
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStable and color tunable emission properties based on non-cyclometalated gold(III) complexes†
Michael
Bachmann
,
Olivier
Blacque
and
Koushik
Venkatesan
*
Department of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. E-mail: venkatesan.koushik@chem.uzh.ch
First published on 21st April 2015
Abstract
Stable and emission tunable non-cyclometalated gold(III) triaryl complexes of the type [(L)Au(C6F5)3] [L = 2-(2,4-difluorophenylpyridine) (1), 4-phenylpyridine (2), 2-phenylpyridine (3), 2-phenylisoquinoline (4), 2-thienylpyridine (5)] were synthesized starting from a common precursor complex [(THT)Au(C6F5)3] [THT = tetrahydrothiophene] in good to modest yields. Extensive characterization of the complexes by various nuclear magnetic resonance spectroscopy techniques and elemental analysis further corroborated the single-crystal X-ray diffraction studies. The complexes displayed room temperature phosphorescence in the neat solid and in 2-MeTHF at 77 K. Detailed photophysical investigations of the complexes in the neat solid and at 77 K revealed the successful tuning of the emission maxima with modest quantum yields across the visible part of the electromagnetic spectrum depending on the electronic properties of the heterocyclic ligands. DFT (Density Functional Theory) and TDDFT (Time Dependent Density Functional Theory) calculations were performed to discern the composition of the excited state as well as confirm the obtained relative emission energies upon substitution with electronically different ligands. The obtained diverse emissive behavior of the complexes combined with the ease of synthesis illustrate the generality and applicability of the design approach to obtain emissive gold(III) complexes devoid of cyclometalation.
Introduction
Luminescent gold(III) complexes have been gaining increasing attention in recent years as next generation triplet phosphors for applications in phosphorescent organic light emitting devices (PhOLEDs).1 In the recent past, poor stability and non-radiative deactivation arising due to reductive elimination and low-lying d–d states that are energetically close to the potentially emissive intraligand (IL) or metal-to-ligand charge transfer (MLCT) states, respectively, have hindered the investigation of Au(III) complexes in contrast to the extensively explored isoelectronic Pt(II) complexes.1a,2 However, such deficiencies have been overcome by Yam and co-workers using strong σ-donating ligands and a tridentate ligand core resulting in stable and luminescent Au(III) complexes. The majority of the strategies to obtain emissive Au(III) complexes so far have relied on a tridentate biscyclometalated or a bidentate monocyclometalated ligand scaffold appended to the gold(III) center.1,3 Despite the recent progress, very limited success has been achieved on luminescent Au(III) complexes devoid of cyclometalation and the only known ones are cationic in nature.1a,4 To the best of our knowledge, there are no luminescent neutral Au(III) complexes that are devoid of cyclometalation. It was hypothesized that the combination of conjugated heterocyclic ligands with the previously known Au(III) pentafluorophenyl motif would not only render the resulting Au(III) complexes emissive through better spatial orientation and electronic overlap of the frontier orbitals of the gold centre and the conjugated heterocyclic ligand, but also was expected to result in stable compounds owing to the presence of the pentafluorophenyl ligands due to increased π-back bonding. The chosen design strategy was also anticipated to allow further tuning of the emission properties by utilizing various conjugated heterocycles with different electronic properties. The introduction of different ligands such as 2-phenylpyridine, 4-phenylpyridine, 2-thienylpyridine, 2-(2,4-difluorophenylpyridine), and 2-phenylisoquinoline was chosen to evaluate the effect of highly efficient interligand charge transfer on the emission quantum yields as well as on the phosphorescent emission maxima of the resulting complexes. Such a general approach to harness room temperature phosphorescence from Au(III) complexes bearing no cyclometalating ligands is demonstrated for the first time.In this work, we report on the synthesis, structural and photophysical investigations of a series of stable Au(III) complexes devoid of cyclometalation that exhibit room temperature phosphorescence in the neat solid and in 2-MeTHF at 77 K. The introduction of the different conjugated heterocycles results in widely tuneable emission properties across the visible part of the electromagnetic spectrum, making the compounds amenable for light emitting applications.
Results and discussion
The different aryl pyridine Au(III)triaryl complexes 1–5 were synthesized using an already reported procedure that involves the treatment of 2 equiv. of the appropriate aryl pyridine ligand with the (THT)Au(III)triaryl complex in Et2O (Scheme 1).5 The complexes were obtained as amorphous powders in 52–71% yield after purification by column chromatography. 1H NMR studies carried out on the final products revealed characteristic resonances for the α-proton on the pyridine in the region 8.06–8.87 ppm strongly indicative of the coordinated aryl pyridine ligand to the Au(III) trispentafluoro phenyl center. Extensive characterization of the complexes has been carried out by 1H, 13C and 19F NMR studies as well as elemental analysis. | ||
| Scheme 1 Synthesis of complexes 1–5. | ||
Single crystal X-ray diffraction studies performed for all Au(III) complexes further corroborated the structural assignment from the NMR studies. The perspective views of all the complexes are shown in Fig. 1. Crystallographic details are provided in Table 1. The analysis of the molecular structures reveals a distorted square-planar geometry around the metal center. The distortion attributed to the steric encumbrance of the ligands is in fact rather small since all trans N–Au–C and C–Au–C bond angles were found close to 180° in the range between 174.61(15) and 179.43(10)°, except the C–Au–C bond angle in 4 which is slightly smaller with 171.30(9)° (Table 2). The Au–N bond distance among the different complexes is not significantly altered by the presence of the electronically different pyridine ligands. Nevertheless, it is shorter in the crystal structure of 2 with 2.068(7) and 2.069(7) Å (two crystallographically independent molecules in the asymmetric unit) than in the other structures for which the bond distance lies in the range of 2.101(2)–2.115(2) Å. Interestingly, the Au–C bond distance is always shorter for the pentafluorophenyl ligand trans to the heterocycle (1.990(8)–2.017(4) Å) than for the other two pentafluorophenyl ligands which are trans to each other (2.040(9)–2.081(10) Å) with an average difference of about 0.05 Å within the same structure. It is worth noting that when the second ring of the heterocycle is in the ortho position relative to the nitrogen atom, which is the case for complexes 1, 3, 4 and 5, that ring is involved in an intramolecular π⋯π interaction with a pentafluorophenyl ligand characterized by a centroid–centroid distance in the range of 3.888(2)–4.124(3) Å and almost parallel rings making dihedral angles in the range of 12.9(3)–17.12(16)° between the corresponding mean planes (Tables S16–S20, ESI†).
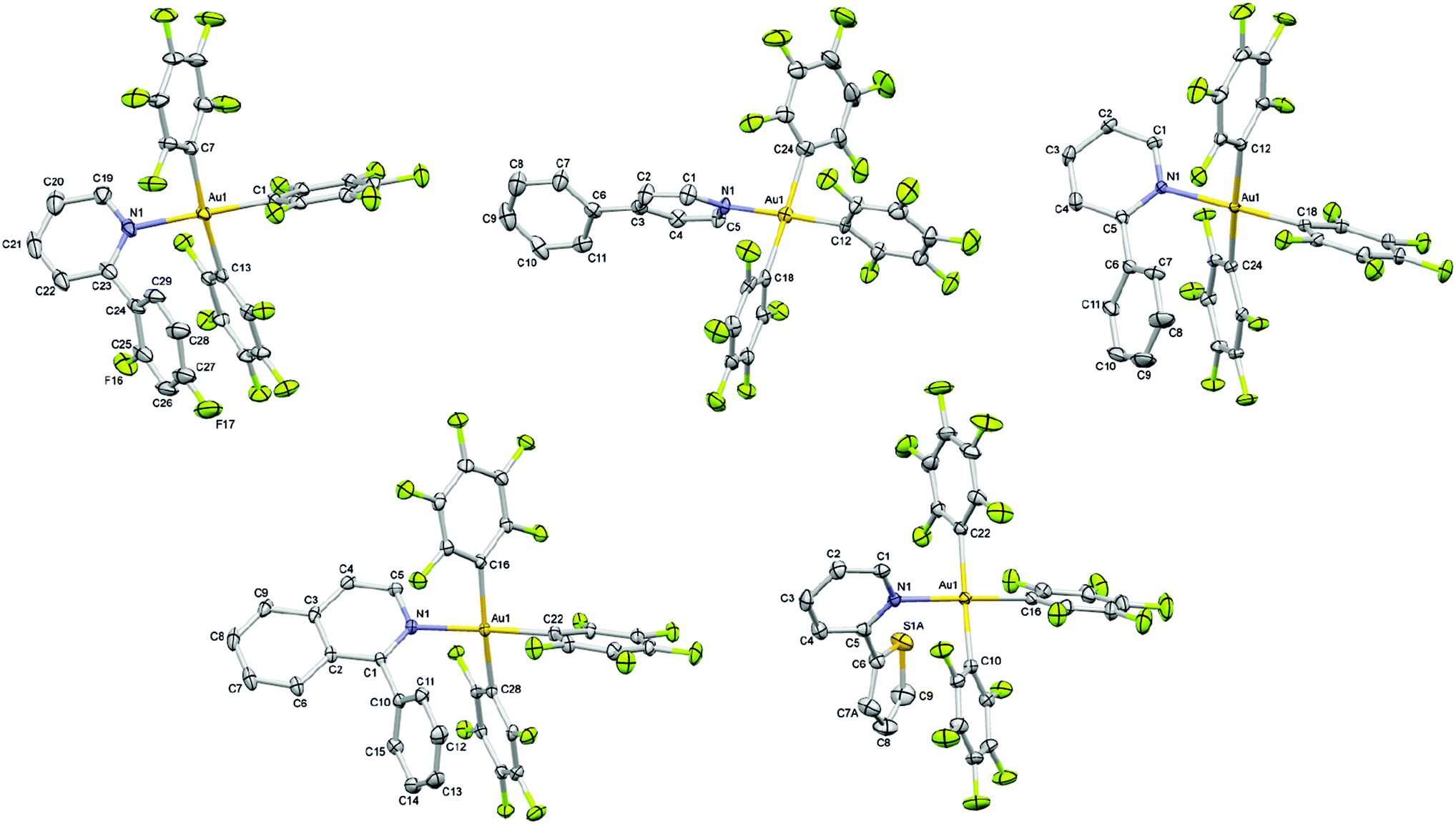 | ||
| Fig. 1 X-ray crystal structures of 1–5 with the selective atomic numbering scheme. Thermal displacement ellipsoids are drawn at the 30% probability level. Hydrogen atoms and solvent molecules are omitted for clarity. Only one of the two crystallographically independent molecules of 2 is presented. | ||
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| CCDC number | 1024826 | 1024827 | 1024828 | 1024829 | 1024830 |
| Empirical formula | C59H16Au2Cl2F34N2 | C119H43Au4F60N4 | C29H9AuF15N | C70H32Au2F30N2O | C27H7AuF15NS |
| Formula weight | 1863.57 | 3456.44 | 853.34 | 1880.91 | 859.36 |
| Temperature/K | 183(1) | 183(1) | 183(1) | 183(1) | 183(1) |
| Crystal system | Triclinic | Triclinic | Monoclinic | Triclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/c |
P |
P21/c |
| a/Å | 8.4911(4) | 13.4052(8) | 7.9254(4) | 11.2092(3) | 7.9119(2) |
| b/Å | 10.3695(4) | 13.8638(6) | 18.503(4) | 12.1878(3) | 18.0726(4) |
| c/Å | 17.1054(11) | 16.8973(12) | 18.4895(11) | 12.5527(3) | 18.4278(4) |
| α/° | 98.487(4) | 83.044(4) | 90 | 104.977(2) | 90 |
| β/° | 100.945(5) | 68.830(6) | 97.579(5) | 106.125(2) | 97.163(2) |
| γ/° | 92.939(4) | 80.730(4) | 90 | 98.915(2) | 90 |
| Volume/Å3 | 1457.63(13) | 2883.4(3) | 2687.7(6) | 1543.23(7) | 2614.40(11) |
| Z | 1 | 1 | 4 | 1 | 4 |
| ρ calc/g cm−3 | 2.123 | 1.991 | 2.109 | 2.024 | 2.183 |
| μ/mm−1 | 5.271 | 5.222 | 5.601 | 4.889 | 5.835 |
| F(000) | 882.0 | 1641.0 | 1616.0 | 902.0 | 1624.0 |
| Crystal size/mm3 | 0.33 × 0.26 × 0.13 | 0.27 × 0.15 × 0.03 | 0.47 × 0.31 × 0.25 | 0.47 × 0.19 × 0.15 | 0.25 × 0.15 × 0.14 |
| Radiation | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) | MoKα (λ = 0.71073) |
| 2θ range for data collection/° | 5.8 to 56.6 | 5.6 to 50.7 | 5.6 to 61.0 | 5.7 to 61.0 | 5.7 to 52.7 |
| Index ranges | −11 ≤ h ≤ 11, −13 ≤ k ≤ 13, −22 ≤ l ≤ 22 | −16 ≤ h ≤ 17, −17 ≤ k ≤ 18, −22 ≤ l ≤ 22 | −11 ≤ h ≤ 11, −26 ≤ k ≤ 26, −26 ≤ l ≤ 26 | −16 ≤ h ≤ 16, −17 ≤ k ≤ 17, −17 ≤ l ≤ 17 | −9 ≤ h ≤ 9, −22 ≤ k ≤ 22, −23 ≤ l ≤ 23 |
| Reflections collected | 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 202 202 |
33922 |
48393 |
27853 |
36456 |
| Independent reflections | 7227 [Rint = 0.049, Rsigma = 0.053] | 10536 [Rint = 0.066, Rsigma = 0.099] |
8205 [Rint = 0.050, Rsigma = 0.032] | 9409 [Rint = 0.038, Rsigma = 0.037] | 5337 [Rint = 0.054, Rsigma = 0.028] |
| Data/restraints/parameters | 7227/39/470 | 10536/400/872 |
8205/0/415 | 9409/0/496 | 5337/2/407 |
| Goodness-of-fit on F2 | 1.049 | 1.031 | 1.036 | 1.066 | 1.039 |
| Final R indexes [I ≥ 2σ(I)] | R 1 = 0.0349, wR2 = 0.0731 | R 1 = 0.0552, wR2 = 0.1197 | R 1 = 0.0246, wR2 = 0.0568 | R 1 = 0.0242, wR2 = 0.0571 | R 1 = 0.0218, wR2 = 0.0536 |
| Final R indexes [all data] | R 1 = 0.0448, wR2 = 0.0775 | R 1 = 0.0800, wR2 = 0.1326 | R 1 = 0.0310, wR2 = 0.0597 | R 1 = 0.0270, wR2 = 0.0587 | R 1 = 0.0241, wR2 = 0.0549 |
| Largest diff. peak/hole/e Å−3 | 1.35/−0.88 | 2.27/−1.08 | 1.85/−0.93 | 1.33/−0.93 | 1.40/−0.68 |
| Distance | Angle | |||||
|---|---|---|---|---|---|---|
| Complex 1 | ||||||
| Au1 | C1 | 2.017(4) | C1 | Au1 | N1 | 177.55(15) |
| Au1 | C7 | 2.078(4) | C7 | Au1 | C13 | 174.61(15) |
| Au1 | C13 | 2.071(4) | ||||
| Au1 | N1 | 2.113(4) | ||||
| Complex 2 | ||||||
| Au1 | C12 | 1.990(8) | C12 | Au1 | N1 | 177.7(3) |
| Au1 | C18 | 2.065(9) | C18 | Au1 | C24 | 175.0(3) |
| Au1 | C24 | 2.081(10) | ||||
| Au1 | N1 | 2.068(7) | ||||
| Au2 | C41 | 2.050(9) | C47 | Au2 | N2 | 177.4(3) |
| Au2 | C53 | 2.040(9) | C41 | Au2 | C53 | 175.1(3) |
| Au2 | C47 | 2.012(9) | ||||
| Au2 | N2 | 2.069(7) | ||||
| Complex 3 | ||||||
| Au1 | C12 | 2.074(2) | C18 | Au1 | N1 | 179.43(10) |
| Au1 | C18 | 2.011(2) | C12 | Au1 | C24 | 175.89(10) |
| Au1 | C24 | 2.054(2) | ||||
| Au1 | N1 | 2.109(2) | ||||
| Complex 4 | ||||||
| Au1 | C16 | 2.077(2) | C22 | Au1 | N1 | 176.97(8) |
| Au1 | C22 | 2.012(2) | C16 | Au1 | C28 | 171.30(9) |
| Au1 | C28 | 2.059(2) | ||||
| Au1 | N1 | 2.101(2) | ||||
| Complex 5 | ||||||
| Au1 | C10 | 2.058(3) | C16 | Au1 | N1 | 178.84(11) |
| Au1 | C16 | 2.016(3) | C10 | Au1 | C22 | 174.81(12) |
| Au1 | C22 | 2.069(3) | ||||
| Au1 | N1 | 2.115(2) | ||||
The complexes were found to be stable in air and in common organic solvents for extended periods and no signs of degradation were observed. This is consistent with the behaviour observed from the thermogravimetric analysis (TGA) studies carried out on an exemplary compound 3 that revealed the onset of total degradation (Td) at 260 °C. In spite of the monodentate ligands coordinated to the gold(III) centre, the thermal stability exhibited by this complex is quite comparable to the monocyclometalated complexes reported earlier.6 This stability can be attributed to the electron withdrawing pentafluorophenyl ligands coordinated to the Au(III) centre that tends to increase the metal–π back-bonding and thereby strengthen the gold–carbon bond. Cyclic voltammetry studies for all the complexes showed irreversible reduction peaks in the range of −1.97 to −1.71 V (vs. Fc0/+ couple) in CH2Cl2 at room temperature (Table 3). No oxidation peak was observed. Due to the similarity of the reduction peak potentials among the different complexes, the reduction process is attributed to be a mostly ligand centered electrochemical event.
| Complex | Reduction Epc/V vs. Fc0/+ |
|---|---|
| 1 | −1.88 |
| 2 | −1.89 |
| 3 | −1.97 |
| 4 | −1.79 |
| 5 | −1.71 |
The photophysical data for the complexes are summarized in Table 4. The UV/Vis profiles of complexes 1–5 are shown in Fig. 2. The shapes of the bands closely resemble those of the free ligands, but exhibit a bathochromic shift in comparison with the free ligand transitions (Fig. S6 in the ESI†). While for complexes 1–3, a single low-energy band was found between 277 and 281 nm, 4 and 5 exhibit a set of bands with the lowest energy band found at 335 and 317 nm, respectively. Such significant differences in the energy of the bands between the complexes can be attributed to the different π-delocalization nature of the given ligand. The complexes have molar extinction coefficients in the range of 0.4 × 104–2.7 × 104 M−1 cm−1.
 | ||
| Fig. 2 UV/Vis absorption spectra of complexes 1–5 in CH2Cl2. | ||
| Complex | Absorption (CH2Cl2) | Emission | ||||
|---|---|---|---|---|---|---|
| λ max/nm (ε/M−1 cm−1) | Medium (T/K) | λ max/nm (τ0/μs) | ϕ em (%) | k r [×103 s−1] | k nr [×105 s−1] | |
| 1 | 277 (8112) | Glass (77) | 440 (0.65) | — | ||
| Solid (298) | 435 (0.29) | 0.1 | 3.45 | 34.46 | ||
| 2 | 279 (27373) |
Glass (77) | 435, 463 sh, 490 sh (0.63) | — | ||
| Solid (298) | 439, 468, 489 sh (0.27) | 1.1 | 40.74 | 36.62 | ||
| 3 | 281 (8449) | Glass (77) | 464 (0.66) | — | ||
| Solid (298) | 504, 540, 577 sh (0.34) | 8.6 | 252.94 | 26.88 | ||
| 4 | 267 (11766) |
Glass (77) | 488, 524, 564 sh (0.6) | — | ||
| 335 (5406) | Solid (298) | 489, 525, 560 sh (0.55) | 1.5 | 27.27 | 17.9 | |
| 5 | 255 (6080) | Glass (77) | 529 (0.62) | — | ||
| 317 (4054) | Solid (298) | 541 (0.21) | 1.0 | 47.62 | 47.14 | |
All the synthesized complexes exhibit intense emission at room temperature in the neat solid and at 77 K (2-MeTHF) rigidified media. No discernible emission was observed at room temperature. The emission profiles of all the complexes in the neat solid and in 2-MeTHF at 77 K are shown in Fig. 3 and 4, respectively. Complexes 1 and 5 exhibit broad non-vibronically structured luminescence in the neat solid with a wavelength maxima found at the 436 and 535 nm, respectively. The emission profiles of complexes 2, 3 and 4 appear vibronically structured with the corresponding λmax at 468, 540 and 525 nm. The emission energies of the complexes reflect the different π–π* energies of the aryl pyridine ligand and are indicative of its greater involvement in the excited state as further supported by DFT and TDDFT calculations. The emission wavelength maxima of complexes 2–5 in 2-MeTHF at 77 K were found to be hypsochromically shifted by approximately 1–40 nm. This blue shift can be attributed to arise from the rigidochromic behaviour of the complexes at 77 K. The excited state lifetimes for the complexes were found in the range 0.21–0.55 μs at room temperature in the neat solid. The large Stokes shifts displayed by the complexes in conjunction with the μs excited-state lifetimes are strongly indicative of the phosphorescence nature of the emission. The reason for the absence of the long-lived emission in solution at room temperature can be attributed to the fast non-radiative decay of the excited state which might be caused primarily by the unhindered rotation of the monodentate ligands around its axis and secondarily to the slightly distorted conformation of the Au(III) complex upon absorption of the photon. The structural distortion could be due to the high flexibility imparted to the molecule not only because of the presence of monodentate ligands, but also due to the participation of the entire molecule in the excited state. This excited-state behaviour is quite in contrast to the previously reported cyclometalated Au(III) complexes, where only the involvement of the cyclometalated ligand was found in most cases. This hypothesis is further supported by DFT and TDDFT calculations. In comparison with the behaviour of complexes 1–5 in solution, we suggest that the additional rigidity provided by the crystal packing effect through intermolecular interactions in the solid state minimizes the structural distortion of the molecules and results in emission for the complexes. This suggestion is consistent with the non-emissive nature of the complexes found when they are spin-coated as thin films in a PMMA matrix. The thin films start to emit only after 90 wt% doping of the complex in the PMMA matrix. Although the PMMA matrix provides a rigid environment, the interactions provided by the PMMA are not sufficient enough in comparison with the tight packing present in the single crystal environment that reduces the distortion to a significant extent.
 | ||
| Fig. 3 Normalized emission spectra of 1–5 in the neat solid. | ||
 | ||
| Fig. 4 Emission spectra of complexes 1–5 in 2-MeTHF at 77 K. | ||
The luminescence properties of our series of compounds were investigated using DFT and TDDFT calculations using the hybrid functional PBE1PBE in conjunction with the Stuttgart/Dresden effective core potential (SDD) basis set for the Au center and the standard 6-31+G(d) basis set for the remaining atoms, the same level of theory used in our previous studies1l,3,6 (see Computational details). The DFT optimized ground state (S0) geometries are in good agreement with the X-ray structures. The square-planar coordination of the metal center is retained and the Au–C bond distance for the pentafluorophenyl ligand trans to the heterocycle is calculated to be shorter than that for the two other pentafluorophenyl ligands trans to each other, as confirmed by the average values of 2.009 Å vs. 2.073 Å, respectively. As expected, the calculated Au–N bond distance is longer than the Au–C bond distances within the same molecule with an average value of 2.143 Å. The latter values do not take into account the bond distance of 2.115 Å found for complex 2 which is about 0.03 Å shorter than those computed for the other complexes, again consistent with the structural observations from the X-ray diffraction studies. The frontier molecular orbitals involved in the calculated singlet–singlet transitions (Table 5, Fig. 5 and 6, and Fig. S7–S11 in the ESI†) reveal that the lowest energy bands found at 335 and 317 nm in the UV/Vis profiles of 4 and 5 have predominantly originated from a HOMO → LUMO excitation corresponding to an intra-ligand charge transfer located at the pyridine aryl ligand denoted as 1ILCT[πpyrid → π*pyrid]. Despite being at a higher energy of 279 nm, the intense band observed for 2 is more comparable to the low energy bands of 4 and 5 with a 1ILCT[πpyrid → π*pyrid] character originated from a HOMO → LUMO excitation, than to the ones of 1 and 3 (however observed at similar energies of 277 nm for 1 and 281 nm for 3) which originated from several excitations and transitions involving molecular orbitals where most of the electron density is not only located on the aryl pyridine rings but also on pentafluorophenyl ligands, leading to an admixture of intra-ligand and ligand-to-ligand π → π* characters. The differences in the energy of the bands between complexes 1–3 and 4–5 (about 20–25 nm, Table S21 in the ESI†) can be substantiated from the calculated HOMO–LUMO gap which is significantly smaller for 4 and 5 due to a higher lying HOMO for these two complexes, probably destabilized by the presence of thienyl pyridine and phenyl isoquinoline, ligands with a strong ability for π-delocalization. Unlike the TDDFT singlet–singlet vertical excitations, the lowest singlet–triplet T1–S0 energies calculated from the ground state structures are not really in agreement with the experimental data. The emission maxima estimated by the energy difference between the DFT optimized singlet ground state S0 and the emitting triplet state T1 are more in line with the experimental emission wavelengths (Table 5). Nevertheless, both TDDFT and DFT calculations lead to the same conclusion that the emission originates from a transition with an intra-ligand 3ILCT [πpyrid → π*pyrid] character for all complexes. The spin density surfaces of the lowest triplet states and the SOMOs obtained from restricted open shell calculations visually identify the origin of the emission (Fig. 5 and 6) but the absence of long-lived emission in solution at room temperature might bias our interpretation of the emission process based on solvent-corrected gas-phase calculations. We assume that the unhindered rotation of the monodentate ligands around its axis and the flexibility of the molecules due to the presence of only monodentate ligands can cause the absence of emission in solution at room temperature. At the gas-phase DFT level, structural distortions on going from S0 to T1 are visible on the heterocycles as shown in the overlay plots of the optimized ground and triplet state structures but no significant differences in the square-planar geometry are present around the metal center (Fig. S12, ESI†). The absence of emission in solution, powder and in spin-coated PMMA films is strongly suggestive of the additional rigidity provided by the packing arrangement in crystals or in the solid state responsible for the radiative decay from the complexes. In this context, we delineate various interactions that could be responsible for that rigidity in the crystal structures in the following section.
 | ||
| Fig. 5 Spin density surfaces for the optimized triplet states of 1–5 (the positive spin densities are shown in blue and the negative ones are shown in green). | ||
 | ||
| Fig. 6 Singly occupied molecular orbitals (top: SOMO, bottom: SOMO−1) obtained by restricted open-shell single point calculations on the optimized triplet state geometries of 1–5. | ||
| Exp. abs. λmaxa |
1 | 2 | 3 | 4 | 5 |
|---|---|---|---|---|---|
| 277 | 279 | 281 | 335 | 317 | |
| a Recorded at room temperature in CH2Cl2. b H = HOMO, L = LUMO. c Recorded at 77 K in glass medium. d Recorded at room temperature in the solid state. e Calculated as the energy difference between the DFT optimized ground state and low-lying triplet state. | |||||
| S0–Snb |
n = 1 | n = 1 | n = 1 | n = 1 | n = 1 |
| 288.6 (f = 0.063) | 281.6 (f = 0.785) | 288.8 (f = 0.061) | 316.8 (f = 0.180) | 315.0 (f = 0.175) | |
| H → L (c = 0.58) | H → L (c = 0.65) | H → L (c = 0.62) | H → L (c = 0.66) | H → L (c = 0.68) | |
| n = 2 | n = 4 | n = 3 | n = 6 | n = 2 | |
| 283.4 (0.025) | 274.9 (0.043) | 281.2 (0.056) | 278.8 (0.039) | 299.0 (0.078) | |
| H−4 → L (0.33) | H−2 → L+1 (0.44) | H−2 → L (0.36) | H−4 → L+1 (0.39) | H → L+1 (0.68) | |
| H−2 → L (0.27) | H−5 → L+1 (0.26) | H−4 → L (0.26) | H−5 → L+1 (0.34) | ||
| H−5 → L (0.21) | H−6 → L (0.22) | ||||
| n = 3 | n = 5 | n = 4 | n = 7 | n = 4 | |
| 283.4 (0.028) | 273.4 (0.026) | 279.7 (0.124) | 277.6 (0.022) | 282.9 (0.028) | |
| H−1 → L (0.44) | H−4 → L+1 (0.49) | H → L+1 (0.51) | H−2 → L+1 (0.53) | H−2 → L (0.29) | |
| H−3 → L+1 (0.26) | H−2 → L+1 (0.26) | H → L (0.25) | H−6 → L (0.21) | H−2 → L+1 (0.29) | |
| H−5 → L+1 (0.20) | H−4 → L+1 (0.26) | ||||
| n = 6 | n = 7 | n = 7 | n = 8 | n = 7 | |
| 278.5 (0.028) | 272.8 (0.026) | 270.6 (0.048) | 277.4 (0.024) | 275.8 (0.040) | |
| H−2 → L (0.28) | H−6 → L (0.60) | H−7 → L (0.32) | H−6 → L (0.44) | H−4 → L (0.37) | |
| H−3 → L (0.27) | H−7 → L (0.27) | H−4 → L (0.28) | H−7 → L (0.21) | H−4 → L+1 (0.33) | |
| H−4 → L+1 (0.25) | H−1 → L (0.21) | H−5 → L (0.28) | |||
| H → L+1 (0.24) | H−5 → L+1 (0.25) | ||||
| n = 7 | n = 15 | n = 12 | n = 14 | n = 9 | |
| 271.0 (0.029) | 254.6 (0.024) | 256.9 (0.083) | 266.9 (0.061) | 270.5 (0.021) | |
| H−5 → L (0.40) | H → L+1 (0.67) | H−4 → L+1 (0.38) | H−7 → L+1 (0.45) | H−7 → L (0.47) | |
| H−4 → L (0.25) | H−5 → L+1 (0.36) | H−7 → L+1 (0.38) | |||
| H−6 → L (0.21) | |||||
| n = 11 | n = 10 | ||||
| 258.2 (0.078) | 268.5 (0.027) | ||||
| H−4 → L+1 (0.46) | H−6 → L (0.57) | ||||
| H−4 → L (0.30) | H−5 → L (0.28) | ||||
| H−6 → L+1 (0.23) | |||||
| n = 11 | |||||
| 265.9 (0.051) | |||||
| H → L+2 (0.65) | |||||
| n = 13 | |||||
| 263.5 (0.059) | |||||
| H−4 → L+1 (0.29) | |||||
| H−5 → L+1 (0.26) | |||||
| H−6 → L (0.21) | |||||
| Exp. em. λmaxc |
440 | 435 | 464 | 488 | 529 |
| Exp. em. λmaxd |
435 | 439 | 504 | 489 | 541 |
| Calc. em. λmaxe |
423.6 | 445.8 | 470.9 | 514.2 | 522.4 |
| T1–S0 | 370.9 | 395.2 | 377.0 | 477.6 | 430.5 |
| H−7 ← L+3 (0.30) | H ← L (0.65) | H ← L+1 (0.36) | H ← L (0.69) | H ← L (0.53) | |
| H ← L (0.28) | H−6 ← L+7 (0.27) | H ← L (0.30) | H ← L+2 (0.21) | ||
| H ← L+1 (0.27) | H ← L+12 (0.23) | H−5 ← L+3 (0.21) | |||
| H−5 ← L+7 (0.20) | |||||
Detailed investigations of the X-ray crystal structures of 1–5 gave no evidence for the presence of Au⋯Au interactions. The shortest Au⋯Au distances are observed in the crystal structures of 4 and 2 with 6.4756(4) and 6.8626(6) Å, respectively. In the other structures, the Au⋯Au distances are not smaller than 7.9119(3) Å for 5, 7.9254(5) Å for 3 and 8.4911(5) Å for 1. The packing studies confirm the presence of weak intermolecular C–H⋯F–C hydrogen bonding, C–F⋯π interactions and π⋯π stacking interactions besides the already mentioned π⋯π intramolecular interactions (Fig. 7, Tables S16–S20, ESI†).7 The shortest intermolecular H⋯F distance dHF is observed in the crystal structure of 2 with 2.34 Å (C⋯F = 3.145(10) Å) and a corresponding C–H⋯F angle θ of 144.3°. Very similar C–H⋯F–C hydrogen bonds are found in the other crystals characterized by dHF distances in the range of 2.43–2.50 Å (C⋯F distances in the range of 3.192(4)–3.263(4) Å) and θ angles in the range of 129.0–149.2°. Such interactions can reasonably be interpreted as weak hydrogen bonds having an effect on the crystal packing. In the crystal structures, the molecules are also linked by weak C–F⋯π interactions and π⋯π stacking interactions completing a sort of three-dimensional framework. The shortest distances between the involved fluorine atom and the centroid (Ct) of the ring vary from 2.983(3) Å in 5 to 3.407(2) Å in 4 with C–F⋯Ct angles in the range of 131.98(17)–151.0(8)°, while the π⋯π interactions are characterized by the shortest Ct⋯Ct separations observed between 3.7820(18) in 4 and 4.369(3) in 1 with dihedral angles between the mean planes which vary from 0 to 26.5(6)°.
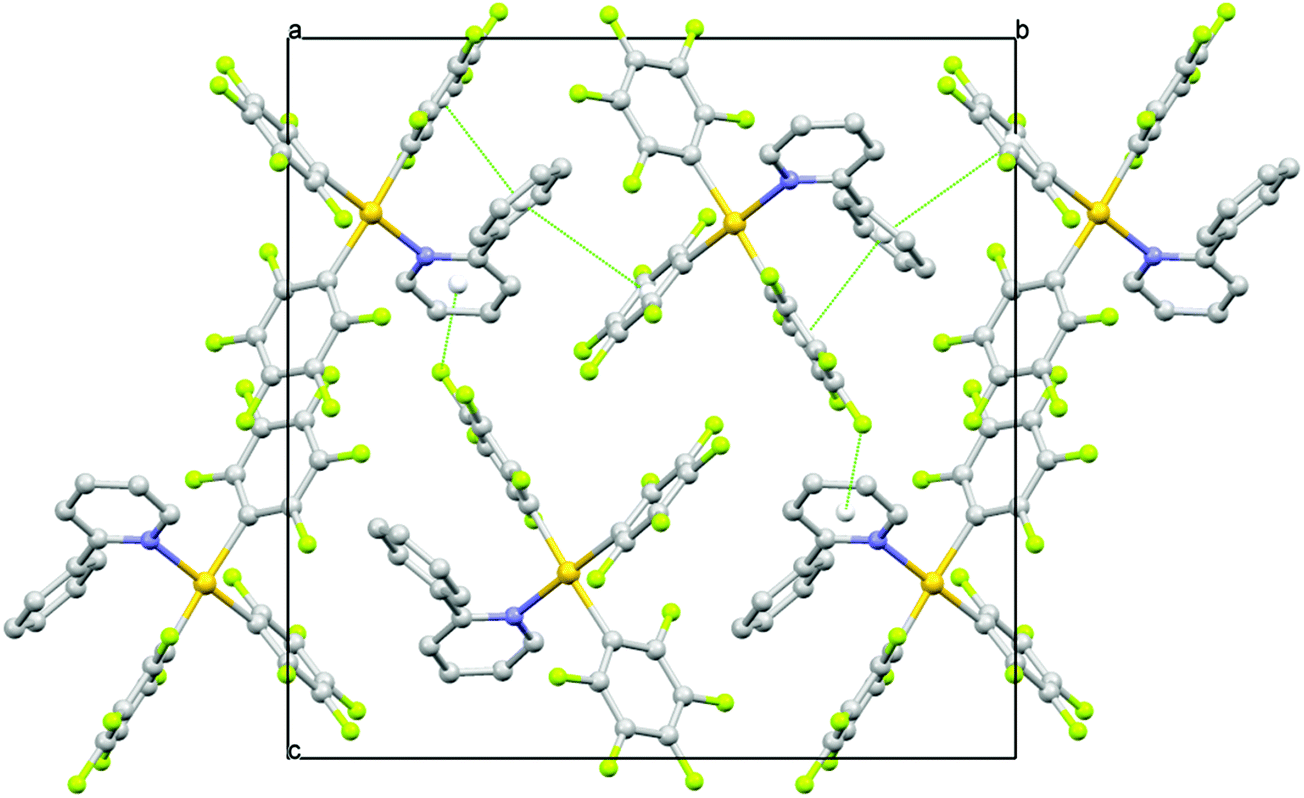 | ||
| Fig. 7 A view along the a axis of the crystal packing of 3 showing π⋯π and C–F⋯π interactions as green dashed lines. | ||
Based on the above discussion, it can be strongly presumed that the presence of a number of several weak interactions that contribute to the rigidity of the molecules in the crystal phase is partially responsible for reducing the flexibility and thereby resulting in emission from the complexes.
Conclusions
In conclusion, a new design concept to achieve room temperature emission from Au(III) complexes devoid of cyclometalation has been successfully realized using electronically different pyridine aryl ligands in conjunction with an Au(III) triaryl motif. The emission energies are readily tunable across the visible range of the electromagnetic spectrum with the electronic nature of the heterocycle allowing to even achieve deep-blue emission at room temperature in the neat solid and in 2-MeTHF at 77 K. While the highest quantum yield obtained is around 8.6%, it is expected that this design concept can be further extended to achieve non-cyclometalated Au(III) compounds with finely tunable luminescence properties by using other kinds of conjugated heterocyclic ligands that could have potential in optoelectronic and other light emitting related applications.Experimental section
General procedures and instrumentation
All starting materials were purchased from commercial sources and used as received unless otherwise stated. All chemicals were of reagent grade and the solvents used for synthesis were of analytical grade. The reagents Ag(C6F5),8 TlCl(C6F5)2,9 (THT)AuCl10 and (THT)AuC6F55a were synthesized based on methods reported in the literature.All manipulations requiring an inert atmosphere were carried out using standard Schlenk techniques under dinitrogen. 1H, 13C{1H} and 19F NMR spectra were recorded on Bruker 400 MHz and 500 MHz or Varian 200 MHz and 300 MHz spectrometers. Chemical shifts (δ) are reported in parts per million (ppm) referenced to tetramethylsilane (δ 0.00) ppm using the residual proton solvent peaks as internal standards (1H NMR experiments) or the characteristic resonances of the solvent nuclei (13C NMR experiments). 19F NMR was referenced to CFCl3 (δ 0.00) ppm. Coupling constants (J) are quoted in Hertz (Hz) and the following abbreviations are used to describe the signal multiplicities: s (singlet); d (doublet); t (triplet); q (quartet); quint (quintet); sext (sextet); sept (septet); m (multiplet). Proton and carbon assignments have been made using routine one and two dimensional NMR spectroscopy where appropriate. Infrared (IR) spectra were recorded on a SpectrumTwo FT-IR spectrometer (Perkin-Elmer) equipped with a Specac Golden Gate™ ATR (attenuated total reflection) accessory, applied as neat samples with frequencies (νmax) quoted in wavenumbers (cm−1). Elemental microanalysis was carried out with a Leco CHNS-932 analyzer. TLC analysis was performed on precoated Merck Silica Gel60F254 slides and visualized by luminescence quenching either at (short wavelength) 254 nm or (long wavelength) 365 nm. Chromatographic purification of products was performed on a short column (length 15.0 cm; diameter 1.5 cm) using a silica gel 60, 230–400 mesh using a forced flow of the eluent. UV/Vis absorption measurements were carried out on a Perkin-Elmer Lambda 35 UV-Vis spectrophotometer. Emission spectra were acquired on a Perkin-Elmer spectrophotometer using 450 W xenon lamp excitation by exciting at the longest-wavelength absorption maxima with the excitation slit width of 5 nm and the emission slit width of 10 nm. 77 K emission spectra were acquired in frozen 2-methyltetrahydrofuran (2-MeTHF) glass. Luminescence quantum yields ϕem of the complexes in solution were determined at 298 K (estimated uncertainty ± 15%) using standard methods, and wavelength-integrated intensities (I) of the corrected emission spectra were compared to iso-absorptive spectra of quinine sulfate standard (ϕref = 0.55 in a 1 N H2SO4 air-equilibrated solution) and were corrected for solvent refractive index. Absolute quantum yields were measured in the solid using an integrating sphere on the Edinburgh spectrophotometer FLS920. YAG:Ce (powder) was used as a calibration reference with ϕem = 97%. Phosphorescence lifetimes in thin films were measured using the Edinburgh laser flash photolysis spectrophotometer LP920 with a Nd:YAG 355 nm laser as an excitation source fitted with a single monochromator. Cyclic voltammograms were measured with a Metrohm 757 VA Computrace with a glassy carbon electrode (d = 2 mm) or a gold electrode with a Pt counter electrode versus an Ag/AgCl reference electrode. The following ligand abbreviations have been used: 2-phenylpyridine (2-ppy), 4-phenylpyridine (4-ppy), 2-thienylpyridine (2-thpy), 2-phenylisoquinoline (2-pqu) and 2-(2,4-difluorophenyl)pyridine (2-fppy).
Synthesis of complexes 1–5
Tris(pentafluorophenyl)(2-(2,4-difluorophenylpyridine)gold(III) (1). Yield: 71%, 40.15 mg, 0.0452 mmol. 1H NMR (500 MHz, CD2Cl2, 25 °C): δ (ppm) = 8.87 (d, 3J(H–H) = 5.8 Hz, 1H), 8.19–8.14 (m, 2H), 7.71–7.69 (m, 2H), 7.36–7.32 (m, 1H), 6.90–6.85 (m, 1H). 13C{1H} NMR (125.8 MHz, CD2Cl2, 25 °C): δ (ppm) = 166.7 (s), 164.6 (s), 161.0 (s), 159.0 (s), 155.0 (s), 151.9 (s), 148.2–148.0 (m, Ar–C–F), 145.9–146.1 (m, Ar–C–F), 141.7 (s), 139.4–139.0, (m, Ar–C–F), 138.9–138.7 (m, Ar–C–F), 137.1–137.5 (m, Ar–C–F), 136.7–136.6 (m, Ar–C–F), 131.7 (s), 130.9 (s), 126.6 (s), 122.6 (s), 118.6 (s), 114.0–113.8 (m, Ar–C–F), 105.2 (t, 26.4 Hz, Ar–Au). 19F NMR (282.4 MHz, CD2Cl2, 25 °C): δ (ppm) = −106.1 to −106.2 (m, 1F), −114.4 to −114.5 (m, 1F), −123.6 to −124.2 (m, 6Fortho), −158.3 (t, 3J(F–F) = 19.8 Hz, 2Fpara), −159.1 (t, 3J(F–F) = 19.8 Hz, 1Fpara), −163.0 to −163.2 (m, 4Fmeta), −163.9 to −164.1 (m, 2Fmeta). Anal. Calc. for C29H7AuF17N: C, 39.17; H, 0.79; N, 1.58 found: C, 39.12; H, 0.82; N, 1.62.
Tris(pentafluorophenyl)(4-phenylpyridine)gold(III) (2). Yield: 66%, 36.0 mg, 0.0422 mmol. 1H NMR (500 MHz, CD2Cl2, 25 °C): δ (ppm) = 8.52 (d, 3J(H–H) = 6.6 Hz, 2H), 7.79 (d, 3J(H–H) = 6.8 Hz, 2H), 7.69–7.67 (m, 2H), 7.58–7.53 (m, 3H). 13C{1H} NMR (125.8 MHz, CD2Cl2, 25 °C): δ (ppm) = 149.7 (s), 131.6 (s), 129.7 (s), 127.3 (s), 125.0 (s), 121.0–119.5 (m, Ar–C–F), 110.5–110.3, (m, Ar–C–F). 19F NMR (282.4 MHz, CD2Cl2, 25 °C): δ (ppm) = −123.7 to −123.8 (m, 2Fortho), −125.6 to −125.8 (m, 4Fortho), −158.5 (t, 3J(F–F) = 19.8 Hz, 2Fpara), −159.2 (t, 3J(F–F) = 19.8 Hz, 1Fpara), −162.8 to −162.9 (m, 4Fmeta), −163.9 to −164.0 (m, 2Fmeta). Anal. Calc. for C29H9AuF15N: C, 40.82; H, 1.06; N, 1.64 found: C, 41.18; H, 0.99; N, 1.64.
Tris(pentafluorophenyl)(2-phenylpyridine)gold(III) (3). Yield: 62%, 33.6 mg, 0.0394 mmol. 1H NMR (500 MHz, CD2Cl2, 25 °C): δ (ppm) = 8.75 (d, 3J(H–H) = 6.0 Hz, 1H), 8.10 (t, 3J(H–H) = 7.5 Hz, 1H), 7.67 (d, 3J(H–H) = 8.0 Hz, 1H), 7.64–7.51 (m, 6H). 13C{1H} NMR (125.8 MHz, CD2Cl2, 25 °C): δ (ppm) = 161.6 (s, Ar–C), 150.9 (s), 147.7–145.6 (m, Ar–C–F), 145.8–143.8 (m, Ar–C–F), 141.7 (s), 141.0–138.8 (m, Ar–C–F), 139.1–136.9 (m, Ar–C–F), 131.0 (s), 130.0 (s), 128.8 (s), 127.6 (s), 127.4 (s), 125.3, 118.7 (t, 3J(C–F) = 42.8 Hz, Ar–Au), 95.6 (t, 3J(C–F) = 35.2 Hz, Ar–Au). 19F-NMR (282.4 MHz, CD2Cl2, 25 °C): δ (ppm) = −124.5 to −124.0 (m, 6Fortho), −159.0 (t, 3J(F–F) = 19.8 Hz, 2Fpara), −159.4 (t, 3J(F–F) = 19.8 Hz, 1Fpara), −163.4 to −163.6 (m, 4Fmeta), −164.2 to −164.3 (m, 2Fmeta). Anal. Calc. for C29H9AuF15N: C, 40.82; H, 1.06; N, 1.64 found: C, 40.49; H, 0.99; N, 1.82.
Tris(pentafluorophenyl)(2-phenylisoquinoline)gold(III) (4). Yield: 71%, 40.9 mg, 0.0453 mmol. 1H NMR (500 MHz, CD2Cl2, 25 °C): δ (ppm) = 8.61–8.60 (d, 3J(H–H) = 4.2 Hz, 1H), 8.02–8.01 (d, 3J(H–H) = 6.3 Hz, 1H), 7.93 (d, 3J(H–H) = 3.1 Hz, 2H) 7.74–7.64 (m, 3H), 7.57 (t, 3J(H–H) = 5.9 Hz, 2H), 7.48 (d, 3J(H–H) = 5.1 Hz, 2H). 13C{1H} NMR (125.8 MHz, CD2Cl2, 25 °C): δ (ppm) = 164.2 (s), 148.1–147.8 (m, Ar–C–F), 146.2–145.9 (m, Ar–C–F), 141.7 (s) 141.3–140.8 (m, Ar–C–F), 139.3–138.8 (m, Ar–C–F), 141.0–138.8 (m, Ar–C–F), 137.5 (s) 137.2–136.8 (m, Ar–C–F), 136.2 (s), 134.6 (s), 131.5 (s), 130.3 (s), 130.1 (s), 129.9 (s), 129.7 (s), 129.3 (s), 127.6 (s), 124.2 (s). 19F NMR (282.4 MHz, CD2Cl2, 25 °C): δ (ppm) = −123.4 to −123.9 (m, 6Fortho), −159.3 (t, 3J(F–F) = 19.8 Hz, 2Fpara), −159.6 (t, 3J(F–F) = 19.8 Hz, 1Fpara), −163.5 to −163.7 (m, 4Fmeta), −164.2 to −164.3 (m, 2Fmeta). Anal. Calc. for C33H11AuF15N·Et2O: C, 45.46; H, 2.17; N, 1.43; found: C, 45.38; H, 1.71; N, 1.49.
Tris(pentafluorophenyl)(2-thienylpyridine)gold(III) (5). Yield: 52%, 29.2 mg, 0.034 mmol. 1H NMR (500 MHz, CD2Cl2, 25 °C): δ (ppm) = 8.66 (d, 3J(H–H) = 5.8 Hz, 1H), 8.06 (t, 3J(H–H) = 7.9 Hz, 1H), 7.98 (d, 3J(H–H) = 3.7 Hz, 1H), 7.72 (d, 3J(H–H) = 7.5 Hz, 1H), 7.61 (d, 3J(H–H) = 5.1 Hz, 1H), 7.53 (t, 3J(H–H) = 6.5 Hz, 1H), 7.26 (t, 3J(H–H) = 4.6 Hz, 1H). 13C{1H} NMR (125.8 MHz, CD2Cl2, 25 °C): δ (ppm) = 154.9 (s), 151.4 (s), 147.9–147.7 (m, Ar–C–F), 146.1–145.8 (m, Ar–C–F), 144.6–144.1 (m, Ar–C–F), 141.9 (s), 141.4–140.9 (m, Ar–C–F), 140.0 (s), 139.5–138.8 (m, Ar–C–F), 138.7–138.4 (m, Ar–C–F), 137.1–136.8 (m, Ar–C–F), 136.7–136.3 (m, Ar–C–F), 131.1 (s), 129.8 (s), 129.2 (s), 125.5 (s), 118.7 (t, 3J(C–F) = 44.0 Hz, Ar–Au), 95.8 (t, 3J(C–F) = 39.0 Hz, Ar–Au). 19F NMR (282.4 MHz, CD2Cl2, 25 °C): δ (ppm) = −123.5 to −123.6 (m, 2Fortho), −123.8 to −123.9 (m, 4Fortho), −158.9 (t, 3J(F–F) = 19.5 Hz, 2Fpara), −159.3 (t, 3J(F–F) = 19.8 Hz, 1Fpara), −163.4 to −163.6 (m, 4Fmeta), −164.1 to −164.4 (m, 2Fmeta). Anal. Calc. for C27H7AuF15NS: C, 37.74; H, 0.82; N, 1.63 found: C, 37.84; H, 0.90; N, 1.65.
X-ray crystallography
Single-crystal X-ray diffraction data were collected at 183(1) K on an Agilent Technologies Xcalibur Ruby area-detector diffractometer using a single wavelength Enhance X-ray source with MoKα radiation (λ = 0.71073 Å)11 from a micro-focus X-ray source and an Oxford Instruments Cryojet XL cooler. The selected suitable single crystal was mounted using polybutene oil on a flexible loop fixed on a goniometer head and immediately transferred to the diffractometer. Pre-experiment, data collection, data reduction and analytical absorption correction12 were performed with the program suite CrysAlisPro.13 Using Olex2,14 the structure was solved with the SHELXS9715 structure solution program using direct methods and refined with the SHELXL201315 program package by full-matrix least-squares minimization on F2. PLATON16 was used to check the result of the X-ray analyses. For more details of the data collection and refinement parameters, see the Crystallographic Information Files. CCDC 1024826 (for 1), CCDC 1024827 (for 2), CCDC 1024828 (for 3), CCDC 1024829 (for 4) and CCDC 1024830 (for 5) contain the supplementary crystallographic data for this paper.
Computational details
The absorption and emission properties of our series of compounds were investigated by density functional theory (DFT) calculations using the Gaussian 03 program package.17 The hybrid functional PBE1PBE18 in conjunction with the Stuttgart/Dresden effective core potential (SDD) basis set19 for the Au center augmented with one f-polarization function (α = 1.050) and the standard 6-31+G(d) basis set20 for the remaining atoms was applied. The molecular structures of the electronic ground states and the lowest triplet states of compounds 1–5 were studied. On the basis of the ground-state optimized geometries, time-dependent DFT (TD-DFT) calculations21–23 combined with the conductive polarizable continuum model (CPCM)24,25 were used to produce the fifteen lowest singlet–singlet and singlet–triplet vertical excitations in the dichloromethane media with the corresponding energies, transition coefficients, and oscillator strengths. Full geometry optimizations without symmetry constraints were carried out in the gas phase for the singlet ground states (S0) and the lowest triplet states (T1). The optimized geometries S0 and T1 were confirmed to be potential energy minima by vibrational frequency calculations at the same level of theory, as no imaginary frequency was found.Acknowledgements
This work was supported by the Swiss National Science Foundation (grant no. 200021_135488) and University of Zurich. K.V. is grateful to Prof. R. Alberto for his generous support.References
- (a) V. W.-W. Yam, S. W.-K. Choi, T.-F. Lai and W.-K. Lee, J. Chem. Soc., Dalton Trans., 1993, 1001–1002 RSC; (b) C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409–12420 RSC; (c) W.-P. To, K. T. Chan, G. S. M. Tong, C. Ma, W.-M. Kwok, X. Guan, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 6648–6652 CrossRef CAS PubMed; (d) V. K.-M. Au, K. M.-C. Wong, D. P.-K. Tsang, M.-Y. Chan, N. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2010, 132, 14273–14278 CrossRef CAS PubMed; (e) V. W.-W. Yam, K. M.-C. Wong, L.-L. Hung and N. Zhu, Angew. Chem., Int. Ed., 2005, 44, 3107–3110 CrossRef CAS PubMed; (f) K. M.-C. Wong, X. L. Zhu, L.-L. Hung, N. Y. Zhu, V. W.-W. Yam and H.-S. Kwok, Chem. Commun., 2005, 2906–2908 RSC; (g) V. K.-M. Au, K. M.-C. Wong, N. Y. Zhu and V. W.-W. Yam, J. Am. Chem. Soc., 2009, 131, 9076–9085 CrossRef CAS PubMed; (h) V. K. M. Au, K. M. C. Wong, N. Y. Zhu and V. W. W. Yam, Chem. – Eur. J., 2011, 17, 130–142 CrossRef CAS PubMed; (i) V. K. M. Au, W. H. Lam, W. T. Wong and V. W. W. Yam, Inorg. Chem., 2012, 51, 7537–7545 CrossRef CAS PubMed; (j) V. K. M. Au, N. Y. Zhu and V. W. W. Yam, Inorg. Chem., 2013, 52, 558–567 CrossRef CAS PubMed; (k) M. C. Tang, D. P. K. Tsang, M. M. Y. Chan, K. M. C. Wong and V. W. W. Yam, Angew. Chem., Int. Ed., 2013, 52, 446–449 CrossRef CAS PubMed; (l) A. Szentkuti, M. Bachmann, J. A. Garg, O. Blacque and K. Venkatesan, Chem. – Eur. J., 2014, 20, 2585–2596 CrossRef CAS PubMed; (m) G. Cheng, K. T. Chan, W. P. To and C. M. Che, Adv. Mater., 2014, 26, 2540–2546 CrossRef CAS PubMed.
- (a) V. W.-W. Yam, T.-F. Lai and C.-M. Che, J. Chem. Soc., Dalton Trans., 1990, 3747–3752 RSC; (b) P.-T. Chou and Y. Chi, Chem. – Eur. J., 2007, 13, 380–395 CrossRef CAS PubMed; (c) M. Krikorian, S. Liu and T. M. Swager, J. Am. Chem. Soc., 2014, 136, 2952–2955 CrossRef CAS PubMed.
- J. A. Garg, O. Blacque, T. Fox and K. Venkatesan, Inorg. Chem., 2010, 49, 11463–11472 CrossRef CAS PubMed.
- V. R. Bojan, J. M. López-de-Luzuriaga, E. Manso, M. Monge and M. E. Olmos, Organometallics, 2011, 30, 4486–4489 CrossRef CAS.
- (a) R. Uson, A. Laguna, M. Laguna, E. Fernandez, P. G. Jones and G. M. Sheldrick, J. Chem. Soc., Dalton Trans., 1982, 1971–1976 RSC; (b) R. Usón, A. Laguna, M. Laguna and E. Fernandez, Inorg. Chim. Acta, 1980, 45, 177–178 CrossRef.
- J. A. Garg, O. Blacque and K. Venkatesan, Inorg. Chem., 2011, 50, 5430–5441 CrossRef CAS PubMed.
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond in Structural Chemistry and Biology, Oxford University Press Inc., New York, 1999 Search PubMed.
- R. Usón, A. Laguna, E. J. Fernández, A. Mendia and P. G. Jones, J. Organomet. Chem., 1988, 350, 129–138 CrossRef.
- A. Laguna, E. J. Fernández, A. Mendía, M. E. Ruiz-Romero and P. G. Jones, J. Organomet. Chem., 1989, 365, 201–206 CrossRef CAS.
- R. Usón, A. Laguna, M. Laguna and E. Fernandez, Inorg. Chim. Acta, 1980, 45, L177–L178 CrossRef.
- Agilent Technologies (formerly Oxford Diffraction), Oxfordshire, England, 2012.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 887–897 CrossRef.
- CrysAlisPro, Agilent Technologies, Yarnton, Oxfordshire, England, 2012 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Rob, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Gaussian, Inc., Wallingford, CT, 2003 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef CAS PubMed.
- T. H. Dunning Jr. and P. J. Hay, Modern Theoretical Chemistry, Plenum, New York, 1976, vol. 3 Search PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. J. Salahub, Chem. Phys., 1998, 108, 4439 CAS.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS PubMed.
- V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995 CrossRef CAS.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1024826–1024830. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00405e |
| This journal is © The Royal Society of Chemistry 2015 |