
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of [Fe(BPMEN)ACC]SbF6: a structural and functional mimic of ACC-oxidase†
Y.
Roux‡
a,
W.
Ghattas‡
a,
F.
Avenier
a,
R.
Guillot
a,
A. J.
Simaan
*b and
J.-P.
Mahy
*a
aInstitut de Chimie Moléculaire et des Matériaux d'Orsay, Université Paris Sud, CNRS (UMR 8182), Orsay 91405 CEDEX, France. E-mail: jean-pierre.mahy@u-psud.fr
bAix Marseille Université, Centrale Marseille, CNRS, iSm2 UMR 7313, 13397, Marseille, France. E-mail: jalila.simaan@univ-amu.fr
First published on 24th February 2015
Abstract
A mononuclear Fe(II) complex bearing 1-aminocyclopropane-1-carboxylic acid (ACCH) was synthesized and characterized. X-ray crystallography demonstrated that ACC binds to the Fe(II) ion in a bidentate mode constituting the first structural mimic of the expected binding of ACC to the Fe(II) center of the ethylene forming enzyme ACC-oxidase (ACCO). [Fe(BPMEN)ACC]SbF6 also constitutes a functional biomimetic complex of ACCO, as it reacts with hydrogen peroxide producing ethylene.
The final step in the biosynthesis of the phytohormone ethylene1 is the oxidation of 1-aminocyclopropane-1-carboxylic acid (ACCH)2 catalyzed by ACC-oxidase (ACCO).3 The X-ray crystal structure of substrate-free ACCO4 has confirmed the anticipated makeup of its active site i.e. a non-heme Fe(II) cation coordinated by the classical N,N,O facial triad.5,6 In contrast, the ambiguous binding of 1-aminocyclopropane-1-carboxylate (ACC) in the active site of ACCO has only been probed by spectroscopic studies, which have nonetheless concluded that ACC binds to the Fe(II) ion of the active site in a bidentate mode via both its amine and carboxylate functions.7 Accordingly, the structural characterization of a dinuclear ACC-containing Fe(III) complex, [Fe2(TACN)2(μ-O)(μ-ACCH)2]4+ (TACN = 1,4,7-triazacyclononane) has shown that ACC can bind iron centres. In this case however, two ACCH fragments are bridging two Fe(III) cations by their carboxylate functions.8 Bidentate ACC has only been reported in a few Cu(II)–ACC complexes,9 and has never been observed in the case of mononuclear Fe(II) complexes. Indeed, the good water solubility of amino acids is inappropriate for the synthesis of Fe(II) complexes, which are best obtained in aprotic poorly-coordinating solvents that conversely do not dissolve amino acids.10 To the best of our knowledge, structural characterization of amino acid-containing Fe(II) complexes has only been reported in the case of a proline-containing complex.11 Proline is structurally distinguished from other natural α-amino acids by a secondary amine function engaged in a 5-membered ring and therefore is inappropriate to structurally mimic the coordination of other natural α-amino acids. Furthermore, the synthesis of the proline-containing Fe(II) complex relied on the solubilization of proline in DMSO, which did not allow to get the structural information when either phenylalanine, tryptophan or valine was used instead of proline.11
Here, in order to overcome the solubility limitations, we treated an aqueous solution of ACCH with one equivalent of tetra-n-butylammonium hydroxide (N(n-Bu)4OH). Subsequent water evaporation provided an ionic liquid fully miscible with acetonitrile, which allowed its combination with an acetonitrile solution of the previously described [Fe(BPMEN)(CH3CN)2](SbF6)2 complex (1) (BPMEN = N,N′-dimethyl-N,N′-bis(pyridylmethyl)ethane-1,2-diamine) (Scheme 1).12
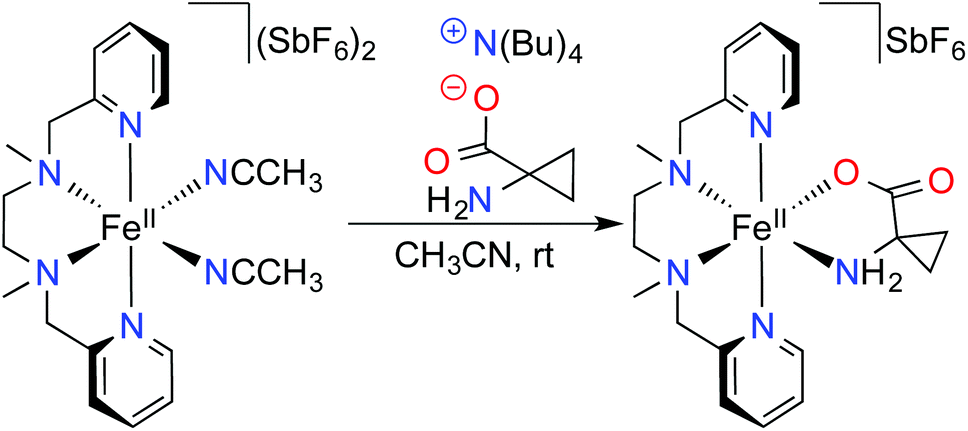 | ||
| Scheme 1 Preparation of the [Fe(BPMEN)ACC]SbF6 complex (2) from [Fe(BPMEN)(CH3CN)2](SbF6)2 (1) under an inert atmosphere. | ||
When one equivalent of N(n-Bu)4ACC was added to an acetonitrile solution of complex 1, the solution turned from purple to a pale yellow color. The monitoring of the UV-vis absorbance as a function of the increasing amounts of N(n-Bu)4ACC added to 1 (Fig. 1) showed the progressive evolution of the spectrum of 1 (λmax = 373 nm, ε = 3340 M−1 cm−1, MLCT band)13 into a new spectrum (λmax = 395 nm, ε = 860 M−1 cm−1). The occurrence of an isosbestic point at 410 nm clearly indicated a single transformation of the starting material into a new species. The transformation was optimal for one equivalent of N(n-Bu)4ACC added. Further addition of N(n-Bu)4ACC led to a decrease of the characteristic MLCT band at 395 nm that completely disappeared after the addition of three equivalents of N(n-Bu)4ACC (Fig. S1†).
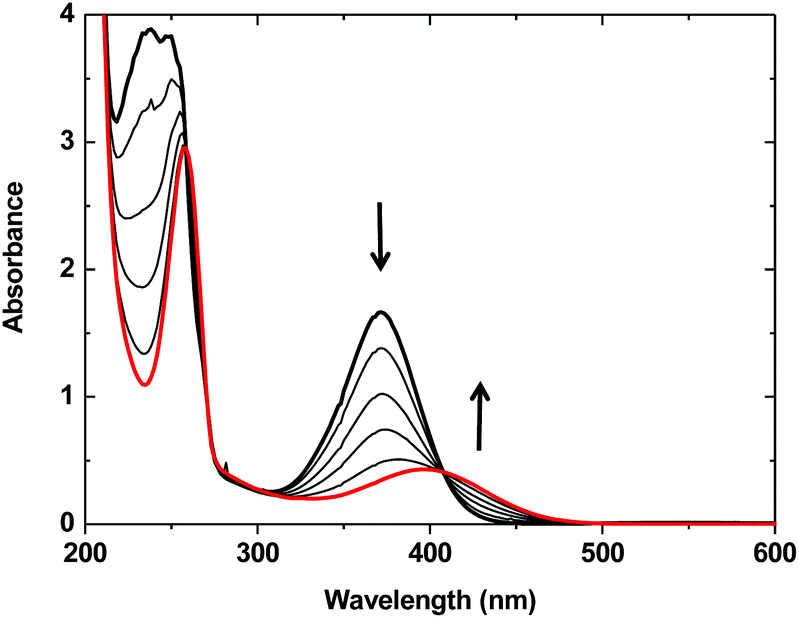 | ||
| Fig. 1 Evolution of the UV-vis spectrum of a 0.5 mM acetonitrile solution of [Fe(BPMEN)(CH3CN)2](SbF6)2 (1) (bold black line) upon successive additions of upto 1 equiv. (red line) of N(n-Bu)4ACC. | ||
High resolution electrospray ionization mass spectrometry (HR ESI-MS) analysis was carried out on the pale yellow solution obtained after the addition of one equivalent of the amino acid (Fig. 2). The results revealed the formation of a single new compound characterized by a peak at m/z 426.1604, which is in agreement with the complexation of one ACC molecule to the Fe(II) ion of complex 1 in the place of two acetonitrile molecules observed in the X-ray crystal structure (Fig. S2†).
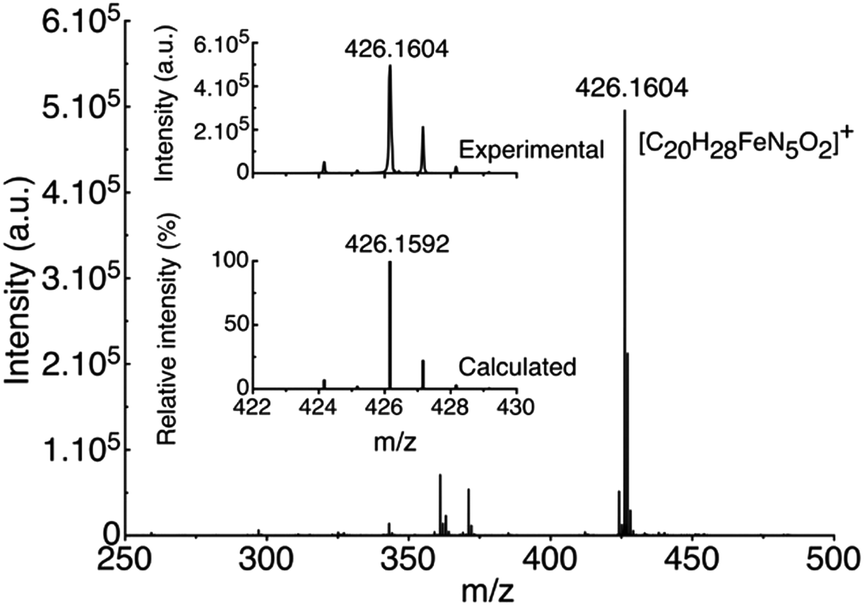 | ||
| Fig. 2 HR ESI-MS spectrum obtained upon additions of 1 equiv. of N(n-Bu)4ACC onto [Fe(BPMEN)(CH3CN)2](SbF6)2 (1). | ||
The new pale yellow species was then precipitated by the addition of ether in the acetonitrile solution and recrystallized from slow ether diffusion in acetonitrile to afford monocrystals suitable for X-ray diffraction analysis. The resulting diffraction pattern was in agreement with a [Fe(BPMEN)ACC]SbF6 molecular formula for complex 2 and a structure in which ACC is bound to the Fe(II) center in a bidentate mode via both its amine and carboxylate functions, as projected for the enzymatic active site (Fig. 3, Tables S1 and S2†).5 In addition, both the UV-vis spectrum and the mass spectrum of complex 2 obtained from the solid state matched those of the species formed in solution. In comparison with the X-ray structure of complex 1, the distorted octahedral geometry is maintained in complex 2, but the average metal–ligand distance has increased from 1.98 Å in 1 to 2.18 Å in 2. It is noteworthy that average Fe–N distances below 2.0 Å in 1 and above 2.1 Å in 2 suggest a low spin to high spin transition upon complexation of the amino acid to the Fe(II) center.14
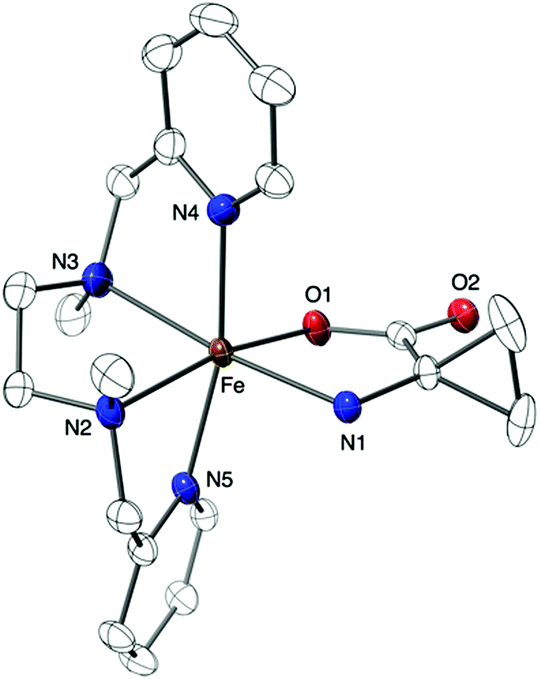 | ||
| Fig. 3 X-ray crystal structure of the [Fe(BPMEN)ACC]SbF6 complex (2) (thermal ellipsoids are set at 50% probability). The hydrogen atoms and the counter ion are omitted for the sake of clarity. Selected bond lengths: Fe–N1 2.203(2) Å, Fe–N2 2.232(2) Å, Fe–N3 2.242(3) Å, Fe–N4 2.203(2) Å, Fe–N5 2.211(2) Å, Fe–O1 2.0164(19) Å. | ||
Bulk magnetization data were collected from the crystalline samples of complex 2. The corresponding χmT vs. T plot (Fig. S3†) showed an initial sharp increase (upto ca. 50 K) followed by a slight monotonic increase of χmT with the increasing temperature. Both the overall shape of χmT vs. T and a χmT value reaching 3.48 cm3 K mol−1 at 400 K concur with a high spin mononuclear Fe(II) center (S = 2, g = 2.1). Therefore, complex 2 is in the high spin state as suggested by the bond lengths obtained from the crystal structure. The coordination of the amino acid on the Fe(II) ion stabilizes the high spin state whereas, complex 1 is known to be in the low spin state at low temperature, in spin transition at room temperature and at the high spin state only above 400 K.15
Although the enzymatic system contains an Fe(II) ion in its active site, no functional mimic of ACCO reported so far involves Fe(II). Therefore, we tested complex 2 in the oxidation of ACC into ethylene, first using O2 and then in the presence of H2O2. The UV-vis spectrum of acetonitrile solutions of complex 2 did not change when O2 was introduced. In contrast, when 10 equivalents of H2O2 were added to an acetonitrile solution of complex 2, its UV-vis spectrum changed drastically, however, no clean transformation with isosbestic points could be observed (Fig. S4†). The addition of up to 100 equivalents of H2O2 to complex 2 was then performed in sealed tubes and GC analysis of the resulting gas revealed that the formation of ethylene reached ca. 23% yield when 5 to 10 equivalents of H2O2 were added, compared to a 15% yield in the blank experiment using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of iron(II) triflate and N(n-Bu)4ACC. The rather low ACC oxidation yield is not surprising considering the fact that complex 2 is hexacoordinated and thus, a direct interaction between the iron cation and hydrogen peroxide requires the de-coordination of one of the six ligands of iron. The formation of ethylene suggests that one of these six ligands is indeed labile enough to allow hydrogen peroxide activation at the metal center.
:1 mixture of iron(II) triflate and N(n-Bu)4ACC. The rather low ACC oxidation yield is not surprising considering the fact that complex 2 is hexacoordinated and thus, a direct interaction between the iron cation and hydrogen peroxide requires the de-coordination of one of the six ligands of iron. The formation of ethylene suggests that one of these six ligands is indeed labile enough to allow hydrogen peroxide activation at the metal center.
In summary, our work describes the synthesis, the reactivity and the characterization in solution and in the solid state of the first mononuclear Fe(II) complex bearing an ACC ligand. This complex demonstrates that ACC can bind to the Fe(II) ion in a bidentate mode, constituting a structural mimic of the binding of ACC to the Fe(II) center of ACCO.
We are grateful to Dr Eric Rivière for SQUID measurements and to the ‘Agence Nationale de la Recherche’ (ANR BIOXICAT and REBAR) and ‘Région île de France’ (DIM développement soutenable) for financial support.
Notes and references
- (a) G.-P. L. Girardin, Fortschr. Gesamtgeb. Agrikulturchem., 1864, 7, 199–200 Search PubMed; (b) D. Neljubow, Beih. Bot. Zentralbl., 1901, 10, 128–139 Search PubMed.
- (a) T. Boller, R. Herner and H. Kende, Planta, 1979, 145, 293–303 CrossRef CAS PubMed; (b) D. O. Adams and S. F. Yang, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 170–174 CrossRef CAS.
- (a) M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr., Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed; (b) S. F. Yang and N. E. Hoffman, Annu. Rev. Plant Physiol., 1984, 35, 155–189 CrossRef CAS; (c) D. J. McGarvey and R. E. Christoffersen, J. Biol. Chem., 1992, 267, 5964–5967 CAS.
- Z. Zhang, J.-S. Ren, I. J. Clifton and C. J. Schofield, Chem. Biol., 2004, 11, 1383–1394 CrossRef CAS PubMed.
- (a) V. J. Lay, A. G. Prescott, P. G. Thomas and P. John, Eur. J. Biochem., 1996, 242, 228–234 CAS; (b) Z. Zhang, J. N. Barlow, J. E. Baldwin and C. J. Schofield, Biochemistry, 1997, 36, 15999–16007 CrossRef CAS PubMed.
- (a) E. L. Hegg and L. Que Jr., Eur. J. Biochem., 1997, 250, 625–629 CAS; (b) K. Koehntop, J. Emerson and L. Que Jr., J. Biol. Inorg. Chem., 2005, 10, 87–93 CrossRef CAS PubMed.
- (a) A. M. Rocklin, D. L. Tierney, V. Kofman, N. M. W. Brunhuber, B. M. Hoffman, R. E. Christoffersen, N. O. Reich, J. D. Lipscomb and L. Que Jr., Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 7905–7909 CrossRef CAS; (b) A. Rocklin, K. Kato, H. Liu, L. Que Jr. and J. Lipscomb, JBIC, J. Biol. Inorg. Chem., 2004, 9, 171–182 CrossRef CAS PubMed; (c) J. Zhou, A. M. Rocklin, J. D. Lipscomb, L. Que Jr. and E. I. Solomon, J. Am. Chem. Soc., 2002, 124, 4602–4609 CrossRef CAS PubMed; (d) D. L. Tierney, A. M. Rocklin, J. D. Lipscomb, L. Que Jr. and B. M. Hoffman, J. Am. Chem. Soc., 2005, 127, 7005–7013 CrossRef CAS PubMed.
- W. Ghattas, Z. Serhan, N. El Bakkali-Taheri, M. Réglier, M. Kodera, Y. Hitomi and A. J. Simaan, Inorg. Chem., 2009, 48, 3910–3912 CrossRef CAS PubMed.
- (a) W. Ghattas, C. Gaudin, M. Giorgi, A. Rockenbauer, A. J. Simaan and M. Reglier, Chem. Commun., 2006, 1027–1029 RSC; (b) W. Ghattas, M. Giorgi, C. Gaudin, A. Rockenbauer, M. Reglier, Y. Hitomi and A. J. Simaan, Bioinorg. Chem. Appl., 2007, 43324 Search PubMed; (c) W. Ghattas, M. Giorgi, Y. Mekmouche, T. Tanaka, A. Rockenbauer, M. Réglier, Y. Hitomi and A. J. Simaan, Inorg. Chem., 2008, 47, 4627–4638 CrossRef CAS PubMed; (d) N. Judaš and N. Raos, Inorg. Chem., 2006, 45, 4892–4894 CrossRef PubMed.
- (a) M. Costas, K. Chen and L. Que Jr., Coord. Chem. Rev., 2000, 200–202, 517–544 CrossRef CAS; (b) E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS PubMed.
- C. P. Magill, C. Floriani, A. Chiesi-Villa and C. Rizzoli, Inorg. Chem., 1994, 33, 1928–1933 CrossRef CAS.
- (a) K. Chen and L. Que Jr., Chem. Commun., 1999, 1375–1376 RSC; (b) M. C. White, A. G. Doyle and E. N. Jacobsen, J. Am. Chem. Soc., 2001, 123, 7194–7195 CrossRef CAS.
- G. J. P. Britovsek, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS PubMed.
- A. J. Simaan, S. Poussereau, G. Blondin, J.-J. Girerd, D. Defaye, C. Philouze, J. Guilhem and L. Tchertanov, Inorg. Chim. Acta, 2000, 299, 221–230 CrossRef.
- (a) K. P. Bryliakov, E. A. Duban and E. P. Talsi, Eur. J. Inorg. Chem., 2005, 299, 72–76 CrossRef; (b) G. J. P. Britovesk, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data, UV-Vis measurements, magnetization measurements. CCDC 1029372 and 1029373. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00347d |
| ‡ Equally contributed to the work reported. |
| This journal is © The Royal Society of Chemistry 2015 |