of high conductivities in low polarity media.12 The numerous possibilities of an exchange of the uninegative ligand ORF with a more general definition of RF as the electronegative fluorinated residue allows for the construction of a large variety of WCAs comprising amongst others the teflatometallates [M(OTeF5)n]− (n = 4, M = B;13n = 6, M = As,14 Sb,15,16 Bi,14 Nb15,17), as well as the trifluorosulfonatometallates [M(OSO2RF)n]−.18
This article is devoted to the chemistry of the uninegative difluorophosphato ligand (O2PF2) starting from difluorophosphoric acid and its derivatives. Simple inorganic [O2PF2]− salts (K+, Cs+;19 Li+, Na+, Rb+, [NH4]+ (ref. 20)) are known. Structures containing difluorophosphate display a structural variety and different coordination modes. The expected simple ionic form with two almost equal P–O bonds is supplemented by a structure type with covalent bonding to one O atom of the ligand, leading to a monofunctional OP(O)F2 group with a clear distinction of the two P–O bonds in the molecule, i.e. in the compounds P2O3F2,21 (H3C)3Si(O2PF2)22 and Xe(O2PF2)2.23 A third structural type, in which the anion acts as a bridging ligand between two metal atoms, is realized in the compounds Cl2Ti(O2PF2)2,24 [(H3C)2Ga(O2PF2)]2,25,26 [(H3C)2Al(O2PF2)]3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 25,26 and R2Sn(O2PF2)2.27 In 1984, Dove et al. reported the existence of (difluorophosphato)borates in NMR tube experiments, delivering first spectroscopic evidence for the existence of [B(O2PF2)4]− and [FB(O2PF2)3]− anions in solution in equilibrium with other species.1
25,26 and R2Sn(O2PF2)2.27 In 1984, Dove et al. reported the existence of (difluorophosphato)borates in NMR tube experiments, delivering first spectroscopic evidence for the existence of [B(O2PF2)4]− and [FB(O2PF2)3]− anions in solution in equilibrium with other species.1
Results and discussion
Synthesis of Li3[Al(O2PF2)6] and Al(O2PF2)3
A tetracoordinate aluminate – [Al(OP(O)F2)4]− – was sought for by protolysis of the intermediately formed organometallic aluminate Li[Al(n-Bu)Et3]28 with difluorophosphoric acid (eqn (1) and (2)).| n-BuLi + AlEt3 → Li[Al(n-Bu)Et3] | (1) |
| Li[Al(n-Bu)Et3] + 4HO2PF2 → Li[Al(O2PF2)4] + 3EtH + n-BuH | (2) |
A colorless powder, slightly soluble in diethyl ether, dimethyl carbonate and acetonitrile was obtained. In disagreement to the sought Al-coordination number of four, the 27Al-NMR spectrum in dimethyl carbonate showed a signal at δ = −18.1 ppm in the chemical shift range of sixfold coordinated aluminum. A doublet signal in the 19F-NMR spectrum at δ = −86.2 ppm and a triplet signal in the 31P-NMR spectrum at δ = −17.7 ppm, each with a coupling constant of 1JFP = 927 Hz, confirmed the sole existence of difluorophosphato ligands for the coordination of the aluminum. Crystallization of the product in diethyl ether at −20 °C led to colorless crystals, which were measured at 110 K. The structure was determined as ether solvated lithium hexakis-(difluorophosphato)-aluminate [Li(Et2O)]3[Al(O2PF2)6]. The coordination of aluminum by six difluorophosphato ligands corresponded with the NMR spectroscopic results. Three oxygen atoms of the difluorophosphate ligands and one additional solvent molecule diethyl ether each coordinate the lithium cation. With these findings, the reaction scheme had to be modified according to eqn (3) towards the formation of a product mixture comprising Li3[Al(O2PF2)6] and the Lewis acid Al(O2PF2)3.
 | (3) |
Alterations in the reaction scheme regarding reactants and stoichiometries showed no success concerning the preparation of Li3[Al(O2PF2)6] without the Lewis acid as a byproduct and therefore were abandoned.
29 was considered a powerful building unit on the way to heteroleptic anions [XAl(O2PF2)3]−. Thus, synthetic access to this compound was worked out and found in the equimolar reaction of triethylaluminum with difluorophosphoric acid in pentane. | (4) |
The compound was obtained as a colorless powder insoluble in organic solvents, therefore MAS-NMR and IR-/Raman-spectroscopy were used to determine the structure. Formation of a uniform aluminum species was proven by the 27Al-MAS-NMR spectrum, which showed a signal at the chemical shift of δ = −16.2 ppm indicating a sixfold coordinated aluminum. 31P-MAS-NMR spectroscopy displayed the existence of two inequivalent difluorophosphates at δ = −29.0 ppm and −37.0 ppm, which suggested a coordination of the aluminum in two different ways. Vibrational spectroscopy delivered further structural indications. Monofunctional bonding of the difluorophosphates should lead to the fixation of a P–O single bond which results in a terminal formal P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond and therefore a growing difference in the vibrational frequencies of the PO2 stretching frequencies. Deviating from this, the very low difference of 94 cm−1 in Al(O2PF2)3 (cf. Δ
O double bond and therefore a growing difference in the vibrational frequencies of the PO2 stretching frequencies. Deviating from this, the very low difference of 94 cm−1 in Al(O2PF2)3 (cf. Δ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 183 cm−1 in [EMIm][O2PF2]30 (EMIM = 1-ethyl-3-methylimidazolium)) indicated a bifunctional coordination of the difluorophosphate groups, which led to similar PO bonds and therefore close vibrational frequencies. As a conclusion of the different spectroscopic measurements, a polymeric structure of the Lewis acid was postulated, containing bridging and η2-chelating ligands (Fig. 1).
= 183 cm−1 in [EMIm][O2PF2]30 (EMIM = 1-ethyl-3-methylimidazolium)) indicated a bifunctional coordination of the difluorophosphate groups, which led to similar PO bonds and therefore close vibrational frequencies. As a conclusion of the different spectroscopic measurements, a polymeric structure of the Lewis acid was postulated, containing bridging and η2-chelating ligands (Fig. 1).
 | ||
| Fig. 1 Proposed polymeric structure of Al(O2PF2)3. | ||
Further verification of this assignment was obtained by the calculated IR-spectra of a cyclic trimer (Al(O2PF2)3)3, constituting a compromise between computational cost and accuracy on the structural level, as it possesses all the aforementioned difluorophosphate coordination modes, while still being manageable with quantum chemical methods. The experimental spectrum is in fair agreement with the calculation and is shown in the ESI.† Due to the sixfold coordination of aluminum, the Lewis acidity of this compound and its suitability as a building block for heteroleptic aluminates are diminished leading to the abandoning of this approach.
From attempts to prepare the Lewis acid B(O2PF2)3 to the successful syntheses of borates [B(O2PF2)4]−
The poor solubilities of these aluminates motivated us to turn towards boron based systems. The smaller boron atom should allow the reduction of the coordination number from six in aluminum to four, which should lead to lighter anions with lower charge and thus increased solubilities in organic solvents. | (5) |
The optimized synthesis of the homoleptic [B(O2PF2)4]− anion was realized by the reaction of one equivalent of the dimethyl sulfide complex of BH3 and four equivalents of difluorophosphoric acid in dichloromethane. It yielded the pure room temperature IL [H(S(CH3)2)]+[B(O2PF2)4]− that could be purified from the solvent and residual unreacted reactants by applying high vacuum. FT-IR spectroscopy showed the expected vibrational bands for both [H(S(CH3)2]+ cation and [B(O2PF2)4]− anion (see Table 2 below). In the area above 1450 cm−1, the bands of the [H(S(CH3)2]+ cation could be found and assigned to the respective vibrational modes. The stretching mode of the S–H bond ( = 2851 cm−1) as well as the bending ( = 1436 cm−1) and stretching vibrations ( = 2921 cm−1 and 3042 cm−1) of the CH3 groups have been identified in the spectra. Only a deformation band of the CH3 groups at = 1082 cm−1 is overlapped by the symmetric stretch of the anion PO2 groups. Multinuclear NMR spectroscopy is in complete agreement with the assigned structure: the integration of the 1H-NMR signals of the [H(S(CH3)2]+ cation is in a ratio of 6:1 for the methyl protons (δ = 2.71 ppm) and the H–S proton (δ = 9.14 ppm) and thus confirms the complete protonation of the dimethyl sulfide. The structure of the anion was assigned by 11B-NMR spectroscopy. A singlet at −4.1 ppm well in the chemical shift range of tetracoordinate boron proved the borate formation. As these NMR measurements were carried out with the neat liquid substance, the hyperfine structure of the spectrum was not well resolved in these experiments. However, the hyperfine structure of the anion was observed in the more fluid solution of the lithium salt Li[B(O2PF2)4] (see below). With the 19F-NMR spectrum showing a doublet signal at a chemical shift of δ = −84.3 ppm and the 31P-NMR spectrum a triplet signal at δ = −30.9 ppm, the presence of difluorophosphato ligands in the anion was confirmed; potentially remaining B–H species were excluded by 1H-NMR spectroscopy giving definite proof of the existence of the [B(O2PF2)4]− anion. In addition, 19F–11B HSQC spectra confirmed the structure of the anion by showing a correlation of the fluorine atoms in the difluorophosphato ligands and the central boron atom.
 | (6) |
Again, a 1:3 stoichiometry did not lead to a Lewis acid, but rather a Brønsted acid. The experimental FT-IR spectrum of H[B(O2PF2)4] was in agreement with the DFT calculations for the [B(O2PF2)4]− anion. All vibrations were assigned to the [B(O2PF2)4]− anion, except for a weak band at = 1567 cm−1. This broad band in the FT-IR spectrum provided first spectroscopic evidence of the coordination of a (bridging) proton in H[B(O2PF2)4] (see Stoyanov et al.31). In agreement with this assignment, this band disappeared after synthesis and purification of the lithium salt. As expected, the 1H-NMR spectrum showed a singlet at a chemical shift of δ = 16.57 ppm for the proton. The significant downfield shift in this compound compared to the signal of the protons in the reactant difluorophosphoric acid (δ = 14.61 ppm) may be interpreted as an explicit increase of the acidity of the system. The doublet signal in the 19F-NMR spectrum at δ = −84.5 ppm (δ = −85.1 ppm; ref. 27) possessed a coupling constant of 1JFP = 991 Hz (1JFP = 984 Hz; ref. 27) and corresponded to the triplet signal at δ = −32.3 ppm (δ = −31.5 ppm; ref. 27) in the 31P-NMR spectrum. 11B-NMR spectroscopy displayed a singlet signal (δ = −4.0 ppm) belonging to the [B(O2PF2)4]− anion. As this sample was measured as a neat compound, the high concentration prevented the resolution of the hyperfine structure of the anion, which was analyzed in detail from the dissolved Li[B(O2PF2)4] salt. Nevertheless, the assignment by Dove et al.1 in their NMR experiments was confirmed without a doubt. Furthermore, Dove et al. reported reactions of [B(O2PF2)4]− with fluorine atoms present in their samples yielding heteroleptic borates of the [FxB(O2PF2)4−x]−-type, out of which they were able to characterize the [FB(O2PF2)3]− anion. In our studies, we also found a certain instability of [B(O2PF2)4]− in the presence of fluorine sources, such as [BF4]−, where we could identify all heteroleptic [FxB(O2PF2)4−x]− anions (x = 0–3; see Table 1; elaborate discussion on these compounds will be published separately). Apart from these cases, the symmetrical borate is stable towards decomposition for weeks.
| Li[B(O2PF2)4] | Li[FB(O2PF2)3] | Li[F2B(O2PF2)2] | Li[F3B(O2PF2)] | |
|---|---|---|---|---|
| LiH | 69 | 25 | 3 | 3 |
| n-Butyllithium | 83 | 1 | 10 | 6 |
| t-Butyllithium | 92 | 4 | 2 | 2 |
Synthesis of Li[B(O2PF2)4]
The large scale synthesis of electrochemically pure Li[B(O2PF2)4] was developed starting from different compounds containing the [B(O2PF2)4]− anion.The main product Li[B(O2PF2)4] was identified clearly by multinuclear NMR spectroscopy. It showed a signal at δ = −4.2 ppm with the hyperfine structure of a quintet of nonets in the 11B-NMR spectrum (Fig. 2; 2JPB = 8.0 Hz and 3JFB = 1.5 Hz). In the 19F-NMR spectrum the doublet of quartet signal at δ = −83.7 ppm contained a 1JFP coupling constant of 983 Hz and the previously mentioned 3JFB coupling of 1.5 Hz. 31P-NMR spectroscopy provided a signal at δ = −30.2 ppm with the multiplicity of a triplet of quartets, containing coupling constants of 981 Hz (1JFP) and 8.0 Hz (2JPB). The [FxB(O2PF2)4−x]− byproducts were identified in the 11B-NMR spectrum with the knowledge of further, separately performed investigations on this system that will be published separately. An increase of x in [FxB(O2PF2)4−x]− (x = 0–4) caused a definite low-field shift of the 11B-NMR signals of the respective borate, indicating a better electronic shielding of the boron center by difluorophosphato ligands compared to the fluoride ligands.
 | ||
| Fig. 2
11B-NMR spectrum of Li[B(O2PF2)4] (128.39 MHz, in ethylene carbonate–dimethyl carbonate (EC–DMC (1:1)), external toluene-d8 lock, 298 K). | ||
 | (7) |
The exclusion of any other nucleophiles than [O2PF2]− led to a clean preparation of Li[B(O2PF2)4] without noteworthy impurities. Work up of the lithium salt only includes simple washing steps with dichloromethane to get rid of the formed difluorophosphoric acid. The exclusive formation of the Li[B(O2PF2)4] was verified by the recorded NMR spectra. A solitary multiplet in the 11B-NMR spectrum (Fig. 2) at δ = −4.2 ppm (quintet of nonets) demonstrated the stability of the anion under the chosen conditions; the matching 19F- and 31P-NMR spectra showed the expected doublet (δ = −84.0 ppm; 19F) and triplet (δ = −30.2 ppm; 31P) signals for the difluorophosphato ligands. Complete conversion to the lithium salt was confirmed by the 7Li-NMR spectrum (singlet at δ = −2.0 ppm) and the 1H-NMR spectrum displaying the absence of a proton signal corresponding to the Brønsted acid.
The electrochemical stability of the lithium salt was tested by cyclic voltammetry (Fig. 3) to investigate the ability for usage as a conducting salt in lithium ion batteries (LIBs). The fifth cycle of the anodic measurement showed a smooth course of the current with a large plateau-like potential range of 3.3 to 4.3 V devoid of signs of redox processes in the electrolyte solution and with current values below 5 × 10−7 A. At potentials above 4.3 V, the inevitable oxidation of ethylene carbonate and dimethyl carbonate occurred, which led to an increase in the current values up to 1.5 × 10−6 A. For the cathodic measurement, a wide potential range of 2.8 to 0.9 V was observed in the forward sweep displaying current values of −2.0 × 10−7 to −6.0 × 10−7 A with peak current values of −1.0 × 10−5 A reaching at a potential of 0 V.
 | ||
| Fig. 3 Cyclic voltammogram of Li[B(O2PF2)4] (1.0 mol L−1 in ethylene carbonate–dimethyl carbonate (1:1)); fifth cycles of anodic (3 to 5 V vs. Li/Li+) and cathodic measurement (3 to 0 V vs. Li/Li+) combined. | ||
Therefore, it was concluded that the electrochemical window of Li[B(O2PF2)4] exceeds the stability of the electrolyte solvents. Further investigations with respect to the application of the compound in LIBs were undertaken and will be discussed in a follow-up paper with focus on the electrochemical properties.
Crystal structures
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) with the unit cell dimensions of a = 17.4058(4) Å, b = 17.4058(4) Å, c = 21.4947(6) Å, α = β = 90°, γ = 120° and Z = 6. The final R-index [(I > 2σ(I)] of the refined structure converged to R1 = 0.0337 (wR2 = 0.0824). The asymmetric unit of [Li(Et2O)]3[Al(O2PF2)6] is constituted of a lithium cation coordinated by a diethyl ether molecule and by two difluorophosphato ligands bound to a central aluminum atom, one of them being disordered with a population of the two disordered sites in a ratio of 60:40. Fig. 4, left displays a section of the crystal structure with an emphasis on the aluminate part of the compound. The central aluminum atom shows an octahedral coordination with six difluorophosphato ligands with Al–O bond lengths of 187.4(1) pm for the ordered difluorophosphato ligands and 187.9(1) pm for the disordered ones.
with the unit cell dimensions of a = 17.4058(4) Å, b = 17.4058(4) Å, c = 21.4947(6) Å, α = β = 90°, γ = 120° and Z = 6. The final R-index [(I > 2σ(I)] of the refined structure converged to R1 = 0.0337 (wR2 = 0.0824). The asymmetric unit of [Li(Et2O)]3[Al(O2PF2)6] is constituted of a lithium cation coordinated by a diethyl ether molecule and by two difluorophosphato ligands bound to a central aluminum atom, one of them being disordered with a population of the two disordered sites in a ratio of 60:40. Fig. 4, left displays a section of the crystal structure with an emphasis on the aluminate part of the compound. The central aluminum atom shows an octahedral coordination with six difluorophosphato ligands with Al–O bond lengths of 187.4(1) pm for the ordered difluorophosphato ligands and 187.9(1) pm for the disordered ones.
 | ||
| Fig. 4 Hydrogen atoms were omitted for reasons of clarity; thermal ellipsoids are shown at a probability of 50%. Left: section of the molecular structure of [Li(Et2O)]3[Al(O2PF2)6] with focus on the structure of the [Al(O2PF2)6]3− anion; contacts to the lithium cations are displayed with dashed bonds. Right: coordination of the lithium cations by the diethyl ether molecules and the [Al(O2PF2)6]3− anions shown in an excerpt of the crystal structure of [Li(Et2O)]3[Al(O2PF2)6]; coordinative bonds are displayed as dashed bonds. | ||
The P–O distances in the difluorophosphato ligands are 145.1(5) to 148.7(5) pm (cf.: dP–O(K[O2PF2]) = 147.0 pm; ref. 32), whereas the corresponding P–F distances range from 152.2(8) pm up to 153.3(1) pm (cf.: dP–X(K[O2PF2]) = 157.5 pm; ref. 32). The shortening of the P–F bonds in the [Al(O2PF2)6]3− anion compared to the ionic difluorophosphate in the potassium salt is probably induced by the increased electrophilicity due to the direct bonding of the ligand to the aluminum center resulting in strengthened P–F bonds. The coordination of the lithium cations is illustrated in Fig. 4, right. It includes a dimeric structure of two lithium cations each coordinated by one diethyl ether molecule and three difluorophosphato ligands. With a Li–O distance of 197.6(3) pm, the four-membered ring substructure is composed of two lithium cations and the terminal μ2-coordinated oxygen atoms of two difluorophosphato ligands. In each case, another disordered difluorophosphato ligand is coordinated μ1 to the lithium cation at a distance of 186.4(3) pm and the fourth coordination site is occupied by the oxygen atom of a diethyl ether molecule with a distance of 194.5(3) pm. The compound [Li(Et2O)]3[Al(O2PF2)6] forms a layered structure along the c-axis of the unit cell with a two dimensional network in the a–b plane built from [Al(O2PF2)6]3− anions interconnected by solvent coordinated lithium cations (see ESI† for figures).
:20.
The fourfold coordination of the central boron atom of the [B(O2PF2)4]− anion is illustrated in Fig. 5, left displaying B–O bond lengths between 145.6(1) and 146.1(1) pm. The difluorophosphato ligands include P–O distances in the ordered ligands of 152.1(1) to 152.2(1) pm for the oxygen atoms bound covalently to the central boron atom and P–O bond lengths of 144.0(1) to 145.0(1) pm for the terminal oxygen atoms coordinated to the lithium cation. In the disordered ligand, the P–Oboron distances were determined as 149.9(1) and 155.6(2) pm, respectively, whereas the P–OLithium bond lengths measured 144.2(4) and 147.7(13) pm respectively (cf.: dP–O(K[O2PF2]) = 147.0 pm; ref. 32). The corresponding P–F distances range from 150.6(1) to 152.9(1) pm for the ordered difluorophosphato ligands and from 146.3(5) to 153.7(5) pm for the disordered ligands (cf.: dP–X(K[O2PF2]) = 157.5 pm; ref. 32). As it was shown in the case of the [Al(O2PF2)6]3− anion in the previous section, the direct bonding of the ligands to the electrophilic boron center resulted in a decrease of negative charge in the ligand in comparison with the ionic difluorophosphate and therefore, stronger bonds to the fluorine atoms. The embedding of the lithium cations into the structure of Li[B(O2PF2)4] is illustrated in Fig. 5 right. It shows the fourfold coordination of lithium by the terminal oxygen atom of difluorophosphato ligands belonging to four different [B(O2PF2)4]− anions. In this way, the lithium cation interconnects the anions to three-dimensional networks showing Li–O distances of 191.7(2) pm to 194.2(2) pm for the ordered ligands and 187.0(20) pm to 194.5(4) pm for the disordered ligands with an approximately tetrahedral coordination geometry displaying O–Li–O bond angles from 105.6(1)° to 114.4(1)° (Fig. 6).
 | ||
| Fig. 5 Thermal ellipsoids are shown at a probability of 50%; coordinative bonds are displayed using dashed bonds. Left: section of the molecular structure of Li[B(O2PF2)4] with focus on the structure of the [B(O2PF2)4]− anion. Right: coordination of the lithium cation by four different [B(O2PF2)4]− anions, in each case with one terminal oxygen atom, shown in a section of the crystal structure of Li[B(O2PF2)4]. | ||
 | ||
| Fig. 6 Contents of one unit cell of Li[B(O2PF2)4], displaying the interconnection of the [B(O2PF2)4]− anions via the fourfold coordinated lithium cation; coordinative bonds are displayed using dashed bonds; thermal ellipsoids are shown at a probability of 50%. | ||
Vibrational spectroscopy of the difluorophosphates
This section provides a more in-depth insight into the coordination of the difluorophosphates in the previously mentioned compounds. The method of choice to determine structural motifs in the bonding of the difluorophosphates is vibrational spectroscopy due to the high sensitivity of their stretching vibrations. Several structural types of coordination are well known and described in the literature,33 such as ionic [O2PF2]− in alkali metal salts,20 monofunctional OP(O)F2 groups in (H3C)3Si(O2PF2)34 or bridging difluorophosphates in [(H3C)2Ga(O2PF2)]2.26 As a reference state for an undisturbed [O2PF2]− anion with small interactions between the cation and anion, the literature data of the imidazolium based ionic liquid [EMIm][O2PF2]30 were used in this work. The difluorophosphates in all the complex ions shown in this work are expected to possess four clearly recognizable stretching vibrations based on the asymmetric and symmetric vibrational modes of both the PO and PF bonds. Depending on the type of coordination, the corresponding vibrational frequencies can vary considerably, especially for the directly bound OP(O)F2 groups. A summary of the IR spectroscopic data of the investigated compounds is given in Table 2. The stretching vibrations of the PO2 group in [EMIm][O2PF2] ( = 1323 cm−1 and 1140 cm−1) displayed a difference of Δν = 183 cm−1, which is considered a rather small value due to the good resonance of the two oxygen atoms in the undisturbed ion (I in Fig. 7). Stronger bonding of one oxygen atom led to a monofunctional OP(O)F2 group with a fixed P–O–σ bond and a formal terminal PO double bond (II in Fig. 7). The [B(O2PF2)4]− anion includes this coordination mode II and therefore all of its presented compounds featured a much higher Δν of 254–260 cm−1, with the terminal PO double bond blueshifted by 11 to 25 cm−1, whereas the P–O–σ bond exhibited a redshift of 47–60 cm−1. Coordination of the difluorophosphates in the aluminum species Al(O2PF2)3 was realized differently: the aluminum centers in this structure were bridged by the two oxygen atoms of the phosphates. This structural variation (III in Fig. 7) led to a smaller difference between the asymmetric and symmetric stretching vibration of the PO2 group, which decreased to Δ = 95 cm−1 for the Lewis acid Al(O2PF2)3, underlining the similarity of the bonding situation for both coordinated oxygen atoms.
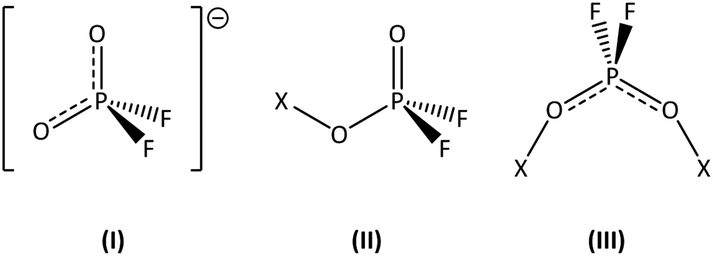 | ||
| Fig. 7 Modes of coordination of the difluorophosphato ligand: ionic (I), monofunctional OP(O)F2 groups (II), symmetrically bridging between two metal atoms (III). The terminal PO bond is written as a formal double bond, although it is clear that its Lewis structure is better represented by a P+–O− notation. However, the formal double bond PO intuitively accounts for the observed blue shift in the vibrational frequency. | ||
| [EMIm][O2PF2]30 | Li3[Al(O2PF2)6] | Al(O2PF2)3 | [H(S(CH3)2)][B(O2PF2)4] | H[B(O2PF2)4] | [H(N(Oc)3][B(O2PF2)4] | Li[B(O2PF2)4] | Assignmenta |
|---|---|---|---|---|---|---|---|
|
a M stands for the group XIII elements boron or aluminum.
b The denominations ν(P–O) and ν(PO) apply to the monofunctional OP(O)F2 groups in the [M(O2PF2)x]y− anion; the denominations νs(PO2) and νas(PO2) are for the aluminum species Al(O2PF2)3 and the free [O2PF2]− anion.
|
|||||||
| 498 (s) | 417 (w) | 402 (w) | 472 (w) | 469 (w) | 472 (w) | 479 (w) | δ(PF2) |
| — | 503 (w) | 495 (w) | 511 (w) | 502 (w) | 514 (w) | 502 (sh) | δ(PO2) |
| — | 536 (w) | 512 (sh) | 555 (w) | 552 (w) | 560 (w) | 568 (w) | δ(MO2) |
| — | 624 (w) | 622 (w) | 648 (vw) | 647 (w) | 724 (w) | — | δ(MO2) |
| — | 723 (w) | — | — | — | — | — | — |
| — | — | 847 (w) | — | — | — | — | — |
| 820 (s) | 891 (w)/922 (m) | 916 (m) | 832 (m) | 836 (m) | 832 (m) | 833 (m) | ν s(PF2) |
| 964 (s) | 956 (m) | 933 (m) | 940 (m) | 931 (m) | 945 (m) | ν as(PF2) | |
| — | — | — | 993 (w) | 994 (w) | 983 (w) | 1002 (w) | ν(MO2) |
| 1140 (s) | 1175 (s)/1204 (s) | 1192 (m) | 1083 (s) | 1093 (s) | 1086 (s) | 1080 (s) | ν s(PO2)/ν(P–O)b |
| 1323 (s) | 1283 (s) | 1287 (s) | 1337 (s) | 1348 (m) | 1346 (s) | 1334 (s) |
ν
as(PO2)/ν(PO)b |
| — | — | — | 1436 (vw) | — | 1468 (w) | — | — |
| — | — | — | — | 1567 (w) | — | — | — |
On the nature of the Lewis acid B(O2PF2)3
The goal of synthesizing unsymmetric borates of the type [XB(O2PF2)3]− should be accomplished by the synthesis of the Lewis acid B(O2PF2)3. This compound is expected to possess high Lewis acidity and therefore large potential as a synthetic building block on the way to the desired unsymmetric borates. However, experimentally in our hands it was impossible to obtain this molecule. Therefore, the Lewis acidity of B(O2PF2)3 was investigated by quantum chemical calculations‡ concerning its fluoride ion affinity (FIA).35,36 The FIA presents a way to classify Lewis acids respective to the energy that is released upon the binding of a fluoride anion and is determined for the respective substances in isodesmic reactions. The reactions taken into account for this particular compound were as follows: | (8) |
 | (9) |
The combination of both reactions resulted in the basic equation to establish the FIA(B(O2PF2)3):
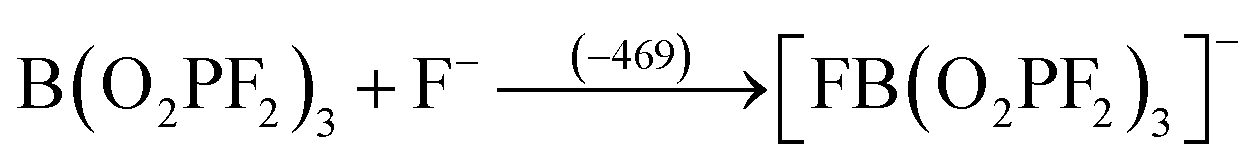 | (10) |
According to the calculations, B(O2PF2)3 displays a very high FIA of 469 kJ mol−1 lying within the range of the system SbF5/[SbF6]− (493 kJ mol−1) that represents the limit to superacidity.36,37 As shown above, the reactions of the dimethyl sulfide complex of BH3 and difluorophosphoric acid led to the formation of the symmetric borate [B(O2PF2)4]−. Alterations in the stoichiometry of the acid to two or three equivalents showed no influence on the resulting boron species, as the symmetric [B(O2PF2)4]− anion obviously constitutes the thermodynamic minimum in these reactions. The synthesis of B(O2PF2)3 probably failed because the tricoordinate central boron atom is not stable in the chosen environments due to its high Lewis acidity.
The aforementioned Lewis acid Al(O2PF2)3 showed a similar behavior with respect to the threefold coordination of the central atom in these structures. As previously mentioned, the aluminum compound avoids the coordination number of three by the formation of a polymeric structure with a sixfold coordination of the central aluminum. The FIA of monomeric, gaseous Al(O2PF2)3 gives an explanation for this behavior as well. It was calculated analogous to eqn (8)–(10) using the respective aluminum containing species on the same quantum chemical level.
 | (11) |
In the case of Al(O2PF2)3, the FIA was determined to be 494 kJ mol−1. Thus, its very acidic character underlines the very pronounced tendency towards higher coordination numbers for aluminum.
The high Lewis acidity of B(O2PF2)3 is reflected in the energetic level of its LUMO (lowest unoccupied molecular orbital) at −0.92 eV.§ According to G. Frenking et al.,38 the energy of the LUMO dictates the interaction with a Lewis base, having a decisive influence on the acidity of a Lewis acid. Compared to BF3 (ELUMO = +0.46 eV§), the lower LUMO level of B(O2PF2)3 corresponds with the abovementioned increase of the Lewis acidity. For the analogous aluminum system (ELUMO(AlF3) = −1.49 eV§; ELUMO(Al(O2PF2)3) = −1.01 eV§), the orbital based impact on the acidity is less than the ionic contributions, which may relativize the inverse situation of the LUMO energy levels, therefore still being consistent with the high Lewis acidity of 494 kJ mol−1 for Al(O2PF2)3.
Evaluation of the WCA-performance of [B(O2PF2)4]−: synthesis of [H(N(Oct)3)][B(O2PF2)4]
For an experimental evaluation of the basicity of the [B(O2PF2)4]− anion, its tri-n-octylammonium salt was synthesized. C. A. Reed et al.39 established a ν(NH) scale for weakly basic anions, which allows for a comparison of the before mentioned anions (A−), as well as their conjugate acids (HA). The compound [H(N(Oct)3)][B(O2PF2)4] was obtained as a colorless room temperature ionic liquid in a reaction of the Brønsted acid H[B(O2PF2)4] and tri-n-octylamine in dichloromethane, according to eqn (9): | (12) |
The tri-n-octylammonium cation caused the appearance of vibrational bands in the area of 2850 cm−1 to 3100 cm−1, additional to the bands of the anion (Table 2). Those were assigned to the C–H stretching vibrations ( = 2859 cm−1; 2928 cm−1 and 2957 cm−1) and the N–H stretching vibration at = 3092 cm−1. The ν(NH) frequency being slightly below the region of carboranates, but yet above the values for the least basic oxyanions (Table 3) displayed the very low basicity of the [B(O2PF2)4]− anion and therefore the high acidity of its conjugate acid H[B(O2PF2)4].
| Anion | ν(NH) frequency | Anion | ν(NH) frequency |
|---|---|---|---|
| [B(C6F5)4]− | 3241 | [CHB11Me5I6]− | 3100 |
| [B12F12]2− | 3226 | [B(O 2 PF 2 ) 4 ] − | 3092(4) |
| [CMeB11F11]− | 3219 | [N(SO2CF3)2]− | 3086 (in CCl4) |
| [SbF6]− | 3201 | [(HSO4)2]2− | 3080 |
| [B12I12]2− | 3116 | [C5H(CN)4]− | 3070 |
| [CHB11H5I6]− | 3106 | [F3CSO3]− | 3056, 2815 |
| [C5(CN)5]− | 3105 | [FSO3]− | 3045 |
Conclusion
A new class of WCAs with the difluorophosphato ligand was investigated. Tetracoordinate [Al(O2PF2)4]−-type structures were aspired. However, the originally sought after tetracoordinate Li[Al(O2PF2)4] proved to be unstable in solution, leading to dismutation and formation of hexacoordinate aluminum species, i.e. the homoleptic aluminate Li3[Al(O2PF2)6] and the Lewis acid Al(O2PF2)3. In contrast to the results with Al as the central atom, the intended use as a WCA implies the realization of a singly charged anion. Thus, the preferred WCA-coordination number of four led to the exchange of aluminum by boron. The synthesis of such borates was attempted by the preparation of the Lewis acid B(O2PF2)3 as an important building block. However, this was never successful. Syntheses of the tricoordinated boron compound B(O2PF2)3 always yielded the homoleptic anion [B(O2PF2)4]−, regardless of the stoichiometry of the reactants. Thus, the room temperature IL [H(S(CH3)2)][B(O2PF2)4] was prepared in the reaction of BH3·S(CH3)2 and difluorophosphoric acid, whereas the route with boron tribromide as the reactant led to the room temperature liquid Brønsted acid H[B(O2PF2)4]. The basicity of the [B(O2PF2)4]− WCA was assigned to be in between the range of carboranates and oxyanions by an IR spectroscopic analysis of the tris-n-octylammonium salt of this anion.The preparation of lithium tetrakis(difluorophosphato)borate was investigated based on the two routes starting from BH3·S(CH3)2 and boron tribromide. Heterogeneous reactions of [H(S(CH3)2)][B(O2PF2)4] and lithium metal or lithium hydride delivered poor results, whereas homogeneous reactions using butyllithium led to purities up to 92%. The reaction of the Brønsted acid H[B(O2PF2)4] with lithium difluorophosphate led to excellent purities above 99% in large batches (up to 25 g).
Cyclovoltammetry in EC/DMC showed that the electrochemical window of Li[B(O2PF2)4] exceeds the stability of the electrolyte solvents leading to further investigations towards a potential application in LIBs as conducting salts or additives which will be discussed in a separate follow-up paper.
Acknowledgements
This work was supported by Merck KGaA and BASF SE in the context of the Battery Materials Laboratory at the Freiburger Materialforschungszentrum (FMF). We thank Dr Harald Scherer and Fadime Bitgül for the measurement of the NMR spectra, Dr Daniel Kratzert for his support regarding single X-ray crystallography, and Dr Daniel Himmel for valuable discussions of the DFT calculations.References
- M. F. Dove, R. C. Hibbert and N. Logan, J. Chem. Soc., Dalton Trans., 1984, 2719 RSC.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066–2090 CrossRef CAS PubMed.
- I. Krossing and A. Reisinger, Coord. Chem. Rev., 2006, 250, 2721–2744 CrossRef CAS.
- A. R. Pitochelli and F. M. Hawthorne, J. Am. Chem. Soc., 1960, 82, 3228–3229 CrossRef CAS.
- K. Shelly, D. C. Finster, Y. J. Lee, W. R. Scheidt and C. A. Reed, J. Am. Chem. Soc., 1985, 107, 5955–5959 CrossRef CAS.
- (a) S. V. Ivanov, S. M. Miller, O. P. Anderson, K. A. Solntsev and S. H. Strauss, J. Am. Chem. Soc., 2003, 125, 4694–4695 CrossRef CAS PubMed; (b) A. Avelar, F. S. Tham and C. A. Reed, Angew. Chem., Int. Ed., 2009, 48, 3491–3493 CrossRef CAS PubMed; (c) W. Gu and O. V. Ozerov, Inorg. Chem., 2011, 50, 2726–2728 CrossRef CAS PubMed; (d) J. Derendorf, M. Kessler, C. Knapp, M. Rühle and C. Schulz, Dalton Trans., 2010, 39, 8671–8678 RSC; (e) C. Bolli, J. Derendorf, M. Kessler, C. Knapp, H. Scherer, C. Schulz and J. Warneke, Angew. Chem., Int. Ed., 2010, 49, 3536–3538 CrossRef CAS PubMed; (f) M. Kessler, C. Knapp, V. Sagawe, H. Scherer and R. Uzun, Inorg. Chem., 2010, 49, 5223–5230 CrossRef CAS PubMed.
- I. Krossing, in Comprehensive Inorganic Chemistry II (Second Edition), ed. J. Reedijk and K. Poeppelmeier, Elsevier, Amsterdam, 2013, pp. 681–705 Search PubMed.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS.
- U. P. Preiss, G. Steinfeld, H. Scherer, A. M. Erle, B. Benkmil, A. Kraft and I. Krossing, Z. Anorg. Allg. Chem., 2013, 639, 714–721 CrossRef CAS.
- (a) J. Schaefer, D. Himmel and I. Krossing, Eur. J. Inorg. Chem., 2013, 2013, 2712–2717 CrossRef CAS; (b) J. Schaefer, A. Kraft, S. Reininger, G. Santiso-Quinones, D. Himmel, N. Trapp, U. Gellrich, B. Breit and I. Krossing, Chem. – Eur. J., 2013, 19, 12468–12485 CrossRef CAS PubMed; (c) A. Higelin, S. Keller, C. Göhringer, C. Jones and I. Krossing, Angew. Chem., Int. Ed., 2013, 52, 4941–4944 CrossRef CAS PubMed; (d) J. Schaefer, A. Steffani, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6009–6012 CrossRef CAS PubMed; (e) A. Reisinger, N. Trapp, C. Knapp, D. Himmel, F. Breher, H. Rüegger and I. Krossing, Chem. – Eur. J., 2009, 15, 9505–9520 CrossRef CAS PubMed; (f) G. Santiso-Quiñones, R. Brückner, C. Knapp, I. Dionne, J. Passmore and I. Krossing, Angew. Chem., Int. Ed., 2009, 48, 1133–1137 CrossRef PubMed.
- (a) T. Köchner, T. A. Engesser, H. Scherer, D. A. Plattner, A. Steffani and I. Krossing, Angew. Chem., Int. Ed., 2012, 51, 6529–6531 CrossRef PubMed; (b) A. J. Lehner, N. Trapp, H. Scherer and I. Krossing, Dalton Trans., 2011, 40, 1448–1452 RSC; (c) T. Köchner, S. Riedel, A. J. Lehner, H. Scherer, I. Raabe, T. A. Engesser, F. W. Scholz, U. Gellrich, P. Eiden, R. A. Paz-Schmidt, D. A. Plattner and I. Krossing, Angew. Chem., Int. Ed., 2010, 49, 8139–8143 CrossRef PubMed; (d) I. Raabe, D. Himmel, S. Müller, N. Trapp, M. Kaupp and I. Krossing, Dalton Trans., 2008, 946–956 RSC; (e) M. Gonsior, I. Krossing and E. Matern, Chem. – Eur. J., 2006, 12, 1986–1996 CrossRef CAS PubMed; (f) M. Gonsior, I. Krossing and E. Matern, Chem. – Eur. J., 2006, 12, 1703–1714 CrossRef CAS PubMed; (g) I. Krossing, A. Bihlmeier, I. Raabe and N. Trapp, Angew. Chem., Int. Ed., 2003, 42, 1531–1534 CrossRef CAS PubMed.
- I. Raabe, K. Wagner, K. Guttsche, M. Wang, M. Grätzel, G. Santiso-Quiñones and I. Krossing, Chem. – Eur. J., 2009, 15, 1966–1976 CrossRef CAS PubMed.
- D. M. Van Seggen, P. K. Hurlburt, M. D. Noirot, O. P. Anderson and S. H. Strauss, Inorg. Chem., 1992, 31, 1423–1430 CrossRef CAS.
- H. P. Mercier, J. C. Sanders and G. J. Schrobilgen, J. Am. Chem. Soc., 1994, 116, 2921–2937 CrossRef CAS.
- D. M. Van Seggen, P. K. Hurlburt, O. P. Anderson and S. H. Strauss, Inorg. Chem., 1995, 34, 3453–3464 CrossRef CAS.
- S. T. Cameron, I. Krossing and J. Passmore, Inorg. Chem., 2001, 40, 4488–4490 CrossRef PubMed.
- K. Moock and K. Seppelt, Z. Anorg. Allg. Chem., 1988, 561, 132–138 CrossRef CAS.
- (a) G. A. Olah, K. Laali and O. Farooq, J. Org. Chem., 1984, 49, 4591–4594 CrossRef CAS; (b) W. V. Cicha, A. J. Kornath, R. J. McKinney, V. N. M. Rao, J. S. Thrasher and A. Waterfeld, U.S. Patent5,773,637, 1998 Search PubMed.
- W. Lange, Ber. Dtsch. Chem. Ges. A/B, 1929, 62, 786–792 CrossRef.
- R. C. Thompson and W. Reed, Inorg. Nucl. Chem. Lett., 1969, 5, 581–585 CrossRef CAS.
- U. Wannagat and J. Rademachers, Z. Anorg. Allg. Chem., 1957, 289, 66–79 CrossRef CAS.
- U. Biermann and O. Glemser, Chem. Ber., 1969, 102, 3342–3346 CrossRef CAS.
- M. Eisenberg and D. D. Desmarteau, Inorg. Chem., 1972, 11, 1901–1904 CrossRef CAS.
- J. R. Dalziel, R. D. Klett, P. A. Yeats and F. Aubke, Can. J. Chem., 1974, 52, 231–239 CrossRef CAS.
- B. Schaible, W. Haubold and J. Weidlein, Z. Anorg. Allg. Chem., 1974, 403, 289–300 CrossRef CAS.
- B. Schaible and J. Weidlein, Z. Anorg. Allg. Chem., 1974, 403, 301–309 CrossRef CAS.
- T. H. Tan, J. R. Dalziel, P. A. Yeats, J. R. Sams, R. C. Thompson and F. Aubke, Can. J. Chem., 1972, 50, 1843–1851 CrossRef CAS.
- T. Hagiwara, M. Takeda, H. Hamana and T. Narita, Makromol. Chem., Rapid Commun., 1987, 8, 167–170 CrossRef CAS.
- A.-F. Shihada, B. K. Hassan and A. T. Mohammed, Z. Anorg. Allg. Chem., 1980, 466, 139–144 CrossRef CAS.
- K. Matsumoto and R. Hagiwara, Inorg. Chem., 2009, 48, 7350–7358 CrossRef CAS PubMed.
- D. Stasko, S. P. Hoffmann, K.-C. Kim, N. L. P. Fackler, A. S. Larsen, T. Drovetskaya, F. S. Tham, C. A. Reed, C. E. F. Rickard, P. D. W. Boyd and E. S. Stoyanov, J. Am. Chem. Soc., 2002, 124, 13869–13876 CrossRef CAS PubMed.
- R. W. Harrison, R. C. Thompson and J. Trotter, J. Chem. Soc. A, 1966, 1775 RSC.
- K. Dehnicke and A.-F. Shihada, in Electrons in Oxygen- and Sulphur-Containing Ligands, Springer, Berlin, Heidelberg, 1976, vol. 28, pp. 51–82 Search PubMed.
- H. W. Roesky, Chem. Ber., 1967, 100, 2147–2150 CrossRef CAS.
- (a) T. E. Mallouk, G. L. Rosenthal, G. Mueller, R. Brusasco and N. Bartlett, Inorg. Chem., 1984, 23, 3167–3173 CrossRef CAS; (b) K. O. Christe, D. A. Dixon, D. McLemore, W. W. Wilson, J. A. Sheehy and J. A. Boatz, J. Fluorine Chem., 2000, 101, 151–153 CrossRef CAS; (c) I. Krossing and I. Raabe, Chem. – Eur. J., 2004, 10, 5017–5030 CrossRef CAS PubMed; (d) H. D. B. Jenkins, I. Krossing, J. Passmore and I. Raabe, J. Fluorine Chem., 2004, 125, 1585–1592 CrossRef CAS.
- L. O. Müller, D. Himmel, J. Stauffer, G. Steinfeld, J. Slattery, G. Santiso-Quiñones, V. Brecht and I. Krossing, Angew. Chem., Int. Ed., 2008, 47, 7659–7663 CrossRef PubMed.
- H. Böhrer, N. Trapp, D. Himmel and I. Krossing, Dalton Trans., 2015 Search PubMed , accepted.
- F. Bessac and G. Frenking, Inorg. Chem., 2003, 42, 7990–7994 CrossRef CAS PubMed.
- E. S. Stoyanov, K.-C. Kim and C. A. Reed, J. Am. Chem. Soc., 2006, 128, 8500–8508 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental details, the discussed 1D- and 2D-NMR spectra, as well as IR spectra of the reactions are deposited. The complete crystal structure data including the CCSD deposition numbers are displayed. CCDC 1033591 and 1033599. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00469a |
| ‡ The components of reaction (9) were calculated at the G3-level; M(O2PF2)3 and [FM(O2PF2)3]− (M = B, Al) were calculated at the PBE0/def2-TZVP(P)-level. |
| § All LUMO energies were calculated at the PBE0/def2-TZVP(P)-level. |
| This journal is © The Royal Society of Chemistry 2015 |