
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
P,O-Phosphinophenolate zinc(II) species: synthesis, structure and use in the ring-opening polymerization (ROP) of lactide, ε-caprolactone and trimethylene carbonate†
Christophe
Fliedel
ab,
Vitor
Rosa
 ab,
Filipa M.
Alves
c,
Ana. M.
Martins
c,
Teresa
Avilés
*a and
Samuel
Dagorne
*b
ab,
Filipa M.
Alves
c,
Ana. M.
Martins
c,
Teresa
Avilés
*a and
Samuel
Dagorne
*b
aLAQV, REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia da Universidade Nova de Lisboa, Caparica, 2829-516, Portugal
bInstitut de Chimie de Strasbourg (UMR 7177), CNRS-Université de Strasbourg, 1 rue Blaise Pascal, 67000 Strasbourg, France. E-mail: dagorne@unistra.fr
cCentro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Av. Rovisco Pais, 1049-001 Lisboa, Portugal
First published on 25th March 2015
Abstract
The P,O-type phosphinophenol proligands (1·H, 2-PPh2-4-Me-6-Me-C6H2OH; 2·H, 2-PPh2-4-Me-6-tBu-C6H2OH) readily react with one equiv. of ZnEt2 to afford in high yields the corresponding Zn(II)-ethyl dimers of the type [(κ2-P,O)Zn-Et]2 (3 and 4) with two μ-OPh bridging oxygens connecting the two Zn(II) centers, as determined by X-ray diffraction (XRD) studies in the case of 3. Based on diffusion-ordered NMR spectroscopy (DOSY), both species 3 and 4 retain their dimeric structures in solution. The alcoholysis reaction of Zn(II) alkyls 3 and 4 with BnOH led to the high yield formation of the corresponding Zn(II) benzyloxide species [(κ2-P,O)Zn-OBn]2 (5 and 6), isolated in a pure form as colorless solids. The centrosymmetric and dimeric nature of Zn(II) alkoxides 5 and 6 in solution was deduced from DOSY NMR experiments and multinuclear NMR data. Though the heteroleptic species 5 is stable in solution, its analogue 6 is instable in CH2Cl2 solution at room temperature to slowly decompose to the corresponding homoleptic species 8via the transient formation of (κ2-P,O)2Zn2(μ-OBn)(μ–κ1:κ1-P,O) (6′). Crystallization of compound 6 led to crystals of 6′, as established by XRD analysis. The reaction of ZnEt2 with two equiv. of 1·H and 2·H allowed access to the corresponding homoleptic species of the type [Zn(P,O)2] (7 and 8). All gathered data are consistent with compound 7 being a dinuclear species in the solid state and in solution. Data for species 8, which bears a sterically demanding P,O-ligand, are consistent with a mononuclear species in solution. The Zn(II) alkoxide species 5 and the [Zn(P,O)2]-type compounds 7 and 8 were evaluated as initiators of the ring-opening polymerization (ROP) of lactide (LA), ε-caprolactone (ε-CL) and trimethylene carbonate (TMC). Species 5 is a well-behaved ROP initiator for the homo-, co- and ter-polymerization of all three monomers with the production of narrow disperse materials under living and immortal conditions. Though species 7 and 8 are ROP inactive on their own, they readily polymerize LA in the presence of a nucleophile such as BnOH to produce narrow disperse PLA, presumably via an activated-monomer ROP mechanism.
Introduction
Aliphatic polyesters and polycarbonates such as polylactic acid (PLA), poly(ε-caprolactone) (PCL) and poly(trimethylene carbonate) (PTMC) are of current importance as biodegradable and bioassimilable materials and, as such, have already found a wide array of applications, spanning from their use in food packaging to biomedicine (as excipients in controlled drug delivery and bone tissue prosthesis components, for example).1 The thermoplastic properties of PLA and PCL also position such materials as possible biodegradable alternatives to petrochemically-sourced thermoplastic polyolefins. The resulting properties of these materials are favorably impacted by their well-defined nature, including controlled polymer chain length and, in the case of PLA, stereoregularity.1,2 In this regard, controlled ring-opening polymerization (ROP) of lactide (LA), ε-caprolactone (ε-CL) and trimethylene carbonate (TMC) by discrete ligand-supported metal alkoxide initiators constitutes the method of choice for the production of well-defined PLA, PCL and PTMC, respectively.2 For instance, PLA is industrially produced via ROP of L-LA, a dimer of the renewable resource L-lactic acid, using Sn(Oct)2 as the ROP initiator.3 Over the past fifteen years, numerous complexes of oxophilic metals, primarily Mg(II)-, Ca(II)-, Ti(IV)-, Zr(IV)-, Zn(II)-, Al(III)-, Ga(III)-, In(III)-, and Bi(III)-based derivatives, have been investigated as ROP initiators for catalytic performance improvement and the production of stereoregular PLA material.2,4 In general, ROP performances of ligand-based metal complexes are greatly impacted by the nature of the ancillary ligand(s). To date, studies on metal-based ROP initiators overwhelmingly involve the use of an N- and/or O-based hard donor polydentate anionic ligand (as supporting/ancillary ligands) for metal stabilization.2,4 In contrast, metal complexes bearing softer heteroatom-based Lewis bases (such as phosphine, thioether donors) have been little explored in cyclic esters/carbonates ROP despite the potential positive influence of such softer donor ligands on polymerization catalysis performance. For instance, earlier studies on various group 4 metal chelates concluded on the beneficial effect of chelating ligands incorporating softer L donors, such as phosphorus and sulfur, on olefin polymerization activity.5 Also, we earlier showed that cationic Al(III) complexes bearing bidentate phosphino-phenolate ligands of type A (Chart 1) are effective initiators in the ROP of propylene oxide and ε-CL,6a while Lamberti, Mazzeo and co-workers reported on the use of thioether-phenolate Zn(II), Mg(II) and Al(III) species for the effective and controlled ROP of ε-CL and lactide.6b–c,7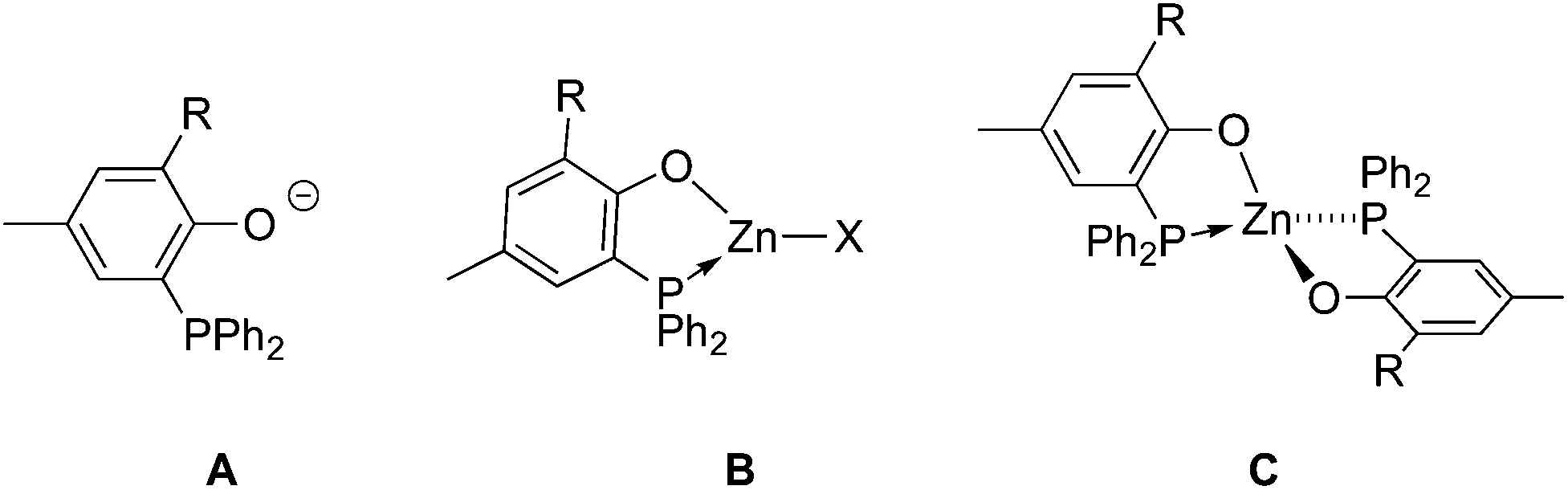 | ||
| Chart 1 P,O-Phosphinophenolate ligands A and associated (P,O)Zn chelates B and C. | ||
As ROP initiators of cyclic esters/carbonates, Zn(II)-based species are of particular interest since Zn is a cheap, readily available and biocompatible metal source, the latter feature being of utmost importance for the application of the derived biomaterials in biomedicine.8 This, together with the suitability of bidentate phosphinophenolate ligands A in Al(III)-mediated ROP catalysis in our previous studies,6a prompted us to extend our investigations to phosphinophenolate organozinc(II) species of type B and C (Chart 1) for use in cyclic esters/carbonates ROP. Unsurprisingly, though a couple of phosphino/phosphido Zn(II) compounds have been reported to mediate the ROP of LA and ε-CL,9 the vast majority of Zn(II) ROP initiators are supported by N- and/or O-based ligands, some of the latter being among the most effective metal-based initiators known so far in the ROP of LA and ε-CL.2f,8 Anionic P,O ligands of type A are readily accessible and their coordination chemistry has been studied with various transition metal centres and the derived P,O metal complexes have been widely studied as olefin oligomerization/polymerization catalysts.10 Type A ligands seem well-suited for the synthesis of ligand-supported Zn(II) ROP initiators as they combine a hard phenolate donor, for an expected strong attachment to the Zn(II) metal center, with a soft phosphine donor that may form a more labile Zn–P coordination bond (yet stabilized through chelate formation) likely to dissociate under ROP conditions, a favorable feature for high catalytic activity. Here we report on the synthesis and structural characterization of phosphino-phenolate Zn(II) complexes and their subsequent use in the controlled and immortal ROP of LA, ε-CL and TMC. Taking advantage of the good ROP activity and control exhibited in homo-polymerisation, the production of PCL/PTMC-co-PLLA co-polymers and PTMC-co-PCL-co-PLLA ter-polymers was also achieved.
Results and discussion
Phosphinophenol protio-ligands (1·H and 2·H)
Phosphinophenol protio-ligands bearing methyl and tBu ortho-substituents, 1·H and 2·H respectively (Scheme 1), were used to sterically hinder the Zn(II) metal centre upon complexation and thus disfavor excessive aggregation, a well-established tendency of Zn(II) alkyl/alkoxide compounds. The phosphines 1·H and 2·H were synthesized according to a literature procedure and their identity was confirmed by NMR analysis.11 In particular, the 31P{1H} NMR spectra of 1·H and 2·H both exhibit a singlet resonance at δ −28.9 and −31.7 ppm (CDCl3), respectively, in line with the presence of a triaryl phosphine group. In the case of 2·H, single crystals suitable for X-ray diffraction (XRD) studies could be grown from a saturated EtOAc–hexane mixture and subsequently analyzed through XRD, confirming its molecular structure (Fig. 1). In the solid state, the P–C–C–O chelating backbone is nearly planar as expected [O1–C2–C1–P1 torsion angle of 3.3(2)°] and the phosphorus center adopts a slightly distorted tetrahedral geometry with similar CPhenyl–P–CPhenyl and CPhenol–P–CPhenyl bond angles [103.33(8)–104.11(8)°]. The structural data for 2·H are similar to those of the other few XRD-characterized P,O phosphinophenol species.10c,12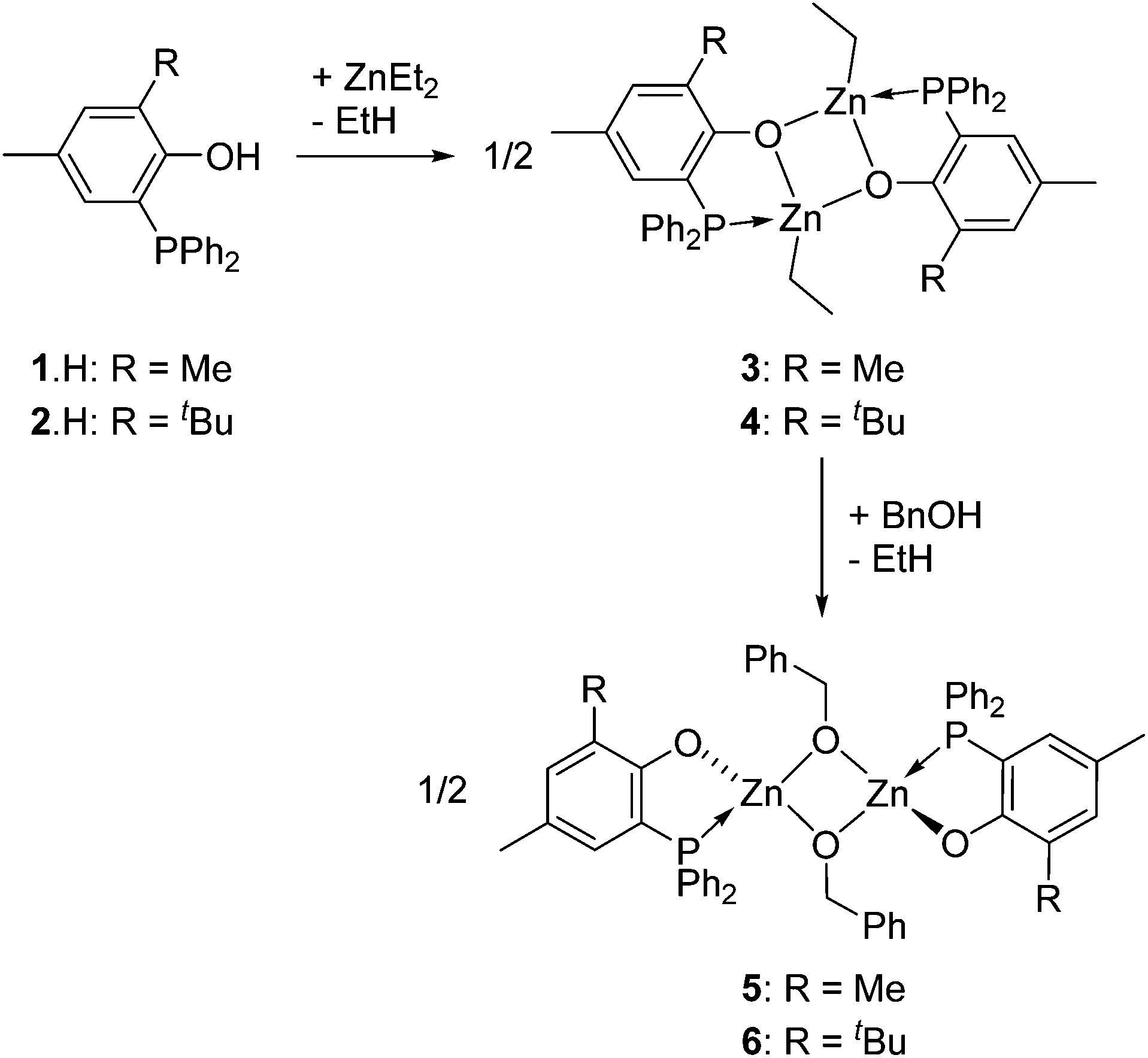 | ||
| Scheme 1 Synthesis of the phosphine-phenolate (P,O)-supported zinc(II) alkyl (3 and 4) and alkoxide (5 and 6) complexes. | ||
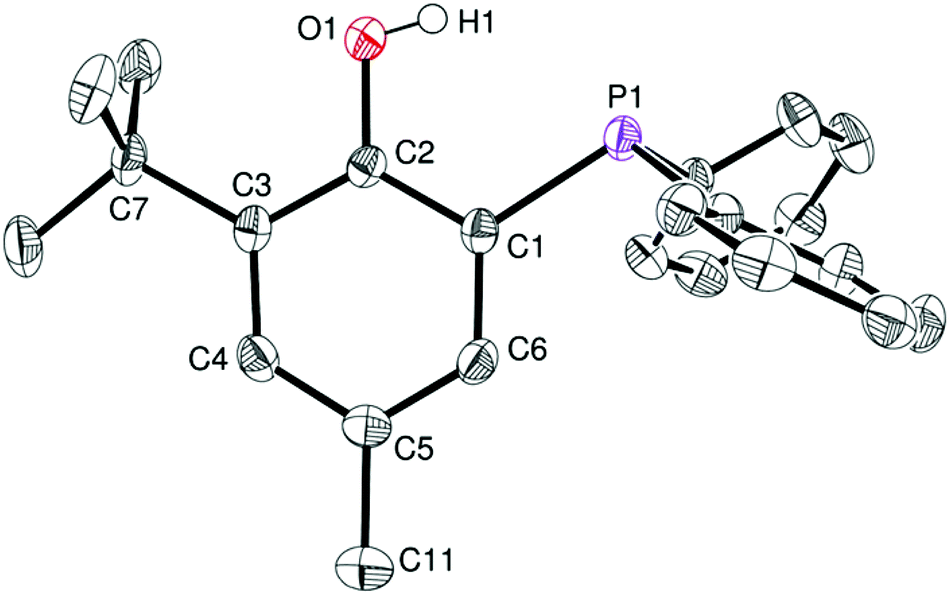 | ||
| Fig. 1 ORTEP drawing of the molecular structure of proligand 2·H. Thermal ellipsoids are set at the 50% probability level. Hydrogens are omitted for clarity except the crystallographically observed O–H1 hydrogen. Selected bond distances (Å): P1–C1 1.827(2), O1–C2 1.372(2), and angles (°): O1–C2–C3 118.1(2), P1–C1–C6 124.0 (1), P1–C1–C2 117.3(1). | ||
Heteroleptic phosphinophenolate Zn(II) alkyls/alkoxides 3–6
Access to the [(P,O)-ZnEt]-type Zn(II) ethyl complexes 3 and 4 (Scheme 1) was readily achieved by reaction of ZnEt2, acting both as a base and a metal source, with a stoichiometric amount of the corresponding ligand precursor (1·H and 2·H, respectively, Scheme 1). Analytically pure compounds 3 and 4 were isolated as colorless solids in good yields (82 and 86% yield, respectively) upon precipitation from the reaction solvent (CH2Cl2) and after a pentane wash of the collected solids. Both Zn(II) alkyl complexes are poorly soluble in common organic solvents (precluding 13C NMR analysis), suggesting that both species 3 and 4 form aggregates in solution. The 1H NMR spectra of 3 and 4 display the expected resonances for one ZnCH2CH3 moiety (δ −0.28 and 0.69 for 3; −0.05 and 0.74 for 4; Fig. S1 and S2, ESI†). The 31P{1H} NMR singlet signals for 3 and 4 [δ −30.3 (3) and −31.0 (4) in CDCl3] resonate at a similar chemical shift to that of the corresponding free proligands [δ −28.9 (1·H) and −31.7 (2·H) ppm], possibly reflecting a weak Zn(II)–P coordination bond due to the oxophilic character of the Zn(II) centers. In the case of complex 3, a dimeric [(P,O)-ZnEt]2 structure is proposed in solution on the basis of diffusion-ordered NMR spectroscopy (DOSY) analysis (CDCl3, room temp.), which yielded a hydrodynamic radius of 6.01 Å for 3 and thus an estimated molecular volume of 909 Å3 in solution (ESI – Fig. S9 and Table S2†). The latter matches well the estimated volume of dimer 3 in the solid state (936 Å3, see ESI – Fig. S14†).13 It is thus probable that species 3 adopts a dimeric structure in solution similar to that observed in the solid state (Fig. 2). The analogous NMR features for 3 and 4 suggest that both complexes adopt a comparable structure in solution.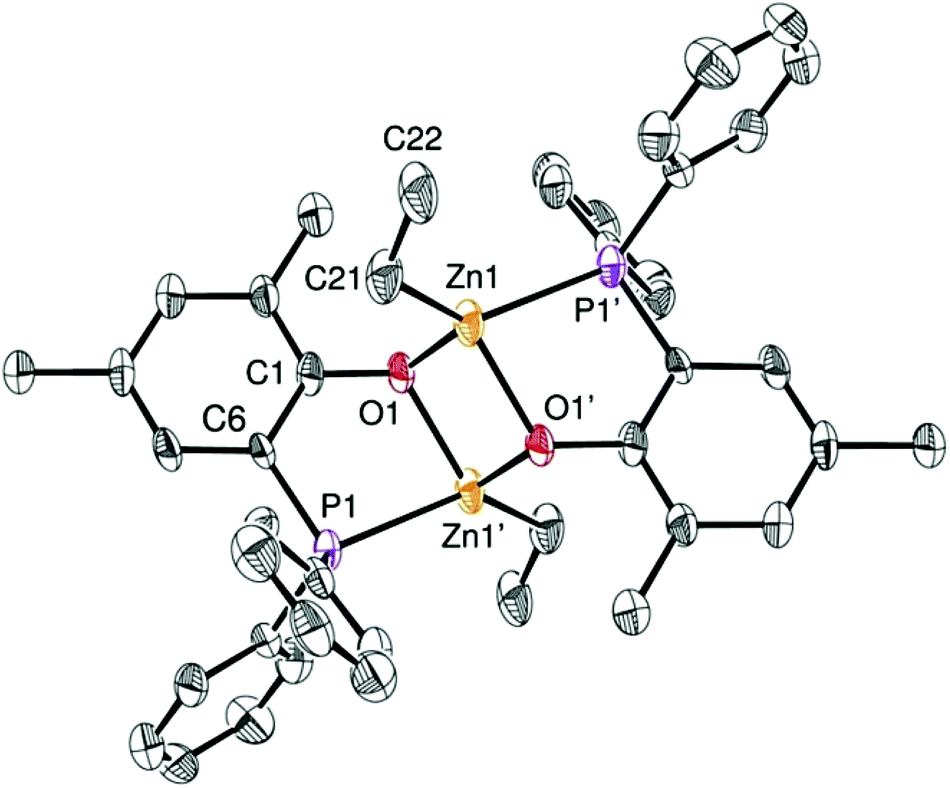 | ||
| Fig. 2 ORTEP drawing of the molecular structure of 3. Thermal ellipsoids are set at the 50% probability level. Hydrogen atoms are omitted for clarity. Selected bond distances (Å): Zn1–P1′ 2.458(3), Zn1–O1′ 2.069(5), Zn1–C21 1.985(8), Zn1–O1 2.058(6), O1–C1 1.358(6), P1–C6 1.796(7) and angles (°): O1–Zn1′–P1 80.31(1), O1–Zn1–C21 113.6(3), P1′–Zn1–C21 133.3(2), O1–Zn1–P1′ 103.8(1), O1′–Zn1–C21 130.0(3), O1–Zn1–O1′ 82.6(2). | ||
The solid state molecular structure of complex 3 could be unambiguously established through XRD studies (Fig. 2; ESI – Table S1†) and constitutes the first P,O-zinc alkyl complex to be structurally characterized.14 Complex 3 crystallizes as a centrosymmetric dimer with each Zn(II) center adopting a significantly distorted tetrahedral geometry. This is due to a rather acute P,O-ligand bite angle [O1–Zn1′–P1 = 80.31(1)°] and the geometrical constraints imposed upon dimer formation. For instance, the small O–Zn–O bond angles [82.6(2)°] of the Zn2O2 core are compensated for by rather large P1′–Zn1–C21 and O1′–Zn1–C21 bond angles [133.3(2)° and 130.0(3)°, respectively]. Each metal center bears a κ2-chelating P,O-phosphino phenolate ligand, a μ-O bridging phenolate connecting the two Zn(II) centers, and an ethyl group. The Zn–P and Zn–Calkyl bond lengths [2.458(3) Å and 1.985(8) Å, respectively] are in the expected ranges for related Zn–phosphine/phosphido compounds [Zn–P: 2.598(2)–2.390(1) Å; Zn–Calkyl: 2.147(2)–1.941(6) Å].14
The corresponding zinc alkoxide derivatives of the type [(P,O)Zn–OCH2Ph]2 (5 and 6, Scheme 1) were prepared in good yield (88–91%) via an alcoholysis reaction between the [(P,O)ZnEt]2 complexes 3 and 4 and 2 equiv. of BnOH at room temperature in CH2Cl2 (Scheme 1). Complexes 5 and 6 were isolated as colorless solids and their formulation was deduced from elemental analysis and NMR data (ESI – Fig. S3 and S4†). The 1H NMR data for these species agree with the presence of one Zn-OBn moiety per phosphinophenolate ligand and with overall centrosymmetric structures (Ci symmetry). In particular, the 1H NMR spectra only contain one set of signals for the P,O ligand while the presence of two 1H NMR resonances for the methylene benzyl group indicates inequivalent Zn-OCHH′Ph methylene protons in 5 and 6. These data are in line with the proposed dimeric structure for 5 and 6 (presumably through two μ-O-benzyloxide bridges) proposed in Scheme 1. DOSY NMR measurements support a dimeric structure for species 5 and 6. Hydrodynamic radii of 6.00 Å and 6.04 Å were estimated for species 5 and 6, respectively (CDCl3, room temp.) corresponding to calculated molecular volumes of 905 Å3 and 921 Å3 (see ESI – Fig. S10, S11 and Table S2†). In the case of species 5, the experimental value matches well with the calculated volume for a model molecule (923 Å3; estimated from a Chem 3D optimized geometry; ESI – Fig. S15 and S16†). Besides, the molecular volumes for 5 and 6 are close to that measured for the [(P,O)-ZnEt]2 dimer 3 (VDOSY = 909 Å3 and VX-ray = 936 Å3), whose molecular structure was XRD-established. Therefore, P,O-supported zinc alkyls (3 and 4) and alkoxides (5 and 6) likely display a similar level of aggregation in solution under the studied conditions.
Though heteroleptic species 5 is stable for days in solution (CD2Cl2, room temp.), the more sterically encumbered analogue 6 is unstable in CD2Cl2 and slowly converts over the course of several days to the corresponding homoleptic Zn species 8 (along with presumed [Zn(OBn)2]n-type aggregate species)15via the formation of dinuclear Zn species 6′ (Scheme 2), as deduced from 1H and 31P{1H} NMR monitoring experiments (31P{1H} NMR data: Fig. 3; 1H NMR data: ESI – Fig. S8B†). Based on 1H NMR data, 50% of complex 6 is converted to 6′ after 2 days. Complex 6 is completely consumed within 4 days with the presence of compound 6′ and homoleptic species 8 as the major product (in a 1/2 6′/8 ratio). The absence of species 8 after 2 days of reaction while it is the major product after 4 days strongly suggests that 8 is produced from dinuclear species 6′. A nearly quantitative formation of 8 is observed after 8 days.
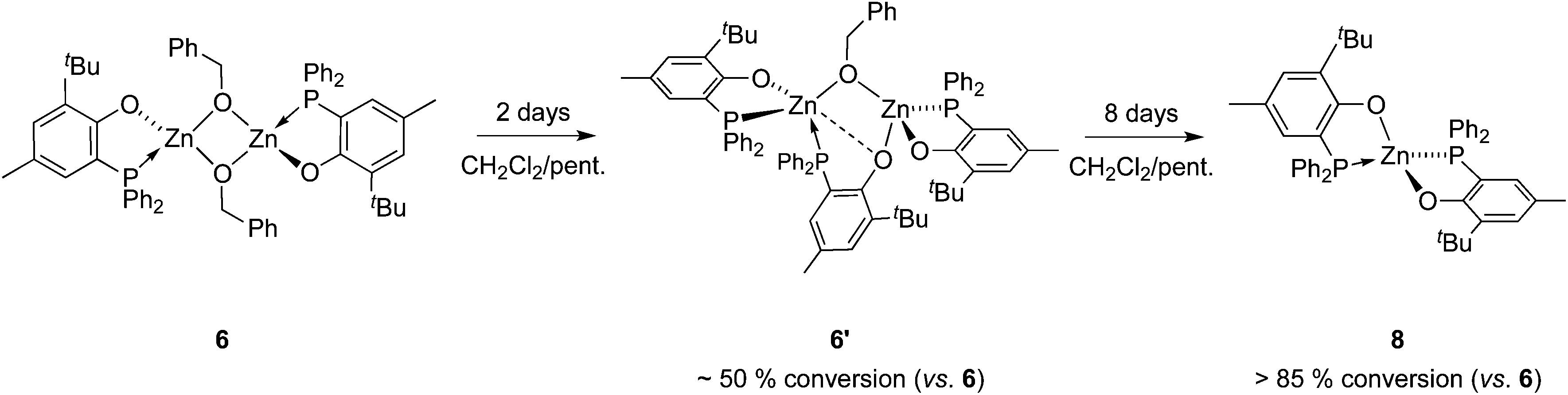 | ||
| Scheme 2 Conversion of heteroleptic species 6 to homoleptic species 8via the transient formation of 6′. | ||
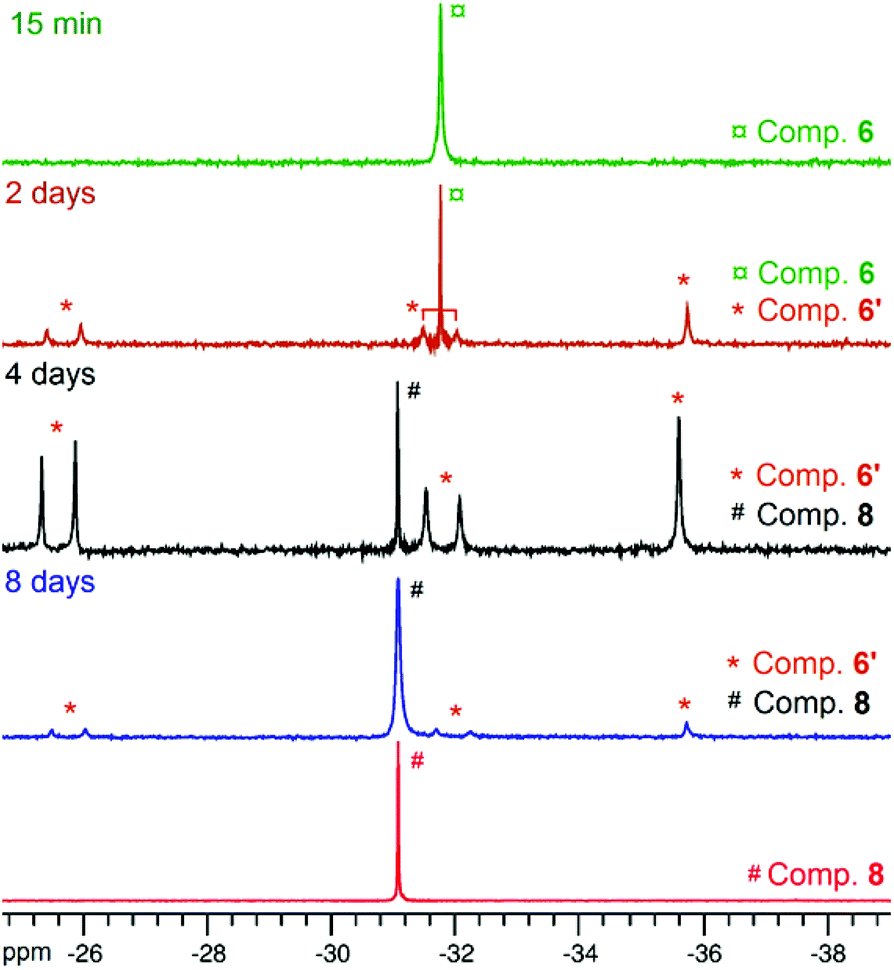 | ||
| Fig. 3 31P{1H} NMR (162 MHz, CD2Cl2, RT) monitoring of the conversion of heteroleptic species 6 to homoleptic 8via the formation of species 6′. Conditions: [6]0 = 10 mM, CD2Cl2, room temperature. For comparison, the 31P{1H} NMR spectrum of analytically pure compound 8 (bottom spectrum) prepared and isolated from the reaction of ZnEt2 and 2 equiv. of ligand 2·H is shown. | ||
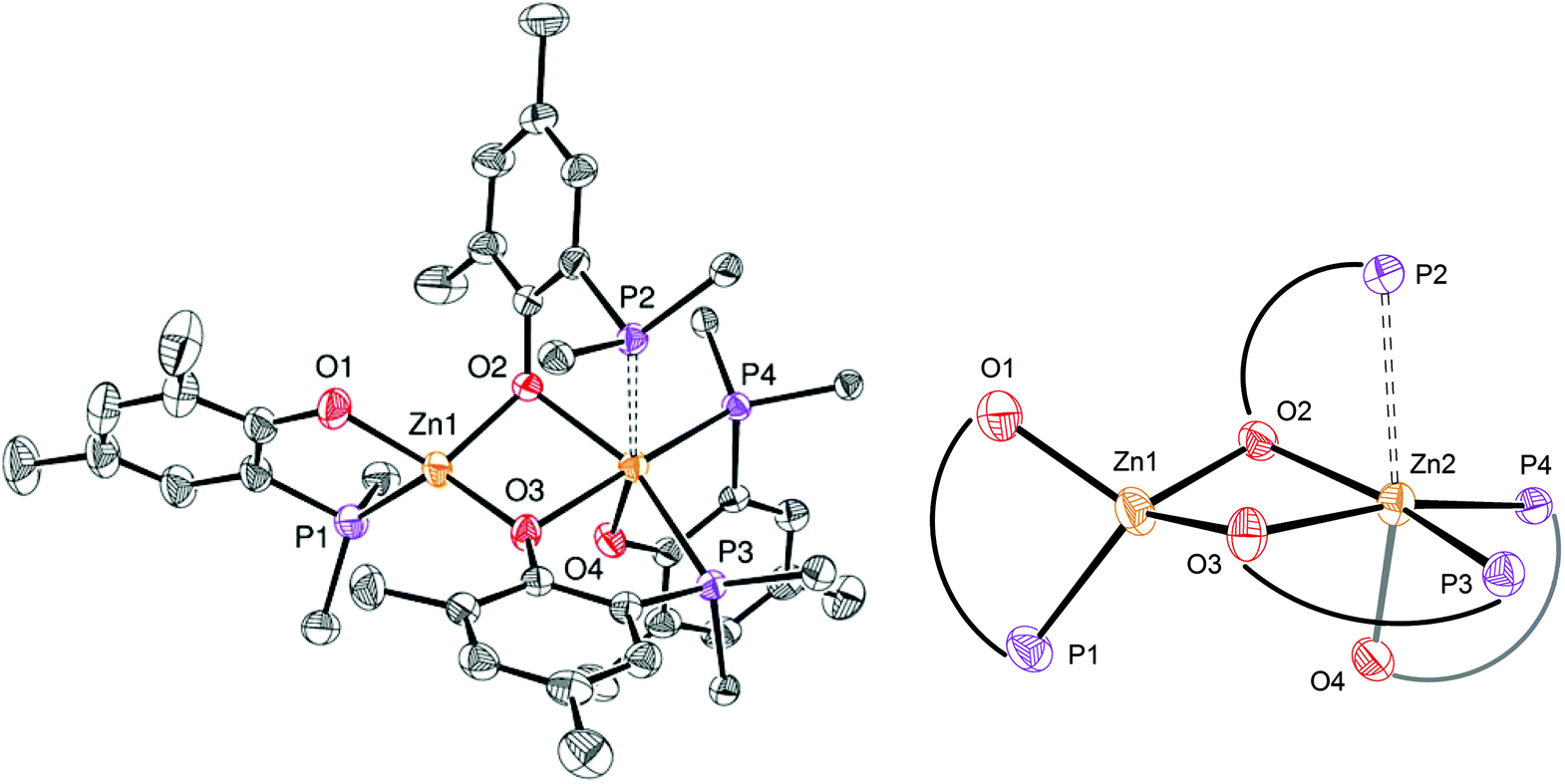
The identity of species 6′ and 8 was established through independent synthesis and characterization data (vide infra for the synthesis of compound 8). Analytically pure complex 6′ was isolated in 41% yield by crystallization of compound 6 (1/1 CH2Cl2–pentane, −35 °C, 2 days) allowing XRD, 1H and 31P{1H} NMR characterization. Note that, according to 1H NMR analysis, the remaining CH2Cl2–pentane mother liquor consisted of complex 8, a trace amount of species 6 and 6′ and [Zn(OBn)2]n species, in line with the conversion of 6 to 8 discussed above. No exploitable 13C{1H} NMR data for 6′ could however be obtained due to its low solubility in common organic solvents. In the solid state and as depicted in Fig. 4, species 6′ consists of a dinuclear Zn(II) species containing only one benzyloxide moiety for three P,O bidentate ligands (vs. a 1/1 ratio in the parent compound 6). The Zn(II) centers in 6′ are connected to one another through a μ-O-benzyloxide moiety and a μ–κ1:κ1-P,O bridging phosphinophenolate ligand. A close Zn2⋯O3 contact [2.465(2) Å] may also be noted with a distance well below the sum of the corresponding VdW radii (2.91 Å), yet too long to be considered as a Zn–O dative bond (typically ranging from 2.10 to 2.25 Å).16 Nevertheless, such a contact possibly explains the observed trigonal pyramidal geometry at Zn(2), with the P2, P3 and O4 atoms occupying the equatorial positions (the central Zn2 atom lies only 0.316 Å outside the P2–P3–O4 mean plane) while the O2 and O3 atoms (from the κ2-P,O- and μ–κ1:κ1-P,O ligands, respectively) are in the apical positions. In contrast, the Zn1 centre may be described as four-coordinate with a distorted tetrahedral geometry due to the bidentate κ2-P,O-chelation of a P,O ligand [bite angle = 84.25(7)°] and the bonding with the μ–κ1:κ1-P,O ligand through O3. Overall, the bonding distances in 6′ are in the expected ranges with Zn–P bond lengths [2.370(9)–2.411(9) Å] comparable to those found in related Zn phosphino complexes [Zn–P: 2.322(1)–2.678(2) Å].16,17 Also, the terminal and bridging Zn–O bond distances in complex 6′ are in line with those found in the literature [Zn–OTerminal: 2.336(5)–1.844(4) Å; Zn–(μ-Oalkoxide)–Zn: 2.148(5)–1.976(5) Å].17
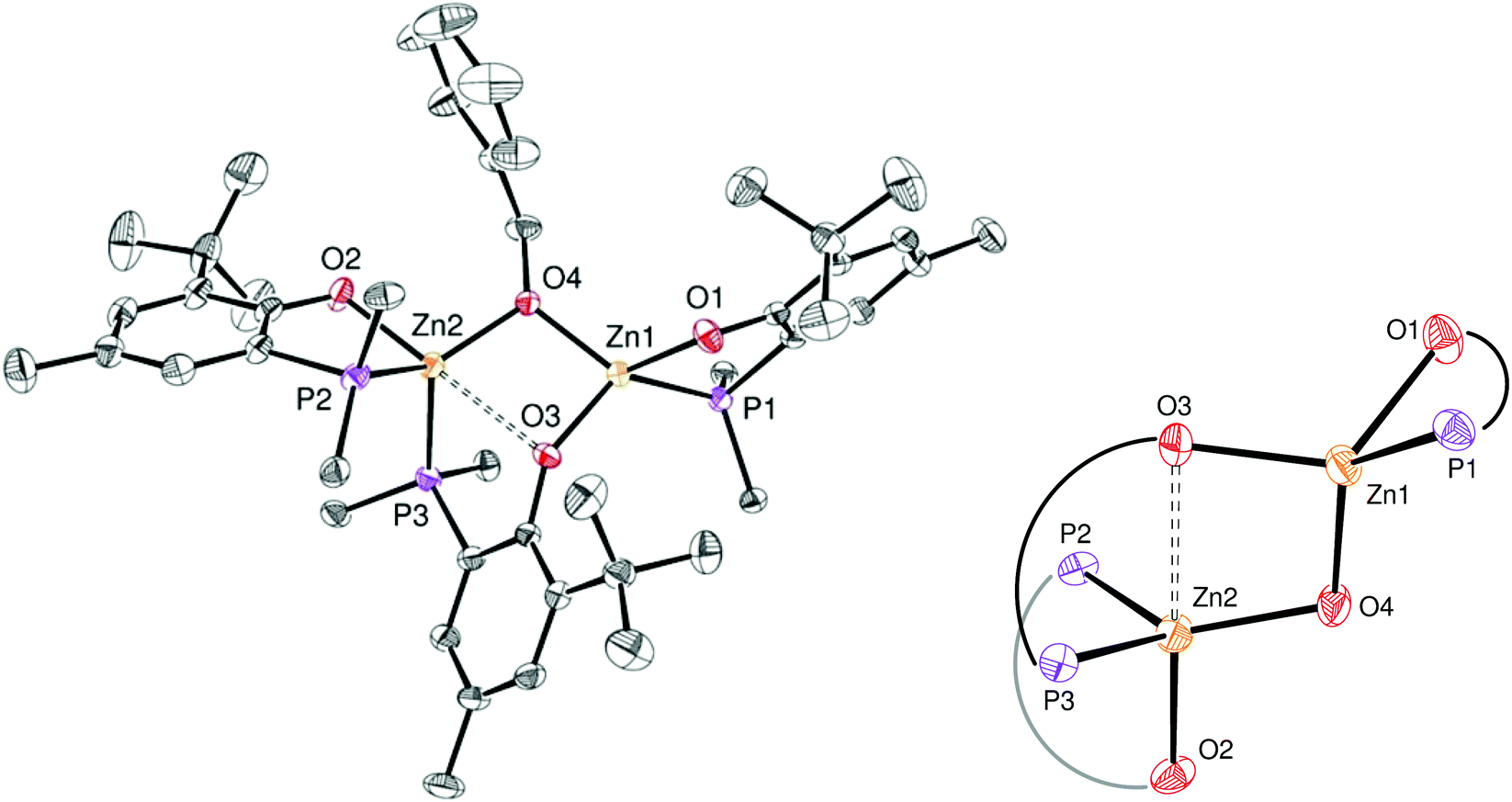 | ||
| Fig. 4 Left: ORTEP drawing of the molecular structure of 6′. Thermal ellipsoids are set at the 50% probability level. Hydrogen atoms are omitted and only the ipso-carbon of each P-phenyl ring is represented for clarity. Selected bond distances (Å): Zn1–P1 2.370(9), Zn1–O1 1.949(2), Zn1–O3 1.934(2), Zn1–O4 1.933(2), Zn2–P2 2.411(9), Zn2–O2 1.983(3), Zn2–P3 2.411(7), Zn2–O3 2.465(2), Zn2–O4 1.941(2) and angles (°): P1–Zn1–O1 84.25(7), O3–Zn1–O4 88.46(9), P1–Zn1–O3 124.9 (7), O1–Zn1–O4 115.9(9), P2–Zn2–O2 81.59(7), P2–Zn2–P3 116.8(3), P2–Zn2–O4 127.3(7), P3–Zn2–O4 109.9(7), O2–Zn2–O3 179.8(8), P3–Zn2–O3 71.27(5). Right: Zoom of the coordination spheres around Zn1 and Zn2. | ||
The NMR data for species 6′ agree with its solid-state structure being retained in CD2Cl2 solution at room temperature (ESI – Fig. S5A–C†). In particular, the 31P{1H} NMR (CD2Cl2) spectrum of 6′ features three sets of resonances (ESI – Fig. S5C†): a sharp singlet (δ −35.6 ppm; P1 in Chart 2) and two doublets assigned to the two P atoms coordinated on the same Zn(II) center and cis to one another (δ −31.8 ppm, 2JP,P = 87 Hz; −25.6 ppm, P2 and P3 in Chart 2). The 1H NMR spectrum of species 6′ is consistent with the presence of one benzyloxide group for three phosphinophenolate entities.
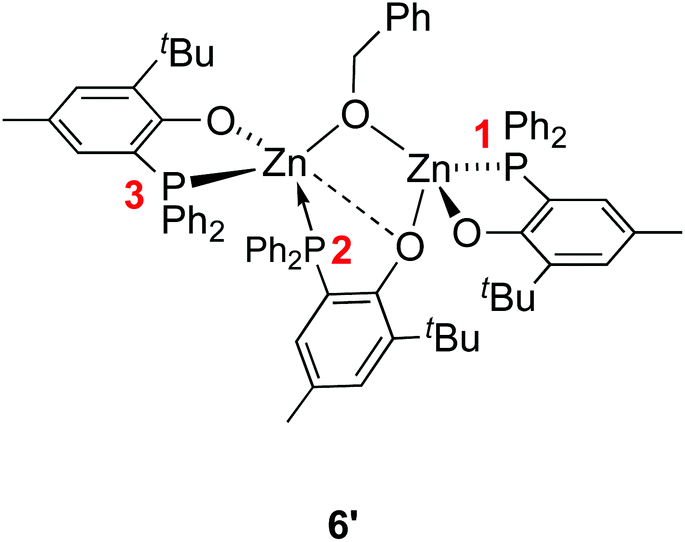 | ||
| Chart 2 Labelling of the phosphorus atoms in the Zn(II) complex 6′. | ||
Homoleptic phosphinophenolate Zn(II) species 7 and 8
The synthesis of the homoleptic compounds [Zn(P,O)2] (7 and 8, Scheme 3) was also performed for their subsequent use in ROP catalysis as well-defined Lewis acids. Thus, the reaction of ZnEt2 with 2 equiv. of phosphinophenols 1·H and 2·H (CH2Cl2, room temp.) led to the quantitative formation of the corresponding bis-(phosphinophenolate) Zn(II) complexes 7 and 8, respectively, which were isolated in high yield as colorless solids. The structures of both species were deduced from NMR data, elemental analysis and, in the case of 7, through XRD studies.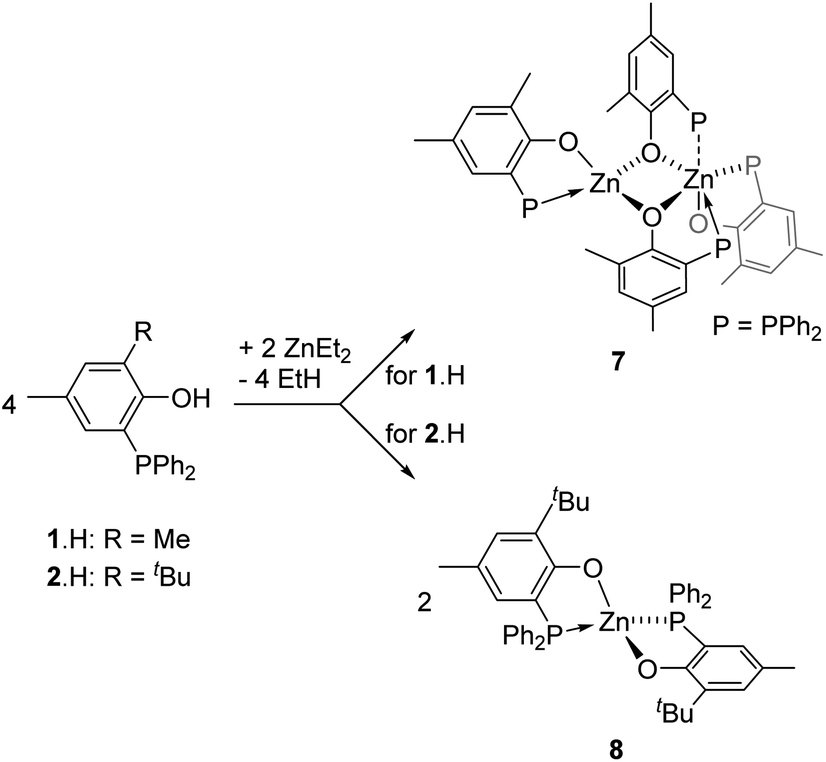 | ||
| Scheme 3 Synthesis of the homoleptic bis(phosphine-phenolate) (P,O)-supported zinc(II) complexes 7 and 8. | ||
In the solid state, as depicted in Fig. 5, the homoleptic Zn(II) complex 7 crystallizes as a dinuclear species with the two Zn(II) centres being connected via two μ-O-phenolate bridges. Though the formation of species 7 formally arises from the assembly of two [Zn(P,O)2] molecules, species 7 is not dimeric as reflected by the different coordination environments of the two Zn(II) centres. While Zn1 adopts a tetrahedral geometry similar to that observed in dimer [(P,O)-ZnEt]2 (3), the Zn2 metal centre lies in a slightly distorted octahedral environment due to the coordination of three bidentate P,O ligands. The three phosphorus ligands P2, P3 and P4 coordinate to Zn2 in a fac-fashion and the corresponding Zn–P bond distances [Zn2–P3 = 2.531(7), Zn2–P4 = 2.543(8), Zn2–P2 = 2.690(6) Å] are significantly longer than the Zn1–P1 bond distance [2.393(9) Å] and those observed in related species 3 and 6′. The Zn–P bond distances in 7 also lie in the upper range of Zn–P bond lengths [Zn–P: 2.322(1)–2.678(2) Å], certainly reflecting the electronic saturation of the Zn2 centre in 7.17 The assembly of two four-coordinate Zn species such as [Zn(P,O)2] entities may be expected to yield two penta-coordinate Zn(II) centres in the solid state: in the present case, the different coordination environment of Zn1 vs. Zn2 in the solid state structure of 7 may relate to the (small) preference of Zn(II) species for a four-coordinate tetrahedral environment.
 | ||
| Fig. 5 Left: ORTEP drawing of the molecular structure of 7. Thermal ellipsoids are set at the 50% probability level. Hydrogen atoms are omitted and only the ipso-carbons of the PPh2 rings are represented for clarity. Selected bond distances (Å) around Zn1: Zn1–P1 2.393(9), Zn1–O1 1.936(1), Zn1–O2 1.973(2), Zn1–O3 1.946(2) and around Zn2: Zn2–P2 2.690(6), Zn2–O2 2.116(1), Zn2–P3 2.531(7), Zn2–O3 2.201(2), Zn2–P4 2.543(8), Zn2–O4 2.005(2) and angles (°) around Zn1: P1–Zn1–O1 86.77(6), O2–Zn1–O3 87.52(7) and around Zn2: P2–Zn2–O2 73.72(5), P2–Zn2–P3 97.43(2), P3–Zn2–O4 94.68(5), O4–Zn2–O2 92.81(7), P4–Zn2–O3 168.05(5). Right: Zoom of the different coordination environment for Zn1 and Zn2. | ||
The 1H NMR data for complex 7 exhibit one set of broadened signals for the P,O ligands (ESI – Fig. S6A†). The 31P{1H} NMR spectrum for 7 contains three resonances: a sharp singlet (δ −30.3 ppm) and two broad resonances (δ −18.7 and −37.2 ppm; ESI – Fig. S6B†). To gain further insight, low temperature 1H and 31P{1H} NMR experiments were performed but the low solubility of species 7 (precipitation as the NMR sample was cooled down) precluded the obtainment of exploitable data. Nevertheless, DOSY NMR data for 7 suggest a comparable level of aggregation in solution (CD2Cl2, room temp.) and in the solid state. As shown in Fig. S12 (ESI),† the molecular volume of 7 in solution (VDOSY = 1358 Å3, hydrodynamic radius = 6.87 Å; ESI – Table S2†) is similar to that estimated from solid state data (VX-ray = 1303 Å3; ESI – Fig. S17†). The 1H, 13C{1H} and 31P{1H} NMR data for complex 8 (CD2Cl2, room temp.) exhibit one set of resonances for the Zn-coordinated P,O ligands and are in line with the proposed structure for 8 (ESI – Fig. S7A–C†). DOSY NMR data for 8 (CD2Cl2, room temp., hydrodynamic radius = 6.04 Å, VDOSY = 922 Å3, ESI – Fig. S13† and Table 2) are consistent with a monomeric structure for species 8: a geometrical optimization of a model molecule of 8 (ESI – Fig. S18†) led to a calculated volume (987 Å3; ESI – Fig. S19†), close to that experimentally determined in solution. Altogether, the solution data for species 7 and 8 at room temperature agree with a dinuclear structure for 7 while species 8 is a [Zn(P,O)2]-type mononuclear species, which is certainly due to a superior steric protection of the Zn(II) centre by the P,O bidentate ligand 2vs. 1.
ROP of rac-LA, ε-CL and TMC by species 5
The Zn(II) benzyloxide dimer 5 efficiently initiates rac-LA, ε-CL and TMC ROP. The results are summarized in Table 1. The combination of Zn alkoxide analogue 6 with lactide also resulted in the production of narrowly disperse and chain-length controlled PLA with a ROP efficiency similar to that observed with 5. However, the lack of stability of 6 in solution (vide supra) casts doubt on the nature of the ROP-active initiator(s) and the ROP results with species 6 are therefore not reported herein.| Run | Init. | Monomer | Zn/M/BnOH | Time (min) | Convb (%) |
M
n![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (corr.) c (corr.) |
M
nd (theo.) |
PDIe |
|---|---|---|---|---|---|---|---|---|
| a Conditions: CH2Cl2, [M]0 = 1 M, room temp., Mn values are given in g mol−1. b Determined by 1H NMR. c Determined by GPC using polystyrene standards and applying the appropriate correcting factor (0.58, 0.56 or 0.88 for PLA, PCL and PTMC, respectively).21 d Calculated according to the conversion (MLA = 144.13 g mol−1, MCL = 114.14 g mol−1, MTMC = 102.09 g mol−1). e Determined by GPC. f ROP run under bulk conditions, 130 °C. | ||||||||
| 1 | 5 | rac-LA | 1/100/0 | 120 | 97 | 13690 |
13980 |
1.02 |
| 2 | 5 | rac-LA | 1/500/4 | 450 | 99 | 14190 |
14270 |
1.02 |
| 3f | 5 | rac-LA | 1/100/0 | 20 | 97 | 50520 |
13980 |
1.63 |
| 4 | 5 | ε-CL | 1/100/0 | 120 | 99 | 10400 |
11300 |
1.03 |
| 5 | 5 | ε-CL | 1/500/4 | 450 | 99 | 13254 |
11300 |
1.11 |
| 6 | 5 | TMC | 1/100/0 | 120 | 87 | 8030 | 8880 | 1.13 |
| 7 | 5 | TMC | 1/500/4 | 450 | 99 | 14560 |
10110 |
1.16 |
| 8 | 7 | rac-LA | 1/100/1 | 180 | 51 | 4140 | 7350 | 1.06 |
| 9 | 8 | rac-LA | 1/100/1 | 180 | 83 | 6900 | 11950 |
1.05 |
Using 5 as a ROP initiator, the quantitative conversion of 100 equiv. rac-LA to extremely narrow disperse and atactic PLA may be achieved within 2 h at room temperature (1 < PDI < 1.05; entry 1, Table 1) as deduced from SEC and NMR data (ESI – Fig. S20†). Good agreement between the expected and experimental chain-lengths [Mn(theo) vs. Mn(corr)] is also observed, in line with a controlled ROP process and the formation of chain-length controlled PLA (Fig. 7). Additional data consistent with a well-behaved ROP process include a first-order dependence on rac-LA concentration (kobs = 0.0282 min−1, Fig. 6) and MALDI-TOF mass spectrometric data consistent with OBn-end-capped PLA materials (ESI – Fig. S23†). A coordination–insertion ROP mechanism is thus assumed for this system with the Zn-OBn group acting as the initiating moiety and, unlike what was earlier observed in related (P,O)Al-based ROP initiators,6a with the apparent non-involvement of the P,O ligand as an initiating group.18 A polymerization run under standard conditions (100 equiv. rac-LA, [M]0 = 1 M, room temp., 2 h) in THF afforded PLA with a 40% conversion (vs. 97% conv. in CH2Cl2), suggesting a monomer/solvent competition. Complex 5 was also found to quantitatively polymerize 100 equiv. of rac-LA under bulk conditions within 20 min (130 °C, no solvent; entry 3, Table 1), yet with substantial loss of PLA chain length control and a broader PDI. The ROP of rac-LA initiated by 5 was also carried out under immortal conditions in the presence of benzyl alcohol acting as an external chain-transfer agent, yielding the nearly quantitative conversion of 500 equiv. of rac-LA within 7.5 h to well-defined and narrow disperse PLA (entry 2, Table 1) as deduced from SEC data (ESI – Fig. S23†).19
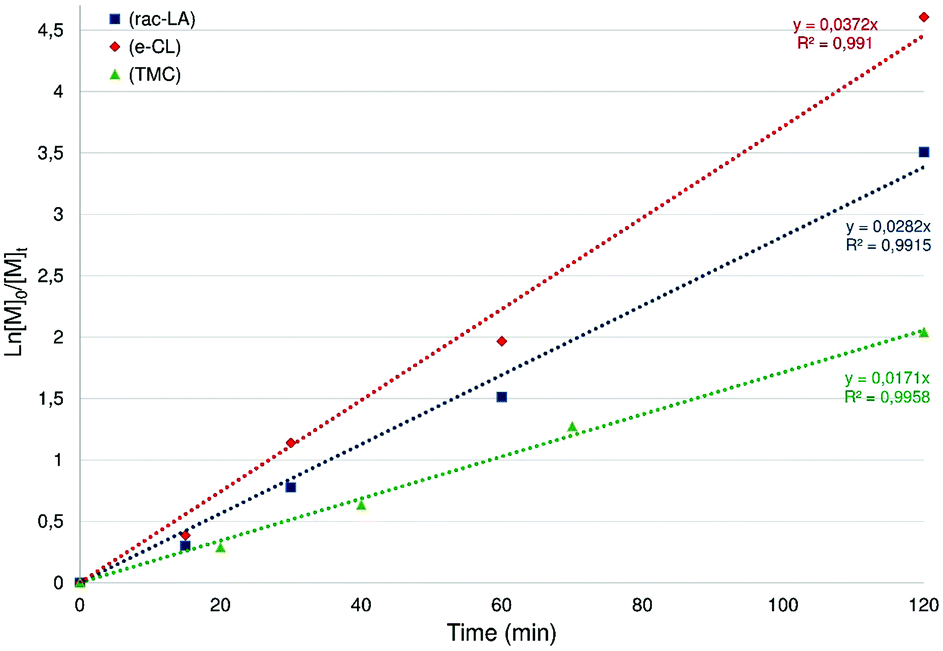 | ||
| Fig. 6 Semilogarithmic plots of rac-LA (blue), ε-CL (red) and TMC (green) conversion versus time for complex 5. Reaction conditions: [M]0/[Zn] = 100, [M]0 = 1 M, CH2Cl2, room temp. | ||
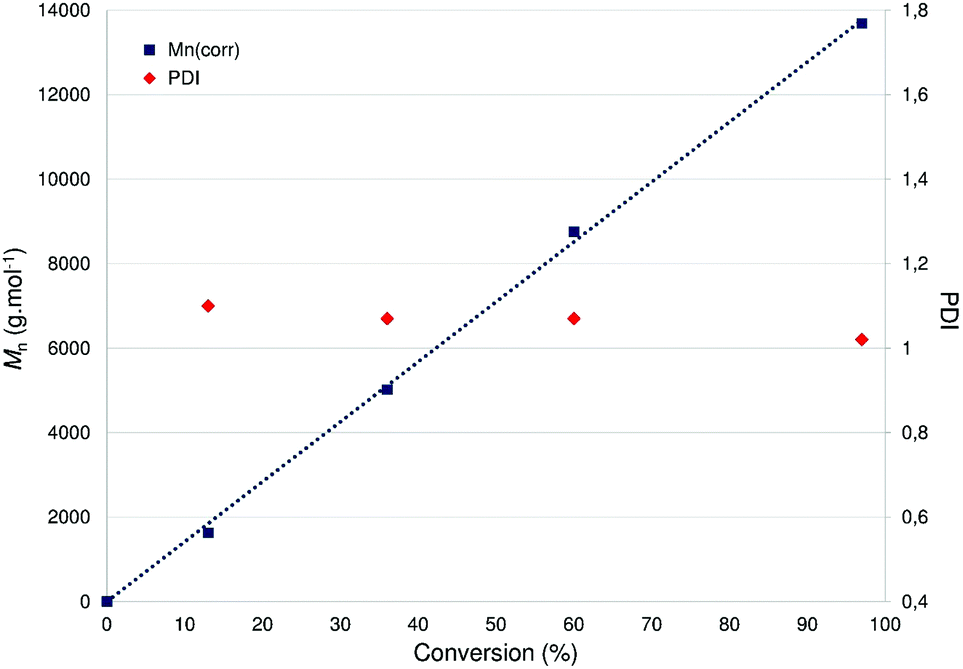 | ||
| Fig. 7 Linear dependence of Mn and PDI [Mw/Mn] of PLA versus monomer (rac-LA) conversion with 5 as a catalyst. Reaction conditions: [LA]0/[Zn] = 100, [LA]0 = 1 M, CH2Cl2, room temp. | ||
Likewise, species 5 also effectively initiates the controlled ROP of ε-CL to afford chain-length-controlled, narrow disperse and OBn-end-capped PCL (entries 4 and 5, Table 1), as deduced from SEC and MALDI-TOF analyses (ESI – Fig. S21 and S25†). Kinetic data for species 5 (Fig. 6, ESI – Fig. S28†) agree with a controlled ROP process proceeding slightly faster than with rac-LA (kobs = 0.0372 min−1).
The use of well-defined metal alkoxide complexes for the controlled ROP of TMC remains rare and, in particular, only a few Zn-based discrete species have been reported so far.4i,8c,20 The performance of Zn(II) alkoxide species 5 for the ROP of TMC was therefore evaluated (entries 6 and 7, Table 1). Compound 5 efficiently polymerizes 100 equiv. TMC at room temperature (2 h, CH2Cl2) with the high yield production of well-defined and OBn-end-capped PTMC material. All data (SEC, MALDI-TOF, kinetic studies: Fig. 6 and ESI – Fig. S22, S26, S29†) agree with controlled polymerization reactions under living and immortal conditions. It may be noted that the ROP of TMC initiated by species 5 is slower than those of rac-LA and ε-CL (kobs = 0.0171 min−1, Fig. 6).
ROP of rac-LA by the Zn(II) species 7 and 8
The homoleptic Zn(II) species 7 and 8 were also tested for lactide ROP activity. Both 7 and 8 are inactive in the ROP of rac-LA under the standard conditions used for ROP initiator 5 (100 equiv., CH2Cl2, room temp., 2 h), further confirming that the Zn-bonded P,O ligand is not an effective initiating group in these systems. However, the combination of an external nucleophile such as BnOH and compound 7/8 initiates the ROP of rac-LA at room temperature to afford narrow disperse and slightly heterotactic-enriched PLA material (Pr = 0.58 and 0.64 for 7 and 8, respectively), albeit with lower Mn values than expected (entries 8 and 9, Table 1). The 7/8 and BnOH initiating mixture is less active than discrete Zn alkoxide species 5 with 51%/83% consumption of 100 equiv. rac-LA after 3 h of reaction (entries 8 and 9 vs. 1, Table 1). Kinetic data for 7/BnOH and 8/BnOH (ESI – Fig. S30†) are consistent with a pseudo-first order reaction relative to monomer and with the ROP mediated by 8/BnOH proceeding roughly twice faster than that by 7/BnOH (kobs = 0.0099 vs. 0.0041 min−1). Such a difference in ROP activity between 7 and 8 is certainly related to the more reactive/accessible Zn centre in mononuclear species 8 (vs. in dinuclear species 7) for monomer activation. MALDI-TOF spectrometric analysis of a PLA sample produced via ROP of rac-LA with a 7/BnOH mixture agrees with the exclusive formation of OBn-end-capped PLA material with equally-separated peaks by 144 u.a. (ESI – Fig. S27†). This, together with the absence of a reaction between species 7 or 8 and 1 equiv. of BnOH as monitored through a NMR scale reaction (CD2Cl2, 2 h, room temp.), rules out a ROP proceeding via a coordination–insertion mechanism from an in situ generated Zn-OBn initiator. It seems likely the 7/8 and BnOH two-component mixture polymerizes lactide via an “activated monomer” mechanism with the Zn compounds 7/8 acting as a well-defined Lewis acid for monomer activation and BnOH as an external nucleophile source.18 The ROP initiation then occurs via a nucleophilic attack of BnOH at a Zn(II)-activated lactide.Co- and ter-polymerization of L-LA, ε-CL and TMC by species 5
The excellent ROP control and activity in the homo-polymerization of rac-LA, ε-CL and TMC of compound 5 were further exploited to access diblock PCL-b-PLLA and PTMC-b-PLLA co-polymers and triblock PTMC-b-PCL-b-PLLA ter-polymers via sequential ROP. Such PLA-based co- and ter-polymers may improve and tune the properties of PLA.22 For instance, the brittleness of PLLA (the commercial form of PLA) may be improved upon incorporation of a softer and flexible PTMC segment, as in PTMC-b-PLLA.Species 5 mediates the sequential ROP of TMC and/or PCL and L-LA to produce in high conversion the corresponding narrow disperse co- and ter-polymers as determined from SEC and NMR data (ESI – Fig. S31–34†). The results are summarized in Table 2. The sequential additions of the desired monomer were carefully NMR monitored to ensure complete or nearly complete consumption prior to the addition of the subsequent monomer. Note that the order of monomer addition is crucial for these ROPs to proceed: when L-LA was first polymerized, the subsequent ROP of TMC or ε-CL did not proceed (CH2Cl2, room temp.). The in situ generated Zn-PLLA chains somehow disfavor PCL and PTMC chain growth and thus L-LA was added last in all ROP runs. Though little is known about the reactivity of TMC vs. cyclic esters under co-polymerization conditions, the present observations with CL and LA are in line with various metal-mediated CL/LA co-polymerization studies where the greater reactivity of LA vs. CL under co-polymerization conditions is well-established.23
| Entry | Init. | Zn/TMC/ε-CL/L-LA | Time (h) | Convb (%) |
M
nc (GPC) |
M
nd (corr.) |
M
ne (theo.) |
PDIf | ||
|---|---|---|---|---|---|---|---|---|---|---|
| TMC | ε-CL | L-LA | ||||||||
| a Conditions: CH2Cl2, [M]0 = 1 M, room temp., Mn values are given in g mol−1. b Determined by 1H NMR. c Determined by GPC using polystyrene standards. d Determined by GPC using polystyrene standards and applying the appropriate correcting factor for the 1H NMR determined (PLA, PCL and PTMC fractions: 0.58, 0.56 and 0.88, respectively)21 according to the conversion of each monomer. e Calculated according to the conversion of each monomer. f Determined by GPC. | ||||||||||
| 1 | 5 | 1/100/0/100 | 2 | — | 4 | 94 | 63770 |
41570 |
23270 |
1.09 |
| 2 | 5 | 1/0/100/100 | — | 2.5 | 4 | 87 | 54050 |
30940 |
22830 |
1.05 |
| 3 | 5 | 1/100/100/100 | 2.5 | 3 | 4 | 97 | 87790 |
54940 |
35070 |
1.13 |
Conclusion
The coordination chemistry of bidentate P,O anionic ligands to Zn(II) allowed access through alkane elimination reactions to structurally diverse (P,O)-Zn species ranging from dimeric Zn(II) alkyl/alkoxide species (3–6) to homoleptic complexes of the type [Zn(P,O)2] (7 and 8). As deduced from combined solid state and solution structural data, the levels of aggregation observed in the solid state (in favor of dinuclear species) and in solution are similar. The use of sterically bulky P,O ligands such as 2·H reduces aggregation in the homoleptic series (mononuclear 8vs. dinuclear 7). Steric hindrance seems to be of importance in the lack of stability of heteroleptic Zn(II) alkoxide complex 6 (to yield homoleptic species 8): as a comparison, the less encumbered analogue 5 is stable under identical conditions. The lability of the Zn–phosphine bond may also favor ligand exchange reactions yielding homoleptic species 8 from [(P,O)Zn-OBn]2 Zn(II) alkoxide 6. When compared to the present [(P,O)ZnOR]2 systems, [(LX)Zn-OBn]2 species (LX = N-/O-based anionic ligand) are typically much more robust.2f Nevertheless, the Zn(II) alkoxide species 5 is a well-behaved ROP initiator of LA, ε-CL and TMC with the production of narrow disperse materials under living and immortal conditions, with catalytic activities comparing well with the majority of Zn(II)-based ROP species thus far developed. Interestingly, the homoleptic species [Zn(P,O)2] 7 and 8 may act as discrete ligand-supported Zn(II) Lewis acids capable of promoting the ROP of lactide in the presence of an external nucleophile such as BnOH. The lability of the Zn–phosphine bonds in 7 and 8 certainly facilitates monomer activation under ROP conditions. Future studies with 7 and 8/alcohol bi-component mixtures may focus on the addition of a Brønsted base for activation of the initiating/propagating alcohol (dual approach of catalytic ROP) for higher performance ROP catalysts.24Experimental section
General procedures
All experiments were performed under N2 using standard Schlenk techniques or in a nitrogen-filled glovebox. Toluene and pentane were collected after passing through drying columns in a solvent purification system and stored over activated molecular sieves (4 Å) for 24 h in a glovebox before use. THF was distilled over the Na–benzophenone complex and stored over activated molecular sieves (4 Å) for 24 h in a glovebox before use. CH2Cl2, CD2Cl2 and CDCl3 were distilled from CaH2, degassed under a N2 flow, and stored over activated molecular sieves (4 Å) in a glovebox before use. Anhydrous BnOH (99.8%) was purchased from Aldrich and stored over activated molecular sieves (4 Å) for 24 h in a glovebox before use. All deuterated solvents were obtained from Eurisotop (Groupe CEA, Saclay, France). rac-Lactide and L-Lactide (98% purity) were purchased from Aldrich and sublimed once before use. Trimethylene carbonate (purchased from Boehringer) was recrystallized twice from dry Et2O and ε-caprolactone was distilled from CaH2 prior to use. All other chemicals were purchased from Aldrich and were used as received. The NMR spectra were recorded on Bruker AC 300, 400 or 500 MHz NMR spectrometers with Teflon-valved J. Young NMR tubes at ambient temperature unless otherwise specified. 1H and 13C chemical shifts were determined by reference to the residual 1H and 13C solvent peaks. The diffusion-ordered NMR spectroscopy (DOSY) experiments were performed with a Bruker 600 MHz NMR spectrometer. Elemental analyses for all compounds were performed at the Service de Microanalyse of the Université de Strasbourg (Strasbourg, France). GPC analyses were performed on a system equipped with a Shimadzu RID10A refractive index detector with HPLC grade THF as an eluent (with molecular masses and PDIs calculated using polystyrene standards). These were adjusted with appropriate correction factors for the Mn values. MALDI-TOF-MS analyses were performed at the Service de Spectrométrie de Masse de l'Institut de Chimie de Strasbourg and run in a positive mode: samples were prepared by mixing a solution of the polymers in CH2Cl2 (0.5 mg 100 mL−1); 2,5-dihydroxybenzoic acid was used as the matrix in a volume ratio of 5:1. Proligands 1·H and 2·H were prepared according to literature methods.11
Synthesis of [(P,O)ZnEt]2 complexes 3 and 4
Synthesis of [(P,O)Zn(OBn)]2 complexes 5 and 6 and of the [(2)2Zn2(μ-OBn)(μ-2)] dinuclear species 6′
Synthesis of [Zn(P,O)2] complexes 7 and 8
General procedure for the ROP of LA, ε-CL and TMC by species 5, 7 and 8
In a glovebox, the initiator was charged in a vial equipped with a Teflon™-tight screw-cap and a monomer solution ([M]0 = 1 M, THF or CH2Cl2) was added via a syringe all at once. The solution was vigorously stirred for the appropriate time and at room temperature. When the desired time was reached, aliquots were taken and analyzed by 1H NMR spectroscopy to estimate the conversion. The reaction mixture was exposed to air and volatiles removed under vacuum; the resulting solid was then washed several times with MeOH, dried in vacuo until constant weight and subsequently analyzed by 1H NMR and SEC. In some cases, a MALDI-TOF-MS analysis was performed.For ROPs under immortal conditions with complex 5, an identical procedure to that above was used but with the initial addition of a monomer solution ([M]0 = 1 M, CH2Cl2) containing the desired amount of benzylic alcohol (BnOH) onto complex 5.
For sequential co- and ter-polymerization runs, a similar general procedure was followed with an addition of the incoming monomer after complete or nearly complete consumption of the previous monomer (as monitored by 1H NMR).
Acknowledgements
We thank Dr Lydia Brelot and Corinne Bailly for X-ray diffraction studies. We are also grateful to the Fundação para a Ciência e a Tecnologia (FCT), Portugal for funding the project PTDC/QUI-QUI/099873/2008 and fellowships SFRH/BPD/73253/2010 (C.F.) and SFRH/BPD/44262/2008 (V.R.). The University of Strasbourg and the CNRS are gratefully acknowledged for financial support.References
- (a) M. S. Lindblad, Y. Liu, A.-C. Albertsson, E. Ranucci and S. Karlsson, Adv. Polym. Sci., 2002, 157, 139 CrossRef CAS; (b) M. Vert, Biomacromolecules, 2005, 6, 538 CrossRef CAS PubMed; (c) L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762 CrossRef CAS PubMed; (d) E. S. Place, J. H. Georges, C. K. Williams and M. M. Stevens, Chem. Soc. Rev., 2009, 38, 1139 RSC.
- Selected reviews: (a) B. J. O'Keefe, M. A. Hillmeyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215 RSC; (b) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147 CrossRef CAS PubMed; (c) J. Wu, T.-L. Yu, C.-T. Chen and C.-C. Lin, Coord. Chem. Rev., 2006, 205, 602 CrossRef PubMed; (d) A. P. Dove, Chem. Commun., 2008, 6446 RSC; (e) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11 CrossRef CAS; (f) C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832 RSC; (g) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801 RSC; (h) J.-C. Buffet and J. Okuda, Polym. Chem., 2011, 2, 2758 RSC; (i) A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007 RSC; (j) S. Dagorne, M. Normand, E. Kirillov and J.-F. Carpentier, Coord. Chem. Rev., 2013, 257, 1869 CrossRef CAS PubMed.
- (a) P. R. Gruber, E. S. Hall, J. J. Kolstad, M. L. Iwen, R. D. Benson and R. L. Borchardt (Cargill Incorporated), US Patent, 5 258 488, 1993 Search PubMed; (b) P. R. Gruber, J. J. Kolstad, E. S. Hall, R. S. Eichen Conn and C. M. Ryan (Cargill Incorporated), US Patent, 6 143 863, 2000 Search PubMed.
- For recent and representative examples of metal-mediated ROPs of cyclic esters/carbonates, see: (a) A. Pilone, K. Press, I. Goldberg, M. Kol, M. Mazzeo and M. Lamberti, J. Am. Chem. Soc., 2014, 136, 2940 CrossRef CAS PubMed; (b) A. Meduri, T. Fuoco, M. Lamberti, C. Pellechia and D. Pappalardo, Macromolecules, 2014, 47, 534 CrossRef CAS; (c) M. D. Jones, S. L. Hancock, P. McKeown, P. Schäfer, A. Buchard, L. H. Thomas, M. F. Mahon and J. P. Lowe, Chem. Commun., 2014, 50, 15967 RSC; (d) N. Maudoux, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Organometallics, 2014, 33, 5630 CrossRef; (e) J. S. Klitzke, T. Roisnel, E. Kirillov, O. de L. Casagrande Jr. and J.-F. Carpentier, Organometallics, 2014, 33, 5693 CrossRef CAS; (f) Y. Huang, W. Wang, C.-C. Lin, M. P. Blake, L. Clark, A. D. Schwarz and P. Mountford, Dalton Trans., 2013, 42, 9313 RSC; (g) R. A. Collins, J. Unruangsri and P. Mountford, Dalton Trans., 2013, 42, 759 RSC; (h) R. L. Webster, N. Noroozi, S. G. Hatzikiriakos, J. A. Thomson and L. L. Schafer, Chem. Commun., 2013, 49, 57 RSC; (i) F. Hild, N. Neehaul, F. Bier, M. Wirsum, C. Gourlaouen and S. Dagorne, Organometallics, 2013, 32, 587 CrossRef CAS; (j) C. Bakewell, A. J. White, N. J. Long and C. K. Williams, Inorg. Chem., 2013, 52, 12561 CrossRef CAS PubMed; (k) V. Balasanthiran, M. H. Chisholm, C. B. Durr and J. C. Gallucci, Dalton Trans., 2013, 42, 11234 RSC; (l) I. Yu, A. Acosta-Ramirez and P. Mehrkhodavanti, J. Am. Chem. Soc., 2012, 134, 2057 CrossRef PubMed; (m) C. Bakewell, T.-P.-A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 2057 CrossRef PubMed; (n) P. Horeglad, G. Szczepaniak, M. Dranka and J. Zachara, Chem. Commun., 2012, 48, 1171 RSC; (o) C. Romain, B. Heinrich, S. Bellemin-Laponnaz and S. Dagorne, Chem. Commun., 2012, 48, 2213 RSC; (p) L. Azor, C. Bailly, L. Brelot, M. Henry, P. Mobian and S. Dagorne, Inorg. Chem., 2012, 51, 10876 CrossRef CAS PubMed; (q) E. L. Whitelaw, G. Loraine, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 11469 RSC; (r) C. Y. Li, C. Y. Tsai, C. H. Lin and B. T. Ko, Dalton Trans., 2011, 40, 1880 RSC; (s) A. Otero, A. Lara-Sánchez, J. Fernández-Baeza, C. Alonso-Moreno, J. A. Castro-Osma, I. Márquez-Segovia, L. F. Sánchez-Barba, A. M. Rodríguez and J. C. Garcia-Martinez, Organometallics, 2011, 30, 1507 CrossRef CAS; (t) D. J. Darensbourg, O. Karroonnirun and S. J. Wilson, Inorg. Chem., 2011, 50, 6775 CrossRef CAS PubMed; (u) M. P. Blake, A. D. Schwarz and P. Mountford, Organometallics, 2011, 30, 1202 CrossRef CAS; (v) W.-H. Sun, M. Shen, W. Zhang, W. Huang, S. Liu and C. Redshaw, Dalton Trans., 2011, 40, 2645 RSC; (w) M. Shen, W. Huang, W. Zhang, X. Hao, W.-H. Sun and C. Redshaw, Dalton Trans., 2010, 39, 9912 RSC; (x) M. Bouyahyi, T. Roisnel and J.-F. Carpentier, Organometallics, 2010, 29, 491 CrossRef CAS; (y) F. Hild, P. Haquette, L. Brelot and S. Dagorne, Dalton Trans., 2010, 39, 533 RSC; (z) A. D. Schwarz, Z. Y. Chu and P. Mountford, Organometallics, 2010, 29, 1246 CrossRef CAS.
- (a) D. C. H. Oakes, B. S. Kimberley, V. C. Gibson, D. J. Jones, A. J. P. White and D. J. Williams, Chem. Commun., 2004, 2174 RSC; (b) L. Lavanant, A. Silvestru, A. Faucheux, L. Toupet, R. F. Jordan and J.-F. Carpentier, Organometallics, 2005, 24, 5604 CrossRef CAS; (c) C. Wang, Z. Ma, X.-L. Sun, Y. Gao, Y.-H. Guo, Y. Tang and L.-P. Shi, Organometallics, 2006, 25, 3259 CrossRef CAS; (d) R. J. Long, V. C. Gibson and A. J. P. White, Organometallics, 2008, 27, 235 CrossRef CAS.
- (a) M. Haddad, M. Laghzaoui, R. Welter and S. Dagorne, Organometallics, 2009, 28, 4584 CrossRef CAS; (b) M. Lamberti, I. D'Auria, M. Mazzeo, S. Milione, V. Bertolasi and D. Pappalardo, Organometallics, 2012, 31, 5551 CrossRef CAS; (c) A. Pilone, M. Lamberti, M. Mazzeo, S. Milione and C. Pellechia, Dalton Trans., 2013, 42, 13036 RSC.
- For a recent overview on the use of Al(III) species in the ROP of cyclic polar monomers, see: S. Dagorne and C. Fliedel, Top. Organomet. Chem., 2013, 41, 125 CrossRef CAS.
- For recent examples of Zn(II) complexes for the controlled ROP cyclic esters/carbonates, see: (a) Z. M. Bo, M. Wang, H. Xie, P. Li, L. Li, S. Li and D. Cui, Chem. Commun., 2014, 50, 11411 RSC; (b) C. Fliedel, D. Vila-Viçosa, M. J. Calhorda, S. Dagorne and T. Avilés, ChemCatChem, 2014, 6, 1357 CrossRef CAS; (c) C. Fliedel, S. Mameri, S. Dagorne and T. Avilés, Appl. Organomet. Chem., 2014, 28, 504 CrossRef CAS; (d) H. Wang and H. Ma, Chem. Commun., 2013, 49, 8686 RSC; (e) G. Schnee, C. Fliedel, T. Avilés and S. Dagorne, Eur. J. Inorg. Chem., 2013, 3699 CrossRef CAS; (f) C. Romain, V. Rosa, C. Fliedel, F. Bier, F. Hild, R. Welter, S. Dagorne and T. Avilés, Dalton Trans., 2012, 41, 3377 RSC; (g) V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2011, 40, 523 RSC.
- (a) I. D'Auria, M. Lamberti, M. Mazzeo, S. Milione, G. Roviello and C. Pellechia, Chem. – Eur. J., 2012, 18, 2349 CrossRef PubMed; (b) L.-C. Liang, W.-Y. Lee, T.-L. Tsai, Y.-L. Hsu and T.-Y. Lee, Dalton Trans., 2010, 39, 8748 RSC.
- Selected examples: (a) P. Tao, H.-L. Mu, J.-Y. Liu and Y.-S. Li, Organometallics, 2013, 32, 4805 CrossRef CAS; (b) R. J. Long, V. C. Gibson, A. J. P. White and D. J. Williams, Inorg. Chem., 2006, 45, 511 CrossRef CAS PubMed; (c) J. Heinicke, M. He, A. Dal, H.-F. Klein, O. Hetche, W. Keim, U. Flörke and H. J. Haupt, Eur. J. Inorg. Chem., 2000, 431 CrossRef CAS; (d) J. Pietsch, P. Braunstein and Y. Chauvin, New J. Chem., 1998, 467 RSC; (e) W. Keim, Angew. Chem., Int. Ed. Engl., 1990, 29, 235 CrossRef.
- (a) T. B. Rauchfuss, Inorg. Chem., 1977, 16, 2966 CrossRef CAS; (b) C. A. Willoughby, R. R. Duff, Jr., W. M. Davis and S. L. Buchwald, Organometallics, 1996, 15, 472 CrossRef CAS.
- (a) J. Heinicke, R. Kadyrov, M. K. Kindermann, M. Koesling and P. G. Jones, Chem. Ber., 1996, 129, 1547 CrossRef CAS; (b) D. J. Jones, V. C. Gibson, S. M. Green, P. J. Maddox, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2005, 127, 11037 CrossRef CAS PubMed.
- N. Giuseppone, J.-L. Schmitt, L. Allouche and J.-M. Lehn, Angew. Chem., Int. Ed., 2008, 47, 2235 CrossRef CAS PubMed.
- For other XRD-established molecular structures of phosphine-stabilized zinc alkyl complexes, see: (a) E. E. Wilson, A. G. Oliver, R. P. Hughes and B. L. Ashfeld, Organometallics, 2011, 30, 5214 CrossRef CAS; (b) A. Murso and D. Stalke, Dalton Trans., 2004, 2563 RSC; (c) L.-C. Liang, W.-Y. Lee and C.-H. Hung, Inorg. Chem., 2003, 42, 5471 CrossRef CAS PubMed; (d) J. Dekker, J. W. Munninghoff, J. Boersma and A. L. Spek, Organometallics, 1987, 6, 1236 CrossRef CAS.
- For well-characterized Zn-OR aggregates, see, for instance: Q. Li, K. Nie, B. Xu, Y. Yao, Y. Zhang and Q. Shen, Polyhedron, 2012, 31, 58 CrossRef CAS PubMed; M. A. Dreher, M. Krumm, C. Lizandara-Pueyo and S. Polarz, Dalton Trans., 2010, 39, 2232 RSC.
- For representative solid state structures of Zn species bearing a Zn–O dative bond, see: (a) A. Ozarowski, H. M. Lee and A. Balch, J. Am. Chem. Soc., 2003, 125, 12606 CrossRef CAS PubMed; (b) S. Aboulkacem, W. Tyrra and I. Pantenburg, Z. Anorg. Allg. Chem., 2003, 629, 1569 CrossRef CAS; (c) D. Wang, K. Wurst and M. R. Buchmeiser, J. Organomet. Chem., 2004, 689, 2123 CrossRef CAS PubMed; (d) Q. Li, K. Nie, B. Xu, Y. Yao, Y. Zhang and Q. Shen, Polyhedron, 2010, 31, 58 CrossRef PubMed.
- For structures of phosphine-stabilized Zn(II) inorganic species, see: (a) L.-C. Liang, H.-Y. Shih, H.-S. Chen and S.-T. Lin, Eur. J. Inorg. Chem., 2012, 298 CrossRef CAS; (b) A. Krezel, R. Latajka, G. D. Bujacz and W. Bal, Inorg. Chem., 2003, 42, 1994 CrossRef CAS PubMed; (c) D. J. Darensbourg, M. S. Zimmer, P. Rainey and D. L. Larkins, Inorg. Chem., 2000, 39, 1578 CrossRef CAS; (d) D. J. Darensbourg, J. R. Wildeson, J. C. Yarbrough and J. H. Reibenspies, J. Am. Chem. Soc., 2000, 122, 12487 CrossRef CAS; (e) D. J. Darensbourg, M. S. Zimmer, P. Rainey and D. L. Larkins, Inorg. Chem., 1998, 37, 2852 CrossRef CAS; (f) D. Weiß, A. Schier and H. Schmidbaur, Z. Naturforsch., B: Chem. Sci., 1998, 53, 1307 Search PubMed; (g) J. Podlahova, B. Kratochvil and J. Podlaha, J. Chem. Soc., Dalton Trans., 1985, 2393 RSC.
- T. Endo, General Mechanisms in Ring-Opening Polymerization, in Handbook of Ring-Opening Polymerization, ed. P. Dubois, O. Coulembier and J.-M. Raquez, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2009, pp. 53–64 Search PubMed.
- For reviews on immortal metal-catalyzed ROP of cyclic esters and carbonates, see: (a) T. Aida and S. Inoue, Acc. Chem. Res., 1996, 29, 39 CrossRef CAS; (b) N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363 RSC.
- For selected recent examples, see ref. 4i and 8c and: (a) B. Liu, T. Roisnel, L. Maron, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2013, 19, 3986 CrossRef CAS PubMed; (b) F. Hild, L. Brelot and S. Dagorne, Organometallics, 2011, 30, 5457 CrossRef CAS.
- (a) I. Barakat, P. Dubois and P. Teyssié, J. Polym. Sci., Part A: Polym. Chem., 1993, 31, 505 CrossRef CAS; (b) D. Delcroix, B. Martin-Vaca, D. Bourissou and C. Navarro, Macromolecules, 2010, 43, 8828 CrossRef CAS.
- (a) A. P. Pêgo, A. A. Poot, D. W. Grijpma and J. Feijen, J. Controlled Release, 2003, 87, 69 CrossRef; (b) T. Tyson, A. Finne-Wistrand and A.-C. Albertsson, Biomacromolecules, 2009, 10, 149 CrossRef CAS PubMed; (c) Z. Zhang, D. W. Grijpma and J. Feijen, J. Controlled Release, 2006, 112, 57 CrossRef CAS PubMed.
- See, for instance: (a) H. R. Kricheldorf, K. Bornhorst and H. Hachmann-Thiessen, Macromolecules, 2005, 38, 5017 CrossRef CAS; (b) F. Stassin and R. Jérôme, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 2777 CrossRef CAS; (c) D. Pappalardo, L. Annunziata and C. Pellecchia, Macromolecules, 2009, 42, 6056 CrossRef CAS.
- E. Piedra-Arroni, A. Amgoune and D. Bourissou, Dalton Trans., 2013, 42, 9024 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data of all Zn complexes, DOSY measurement details for compounds 3, 5, 6, 7 and 8, optimized geometries (Chem 3D) and associated parameters for complexes 6 and 8. SEC traces, MALDI-TOF spectra of polymers, selected ROP kinetic data, 1H NMR spectra of selected isolated polymers and a summary of crystallographic data for compounds 2·H, 3, 6′ and 7. CCDC 1046464–1046467. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00458f |
| This journal is © The Royal Society of Chemistry 2015 |