
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-pressure single crystal X-ray diffraction study of the linear metal chain compound Co3(dpa)4Br2·CH2Cl2†
S. R.
Madsen
a,
J.
Overgaard
a,
D.
Stalke
b and
B. B.
Iversen
*a
aDepartment of Chemistry and iNANO, Aarhus University, Langelandsgade 140, 8000 Aarhus C, Denmark. E-mail: bo@chem.au.dk
bInstitute für Anorganische Chemie, Georg-August-Universität, Tammannstrasse 4, 37077 Göttingen, Germany
First published on 20th March 2015
Abstract
The crystal structure of the linear metal chain compound Co3(dpa)4Br2·CH2Cl2 (1) has been investigated up to a pressure of 13.6(2) GPa in a diamond anvil cell (DAC) using single crystal X-ray diffraction. The structure remains orthorhombic as the unit cell volume is reduced by 30% at 12.8 GPa. At 13.6(2) GPa the diffraction pattern is of very poor quality and not even reliable unit cell parameters can be determined. Peak broadening resulting from non-hydrostatic conditions was avoided by annealing the loaded DAC prior to data collection, allowing reliable structural models to be refined up to a pressure of 11.8(2) GPa. On increasing pressure, the disordered CH2Cl2 crystal solvent molecule gradually becomes redistributed from one site to another. Hirshfeld surface analysis suggests that the redistribution is a result of repulsive H⋯H interactions. Pressure also affects the molecular geometry, in particular the Co–Co and Co–Br bond lengths which decrease by 4% and 12%, respectively, at 11.8(2) GPa.
Introduction
The effect of pressure on transition metal coordination complexes is a new and highly active field of research, providing interesting new knowledge about the geometric flexibility, physical properties, and high-pressure chemistry of a diverse range of compounds.1 Co3(dpa)4Br2·CH2Cl2 (1, dpa− is the 2,2′-dypyridylamide anion) belongs to a family of compounds called extended metal atom chain (EMAC) complexes or linear metal string complexes. The EMACs have been intensely studied for their resemblance to electrical wires and their peculiar metal–metal bonding properties.2 One of the most intensively studied compounds is Co3(dpa)4Cl2·nCH2Cl2 which is found in a symmetrical form when n = 1, and an unsymmetrical form when n = 2.2a,3 In the symmetrical form of the complex the lengths of the two Co–Co bonds are identical, whereas in the unsymmetrical form there is a difference between the two Co–Co bond lengths of 0.15 Å at ambient conditions.4 The bonding properties have been investigated both with theoretical methods and in electron density studies.2d,5 The symmetrical complex is believed to be the ground state, but thermal excitation to an unsymmetrical state causes the chain to become more asymmetric on increasing temperature. In the related Cr3(dpa)4Cl2 complex, the temperature dependence is very similar to that of Co3(dpa)4Cl2·2CH2Cl2, indicating that the phenomenon is found among various members of the EMAC family.6 The situation is complicated by the importance of the crystal solvent, the role of which is still not entirely resolved.2b,5bIn the symmetrical Co3(dpa)4Cl2·CH2Cl2 (2) the crystal solvent is disordered at ambient conditions. On cooling to below 165 K the CH2Cl2 molecule becomes ordered, and the space group symmetry is reduced from Pnn2 (orthorhombic) to Pn (monoclinic). On ordering, the CH2Cl2 molecule arranges itself close to one end of the Cl–Co–Co–Co–Cl chain, causing the two Co–Cl bond lengths to differ by 0.023(1) Å at 109 K.2b Recently, the effect of applying an external pressure to the symmetrical complex in 2 was investigated up to 3.6 GPa.7 It was found that the Co–Cl bond length increased between ambient pressure and 0.32 GPa, and that compression caused the CH2Cl2 solvent molecule to become disordered on four sites instead of two. This behavior is completely different from the ordering of the CH2Cl2 molecule observed on cooling.
In the present study, much higher pressures have been applied to 1 (up to 13.6(2) GPa) using a diamond anvil cell (DAC), and the structural changes have been followed using single crystal X-ray diffraction. A mixture of n-pentane and isopentane was used as the pressure transmitting medium. The mixture is effectively hydrostatic up to 7.4 GPa8 and in order to reduce non-hydrostatic strain at higher pressures, the DAC was annealed at 130 °C.9
Experimental
Crystals of 1 were synthesized as described by Cotton et al. 1997.3 Single crystal X-ray diffraction data were collected at room temperature on a Super Nova diffractometer from Oxford Diffraction using Mo Kα radiation (0.71073 Å). Pressure was generated using a plate type DAC from Almax-Boehler equipped with diamond anvils with culet diameters of 600 μm. The pressure was determined using the ruby fluorescence method.10 The ruby crystals were placed in three positions around the single crystal (Fig. S1 in ESI†) in order to better detect any significant pressure gradients appearing above the hydrostatic limit of the pressure transmitting medium. A steel gasket (initial thickness 250 μm) was indented to 150 μm and a hole was drilled with a diameter of 300 μm. Single crystal X-ray diffraction data were collected on two different crystals of compound 1 (different pressure transmitting media were used for the two crystals). For the first crystal the pressure transmitting medium was a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of n-pentane and isopentane and complete data sets were collected between 1.45(5) GPa and 13.6(3) GPa. After observing the effects of non-hydrostaticity, heating of the DAC was attempted as described in the following. As the pressure reached 10.3 GPa at the 7th pressure point, the crystal quality had deteriorated severely and the resolution was only 1.8 Å. In order to try to make the conditions more hydrostatic and potentially reestablish crystal quality, the DAC was put in an oven at 130 °C for 5 hours. The DAC was allowed to cool to room temperature before the pressure was measured and the DAC was mounted on the diffractometer. Following this first annealing the crystal diffracted to an improved resolution of 1.0 Å. Another consequence of annealing was that the pressure dropped to 9.1 GPa. After collecting data at 9.1 GPa, the pressure was increased to 10.5 GPa and the DAC was annealed as before. After annealing the pressure dropped to 9.7 GPa. Data were collected and the pressure was increased to 11 GPa, after which a third and final annealing was performed. This time the heating caused the gasket hole to expand slightly and the pressure dropped to 9.7 GPa. Instead of measuring at 9.7 GPa, the pressure was increased to 10.6(2) GPa and data were collected without annealing. On subsequently increasing the pressure to 11.8(2) GPa, the crystal quality deteriorated, but annealing was not attempted because it seemed more likely that it would result in the gasket deforming. The pressure was increased to 12.8(3) GPa, and a data set was measured even though the conditions were clearly non-hydrostatic and sample diffraction was significantly reduced. At 13.6 GPa the diffraction from the crystal was very weak and no attempt was made to solve the structure. Ruby crystals placed at different positions in the DAC measured 13.2 GPa and 15.4 GPa two hours after the pressure had been increased, showing that the conditions were far from hydrostatic. After 24 hours the variation over the distance between the ruby crystals was reduced to 0.2 GPa, but still the crystal diffracted to a very poor resolution of 2.3 Å. In summary, the following data were collected on crystal A in the listed order of pressure (GPa), suffix H indicates that the DAC had been heated in the oven prior to data collection: 1.45(5), 2.70(5), 4.22(7), 5.54(9), 7.3(1), 9.2(1), 10.3(2), 9.1(1)H, 9.7(1)H, 10.6(2), 11.8(2), 12.8(3), and 13.6(2).
:1 mixture of n-pentane and isopentane and complete data sets were collected between 1.45(5) GPa and 13.6(3) GPa. After observing the effects of non-hydrostaticity, heating of the DAC was attempted as described in the following. As the pressure reached 10.3 GPa at the 7th pressure point, the crystal quality had deteriorated severely and the resolution was only 1.8 Å. In order to try to make the conditions more hydrostatic and potentially reestablish crystal quality, the DAC was put in an oven at 130 °C for 5 hours. The DAC was allowed to cool to room temperature before the pressure was measured and the DAC was mounted on the diffractometer. Following this first annealing the crystal diffracted to an improved resolution of 1.0 Å. Another consequence of annealing was that the pressure dropped to 9.1 GPa. After collecting data at 9.1 GPa, the pressure was increased to 10.5 GPa and the DAC was annealed as before. After annealing the pressure dropped to 9.7 GPa. Data were collected and the pressure was increased to 11 GPa, after which a third and final annealing was performed. This time the heating caused the gasket hole to expand slightly and the pressure dropped to 9.7 GPa. Instead of measuring at 9.7 GPa, the pressure was increased to 10.6(2) GPa and data were collected without annealing. On subsequently increasing the pressure to 11.8(2) GPa, the crystal quality deteriorated, but annealing was not attempted because it seemed more likely that it would result in the gasket deforming. The pressure was increased to 12.8(3) GPa, and a data set was measured even though the conditions were clearly non-hydrostatic and sample diffraction was significantly reduced. At 13.6 GPa the diffraction from the crystal was very weak and no attempt was made to solve the structure. Ruby crystals placed at different positions in the DAC measured 13.2 GPa and 15.4 GPa two hours after the pressure had been increased, showing that the conditions were far from hydrostatic. After 24 hours the variation over the distance between the ruby crystals was reduced to 0.2 GPa, but still the crystal diffracted to a very poor resolution of 2.3 Å. In summary, the following data were collected on crystal A in the listed order of pressure (GPa), suffix H indicates that the DAC had been heated in the oven prior to data collection: 1.45(5), 2.70(5), 4.22(7), 5.54(9), 7.3(1), 9.2(1), 10.3(2), 9.1(1)H, 9.7(1)H, 10.6(2), 11.8(2), 12.8(3), and 13.6(2).
Crystal B was pressurized using silicone oil as pressure transmitting medium (hydrostatic to <2 GPa) and data were collected at 0.01(5) i.e. ambient pressure but in a closed DAC, 0.4(1), and 0.7(1) GPa.
All the single crystal X-ray diffraction data were integrated using the CrysAlisPro11 software and an empirical absorption correction was performed. The crystal structures were refined using SHELXL-97.12 Crystallographic information is listed in ESI.†
Results and discussion
The unit cell of 1 is orthorhombic with space group Pnn2. The central cobalt atom in the Br–Co–Co–Co–Br chain, Co2, lies on a twofold rotation axis, and the asymmetric unit contains half a molecule (Fig. 1). The CH2Cl2 solvent molecule is disordered on four positions (orange and green in Fig. 1) with occupancies summing to 1. Only two of the four positions are crystallographically independent. They will be referred to as the A and B positions in the following discussion. At ambient conditions, the occupancies of the A and B positions refine to 0.439(7) and 0.061(7), respectively, but the structural model fits the data equally well if component B is removed completely. Up to 2.7 GPa, the occupancy of the B position is less than 0.1, and only the A position is included in the final structural model. Likewise, above 9.7 GPa, it is reasonable to leave out the A position and fix the occupancy of the B position to 0.5. For the disordered CH2Cl2 molecule, the ADPs were refined as isotropic and constrained to equal values for chemically identical atoms. ADP similarity and geometric restraints were used, and hydrogen atoms were refined as riding on their parent atom.11 | ||
| Fig. 1 Co3(dpa)4Br2 and CH2Cl2 molecules at 1.45 GPa with atom labels on selected atoms. H atoms have been omitted except on the CH2Cl2 molecules. The four CH2Cl2 molecules are colored orange or green depending on whether they occupy the A or B positions, respectively. | ||
At 9.2 GPa and 10.3 GPa, the reflections were initially very broad and the resolution was relatively low (1.5–1.8 Å compared to 1.0 Å at lower pressures). The unit cell appeared to have changed to monoclinic and the refinements gave high R-values (>15%). However, after heating the DAC, the reflection profiles were much sharper, and the resolution improved to 1.0 Å. The X-ray diffraction pattern once again complied with the orthorhombic crystal system and space group symmetry Pnn2.
In Fig. 2, the unit cell axes are plotted against pressure. The b-axis decreases more rapidly for the non-hydrostatic data (filled blue squares above 8 GPa) than what is observed in the annealed data (empty blue squares). The opposite behavior is observed for the c-axis which from the non-hydrostatic data seems incompressible beyond 9.9 Å (filled green triangles). For the annealed sample, the c-axis continues to decrease in length (empty green triangles).
 | ||
| Fig. 2 Unit cell axes versus pressure. Empty symbols represent data collected after annealing. Vertical uncertainties are of the same size as the symbols, horizontal uncertainties are given in the Experimental section. Below 10 GPa they are below 0.1 GPa, above 10 GPa, they are below 0.3 GPa. | ||
Annealing
Molecular crystals such as 1 are typically soft and therefore the effects of strain are quickly observed. The presence of strain is easily detected by looking at mosaicity descriptors (Fig. 3(a) and (b)) and data resolution (Fig. 3(c)). In CrysAlisPro the average mosaicity e1, e2, and e3 are obtained from fitting a Gaussian function to the peak shape. The values do not distinguish between beam divergence and crystal mosaicity.13 Therefore, the absolute value does not directly quantify the mosaicity, but since the beam divergence is constant, the evolution of the average mosaicity should represent the actual evolution of mosaic spread with pressure. The average mosaicity and the resolution are directly related to the quality of the single crystal, but also the ruby fluorescence signal can be used as a strain indicator (Fig. 3(d)). Significant increases in all the indicators are observed above 5.5 GPa, which is a lower pressure than the previously determined hydrostatic limit of 7.4 GPa. This is, in part, a result of 1 being much softer than ruby which was the previously used as probe of hydrostaticity.8 Data collected after annealing are shown as empty symbols and it is clear from all five plotted parameters that annealing reduces non-hydrostatic strain very efficiently. | ||
| Fig. 3 Peak shape descriptors e1 (blue squares), e2 (red triangles) (a), and e3 (b), the resolution, dmin (c) and the full width half maximum (FWHM) of the ruby fluorescence line (d). Empty symbols represent the annealed sample. | ||
Effect of pressure on 1
In Fig. 4(a) and (b) the Co1–Co2 and Co1–Br1 bond lengths in 1 are plotted between 0.0001 GPa and 11.8 GPa. Data obtained under very non-hydrostatic conditions (at 9.2 and 10.3 GPa) are not included in the plots due to the low data quality which makes structure refinement results unreliable. The uncertainties are large at 7.4 and 11.8 GPa, which (among the included data) are most affected by non-hydrostaticity. In both the Co–Co and Co–Br distances, there is a very small change in slope above 8 GPa. It is barely significant, but might be a combined effect of pressure and annealing, though the data were collected after cooling to room temperature. | ||
| Fig. 4 Co1–Co2 (a), Co1–Br1 (b), and Co–N (c) bond lengths (Å) and equivalent isotropic atomic displacement parameters for Co1, Co2 and Br1 (Å2). | ||
In Fig. 4(c) the Co–N distances are plotted, and as expected most of them decrease with pressure (Co2–N5 being the exception). Expectedly the metal amine Co1–N distances are longer than the metal amide Co2–N distances, as indicated by the average values (dotted lines) being 0.1 Å longer.14 Above 9 GPa, the Co2–N5 distance starts increasing, and at 11.8 GPa it is the longest of all six Co–N distances. However, it should be kept in mind that the uncertainties are rather large, especially at 11.8 GPa.
The ADPs of the Co1, Co2 and Br1 atoms are plotted in Fig. 4(d) and as previously observed,7 they decrease until the hydrostatic limit, above which they increase. The increasing ADPs are an indirect effect of peak broadening which leads to weaker and more inaccurate high-order data. The errors in the high-order data are absorbed in the ADPs. The increase starts at 5.5 GPa, much lower than the previously reported hydrostatic limit of the n-pentane-isopentane mixture (7.4 GPa). As mentioned, this is most likely a result of 1 being much softer than previously used probe which was ruby,15 therefore being more sensitive to strain gradients.
Disorder
As previously described, the CH2Cl2 solvent molecule is disordered over four positions (Fig. 1) at high pressure. At and below 2.7 GPa, inclusion of the B position in the structural model is not significantly improving the quality of the fit. For these data the B position is excluded, and the occupancy of the A position fixed at 0.5. With increasing pressure above 4.2 GPa, the B position of the CH2Cl2 molecule becomes progressively more occupied (Fig. 5). Above 9.7 GPa, the A position is no longer included in the structural model and the occupancy of the B position is fixed at 0.5. | ||
| Fig. 5 Fractional occupancies of the dichloromethane disorder positions A (orange triangles) and B. Below 4.2 GPa and above 9.7 GPa one position is not included in the model and the occupancy of the other position is fixed at 0.5. | ||
In the previous study of the iso-structural compound Co3(dpa)4Cl2·CH2Cl2 (2), similar disorder was observed.7 In 2, the occupancies were equivalent above 2 GPa, and the B position became the dominant position above 3 GPa. In 1, the occupancies of the A and B positions do not become similar until the pressure reaches at least 4 GPa and the occupancy of the B position does not exceed that of the A position until 9 GPa.
In the study of 2, the change in distribution of the solvent molecule on different disorder sites was explained as a consequence of H⋯H repulsion. In the structure of 1 the same tendency is observed from evaluating fingerprint plots calculated with the program CrystalExplorer (Fig. 6).16
 | ||
| Fig. 6 Fingerprint plots of the Co3(dpa)4Br2 molecule with the dichloromethane molecules placed in the A position at (a) 4.2 GPa and (b) 9.1 GPa or in the B position at (c) 4.2 GPa and (d) 9.1 GPa. | ||
In the fingerprint plots, the lowest (di; de) values correspond to H⋯H contacts. At 4.2 GPa the closest H⋯H distance between the Co3(dpa)4Br2 molecule and the CH2Cl2 molecule is 1.65 Å for the A position and 1.77 Å for the B position. At 9.1 GPa the corresponding distances are 1.40 Å for the A position and 2.07 Å for the B position. At 4.2 GPa the majority of the CH2Cl2 molecules are still in the A position, while at 9.1 GPa the majority occupy the B position. Hydrogen atoms are refined as ‘riding’ on their parent atom, and for low-occupancy atoms in the solvent molecules these positions are not very precisely determined. Therefore care should be taken when evaluating fingerprint plots and void spaces, especially since no uncertainties are calculated by CrystalExplorer. Still, the absolute H⋯H distances might be uncertain, but the trends should be reliable. Wood et al., found that for a model compound, CH4, H⋯H distances shorter than 2.0 Å are repulsive. On further compression, as H⋯H distances become shorter than 1.7 Å, phase transitions are typically observed (at least in crystals of small organic molecules).17 Nevertheless, there are a few exceptions for which very short H⋯H interactions are stabilizing.18
Comparison of iso-structural compounds
A Birch–Murnaghan third-order equation of state has been fitted to the data for 1 using the program EosFit15 (shown in Fig. 7 as the dashed line). Only data points within the quasi-hydrostatic pressure range, 0–5.5 GPa, were fitted. The fitted parameters are listed in Table 1 which also lists the values for the iso-structural compound Co3(dpa)4Cl2·CH2Cl2 (2). | ||
| Fig. 7 Unit cell volume of 1versus pressure. Empty symbols represent data collected after annealing, the dashed line is the third-order Birch–Murnaghan equation of state. | ||
| V 0 (Å3) | K 0 (GPa) | K ′0 | |
|---|---|---|---|
| 1 | 2074.3(3) | 6.4(7) | 14(2) |
| 2 | 2044(6) | 5(1) | 18.2(5) |
Given that 1 and 2 are iso-structural, it is not surprising that the bulk moduli and pressure derivatives are very similar.7 The unit cell parameters are plotted against pressure in Fig. 8(a) and (b) for 1 (green symbols) and 2 (red symbols). In 1 (and 2) the c-axis is the most compressible and the b-axis is the least compressible. The overall pressure dependence of 1 is similar to that of 2. There is, however, a small difference; in 1, the c-axis is slightly longer than it is in 2, and the difference appears to increase on increasing pressure. The opposite trend is observed for the a- and b-axes, which are practically identical.
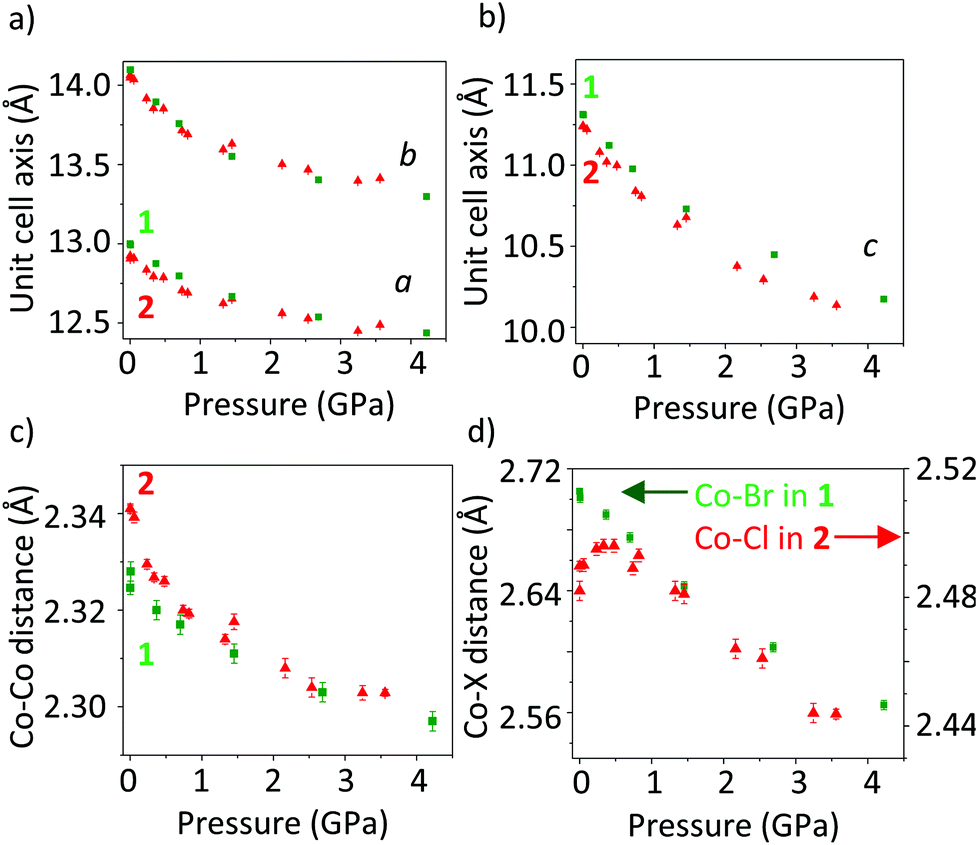 | ||
| Fig. 8 Comparison between 1 (green squares) and 2 (red triangles) up to 4 GPa. (a) Unit cell axes a and b, (b) c-axis, (c) Co–Co distances, and (d) Co–Br and Co–Cl distances. For 2, data were adapted from ref. 7. | ||
Larger differences between 1 and 2 are found when comparing the pressure dependencies of the Co–Co, and Co–X (X is Br or Cl) bond lengths (Fig. 8(c) and (d)). In 1, both bond lengths decrease smoothly on increasing pressure, with the Co–Br bond being more compressible than the Co–Co bond. In 2, the Co–Cl distance initially increases slightly, after which it decreases. DFT calculations using the experimental geometries indicated that the increase in Co–Cl distance may be caused by thermal excitations to a state which is more anti-bonding with respect to the Co–Cl bond.7 The Co–Co bond in 2 is more compressible than in 1, but it is also initially a little longer in 2 (2.341(1)Å) than in 1 (2.325(1)Å).
The Co–Co bonds behave more similarly than the Co–Cl and Co–Br bonds. Exchanging the Br− ion for Cl− clearly makes a difference, and it is not expected that the energy gap to an anti-bonding state is as small in 1 as it is in 2. It follows that a Co–Br bond elongation like the Co–Cl bond elongation in 2 is not expected in 1. Additionally, the larger and softer Br− (ionic radius 1.82 Å) ion is more easily deformed than the harder Cl− ion (ionic radius 1.67 Å), allowing a relatively larger compression of the Co–Br bond. At ambient conditions, the Co–Br and Co–Cl bond lengths are 2.707(1)Å and 2.482(3)Å, respectively.
Conclusions
Co3(dpa)4Br2·CH2Cl2 (1) has been investigated in the pressure range 0–13.6 GPa using single crystal X-ray diffraction. A mixture of n-pentane and isopentane was used as pressure transmitting medium, and above 5.5 GPa the effects of non-hydrostatic conditions are clearly visible in both the raw data and in the refined crystal structure. At 10.3 GPa the resolution was reduced to 1.8 Å (from 1.0 Å at ambient conditions) and the structure could not be reliably refined. After annealing the DAC, the diffraction power of the crystal was restored and the resolution was again 1.0 Å.Between 0.0001 and 12.8 GPa the unit cell volume of 1 decreased by 30%, with the c-axis being the most compressible. This anisotropy of compression is similar to the iso-structural compound Co3(dpa)4Cl2·CH2Cl2 (2) which was recently described.7 The two structures also show similar crystal solvent redistribution at high pressure, though the redistribution occurs at higher pressures in 1 than in 2 (at 4 and 2 GPa, respectively). The CH2Cl2 molecule is distributed on two crystallographically independent positions with occupancy ratios (A:B) changing from 1:0 at ambient conditions, to 0.7:0.3 at 4.2 GPa and 0.3:0.7 at 9.1 GPa. At 9.7 GPa the redistribution appears to be complete and the A:B occupancy ratio is 0:1, though this could be a combined effect of pressure and heating (annealing at 130 °C).
The largest difference in the behavior of 1 and the previously studied 2 is that no Co–Br bond length increase was observed, but rather a smooth decrease of both the Co–Br and Co–Co bond lengths is found. The relative decrease of the Co–Br bond is much more pronounced than that of the Co–Cl bond in 2, even within the 4 GPa-pressure range in which 2 was investigated. This is a result of the Br atom being much softer, and thus the Co–Br bond is more compressible, than the Co–Cl bond.
Acknowledgements
The authors thank Nils Finkelmeier, Jacob Hey, and Reent Michel from the Georg-August Universität in Göttingen, Germany, for help with synthesis and crystal structure refinements and the Danish National Research Foundation for funding (Center for Materials Crystallography, DNRF93).Notes and references
- J. P. Tidey, H. L. S. Wong, M. Schröder and A. J. Blake, Coord. Chem. Rev., 2014, 277–278, 187–207 CrossRef CAS PubMed.
- (a) E. C. Yang, M. C. Cheng, M. S. Tsai and S. M. Peng, J. Chem. Soc., Chem. Commun., 1994,(20), 2377–2378 RSC; (b) F. A. Cotton, R. Clerac, L. M. Daniels, K. R. Dunbar, K. Kirschbaum, C. A. Murillo, A. A. Pinkerton, A. J. Schultz and X. P. Wang, J. Am. Chem. Soc., 2000, 122(26), 6226–6236 CrossRef; (c) F. A. Cotton, R. Clerac, L. M. Daniels, K. R. Dunbar, C. A. Murillo and X. P. Wang, J. Chem. Soc., Dalton Trans., 2001,(4), 386–391 Search PubMed; (d) M. M. Rohmer, A. Strich, M. Benard and J. P. Malrieu, J. Am. Chem. Soc., 2001, 123(37), 9126–9134 CrossRef CAS PubMed.
- F. A. Cotton, L. M. Daniels and G. T. Jordan, Chem. Commun., 1997,(5), 421–422 RSC.
- F. A. Cotton, L. M. Daniels, G. T. Jordan and C. A. Murillo, J. Am. Chem. Soc., 1997, 119(43), 10377–10381 CrossRef CAS.
- (a) D. A. Pantazis and J. E. McGrady, J. Am. Chem. Soc., 2006, 128(12), 4128–4135 CrossRef CAS PubMed; (b) R. D. Poulsen, J. Overgaard, A. Schulman, C. Østergaard, C. A. Murillo, M. A. Spackman and B. B. Iversen, J. Am. Chem. Soc., 2009, 131(22), 7580–7591 CrossRef CAS PubMed.
- (a) F. A. Cotton, L. M. Daniels, C. A. Murillo and I. Pascual, J. Am. Chem. Soc., 1997, 119(42), 10223–10224 CrossRef CAS; (b) J. F. Berry, F. A. Cotton, T. B. Lu, C. A. Murillo, B. K. Roberts and X. P. Wang, J. Am. Chem. Soc., 2004, 126(22), 7082–7096 CrossRef CAS PubMed; (c) L.-C. Wu, M. K. Thomsen, S. R. Madsen, M. S. Schmøkel, M. R. V. Jørgensen, M.-C. Cheng, S.-M. Peng, Y.-S. Chen, J. Overgaard and B. B. Iversen, Inorg. Chem., 2014, 53, 12489–12498 CrossRef CAS PubMed.
- S. R. Madsen, M. K. Thomsen, S. Scheins, Y. S. Chen, N. Finkelmeier, D. Stalke, J. Overgaard and B. B. Iversen, Dalton Trans., 2014, 43(3), 1313–1320 RSC.
- S. Klotz, J. C. Chervin, P. Munsch and G. Le Marchand, J. Phys. D: Appl. Phys., 2009, 42(7) CrossRef CAS , 075413.
- (a) K. Murata, K. Yokogawa, H. Yoshino, S. Klotz, P. Munsch, A. Irizawa, M. Nishiyama, K. Iizuka, T. Nanba, T. Okada, Y. Shiraga and S. Aoyama, Rev. Sci. Instrum., 2008, 79(8) CrossRef CAS PubMed , 085101; (b) S. V. Sinogeikin and J. D. Bass, Phys. Rev. B: Condens. Matter, 1999, 59(22), R14141–R14144 CrossRef CAS.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46(6), 2774–2780 CrossRef CAS PubMed.
- O. V. Dolomanov, R. Gildea and H. Puschmann, CrysAlisPro Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- W. Kabsch, Int. Tables Crystallogr., 2006, F(11.3), 218–225 Search PubMed.
- (a) M. Pfeiffer, A. Murso, L. Mahalakshmi, D. Moigno, W. Kiefer and D. Stalke, Eur. J. Inorg. Chem., 2002,(12), 3222–3234 CrossRef CAS; (b) H. Gornitzka and D. Stalke, Eur. J. Inorg. Chem., 1998, 14(3), 311–317 CrossRef.
- R. J. Angel, J. Gonzalez-Platas and M. Alvaro, Z. Kristallogr., 2014, 229(5), 405–419 CAS.
- (a) M. A. Spackman and P. G. Byrom, Chem. Phys. Lett., 1997, 267(3–4), 215–220 CrossRef CAS; (b) M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 378–392 RSC.
- P. A. Wood, J. J. McKinnon, S. Parsons, E. Pidcock and M. A. Spackman, CrystEngComm, 2008, 10(4), 368–376 CAS.
- (a) C. F. Matta, J. Hernandez-Trujillo, T. H. Tang and R. F. W. Bader, Chem. – Eur. J., 2003, 9(9), 1940–1951 CrossRef CAS PubMed; (b) E. A. Zhurova, C. F. Matta, N. Wu, V. V. Zhurov and A. A. Pinkerton, J. Am. Chem. Soc., 2006, 128(27), 8849–8861 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic information, pictures of crystals in DACs, and fingerprint plots. See DOI: 10.1039/c5dt00447k |
| This journal is © The Royal Society of Chemistry 2015 |