
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of pyranylidene complexes of ruthenium†
Gabriele
Albertin
*a,
Stefano
Antoniutti
a,
Marco
Bortoluzzi
a,
Alessandra
Botter
a and
Jesús
Castro
b
aDipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari Venezia, Dorsoduro 2137, 30123 Venezia, Italy. E-mail: albertin@unive.it
bDepartamento de Química Inorgánica, Universidade de Vigo, Facultade de Química, Edificio de Ciencias Experimentais, 36310 Vigo, Galicia, Spain
First published on 24th March 2015
Abstract
The reaction of the chloro-complex RuCl(η5-C5H5)(PPh3)[P(OMe)3] with alkylpropiolates HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CCOOR1 in alcohol R2OH affords pyranylidene derivatives [Ru(η5-C5H5){
CCOOR1 in alcohol R2OH affords pyranylidene derivatives [Ru(η5-C5H5){![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(COOR1)C(H)C(H)C(OR1)O}(PPh3){P(OMe)3}]BPh4 (1, 3) and alkoxycarbene complexes [Ru(η5-C5H5){C(OR2)(CH2COOR1)}(PPh3){P(OMe)3}]BPh4 (2, 4). A reaction path for the formation of compounds 1–4, involving reactions on a vinylidene intermediate complex, is also discussed. The complexes were characterized spectroscopically (IR and 1H, 13C, 31P NMR) and by X-ray crystal structure determination of [Ru(η5-C5H5){C(COOMe)C(H)C(H)C(OMe)O}(PPh3){P(OMe)3}]BPh4 (1).
C(COOR1)C(H)C(H)C(OR1)O}(PPh3){P(OMe)3}]BPh4 (1, 3) and alkoxycarbene complexes [Ru(η5-C5H5){C(OR2)(CH2COOR1)}(PPh3){P(OMe)3}]BPh4 (2, 4). A reaction path for the formation of compounds 1–4, involving reactions on a vinylidene intermediate complex, is also discussed. The complexes were characterized spectroscopically (IR and 1H, 13C, 31P NMR) and by X-ray crystal structure determination of [Ru(η5-C5H5){C(COOMe)C(H)C(H)C(OMe)O}(PPh3){P(OMe)3}]BPh4 (1).
Introduction
Despite the large number of Fischer-type transition metal carbene complexes reported so far,1 six-membered pyranylidene carbene derivatives (Chart 1) are relatively few and involve mainly Cr, Mo and W central metals.2–5 This fact is somewhat surprising, given the increasing interest in their use in ring-opening reactions,6 Diels–Alder reactions4b,7 and 1,6 addition.4a Several methods for the synthesis of pyranylidene have been developed, mainly including the reaction of preformed carbene complexes with pyridinium ylides,2b,c enol ethers8 and 1,3-diketones,3 or multicomponent species9 such as alkynyl esters and N-methylmorpholine-N-oxide. Alternatively, pyranylidene complexes can be prepared from the reaction of M(CO)5L (M = Cr, Mo, W; L = THF, NEt3) with β-alkynyl α,β-unsaturated carbonyl compounds4 and from the reaction of M(CO)6 with 1-lithio-1,3-dienes.5 However, in only one case the simplest method for preparing a pyranylidene complex, involving dimerization of alkylpropiolate HCCCOOR on a metal fragment, has been reported.9
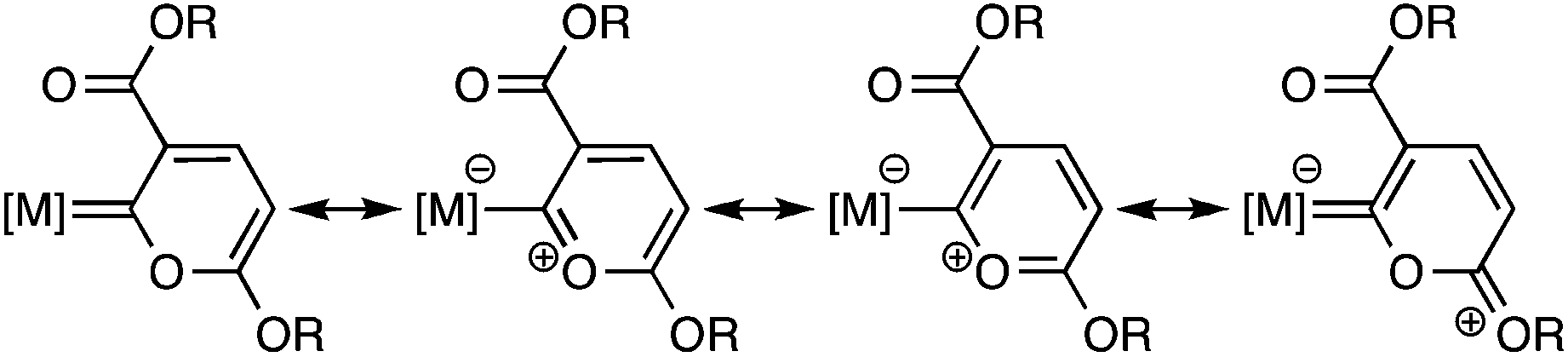 | ||
| Chart 1 | ||
We were interested in the reactivity of half-sandwich complexes containing phosphite ligands10 and, in the course of our studies, found that the reaction of the complex RuCl(η5-C5H5)(PPh3)[P(OMe)3] with alkylpropiolate leads to the first pyranylidene complexes of ruthenium. Our results, including the synthesis and characterization of pyranylidene and alkoxycarbene complexes of Ru(II), are reported here.
Results and discussion
The half-sandwich chloro-complex RuCl(η5-C5H5)(PPh3)[P(OMe)3] reacts with an excess of alkylpropiolate HCCCOOR1 in alcohol R2OH to give a mixture of pyranylidene [Ru(η5-C5H5){C(COOR1)C(H)C(H)C(OR1)O}(PPh3){P(OMe)3}]BPh4 (1, 3) and alkoxycarbene [Ru(η5-C5H5)-{C(OR2)(CH2COOR1)}(PPh3){P(OMe)3}]BPh4 (2, 4) in about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio, which were separated by fractional crystallization and characterized (Scheme 1).
:4 ratio, which were separated by fractional crystallization and characterized (Scheme 1).
 | ||
| Scheme 1 R1 = Me (1, 2), Et (3, 4); R2 = Me (a), Et (b). | ||
Crucial for successful syntheses was the use of alcohol containing the salt NaBPh4 as a solvent, which probably favors substitution of the chloride ligand in the starting complex, yielding the two types of complexes 1, 3 and 2, 4.
The formation of both pyranylidene and alkoxycarbene derivatives in the reaction of RuCl(η5-C5H5)(PPh3)[P(OMe)3] with alkylpropiolate is somewhat surprising, but may be explained according to the reaction path shown in Scheme 2, which involves the initial formation of a vinylidene intermediate [A]. Reaction of this intermediate with alkylpropiolate gives rise to the dimerization of HCCCOOR1, affording pyranylidene derivatives 1, 3. Parallel nucleophilic attack by the oxygen atom of R2OH on the Cα of the vinylidene, followed by proton-transfer, yields the final alkoxycarbene derivatives 2, 4.
 | ||
| Scheme 2 R1 = Me, Et; R2 = Et. | ||
In order to verify this reaction path, we attempted to isolate the vinylidene intermediate [A] or, at least, to identify it in the reaction mixture. At first, we treated the compound RuCl(η5-C5H5)(PPh3)[P(OMe)3] with HCCCOOR1, in the presence of NaBPh4, in a solvent other than alcohol such as dichloromethane, but no reaction was observed, probably owing to the insolubility of NaBPh4 in this solvent. We therefore used a different strategy, involving a reaction of the starting chloro-complex first with silver triflate, to form the triflate intermediate Ru(κ1-OTf)(η5-C5H5)(PPh3)[P(OMe)3], and then with an excess of methylpropiolate in dichloromethane as a solvent (Scheme 3).
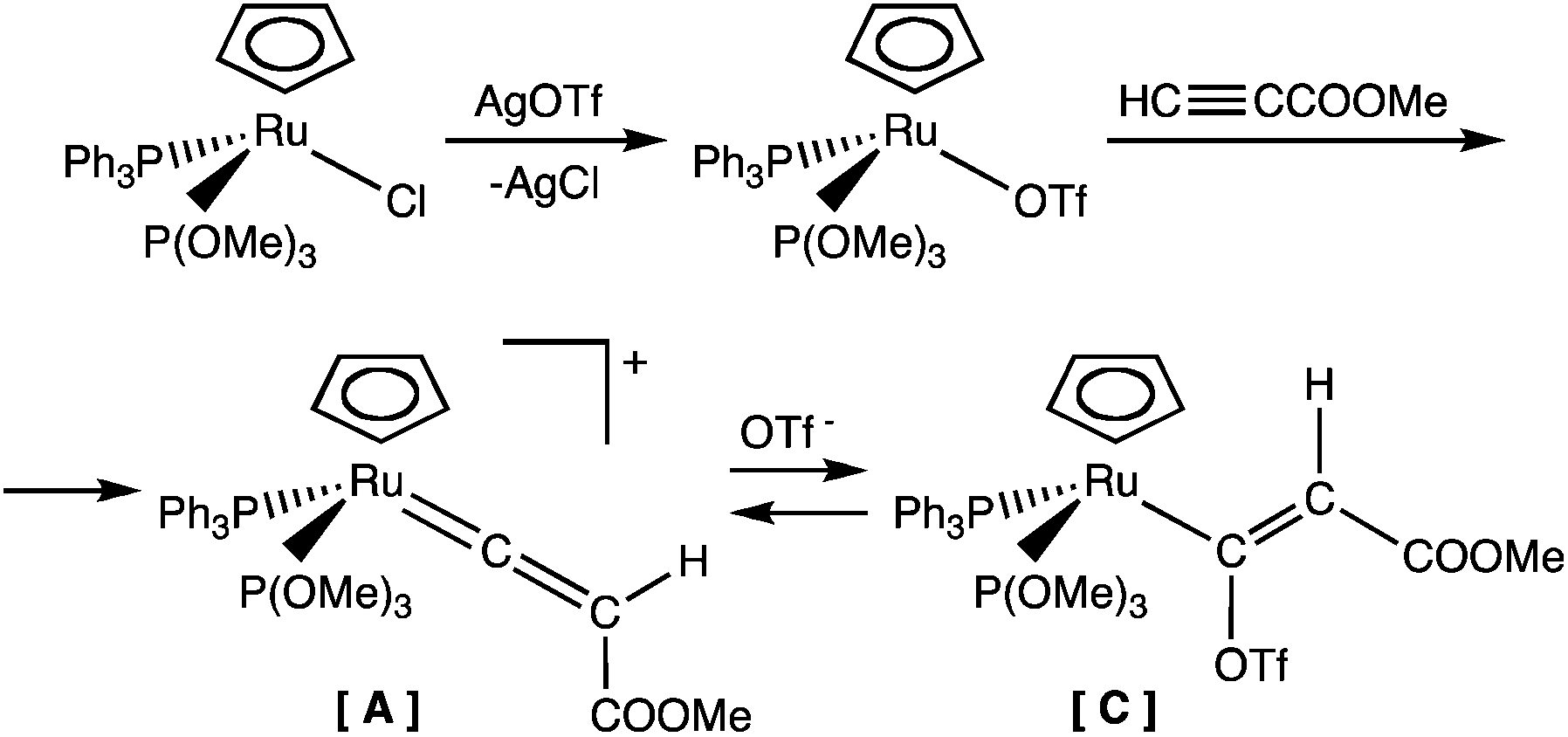 | ||
| Scheme 3 | ||
The triflate complex quickly reacted with methylpropiolate by changing color in the solution, from which we were not able to isolate a solid, but only an oily product. Its 13C NMR spectrum showed a doublet of doublets at 199.71 ppm (JCP = 15.9, JCP = 11.6 Hz), which might be attributed to the Cα carbon atom of a vinylidene species [A]. However, this value is at lower frequency than those observed for the Cα of several known vinylidene derivatives.11 As suggested by a reviewer, this resonance may be attributed to the Cα of the vinyl species [Ru(η5-C5H5){C(OTf)C(H)COOMe}(PPh3)[P(OMe)3] [C], formed by nucleophilic attack of the triflate ion on the carbene carbon atom of the CC(H)COOMe ligand. In an HMBC experiment, the 13C signal at 199.7 ppm is correlated with the multiplet at 5.47 ppm of the 1H spectrum, attributable to the C(H)COOMe of the vinyl ligand, fitting the proposed formulation for [C]. The triflate ligand is labile in the complex Ru(κ1-OTf)(η5-C5H5)(PPh3)[P(OMe)3] and can be substituted by alkyne, which then tautomerizes11 on the metal center to give vinylidene intermediate [A]. Reaction with the triflate ion OTf− can give the vinyl intermediate [C], which may be in equilibrium with [A] (Scheme 3). DFT calculations on model systems, where P-donor ligands are replaced by PH3 and PF3, support this hypothesis, the ΔH difference between [Ru(η5-C5H5){C(OTf)C(H)COOMe}(PH3)(PF3)] and [Ru(η5-C5H5){CC(H)COOMe}(PH3)(PF3)]OTf being only about 1.2 kcal mol−1 in favor of the latter. The fact that vinylidene [A] and/or vinyl [C] complexes are really intermediates of the reaction path proposed in Scheme 2 was confirmed by treatment with ethanol, which gave ethoxycarbene 2b as the final product, the addition of methanol affording methoxycarbene 2a. Instead, addition of alkylpropiolate to vinylidene intermediate [A] only yielded traces of the pyranylidene complex, and it was only with the addition of alcohol to the solution of [A] that the reaction started, affording a mixture of pyranylidene 1 and alkoxycarbene 2 complexes. However, the greater amount of carbene 2 with respect to pyranylidene (4:1 ratio) suggests a faster reaction of vinylidene [A] with alcohol than that with propiolate.
The need for alcohol as a solvent is explained by the fact that the formation of pyranylidene from the reaction of vinylidene [A] with alkylpropiolate must involve a hydrogen shift, which may be a proton transfer strongly favored by protic solvents such as alcohols (Scheme 4).
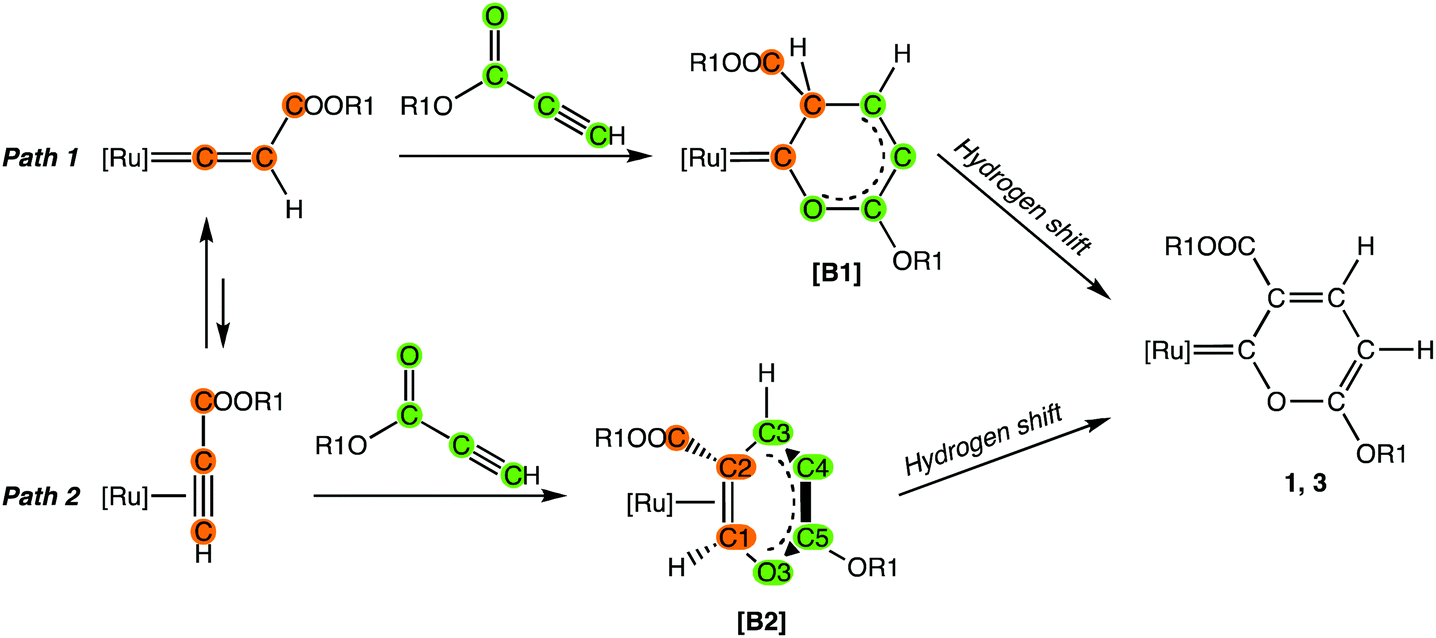 | ||
| Scheme 4 [Ru] = [Ru(η5-C5H5)(PPh3){P(OMe)3}]+; R1 = Me, Et. | ||
In fact, plausible mechanisms for the formation of the pyranylidene probably involve cyclization reactions between an alkylpropiolate molecule and coordinated vinylidene or η2-alkyne, affording intermediates [B1] (path 1) or [B2] (path 2), respectively. Subsequent hydrogen transfers in these intermediates gave the final pyranylidene complexes 1, 3. DFT calculations on a simplified system, in which [Ru] = [Ru(η5-C5H5)(PH3)(PF3)]+ and R1 = Me (see ESI†), indicated the formation of intermediate [B2], which is about 20.7 kcal mol−1 more stable than [B1]. The final formation of the pyranylidene derivative is strongly favored, as its Gibbs energy is lower than that of [B2] by about 53.8 kcal mol−1. DFT calculations ruled out intramolecular pathways for the hydrogen shift from [B2] to the final product, the estimated energy barrier being unreasonably high. On the other hand, the computed proton affinity of [B2], 232 kcal mol−1, enforces the hypothesis that the hydrogen atom could migrate by acid–base equilibria with the solvent.12 Path 2 requires preliminary tautomerization of vinylidene to η2-alkyne, but the energy difference between these species is quite low, about 1.5 kcal mol−1 in favor of the vinylidene complex. Equilibrium mixtures containing η1-vinylidene and η2-alkyne complexes have previously been reported.13 However, the greater stability of vinylidene may partially explain the competitive reaction with alcohols and the formation of the corresponding carbenes as prevailing species.
The reaction path involving [B2] was more deeply investigated from a computational point of view. A coordinate-driving study based on the progressive elongation of the C(2)–C(3) bond of [B2] (see Scheme 4 for numbering) allowed us to find a possible transition-state geometry, which was then fully optimized and characterized by IR simulations (see the ESI†xyz file for details of the atomic coordinates). The activation Gibbs free energy (referred to [Ru]–η2-HCCH + methylpropiolate) was 32.7 kcal mol−1, as depicted in Scheme 5. Quite interestingly, the only imaginary frequency (i340 cm−1) of the transition state mainly concerns the formation of the C(2)–C(3) bond from [Ru]–η2-HCCH and methylpropiolate. The carbon–oxygen bond of the six-membered heterocycle is instead not yet present in the transition state, the O(3)⋯C(1) distance being about 2.5 Å. This result suggests that the carbon–oxygen interaction occurs after the C(2)–C(3) bond formation, in a secondary step having low activation energy. This idea was supported by a further coordinate-driving simulation based on the progressive elongation of the O(3)–C(1) bond of [B2], which did not lead to any C–C bond break.
 | ||
| Scheme 5 Computed reaction pathway for the formation of [B2] from {[Ru]-η2-HCCH + methylpropiolate}. [Ru] = [Ru(η5-C5H5)(PH3)(PF3)]+. | ||
It should be noted that, for the previously reported multicomponent syntheses of Cr(0) and W(0) pyranylidene derivatives9 from the reaction of carbene complexes with alkylpropiolate in the presence of NMO (N-methylmorpholine-N-oxide), the proposed mechanism involved the formation of a propadienylidene intermediate of the type [M]CCC(O−)(OR1), the reaction of which with alkylpropiolate yielded the final pyranylidene complexes. In our case, transformation of vinylidene intermediate [A] in a propadienylidene species, like that proposed for Cr(0) and W(0), is improbable owing to the absence of a base.
Nucleophilic attack of alcohol on the Cα carbon of ruthenium vinylidene complexes affording alkoxycarbene derivatives has previously been reported with unsubstituted [Ru]CCH2 compounds.14 The reaction of our mixed-ligand half-sandwich derivatives with alkylpropiolate highlights a new example of the formation of alkoxycarbene species.
Good analytical data were obtained for both pyranylidene 1, 3 and alkoxycarbene 2, 4 derivatives, which were isolated as yellow-orange solids stable in air and in solution of polar organic solvents, where they behave as 1:1 electrolytes.15 Infrared and NMR data support the proposed formulations, which were further confirmed by X-ray crystal structure determination of [Ru(η5-C5H5){C(COOMe)C(H)C(H)C(OMe)O}(PPh3){P(OMe)3}]BPh4 (1), the ORTEP of which is shown in Fig. 1.
 | ||
| Fig. 1 ORTEP view of the cation of 1. P1 represents a PPh3 ligand and P2 represents a P(OMe)3 ligand. | ||
Compound 1 consists of a BPh4− anion (not shown in the figure) and a ruthenium cation complex. The latter contains a ruthenium atom in a half-sandwich piano-stool structure, coordinated by a η5-cyclopentadienyl ligand having one pyran-2-ylidene ligand and two phosphines [one PPh3 and one P(OMe)3] as legs. Selected bond lengths and angles are shown in Table 1. The overall geometry of the half-sandwich piano-stool complex is octahedral, as demonstrated by near 90° values for angles P–Ru–P and P–Ru–CO, between 88.26(9) and 96.56(3)°. The larger angle is that of P–Ru–P. The η5-coordination mode of the Cp ligand shows Ru–C bond distances between 2.241(3) and 2.265(4) Å. The average value for Ru–C bond lengths [2.257(4) Å] is slightly longer than the usual ones for CpRu moieties,16 and the Ru–Ct distance [1.9136(2) Å] is also longer than that found, for example, at 1.9056(5) Å in Ru(Cp)(GeH3)[P(OMe)3](PPh3).17
| Ru–C(1) | 2.018(3) | Ru–P(2) | 2.2413(8) |
| Ru–P(1) | 2.3244(8) | Ru–CT | 1.9136(2) |
| Ru–C(11) | 2.241(3) | Ru–C(12) | 2.256(4) |
| Ru–C(13) | 2.265(4) | Ru–C(14) | 2.264(3) |
| Ru–C(15) | 2.257(3) | O(1)–C(8) | 1.448(6) |
| O(1)–C(6) | 1.327(5) | O(3)–C(5) | 1.330(4) |
| O(2)–C(6) | 1.192(5) | O(4)–C(5) | 1.307(5) |
| O(3)–C(1) | 1.392(4) | C(1)–C(2) | 1.402(5) |
| O(4)–C(7) | 1.447(5) | C(2)–C(6) | 1.498(6) |
| C(2)–C(3) | 1.381(6) | C(4)–C(5) | 1.350(6) |
| C(3)–C(4) | 1.384(7) | ||
| C(1)–Ru–P(2) | 89.73(10) | C(1)–Ru–P(1) | 88.26(9) |
| P(1)–Ru–P(2) | 96.56(3) | C(5)–O(3)–C(1) | 126.7(3) |
| C(6)–O(1)–C(8) | 116.9(4) | O(3)–C(1)–C(2) | 112.1(3) |
| C(5)–O(4)–C(7) | 118.2(4) | C(2)–C(1)–Ru | 132.6(3) |
| O(3)–C(1)–Ru | 115.2(2) | C(3)–C(2)–C(6) | 116.2(4) |
| C(3)–C(2)–C(1) | 121.3(4) | C(2)–C(3)–C(4) | 122.6(4) |
| C(1)–C(2)–C(6) | 122.5(3) | O(4)–C(5)–O(3) | 109.4(3) |
| C(5)–C(4)–C(3) | 116.2(4) | O(3)–C(5)–C(4) | 120.9(4) |
| O(4)–C(5)–C(4) | 129.7(4) | O(2)–C(6)–C(2) | 125.8(4) |
| O(2)–C(6)–O(1) | 123.6(4) | ||
| O(1)–C(6)–C(2) | 110.6(3) | ||
The Ru–P bond distances [2.3244(8) Å for Ru–PPh3 and 2.2413(8) Å for Ru–P(OMe)3] are similar to those previously found in other phosphite-phosphine Ru(II) compounds, e.g., 2.2945(16) and 2.1933(19) Å in Ru(Cp)(GeH3)[P(OMe)3](PPh3)17 and 2.3113(4) and 2.2239(5) Å in Ru(Cp)Cl[P(OMe)3](PPh3).18 Shorter Ru–P bonds correspond, as usual, to the phosphite ligand.19
The pyran-2-ylidene ligand is bonded to the ruthenium metal with a Ru–C distance of 2.018(3) Å. This is shorter than the usual range for Ru–C(sp2) single bonds [2.03–2.11 Å]20,21 although slightly longer than that found for other Fischer-type carbene complexes.22 The pyran-2-ylidene ring is essentially planar (rms of 0.016 Å). Distances in the ring (see Table 1) indicate a formal (although short) single bond between C(1) and O(3), 1.392(4) Å, and between C(1) and C(2), 1.402(5) Å. Electronic delocalization takes place in the rest of the ring, including the O(3)–C(5) bond, with values between 1.330(4) and 1.384(7) Å.9 The methoxycarbonyl substituent on the pyranylidene ligand is almost perpendicular [dihedral angle between planes of 86.9(2)°] to the plane of the rings, proving the single-bond character of the C(2)–C(6) bond, with a length of 1.498(6) Å.
The IR spectra of pyranylidene complexes 1, 3 show a medium-intensity band at 1731–1733 cm−1, attributed to the νCO of the ester substituent COOR1. Besides signals of the ancillary ligands and BPh4 anions, the 1H NMR spectra show an AB quartet at 7.06 and 5.44 ppm for 1 and at 7.14 and 5.41 ppm for 3 of the H3 and H4 protons of pyranylidene and the signals of methyl or ethyl groups of the COOR1 and OR1 substituents at C2 and C5 atoms, respectively. The methyls of 1 appear as singlets at 3.89 and 3.48 ppm, and the ethyls of 3 as quartets at 4.42 and 4.17 and triplets at 1.02 and 1.26 ppm. However, strong support for the presence of the pyranylidene ligand comes from the 13C NMR spectra, which, at −30 °C, show the characteristic carbene carbon resonance as a broad multiplet at 332 ppm. The spectra show the signals of the C2, C3, C4 and C5 atoms of the heterocycle ligand between 172 and 94 ppm, as well as those of the COOR1 and OR1 substituents, fitting the proposed formulation for pyranylidene complexes.
The NMR proton spectra of alkoxycarbene complexes [Ru(η5-C5H5){C(OR2)(CH2COOR1)}(PPh3){P(OMe)3}]BPh4 (2, 4) show an AB quartet at 4.59–3.89 ppm of the diastereotopic CH2 protons of the CH2COOR1 group of carbene and the signals of the substituents R1 and R2, which appear as singlets for methyls and quartets and triplets for ethyls. However, diagnostic of the presence of alkoxycarbene C(OR2)CH2COOR1 are the 13C NMR spectra, which show a doublet of doublets at 297.67–297.21 ppm, attributed to carbene carbon resonance. Singlets at 61.01 for 2a, 61.42 for 2 and 62.85 ppm for 4a also appear in the spectra which, in an HMQC experiment, were correlated with the AB quartets at 4.59–3.89 ppm in the proton spectra and were attributed to the methylene carbon resonance of the CH2COOR1 group. The signals of the COOR1 and OR2 substituents were also observed, whereas the 31P NMR spectra are AB quartets, matching the proposed formulation.
Conclusions
This paper reports the first pyran-2-ylidene carbene complexes of ruthenium prepared through dimerization of alkylpropiolate on a half-sandwich fragment. An alkoxycarbene derivative was also obtained. A reaction path for the formation of the pyranylidene and ethoxycarbene complexes, involving a vinylidene intermediate, is proposed.Experimental
Materials and physical measurements
All synthetic work was carried out under an appropriate atmosphere (Ar, N2) by standard Schlenk techniques or in an inert atmosphere dry-box. All solvents were dried over appropriate drying agents, degased on a vacuum line, and distilled in vacuum-tight storage flasks. RuCl3·3H2O was a Pressure Chemical Co. (USA) product; other reagents were purchased from commercial sources at the highest available purity and were used as received. Infrared spectra were recorded on a Perkin-Elmer Spectrum-One FT-IR spectrophotometer. NMR spectra (1H, 13C, 31P) were obtained on an AVANCE 300 Bruker spectrometer at temperatures between −90 and +25 °C, unless otherwise noted. 1H and 13C spectra are referred to internal tetramethylsilane. 31P{1H} chemical shifts are reported with respect to 85% H3PO4, with downfield shifts considered positive. COSY, HMQC and HMBC NMR experiments were performed using standard programs. The iNMR software package23 was used to treat NMR data. The conductivity of 10−3 mol dm−3 solutions of the complexes in CH3NO2 at 25 °C was measured on a radiometer CDM 83. Elemental analyses were performed in the Microanalytical Laboratory of the Dipartimento di Scienze del Farmaco, University of Padova (Italy). Melting points (m.p.) were determined in capillary on Büchi 535 apparatus.Synthesis of the complexes
The compound RuCl(η5-C5H5)(PPh3)[P(OMe)3] was prepared following the method previously reported.24![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) C(COOMe)C(H)C(H)C(OMe)O}(PPh3){P(OMe)3}]BPh4 (1) and [Ru(η5-C5H5){C(OR2)(CH2COOMe)}(PPh3){P(OMe)3}]BPh4 (2) [R2 = Me (a), Et (b)].
In a 25 mL three-necked round-bottomed flask were placed solid samples of RuCl(η5-C5H5)(PPh3)[P(OMe)3] (200 mg, 0.34 mmol), an excess of NaBPh4 (0.68 mmol, 0.23 g), 10 mL of THF, 2 mL of the appropriate alcohol (CH3OH or C2H5OH) and an excess of methylpropiolate (1.6 mmol, 144 μL). The reaction mixture was stirred at room temperature for 24 h and then the solvent was removed by evaporation under reduced pressure. The oil obtained was triturated with alcohol (3 mL) until a yellow solid separated out, which was filtered and fractionally crystallized by cooling to −25 °C the solution of the compound in alcohol and enough CH2Cl2 to obtain a saturated solution at room temperature. The first separated solid was pyranylidene complex 1 in 22% average yield (77 mg); ethoxycarbene 2 was the final solid, separated in 62% yield (208 mg) for 2a, 63% (215 mg) for 2b. 1: IR (KBr, cm−1) νCO 1731 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.49–6.87 (m, 35H, Ph), 4.97 (d, JHP = 0.9 Hz, 5H, Cp), AB spin syst., δA 7.06, δB 5.44, JAB = 8.7 Hz (2H, CH), 3.89 (s, 3H, CH3 COOMe), 3.48 (s, 3H, CH3 OMe), 3.43 (d, JHP = 11.4 Hz, 9H, CH3 phos); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 148.33, δB 54.31, JAB = 60.27 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 172.77 (s, C5), 167.93 (s, C6), 165–122 (m, Ph), 140.70 (s, C3), 137.08 (s, C2), 94.57 (s br, C4), 89.15 (s, Cp), 56.73 (s, OCH3), 53.89 (d, JCP = 10.1 Hz, CH3 phos), 53.03 (s, C8); (at −30 °C) δ: 332 (m br, C1); Anal. Calcd for C57H57BO7P2Ru (1027.89): C, 66.60; H, 5.59; Found: C, 66.38; H, 5.71%; ΛM = 53.8 Ω−1 mol−1 cm2; m.p. 155–156 °C (dec). 2a: IR (KBr, cm−1) νCO 1740 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.50–6.87 (m, 35H, Ph), 5.06 (s, 5H, Cp), AB spin syst., δA 4.57, δB 3.92, JAB = 15.6 Hz (2H, CH2), 3.71 (s, 3H, CH3 OMe), 3.60 (s, 3H, CH3 COOMe), 3.43 (d, JHP = 11.4 Hz, 9H, CH3 phos); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 148.83, δB 53.43, JAB = 55.65 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 297.39 (dd, CRu, JCP = 12.3, JCP = 17.7 Hz), 165.05 (s, CO), 165–122 (m, Ph), 91.30 (d, JCP = 1.3 Hz, Cp), 61.98 (s, CH3 COOMe), 61.01 (s, CH2COO), 53.67 (d, JCP = 8.0 Hz, CH3 phos), 53.36 (s, OCH3); Anal. Calcd for C55H57BO6P2Ru (987.87): C, 66.87; H, 5.82; Found: C, 66.66; H, 5.70%; ΛM = 52.5 Ω−1 mol−1 cm2; m.p. 112–113 °C (dec). 2b: IR (KBr, cm−1) νCO 1728 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.49–6.87 (m, 35H, Ph), 5.03 (d, JHP = 1.14 Hz, 5H, Cp), 4.57, 3.94 (d, 2H, CH2COO), 4.23, 3.55 (m, 2H, CH2 OEt), 3.70 (s, 3H, CH3 COOMe), 3.47 (d, JHP = 11.4 Hz, 9H, CH3 phos), 1.12 (t, JHH = 7.0 Hz, 3H, CH3 OEt); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 148.30, δB 53.05, JAB = 56.30 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 297.21 (dd, CRu, JCP = 12.8, JCP = 18.1 Hz), 165.10 (s, CO), 165–122 (m, Ph), 91.05 (dd, JCP = 2.8 Hz, JCP = 1.4 Hz, Cp), 72.97 (d, JCP = 1.0 Hz, CH2 OEt), 61.42 (s, CH2COO), 53.70 (d, JCP = 8.0 Hz, CH3 phos), 53.36 (s, CH3 COOMe), 14.60 (s, CH3 OEt); Anal. Calcd for C56H59BO6P2Ru (1001.89): C, 67.13; H, 5.94; Found: C, 67.02; H, 6.15%; ΛM = 53.2 Ω−1 mol−1 cm2; m.p. 117–118 °C (dec).
C(COOEt)C(H)C(H)C(OEt)O}(PPh3){P(OMe)3}]BPh4 (3) and [Ru(η5-C5H5){C(OMe)(CH2COOEt)}(PPh3){P(OMe)3}]BPh4 (4a).
These complexes were prepared exactly like the related 1 and 2 using ethylpropiolate as a reagent. The two compounds were separated by fractional crystallization by alcohol and CH2Cl2; yield 14% (50 mg) for 3, 65% (221 mg) for 4a. 3: IR (KBr, cm−1) νCO 1733 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.53–6.87 (m, 35H, Ph), 7.14, 5.41 (d, JHH = 9.0 Hz, 2H, CH), 4.91 (d, JHP = 1.0 Hz, 5H, Cp), 4.42, 4.17 (q, JHH = 7.0 Hz, 4H, CH2 OEt), 3.37 (d, JHP = 11.5 Hz, 9H, CH3 phos), 1.26, 1.02 (t, JHH = 7.0 Hz, 6H, CH3 OEt); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 149.86, δB 55.72, JAB = 57.30 Hz; 13C{1H} NMR (CD2Cl2, −30 °C) δ: 355 (br, C1); Anal. Calcd for C59H61BO7P2Ru (1055.94): C, 67.11; H, 5.82; Found: C, 67.23; H, 5.74%; ΛM = 53.9 Ω−1 mol−1 cm2; m.p. 73–74 °C (dec). 4a: IR (KBr, cm−1) νCO 1731 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.85–6.87 (m, 35H, Ph), 5.06 (d, JHP = 1.0 Hz, 5H, Cp), AB spin syst., δA 4.59, δB 3.89, JAB = 15.6 Hz (2H, CH2COO), 4.17 (q, JHH = 7.0 Hz, 2H, CH2 OEt), 3.59 (s, 3H, CH3 OMe), 3.43 (d, JHP = 11.5 Hz, 9H, CH3 phos), 1.28 (t, JHH = 7.0 Hz, 3H, CH3 OEt); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 148.95, δB 53.42, JAB = 55.41 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 297.67 (dd, CRu, JCP = 12.5, JCP = 17.5), 165–122 (m, Ph), 164.51 (s, COOEt), 91.15 (s, Cp), 62.85 (s, CH2COO), 61.97 (s, CH2 OEt), 61.39 (s, OCH3), 53.63 (d, JCP = 8.0 Hz, CH3 phos), 14.26 (s, CH3 OEt); Anal. Calcd for C56H59BO6P2Ru (1001.89): C, 67.13; H, 5.94; Found: C, 66.98; H, 5.86%; ΛM = 51.7 Ω−1 mol−1 cm2; m.p. 114–115 °C (dec).
CC(H)(COOMe)}(PPh3){P(OMe)3}]+OTf− [A].
In a 25 mL three-necked round-bottomed flask were placed 100 mg (0.17 mmol) of RuCl(η5-C5H5)(PPh3)[P(OMe)3], 44 mg (0.17 mmol) of silver triflate, AgOTf, and 7 mL of dichloromethane. The reaction mixture was stirred for 24 h in the dark, filtered to remove solid AgCl, and then an excess of methylpropiolate (0.40 mmol, 36 μL) was added. The resulting solution was stirred for 1 h, and then the solvent was removed under reduced pressure to give an oil, which was characterized as such. 1H NMR (CD2Cl2, 20 °C) δ: 5.27 (s, 5H, Cp), 5.47 (s br, 1H, CH), 3.89 (s, 3H, CH3 COOMe), 3.73 (d, JHP = 11.0 Hz, 9H, CH3 phos); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 138.86, δB 52.20, JAB = 51.28 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 199.71 (dd, Cα), 167.82 (s, CO), 118.50 (s, Cβ), 89.81 (s, Cp), 54.64 (s, CH3 COOMe), 53.11 (d, CH3 phos).
CC(H)(COOMe)}(PPh3){P(OMe)3}]+OTf− [A] with methylpropiolate.
In a 25 mL three-necked round-bottomed flask containing 0.17 mmol of [A] were added 2 mL of dichloromethane, an excess of methylpropiolate (0.40 mmol, 36 μL) and 5 mL of ethanol. The reaction mixture was stirred at room temperature for 24 h and then the solvent was removed under reduced pressure to give an oil, the 1H and 31P NMR data of which indicated the presence of pyranylidene 1 and ethoxycarbene 2b in about 1:4 ratio.
C(COOMe)C(H)C(H)C(OMe)O}(PPh3){P(OMe)3}]BPh4 (1) and [Ru(η5-C5H5){C(OR2)(CH2COOMe)}(PPh3){P(OMe)3}]BPh4 (2) [R2 = Me (a), Et (b)].
In a 25 mL three-necked round-bottomed flask were placed solid samples of RuCl(η5-C5H5)(PPh3)[P(OMe)3] (200 mg, 0.34 mmol), an excess of NaBPh4 (0.68 mmol, 0.23 g), 10 mL of THF, 2 mL of the appropriate alcohol (CH3OH or C2H5OH) and an excess of methylpropiolate (1.6 mmol, 144 μL). The reaction mixture was stirred at room temperature for 24 h and then the solvent was removed by evaporation under reduced pressure. The oil obtained was triturated with alcohol (3 mL) until a yellow solid separated out, which was filtered and fractionally crystallized by cooling to −25 °C the solution of the compound in alcohol and enough CH2Cl2 to obtain a saturated solution at room temperature. The first separated solid was pyranylidene complex 1 in 22% average yield (77 mg); ethoxycarbene 2 was the final solid, separated in 62% yield (208 mg) for 2a, 63% (215 mg) for 2b. 1: IR (KBr, cm−1) νCO 1731 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.49–6.87 (m, 35H, Ph), 4.97 (d, JHP = 0.9 Hz, 5H, Cp), AB spin syst., δA 7.06, δB 5.44, JAB = 8.7 Hz (2H, CH), 3.89 (s, 3H, CH3 COOMe), 3.48 (s, 3H, CH3 OMe), 3.43 (d, JHP = 11.4 Hz, 9H, CH3 phos); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 148.33, δB 54.31, JAB = 60.27 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 172.77 (s, C5), 167.93 (s, C6), 165–122 (m, Ph), 140.70 (s, C3), 137.08 (s, C2), 94.57 (s br, C4), 89.15 (s, Cp), 56.73 (s, OCH3), 53.89 (d, JCP = 10.1 Hz, CH3 phos), 53.03 (s, C8); (at −30 °C) δ: 332 (m br, C1); Anal. Calcd for C57H57BO7P2Ru (1027.89): C, 66.60; H, 5.59; Found: C, 66.38; H, 5.71%; ΛM = 53.8 Ω−1 mol−1 cm2; m.p. 155–156 °C (dec). 2a: IR (KBr, cm−1) νCO 1740 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.50–6.87 (m, 35H, Ph), 5.06 (s, 5H, Cp), AB spin syst., δA 4.57, δB 3.92, JAB = 15.6 Hz (2H, CH2), 3.71 (s, 3H, CH3 OMe), 3.60 (s, 3H, CH3 COOMe), 3.43 (d, JHP = 11.4 Hz, 9H, CH3 phos); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 148.83, δB 53.43, JAB = 55.65 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 297.39 (dd, CRu, JCP = 12.3, JCP = 17.7 Hz), 165.05 (s, CO), 165–122 (m, Ph), 91.30 (d, JCP = 1.3 Hz, Cp), 61.98 (s, CH3 COOMe), 61.01 (s, CH2COO), 53.67 (d, JCP = 8.0 Hz, CH3 phos), 53.36 (s, OCH3); Anal. Calcd for C55H57BO6P2Ru (987.87): C, 66.87; H, 5.82; Found: C, 66.66; H, 5.70%; ΛM = 52.5 Ω−1 mol−1 cm2; m.p. 112–113 °C (dec). 2b: IR (KBr, cm−1) νCO 1728 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.49–6.87 (m, 35H, Ph), 5.03 (d, JHP = 1.14 Hz, 5H, Cp), 4.57, 3.94 (d, 2H, CH2COO), 4.23, 3.55 (m, 2H, CH2 OEt), 3.70 (s, 3H, CH3 COOMe), 3.47 (d, JHP = 11.4 Hz, 9H, CH3 phos), 1.12 (t, JHH = 7.0 Hz, 3H, CH3 OEt); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 148.30, δB 53.05, JAB = 56.30 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 297.21 (dd, CRu, JCP = 12.8, JCP = 18.1 Hz), 165.10 (s, CO), 165–122 (m, Ph), 91.05 (dd, JCP = 2.8 Hz, JCP = 1.4 Hz, Cp), 72.97 (d, JCP = 1.0 Hz, CH2 OEt), 61.42 (s, CH2COO), 53.70 (d, JCP = 8.0 Hz, CH3 phos), 53.36 (s, CH3 COOMe), 14.60 (s, CH3 OEt); Anal. Calcd for C56H59BO6P2Ru (1001.89): C, 67.13; H, 5.94; Found: C, 67.02; H, 6.15%; ΛM = 53.2 Ω−1 mol−1 cm2; m.p. 117–118 °C (dec).
C(COOEt)C(H)C(H)C(OEt)O}(PPh3){P(OMe)3}]BPh4 (3) and [Ru(η5-C5H5){C(OMe)(CH2COOEt)}(PPh3){P(OMe)3}]BPh4 (4a).
These complexes were prepared exactly like the related 1 and 2 using ethylpropiolate as a reagent. The two compounds were separated by fractional crystallization by alcohol and CH2Cl2; yield 14% (50 mg) for 3, 65% (221 mg) for 4a. 3: IR (KBr, cm−1) νCO 1733 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.53–6.87 (m, 35H, Ph), 7.14, 5.41 (d, JHH = 9.0 Hz, 2H, CH), 4.91 (d, JHP = 1.0 Hz, 5H, Cp), 4.42, 4.17 (q, JHH = 7.0 Hz, 4H, CH2 OEt), 3.37 (d, JHP = 11.5 Hz, 9H, CH3 phos), 1.26, 1.02 (t, JHH = 7.0 Hz, 6H, CH3 OEt); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 149.86, δB 55.72, JAB = 57.30 Hz; 13C{1H} NMR (CD2Cl2, −30 °C) δ: 355 (br, C1); Anal. Calcd for C59H61BO7P2Ru (1055.94): C, 67.11; H, 5.82; Found: C, 67.23; H, 5.74%; ΛM = 53.9 Ω−1 mol−1 cm2; m.p. 73–74 °C (dec). 4a: IR (KBr, cm−1) νCO 1731 (s); 1H NMR (CD2Cl2, 20 °C) δ: 7.85–6.87 (m, 35H, Ph), 5.06 (d, JHP = 1.0 Hz, 5H, Cp), AB spin syst., δA 4.59, δB 3.89, JAB = 15.6 Hz (2H, CH2COO), 4.17 (q, JHH = 7.0 Hz, 2H, CH2 OEt), 3.59 (s, 3H, CH3 OMe), 3.43 (d, JHP = 11.5 Hz, 9H, CH3 phos), 1.28 (t, JHH = 7.0 Hz, 3H, CH3 OEt); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 148.95, δB 53.42, JAB = 55.41 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 297.67 (dd, CRu, JCP = 12.5, JCP = 17.5), 165–122 (m, Ph), 164.51 (s, COOEt), 91.15 (s, Cp), 62.85 (s, CH2COO), 61.97 (s, CH2 OEt), 61.39 (s, OCH3), 53.63 (d, JCP = 8.0 Hz, CH3 phos), 14.26 (s, CH3 OEt); Anal. Calcd for C56H59BO6P2Ru (1001.89): C, 67.13; H, 5.94; Found: C, 66.98; H, 5.86%; ΛM = 51.7 Ω−1 mol−1 cm2; m.p. 114–115 °C (dec).
CC(H)(COOMe)}(PPh3){P(OMe)3}]+OTf− [A].
In a 25 mL three-necked round-bottomed flask were placed 100 mg (0.17 mmol) of RuCl(η5-C5H5)(PPh3)[P(OMe)3], 44 mg (0.17 mmol) of silver triflate, AgOTf, and 7 mL of dichloromethane. The reaction mixture was stirred for 24 h in the dark, filtered to remove solid AgCl, and then an excess of methylpropiolate (0.40 mmol, 36 μL) was added. The resulting solution was stirred for 1 h, and then the solvent was removed under reduced pressure to give an oil, which was characterized as such. 1H NMR (CD2Cl2, 20 °C) δ: 5.27 (s, 5H, Cp), 5.47 (s br, 1H, CH), 3.89 (s, 3H, CH3 COOMe), 3.73 (d, JHP = 11.0 Hz, 9H, CH3 phos); 31P{1H} NMR (CD2Cl2, 20 °C) δ: AB spin syst., δA 138.86, δB 52.20, JAB = 51.28 Hz; 13C{1H} NMR (CD2Cl2, 20 °C) δ: 199.71 (dd, Cα), 167.82 (s, CO), 118.50 (s, Cβ), 89.81 (s, Cp), 54.64 (s, CH3 COOMe), 53.11 (d, CH3 phos).
CC(H)(COOMe)}(PPh3){P(OMe)3}]+OTf− [A] with methylpropiolate.
In a 25 mL three-necked round-bottomed flask containing 0.17 mmol of [A] were added 2 mL of dichloromethane, an excess of methylpropiolate (0.40 mmol, 36 μL) and 5 mL of ethanol. The reaction mixture was stirred at room temperature for 24 h and then the solvent was removed under reduced pressure to give an oil, the 1H and 31P NMR data of which indicated the presence of pyranylidene 1 and ethoxycarbene 2b in about 1:4 ratio.
Crystal structure determination
Crystallographic data for 1 were collected at room temperature using a Bruker Smart 6000 CCD detector and Cu-Kα radiation (λ = 1.54178 Å) generated by an Incoatec microfocus source equipped with Incoatec Quazar MX optics. The software APEX225 was used for collecting frames of data, indexing reflections and the determination of lattice parameters, SAINT25 was used for integration of the intensity of reflections, and SADABS25 was used for scaling and empirical absorption correction. The crystallographic treatment was performed with the Oscail program.26 The structure was solved by Patterson methods and refined by full-matrix least-squares based on F2.27 Non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were included in idealized positions and refined with isotropic displacement parameters. Figures were drawn with ORTEP-3 for Windows.28 Details of crystal data and structural refinement are given in Table 2. CCDC 1046060 contains the supplementary crystallographic data for this paper.
| Empirical formula | C58H57BO7P2Ru |
| Moiety formula | C34H37O7P2Ru, C24H20B |
| Formula weight | 1039.86 |
| Temperature | 296(2) K |
| Wavelength | 1.54178 Å |
| Crystal system | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| Unit cell dimensions | a = 10.9219(8) Å |
| b = 14.7181(11) Å | |
| c = 16.2149(12) Å | |
| α = 81.410(4)° | |
| β = 87.130(4)° | |
| γ = 87.354(3)° | |
| Volume | 2572.1(3) Å3 |
| Z | 2 |
| Density (calculated) | 1.343 Mg m−3 |
| Absorption coefficient | 3.474 mm−1 |
| F(000) | 1080 |
| Crystal size | 0.140 × 0.096 × 0.051 mm |
| Theta range for data collection | 2.76 to 67.88° |
| Index ranges | −12 ≤ h ≤ 13 |
| −17 ≤ k ≤ 17 | |
| −18 ≤ l ≤ 19 | |
| Reflections collected | 62732 |
| Independent reflections | 9003 [R(int) = 0.0805] |
| Reflections observed (>2σ) | 7766 |
| Data completeness | 0.963 |
| Absorption correction | Semi-empirical from equivalents |
| Max. and min. transmission | 0.6970 and 0.5239 |
| Refinement method | Full-matrix least-squares on F2 |
| Data/restraints/parameters | 9003/0/627 |
| Goodness-of-fit on F2 | 1.034 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0497 |
| wR2 = 0.1305 | |
| R indices (all data) | R 1 = 0.0577 |
| wR2 = 0.1399 | |
| Largest diff. peak and hole | 1.294 and −0.311 e Å−3 |
Computational details
The computational geometry optimization of the complexes was carried out without symmetry constraints, using the hyper-GGA M06 functional29 in combination with a polarized triple-ζ quality basis set composed of the 6-311G(d,p) set on the light atoms and the ECP-based LANL2TZ(f) set on the ruthenium centre.30 The “restricted” formalism was applied31 and the CPCM implicit solvation model for ethanol was added to all the calculations.32 All the stationary points were characterized by IR simulation (harmonic approximation), from which thermodynamic data were computed, considering T = 298.15 K.31 The software used was Gaussian 09.33 Preliminary geometry optimizations and proton affinity calculations were performed in vacuo with the hybrid DFT functional EDF234 and the polarized double-ζ quality basis set LACVP** (LANL2DZ ECP on ruthenium).35 The software used for EDF2 calculations was Spartan 08.36 All the computational optimizations were performed on an Intel-based x86-64 workstation.
Acknowledgements
Thanks go to Mrs Daniela Baldan from the Università Ca’ Foscari Venezia (Italy) for technical assistance.References
- (a) I. Fernández, F. P. Cossío and M. A. Sierra, Acc. Chem. Res., 2011, 44, 479 CrossRef PubMed; (b) J. W. Herndon, Coord. Chem. Rev., 2010, 254, 103 CrossRef CAS PubMed; (c) K. H. Dötz and J. Stendel Jr., Chem. Rev., 2009, 109, 3227 CrossRef PubMed; (d) M. A. Esteruelas, A. M. López and M. Oliván, Coord. Chem. Rev., 2007, 251, 795 CrossRef CAS PubMed; (e) J. Barluenga, J. Santamaría and M. Tomás, Chem. Rev., 2004, 104, 2259 CrossRef CAS PubMed; (f) M. A. Esteruelas, A. I. González, A. M. López and E. Oñate, Organometallics, 2003, 22, 414 CrossRef CAS; (g) C.-M. Che and J.-S. Huang, Coord. Chem. Rev., 2002, 231, 151 CrossRef CAS; (h) R. R. Schrock, J. Chem. Soc., Dalton Trans., 2001, 2541 RSC; (i) M. A. Sierra, Chem. Rev., 2000, 100, 3591 CrossRef CAS PubMed; (j) A. de Meijere, H. Schirmer and M. Duetsch, Angew. Chem., Int. Ed., 2000, 39, 3964 CrossRef CAS; (k) S. F. Vyboishchikov and G. Frenking, Chem. – Eur. J., 1998, 4, 1428 CrossRef CAS; (l) D. G. Musaev, K. Morokuma and N. Koga, J. Chem. Phys., 1993, 99, 7859 CrossRef CAS PubMed; (m) D. S. Marynick and C. M. Kirkpatrick, J. Am. Chem. Soc., 1985, 107, 1993 CrossRef CAS; (n) T. E. Taylor and M. B. Hall, J. Am. Chem. Soc., 1984, 106, 1576 CrossRef CAS.
- (a) Q.-F. Wang, W. X. Zhang, C. Lin and Z. F. Xi, Chin. Sci. Bull., 2010, 55, 2915 CrossRef CAS; (b) K. N. Juneau, L. S. Hegedus and F. W. Roepke, J. Am. Chem. Soc., 1989, 111, 4762 CrossRef CAS; (c) T. L. Gilchrist, R. Levingston, C. W. Rees, E. von Angerer and R. Robinson, J. Chem. Soc., Perkin Trans. 1, 1973, 2535 RSC.
- (a) N. Szesni, B. Weibert and H. Fischer, Inorg. Chim. Acta, 2006, 359, 617 CrossRef CAS PubMed; (b) R. Aumann, A. G. Meyer and R. Fröhlich, J. Am. Chem. Soc., 1996, 118, 10853 CrossRef CAS; (c) R. Aumann, R. A. G. Meyer and R. Fröhlich, Organometallics, 1996, 15, 5018 CrossRef CAS; (d) L. Jordi, F. Camps, S. Ricart, J. M. Viñas, J. M. Moretó, M. Mejias and E. Molins, J. Organomet. Chem., 1995, 494, 53 CrossRef CAS; (e) F. Camps, J. M. Moretó, S. Ricart, J. M. Viñas, E. Molins and C. Miravitlles, J. Chem. Soc., Chem. Commun., 1989, 1560 RSC.
- (a) H. Kusama, T. Sawada, A. Okita, F. Shiozawa and N. Iwasawa, Org. Lett., 2006, 8, 1077 CrossRef CAS PubMed; (b) N. Iwasawa, M. Shido, K. Maeyama and H. Kusama, J. Am. Chem. Soc., 2000, 122, 10226 CrossRef CAS; (c) K. Ohe, K. Miki, T. Yokoi, F. Nishino and S. Uemura, Organometallics, 2000, 19, 5525 CrossRef CAS.
- (a) Q. Wang, L. Lin, C. Lin, H. Sun, W.-X. Zhang and Z. Xi, Dalton Trans., 2009, 10433 RSC; (b) Q. Wang, W.-X. Zhang and Z. Xi, Organometallics, 2008, 27, 3627 CrossRef CAS.
- R. Aumann, K. Roths, B. Jasper and R. Fröhlich, Organometallics, 1996, 15, 1257 CrossRef CAS.
- (a) H. P. Wu, R. Aumann, R. Fröhlich and B. Wibbeling, Chem. – Eur. J., 2002, 8, 910 CrossRef CAS; (b) S. L. B. Wang and W. D. Wulff, J. Am. Chem. Soc., 1990, 112, 4550 CrossRef CAS.
- L. Jordi, J. M. Moretó, S. Ricart, J. M. Viñas, E. Molins and C. Miravitlles, J. Organomet. Chem., 1993, 444, C28 CrossRef CAS.
- B. Baeza, L. Casarrubios, M. Gómez-Gallego, M. A. Sierra and M. Olivan, Organometallics, 2010, 29, 1607 CrossRef CAS.
- (a) G. Albertin, S. Antoniutti, M. Bortoluzzi, A. Botter and J. Castro, Dalton Trans., 2015, 44, 3439 RSC; (b) G. Albertin, S. Antoniutti, A. Botter, J. Castro and M. Giacomello, Organometallics, 2014, 33, 3570 CAS; (c) G. Albertin, S. Antoniutti, J. Castro and S. Da Lio, Organometallics, 2013, 32, 3651 CrossRef CAS; (d) G. Albertin, S. Antoniutti, D. Baldan, J. Castro and G. Comparin, Organometallics, 2013, 32, 3157 CrossRef CAS; (e) G. Albertin, S. Antoniutti and J. Castro, Organometallics, 2011, 30, 1914 CrossRef CAS; (f) G. Albertin, S. Antoniutti and J. Castro, Organometallics, 2011, 30, 1558 CrossRef CAS; (g) G. Albertin, S. Antoniutti, F. Callegaro and J. Castro, Organometallics, 2009, 28, 4475 CrossRef CAS; (h) G. Albertin, S. Antoniutti, A. Bacchi, G. Pelizzi and G. Zanardo, Organometallics, 2008, 27, 4407 CrossRef CAS.
- (a) J. M. Lynam, Chem. – Eur. J., 2010, 10, 8238 CrossRef PubMed; (b) M. C. Puerta and P. Valerga, Coord. Chem. Rev., 1999, 193–195, 977 CrossRef CAS; (c) M. I. Bruce, Chem. Rev., 1991, 91, 197 CrossRef CAS.
- A. Moser, K. Range and D. M. York, J. Phys. Chem. B, 2010, 114, 13911 CrossRef CAS PubMed.
- (a) I. de los Rios, M. Jiménez Tenorio, M. C. Puerta and P. Valerga, J. Am. Chem. Soc., 1997, 119, 6529 CrossRef CAS; (b) V. Cadierno, M. P. Gamasa, J. Gimeno, C. González-Bernardo, E. Perez-Carreño and S. Garcia-Granda, Organometallics, 2001, 20, 5177 CrossRef CAS.
- M. P. Gamasa, J. Gimeno, B. M. Martin-Vaca, J. Borge, S. Garcia-Granda and E. Perez-Carreño, Organometallics, 1994, 13, 4045 CrossRef CAS.
- W. J. Geary, Coord. Chem. Rev., 1971, 7, 81 CrossRef CAS.
- A search in the CCDC database CSD version 5.35 (updated in November 2013) gives 1512 CpRu compounds. The mean value for the Ru–C bond distance is 2.219 Å.
- G. Albertin, S. Antoniutti, J. Castro and F. Scapinello, J. Organomet. Chem., 2014, 751, 412 CrossRef CAS PubMed.
- F. L. Joslin, J. T. Mague and D. M. Roundhill, Organometallics, 1991, 10, 521 CrossRef CAS.
- C.-W. Chang, Y.-C. Lin, G.-H. Lee, S.-L. Huang and Y. Wang, Organometallics, 1998, 17, 2534 CrossRef CAS.
- V. Cadierno, S. Conejero, M. P. Gamasa, J. Gimeno, L. R. Falvello and R. M. Llusar, Organometallics, 2002, 21, 3716 CrossRef CAS.
- D. J. Armitt, M. I. Bruce, J. C. Morris, B. K. Nicholson, C. R. Parker, B. W. Skelton and N. N. Zaitseva, Organometallics, 2011, 30, 5452 CrossRef CAS.
- See, for example: (a) Y.-H. Lo, T.-H. Wang, C.-Y. Lee and Y.-H. Feng, Organometallics, 2012, 31, 6887 CrossRef CAS; (b) E. Bustelo, M. Jiménez-Tenorio, M. C. Puerta and P. Valerga, Organometallics, 2007, 26, 4300 CrossRef CAS.
- G. Balacco, http://www.inmr.net/.
- G. Albertin, S. Antoniutti, A. Bacchi, G. Pelizzi and G. Zanardo, Organometallics, 2008, 27, 4407 CrossRef CAS.
- Bruker, APEX2, SMART, SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2007 Search PubMed.
- P. McArdle, J. Appl. Crystallogr., 1995, 28, 65 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215 CrossRef CAS.
- (a) A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639 CrossRef CAS PubMed; (b) L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029 CrossRef CAS.
- (a) C. J. Cramer, Essentials of Computational Chemistry, John Wiley and Sons, Chichester, 2nd edn, 2004 Search PubMed; (b) F. Jensen, Introduction to Computational Chemistry, John Wiley and Sons, Chichester, 2nd edn, 2007 Search PubMed.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- C. Y. Lin, M. W. George and P. M. W. Gill, Aust. J. Chem., 2004, 57, 365 CrossRef CAS.
- (a) W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS PubMed; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS PubMed; (c) M. Dolg, Modern Methods and Algorithms of Quantum Chemistry, ed. J. Grotendorst, John Neumann Institute for Computing, NIC Series, Jülich, 2000, vol. 1, p. 479 Search PubMed.
- Spartan ‘08, version 1.1.1, Wavefunction, Inc., Irvine, CA, 2009 RSC Except for molecular mechanics and semi-empirical models, the calculation methods used in Spartan have been documented in: Y. Shao, et al. , Phys. Chem. Chem. Phys., 2006, 8, 3172 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: xyz file for DFT-optimized structures; NMR spectra (pdf). CCDC 1046060. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00418g |
| This journal is © The Royal Society of Chemistry 2015 |