
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand assisted carbon dioxide activation and hydrogenation using molybdenum and tungsten amides†
Subrata
Chakraborty
,
Olivier
Blacque
and
Heinz
Berke
*
Departent of Chemistry, University of Zürich, Winterthurerstrasse 190, 8057 Zürich, Switzerland. E-mail: hberke@chem.uzh.ch
First published on 2nd March 2015
Abstract
The hepta-coordinated isomeric M(NO)Cl3(PNHP) complexes {M = Mo, 1a(syn,anti); W, 1b(syn,anti), PNHP = (iPr2PCH2CH2)2NH, (HN atom of PNHP syn and anti to the NO ligand)} and the paramagnetic species M(NO)Cl2(PNHP) (M = Mo, 2a(syn,anti); W, 2b(syn,anti)) could be prepared via a new synthetic pathway. The pseudo trigonal bipyramidal amides M(NO)(CO)(PNP) {M = Mo, 3a; W, 3b; [PNP]− = [(iPr2PCH2CH2)2N]−} were reacted with CO2 at room temperature with CO2 approaching the M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond in the equatorial (CO,NO,N) plane trans to the NO ligand and forming the pseudo-octahedral cyclic carbamates M(NO)(CO)(PNP)(OCO) (M = Mo, 4a(trans); W = 4b(trans)). DFT calculations revealed that the approach to form the 4b(trans) isomer is kinetically determined. The amine hydrides M(NO)H(CO)(PNHP) {M = Mo, 5a(cis,trans); W, 5b(cis,trans)}, obtained by H2 addition to 3a,b, insert CO2 (2 bar) at room temperature into the M–H bond generating isomeric mixtures of the η1-formato complexes M(NO)(CO)(PNHP)(η1-OCHO), (M = Mo, 6a(cis,trans); M = W, 6b(cis,trans)). Closing the stoichiometric cycles for sodium formate formation the 6a,b(cis,trans) isomeric mixtures were reacted with 1 equiv. of Na[N(SiMe3)2] regenerating 3a,b. Attempts to turn the stoichiometric formate production into catalytic CO2 hydrogenation using 3a,b in the presence of various types of sterically congested bases furnished yields of formate salts of up to 4%.
N double bond in the equatorial (CO,NO,N) plane trans to the NO ligand and forming the pseudo-octahedral cyclic carbamates M(NO)(CO)(PNP)(OCO) (M = Mo, 4a(trans); W = 4b(trans)). DFT calculations revealed that the approach to form the 4b(trans) isomer is kinetically determined. The amine hydrides M(NO)H(CO)(PNHP) {M = Mo, 5a(cis,trans); W, 5b(cis,trans)}, obtained by H2 addition to 3a,b, insert CO2 (2 bar) at room temperature into the M–H bond generating isomeric mixtures of the η1-formato complexes M(NO)(CO)(PNHP)(η1-OCHO), (M = Mo, 6a(cis,trans); M = W, 6b(cis,trans)). Closing the stoichiometric cycles for sodium formate formation the 6a,b(cis,trans) isomeric mixtures were reacted with 1 equiv. of Na[N(SiMe3)2] regenerating 3a,b. Attempts to turn the stoichiometric formate production into catalytic CO2 hydrogenation using 3a,b in the presence of various types of sterically congested bases furnished yields of formate salts of up to 4%.
Introduction
Carbon dioxide is one of the main carbon sources and although it is abundant and inexpensive, its utilization as feedstock for commodity chemicals is insufficiently explored.1 CO2 being a thermodynamically and kinetically stable molecule a promising approach to overcome its often too low reactivity would be its metal mediated “activation”.2,3a In fact recent years have seen tremendous efforts to activate and reduce CO2 and to employ it as a C1 building block for chemical synthesis.3 Activation by binding to a metal center and subsequent reaction modes of the CO2 depend on the type of the CO2 binding. There are three possible ways of CO2 coordination as depicted in Fig. 1.1b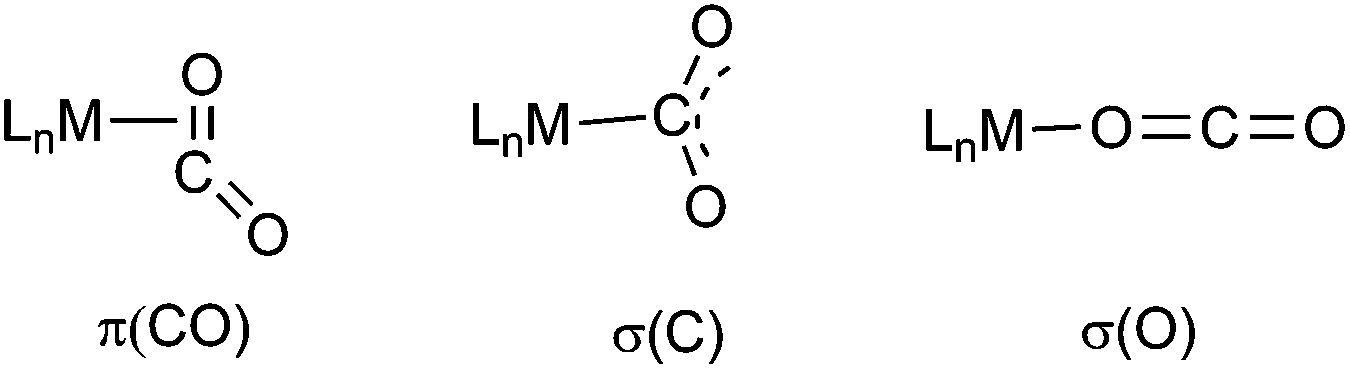 | ||
| Fig. 1 Possible binding modes of CO2 to a metal centre. | ||
Lately metal ligand cooperation (MLC) has been explored as a way to activate H2 or small molecules with C–H, N–H, O–H moieties.4,5 MLC would be a promising approach for CO2 activation, but reported examples of such CO2 activation are scarce. Only recently Sanford and coworkers have demonstrated that (PNN)Ru(H)(CO) pincer systems take up CO2 at room temperature involving C–C bond formation between the unsaturated arm of the pincer ligand and the electrophilic carbon atom of CO2 (Fig. 2).6 Milstein and coworkers investigated related types of reversible CO2 binding employing unsaturated PNP pincer complexes of Ru and Re (Fig. 2).7
 | ||
| Fig. 2 Metal–ligand cooperation in CO2 activation. | ||
“Metal-free” CO2 activation was accomplished using Frustrated Lewis Pair (FLP) type chemistry.8 In this context it seems also worth mentioning that a transition metal based LnRe-H/B(C6F5)3 systems can activate CO2 in a FLP type manner, where the metal hydride species took the role of the FLP Lewis base component.9 Activation of CO2 is anticipated to be preceding its hydrogenation, which may then lead to formic acid, but also can continue in the reduction sequence forming products with higher hydrogen contents, which like formic acid could in principle function as reversible H2 storage materials.10 Though the reaction of H2 with CO2 to obtain formic acid is exothermic (ΔH = −8 kcal mol−1), it is endergonic with ΔG = 8 kcal mol−1 due to gaseous starting materials. Therefore an additional thermodynamic driving force has to be provided, like for instance pressure and the addition of a base inducing salt formation or stabilizing acid–base hydrogen bonding interactions3 causing a shift of the equilibrium towards the formic acid side.
Advances in the efficiency of homogeneous hydrogenation catalysis of CO2 were recently reported with late transition metal complexes,11,12 but somewhat different from these developments it is one of the aims of this research to replace the platinum group metals ruthenium,13–15 rhodium16,17 and iridium18,19 with non-precious metals.20 In this regard molybdenum and tungsten based reductions of CO2 are challenging objectives. Despite some recent achievements in molybdenum and tungsten catalysed homogeneous hydrogenations21 and related hydrosilylations,22 and our group's recent accomplishments in bifunctional imine hydrogenations,23a the stepwise ionic hydrogenation of imines,23b,c Osborn type olefin hydrogenations24 and nitrile hydrogenations,25 the homogeneous hydrogenation of carbon dioxide could not be achieved with compounds of these metals till date.
In this paper a carbon dioxide activation mode supported by MLC could be established as shown in Fig. 2. Furthermore, stoichiometric hydrogenation of CO2 turned out to be accessible using the previously reported molybdenum and tungsten amides 3a,b.23a
Results and discussion
Preparation of M(II) and M(I) (M = Mo, W) complexes bearing (iPr2PCH2CH2)2NH ligand
Reaction of (iPr2PCH2CH2)2NH (PNHP) ligand with the M(NO)Cl3(NCMe)2 complexes (M = Mo, W) led at room temperature in THF to precipitation of the isomeric mixtures of Mo(NO)Cl3(PNHP), 1a(syn,anti) and W(NO)Cl3(PNHP), 1b(syn,anti)) in moderate yields (1a(syn,anti), 55%; 1b(syn,anti), 62%) (Scheme 1). | ||
| Scheme 1 Preparation of the isomeric mixture of M(II)(NO)Cl3(PNHP), 1a,b(syn,anti)) and reduction to M(I)(NO)Cl2(PNHP), 2a,b(syn,anti) complexes (M = Mo,W). | ||
The syn and anti notation for the isomers of 1a,b(syn,anti) correspond to the relative position of the HN atom at the internal N atom of the PNHP ligand with respect to the NO ligand. The spectroscopic yields of syn and anti isomers of both 1a,b(syn,anti) are given in Scheme 1. The isomers of 1a(syn,anti) and 1b(syn,anti) were found to be reasonably soluble only in CH2Cl2. The 31P{1H} NMR spectra of the isomeric mixture of 1a(syn,anti) revealed two singlets at δ 69.3 ppm (1a(syn)) and 71 ppm (1a(anti)) in a ratio of 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 indicating equivalence of both phosphorus atoms of each isomer of 1a(syn,anti) (Fig. 3, left). The 31P{1H} NMR spectra of the isomeric mixture of 1b(syn,anti) exhibited also two singlets, but in this case along with tungsten satellites at δ 56.8 ppm (s, 1JP–W (d, satellite) = 184.8 Hz, 1b(syn)) and 58.3 (s, 1JP–W (d, satellite) = 189.3 Hz, 1b(anti)) assigned to the equivalent phosphorus atoms of each syn and anti isomer (Fig. 3, right).
:3 indicating equivalence of both phosphorus atoms of each isomer of 1a(syn,anti) (Fig. 3, left). The 31P{1H} NMR spectra of the isomeric mixture of 1b(syn,anti) exhibited also two singlets, but in this case along with tungsten satellites at δ 56.8 ppm (s, 1JP–W (d, satellite) = 184.8 Hz, 1b(syn)) and 58.3 (s, 1JP–W (d, satellite) = 189.3 Hz, 1b(anti)) assigned to the equivalent phosphorus atoms of each syn and anti isomer (Fig. 3, right).
 | ||
| Fig. 3 31P{1H} NMR spectra of the isomeric mixture of Mo(NO)Cl3(PNHP), 1a(syn,anti) {left, 1a(syn), 70%; 1a(anti), 30%} and the isomeric mixture of W(NO)Cl3(PNHP), 1b(syn,anti) {right, 1b(syn), 70%; 1b(anti), 30%} at room temperature in CD2Cl2. | ||
In the IR spectra of 1a,b(syn,anti) the isomers could not be distinguished, which showed only one sharp band for the molybdenum and tungsten complexes at 1653 and 1596 cm−1, respectively, attributed to the νNO vibrations. The 1H NMR spectra of the isomeric mixtures of 1a,b(syn,anti) exhibited several doubled signals for the methyl, methyne and methylene protons appearing in the expected region with strong overlap of the signals of the syn and anti isomers impeding their distinct assignment. In the 1H NMR spectra a conspicuous signal appeared at δ 4.4 ppm, which was assigned to the HN atom of 1a(syn). However, the HN atom of 1a(anti) could not be assigned in the 1H NMR spectrum presumably owing to the quite low concentration of this isomer and overlap of the signal with the signal of the CH2Cl2 solvent. The HN atoms of the isomeric mixture of 1b(syn,anti) appeared as broad singlets at δ 4.47 ppm and 5.50 ppm in the 1H NMR spectra and based on the different intensity these could be assigned to the 1b(syn) and 1b(anti) isomers, respectively. The virtual triplet signal in the 13C{1H} NMR at δ 55.9 (t, vJC–P = 3.6 Hz) ppm for isomeric mixture of 1a(syn,anti) was attributed to the carbon atoms of 1a(syn) isomer adjacent to the nitrogen atom of the PNHP ligand and the related signals of 1b(anti) could not be detected. On the other hand the virtual 13C{1H} NMR triplets at δ 55.9 (t, vJC–P = 5.9 Hz) ppm for 1b(syn) and at 55 ppm (t, vJC–P 4.8 Hz) for 1b(anti) could be assigned to the N-adjacent carbon atoms. Several attempts failed to grow suitable single crystals for X-ray diffraction studies of any isomer of the 1a,b(syn,anti) mixture, but 1a,b(syn,anti) could be fully characterized by 1H NMR, 31P{1H} NMR, 13C{1H} NMR, IR and mass spectrometry, and the compositions were established by elemental analyses.
1a,b(syn,anti) are heptacoordinated diamagnetic species with the metal centers in +II oxidation states. Our initial attempts to deprotonate the N–H moiety of 1a(syn,anti) applying NaN(SiMe3)2 to obtain 16 e− unsaturated amido complex23a failed, which led to inseparable mixtures of products and could not be identified. Attempts to reduce 1a,b(syn,anti) by 1-electron reducing agents led to paramagnetic M(+I) species (M = Mo, W). Further reduction to M(0) species seemed difficult with the given strongly electron donating coordinative environment.
The reaction of 1a,b(syn,anti) with approximately one equivalent of 1% Na/Hg in THF at room temperature resulted in the formation of green isomeric mixtures of Mo(I)(NO)Cl2(PNHP) complexes (2a(syn,anti)) or of W(I)(NO)Cl2(PNHP) (2b(syn,anti)) in 74% and 78% yields, respectively (Scheme 1). 2a,b(syn,anti) were found quite soluble in THF and CH2Cl2, but were sparingly soluble in toluene and benzene. In the 31P{1H} and 1H NMR spectra of the isomeric mixtures of 2a,b(syn,anti) no signals could be observed due to the paramagnetic nature of these complexes. The inaccessibility of 1H and 31P{1H} NMR spectra impeded estimates of spectroscopic yields of the syn and anti isomers in solution. Thus, the isomeric mixtures of 2a,b(syn,anti) were characterized by IR and EPR spectroscopy, elemental analyses and X-ray diffraction studies. Sharp bands at 1595 and 1547 cm−1 for the isomeric mixtures of 2a,b(syn,anti) were assigned to νNO vibrations, respectively, which appeared at lower wave numbers than those of 1a,b(syn,anti) indicating a higher degree of back-donation from the metal center to the nitrosyl ligand. Single crystals suitable for X-ray diffraction studies were grown from the isomeric mixtures of 2a,b(syn,anti) from concentrated toluene–pentane mixtures at −30 °C. The crystal structure determination revealed preferential crystallization of the 2a(anti) and 2b(syn) isomers. Both of them crystallized in the orthorhombic space group P212121. The molecular structures revealed neutral pseudo-octahedral complexes. Perspective views with selected bond distances and bond angles of 2a(anti) and 2b(syn) are shown in Fig. 4. The two phosphorus atoms and the nitrogen atom of the PNHP ligand and the two chloride ligands attached to the metal center occupy “equatorial” positions in both cases. The HN proton and the nitrosyl ligand were found to be disposed anti indicating the presence of the anti2a(anti) isomer. No NO/Cl disorder was found in the structure of 2a(anti), but despite this, the presence of isomeric mixtures in solution with one prevailing component could not fully be ruled out.
 | ||
| Fig. 4 Molecular structures of M(NO)Cl2(PNHP), {M = Mo; 2a(anti) (left); W, 2b(syn) (right)} Thermal ellipsoids are drawn at the 50% probability level. Selected bond distances (Å) and bond angles (°) for 2a(anti): Mo1–N2 2.255(2), Mo1–N1 1.855(3), Mo1–Cl1 2.5021(7), Mo1–P1 2.5372(7), N2–H2 0.91, P2–Mo1–P 1156.25(2), P2–Mo1–Cl2 100.64(2), P2–Mo1–Cl1 87.04(2), Mo1–N1–O1 178.7(3). Selected bond distances(Å) and bond angles (°) for 2b(syn): W1–N2 2.232(4), W1–N1B 1.811(19), W1–P1 2.5123(13), W1–Cl2B 2.381(6), N2–H 0.79 (4), P2–W1–P1 157.23(4), P2–W1–N2 78.82(13), W1–N1B–O1B 178(3), N1B–W1–Cl2B 172.2(8). | ||
On the other hand the crystal structure analysis of 2b(syn) revealed that the structure had trans NO/Cl disorder over two positions with site occupancy factors of 0.698(8) and 0.302(8). This NO/Cl disorder again supports the presence of the isomeric mixtures with respect to the HN position and the nitrosyl ligand and the presence of the syn and anti isomer in an approximately 7:3 ratio in the solid state. An inversion at the N atom with prototopic rearrangement at this atom seemed at least not to occur in the solid state. This would be also in accord with the assignment of the same syn/anti ratio for the precursors 1a,b(syn,anti). The isomeric mixtures of 2a,b(syn,anti) are rare paramagnetic complexes with the metal centers in +I oxidation state and of low-spin d5 configurations. To probe the paramagnetic nature, solution EPR measurements were carried out on 2a,b(syn,anti) in toluene at room temperature. The spectra are displayed in the ESI section (Fig. S1†).
We then attempted deprotonation of the HN proton of the PNHP ligand backbone of 2a,b(syn,anti) employing Na[N(SiMe3)2], which led to decomposition and the solution turned black immediately. Moreover, attempts were also undertaken to prepare the previously reported M(NO)(CO)(PNP) {M = Mo, 3a; W, 3b} amides23avia a new synthetic route starting from the 2a,b(syn,anti).
Reduction of 2a,b(syn,anti) in the presence of 1 bar CO and 1% Na/Hg at room temperature showed the formation of several unidentified species instead of the expected unique complex of an isomeric carbonyl chloride M(NO)(CO)Cl(PNHP) (M = Mo, W), which prevented concomitant deprotonation to obtain the requested M(NO)(CO)(PNP) (M = Mo, 3a; W, 3b).
Nevertheless M(NO)(CO)(PNP) 3a,b could be prepared according to our previously published procedure23a starting from M(NO)(CO)4(ClAlCl3) and PNHP ligand followed by deprotonation with NaN(SiMe3)2 and tested their reactivity with CO2 and their hydrogenation capability.
Reactivity of the M(NO)(CO)(PNP) {M = Mo, 3a W, 3b} amide complexes with CO2
Reaction of M(NO)(CO)(PNP) {M = Mo, 3a W, 3b; PNP = N(CH2CH2PiPr2)2} with CO2 (2 bar) at room temperature in THF led to formation of the 2 + 2 addition products across the MN bonds, the carbamate complexes Mo,W(NO)(CO)(PNP)(OCO) 4a,b(trans) (Scheme 2). The specification “trans” refers to the orientation of the CO2 approach transoid to the NO ligand of 3a,b. The orange 4a,b(trans) complexes could be isolated in pure form in quantitative yields. Monitoring the CO2 addition reactions visually, a purple color appeared indicating the presence of short-lived intermediates, which were not isolable and are proposed to be charge transfer complexes. This type of reaction of CO2 with 3a,b to form carbamates 4a,b(trans) is closely related to the 2 + 2 cycloaddition reactions of early transition metal imido complexes with alkynes, olefins and ketones.26 Hessen and coworkers demonstrated an example of a thermodynamically favored insertion of alkynes into the V–amide bond of cationic amido imido vanadium complexes rather than a 2 + 2 cycloaddition reaction.27 Therefore, an insertion reaction of CO2 across the M–amide bond could alternatively be envisaged. Nevertheless, this CO2 activation product is unique in its structure and is therefore in its way of formation presumed to be distinct from the insertion reactions of early transition M–N bonds.
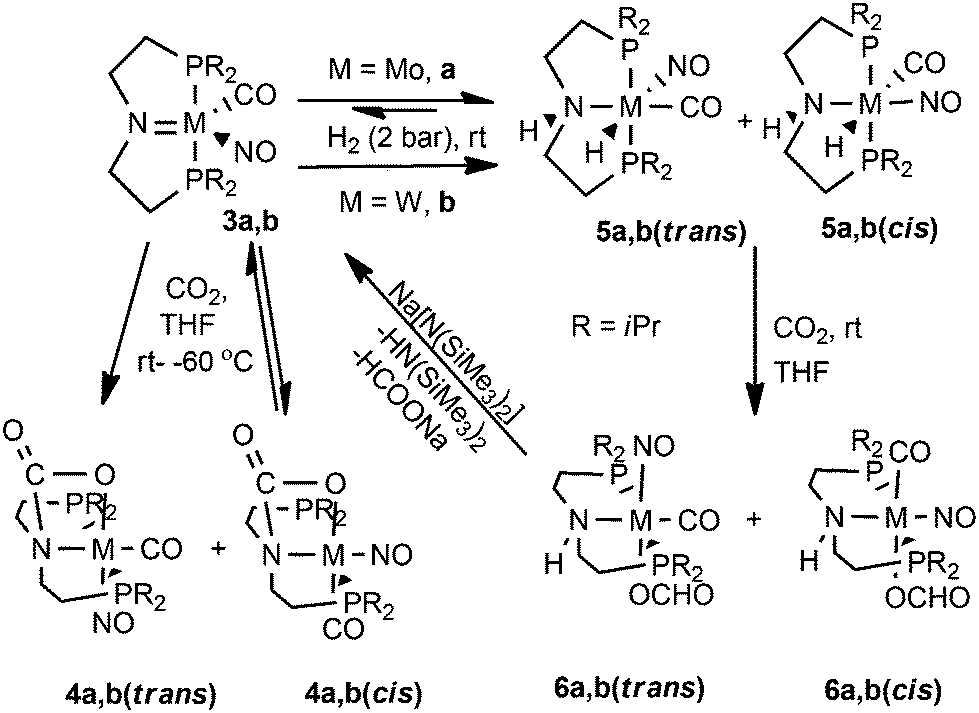 | ||
| Scheme 2 CO2 activation by molybdenum and tungsten amides 3a,b and hydrides 5a,b(cis,trans). | ||
The 31P{1H} NMR spectra of 4a,b(trans) at room temperature exhibited sharp singlets at δ 61.9 and 56.4 ppm, respectively, indicating equivalence of the phosphorus atoms of the metal attached PNP ligand and under the given conditions also the absence of the theoretically possible isomeric products 4a,b(cis) with the CO2 approach from the NO side of 3a,b. When the reactions of 3a,b with CO2 (2 bar) were carried out at −60 °C and pursued by 31P{1H} NMR spectroscopy, additional weak signals were seen at δ 62.7 and at 57.9 ppm besides those of 4a,b(trans) indicating formation of kinetic mixtures of the 4a,b(trans,cis) isomers (7:3 ratios in both cases of compounds). These mixtures equilibrated at room temperature into the thermodynamically more stable 4a,b(trans) as the sole products based on the reversibility of the CO2 additions to 3a,b from the NO side. 4a,b(cis) could thus not be observed by 31P{1H} NMR spectroscopy at room temperature in solution and could also not be isolated (Scheme 2).
The 1H NMR spectra of 4a,b(trans) showed several distinct signals for the methyl, methylene and methyne protons in the expected region supporting also the absence of isomers. The IR spectra of 4a,b(trans) displayed ν(NO) and ν(CO) bands at 1593, 1554 cm−1 and at 1902, 1879 cm−1, respectively. Additional strong absorptions appeared for 4a,b(trans) at 1732 and 1741 cm−1, respectively, and were assigned to the νCO2 vibration of the attached CO2 molecule. In the 13C{1H} NMR spectra of 4a,b(trans) sharp singlets at δ 156.4 and 159.3 ppm were attributed to the CCO2 atoms along with multiplets at δ 247 and 250.6 ppm for the CCO atoms of the carbonyl ligands.
The CCH2 atoms adjacent to the nitrogen atoms of the PNP ligand of 4a,b(trans) were observed as virtual triplets at δ 53.2 (t, vJCP = 3.6 Hz) and at 57.7 (t, vJC–P = 3.6 Hz) ppm, respectively. Single crystals of 4b(trans) suitable for an X-ray diffraction study were obtained by layering pentane over a concentrated THF solution. The structural model obtained from the diffraction study is depicted in Fig. 5, which revealed a pseudo octahedral geometry. The W1–N2 bond distance was found to be 2.2603(19) Å. Upon CO2 addition it became significantly elongated by about 0.2 Å with respect to the approximate WN bond of 3b.23a
 | ||
| Fig. 5 Molecular structure of 4b(trans). Thermal ellipsoids are drawn at the 50% probability level. All hydrogen atoms are omitted for clarity. Selected bond distances [Å] and bond angles [°]: W1–N1 1.789(2), W1–C1 1.969(3), W1–N2 2.2603(19), W1–O3 2.2155(18), C1–O1 1.159(3), C2–O3 1.283(3), C2–O4 1.208(3), N1–W1–C1 88.61(10), N2–W1–O3 172.47(8), N2–C2–O3 108.0 (2) P1–W1–P2 157.60(2). | ||
The transition state of the reaction between W(NO)(CO)(PNP) 3b and CO2 could be modelled by DFT calculations (see ESI†).
Reasonably low activation energies ΔG‡ = 14.4 kcal mol−1 and 14.1 kcal mol−1 were calculated for the CO and NO side approaches of CO2 onto 3b to give 4b(trans) and 4b(cis), respectively. Remarkably, both approaches are kinetically almost not distinct. The free enthalpies of both reactions were determined to be −7.7 kcal mol−1 (4b(trans)) and −5.6 kcal mol−1 (4b(cis)) indicating that 4b(trans) is the thermodynamically more favored and stable product. The calculated lower free energy value for 4b(cis) looks plausible in view of the fact that the CO2 approaches from the NO side are experimentally equilibria. The calculated activation barrier for the reverse reaction from 4b(cis) to form 3b and CO2 seems thus to be still in energetic reach for a thermal process at room temperature (ΔG = 19.8 kcal mol−1). Drawings of the TS structure of the CO2 approach to 3b (Fig. 6 (left)) and the HOMO of the TS (Fig. 6 (right)) are shown in Fig. 6 (see also ESI†). According to the calculations the TS consists of a strongly distorted TS square with W–N, N–C, C–O and O–W contact lengths of 2.144, 2.399, 1.184 and 2.780 Å, respectively. These bond lengths mark an early transition state of the 2e + 2e reaction. The C–N interaction is the main new contact in the transition state, which is also represented in Fig. 6 (right), where large orbital coefficients appear between the C(CO2) and the N atoms. The other new contact of the O(CO2) atom to the W center is still very distant. Despite this feature that asymmetry prevails in the bond forming process, the reaction coordinate was found to be continuous and not two-stage without intermediate generated by consecutive bond formations.
 | ||
| Fig. 6 Geometry of the transition state (TS) of the approach of CO2 to 3b (left) and the TS HOMO−3 orbital for the CO2 2e + 2e addition reaction (right). | ||
Reactivity of the hydride complexes M(NO)H(CO)(PNHP) (M = Mo, 5a(cis,trans); W, 5b(cis,trans)) with CO2
Then we also tested the CO2 reactivity of the previously reported23a isomeric mixtures of the amine hydride compounds M(NO)H(CO)(PNHP) (5a,b(cis,trans)), which were obtained by H2 addition to 3a,b from both sides of the MN bond. The cis and trans notation indicate trans and cis attack of the H2 molecule with respect to the NO ligand leading to the trans H/NO complexes (5a,b(trans)) or to the trans H/CO complexes (5a,b(cis)). However, the reactions to 5a(cis,trans) with H2 were found to be equilibria showing at room temperature an approximate 1:1:1 mixture of 5a(cis)/5a(trans)/3a. Therefore, the reaction with CO2 (2 bar) had to be carried out using the in situ generated mixture of Mo(NO)H(CO)(PNHP) (5a(cis,trans)), which led to the formate products Mo(NO)(CO)(PNHP)(η1-OCHO) 6a(cis,trans) and the cyclic carbamate species 4a(trans) in an approx. 1:1:1 ratio as indicated by 13C{1H} and 31P{1H} NMR spectroscopy (Scheme 2). Since the three different kinds of reactions (2 kinds of CO2 insertions into the M–H bonds and CO2 addition) did not reveal a change in the ratios from the starting materials to the products, we have to assume that all involved reaction rates are about the same. The formate protons of 6a(trans) and 6a(cis) appeared in the 1H NMR spectrum at δ 8.1 and 8.5 ppm, respectively (1:1 ratio). Signals at δ 170 and 168 ppm in the 13C{1H} NMR spectra were assigned to the Cformate nuclei of the 6a(cis,trans) isomers, respectively. 6a(cis,trans) and 4a(trans) were not in equilibrium, but despite this attempts to separate these complexes turned out to be unsuccessful.
Similarly, the reaction of the 5b(cis,trans) mixture, which is not an equilibrium reaction allowing this mixture to be isolated, with CO2 resulted at room temperature in the rapid formation of the isomeric η1-formate complexes W(NO)(CO)(PNHP)(η1-OCHO) (6a,b(trans))28 (Scheme 2) appearing in a 9:1 ratio. A strong signal at δ 51.3 ppm (with tungsten satellites) in the 31P{1H} NMR spectrum was assigned to 6b(trans) along with a weak signal at δ 52.6 ppm for 6b(cis). In the 1H NMR spectrum the formate protons of 6b(trans,cis) were observed at δ 8.5 and 8.0 ppm, respectively, and signals at δ 244.7 and 164.3 ppm in the 13C(1H) NMR spectra were attributed to the carbonyl ligand and the Cformate atom of the major isomer 6b(trans). Supposedly due to a too low concentration of 6b(cis), the 13C NMR resonances of the carbonyl and formate carbon atoms of the minor 6b(cis) isomer could not be observed. In the IR spectrum of the 6b(cis,trans) mixture a band at 1612 cm−1 was attributed to the νas(CO2) vibration in accord with earlier assignments of η1-O-formato tungsten complexes.28 Additional IR bands at 1863 and 1578 cm−1 of the 6b(cis,trans) mixture were assigned to the ν(CO) and ν(NO) ligand vibrations of both compounds showing that both compounds cannot be distinguished in the 1500 to 1900 cm−1 region of the IR spectra.
Despite the fact that 6b(cis) was the minor constituent of the isomeric mixture in solution, tiny crystals of this minor component could precipitated from solution by slow diffusion of pentane into a concentrated THF solution. The X-ray diffraction study revealed a pseudo-octahedral framework with cis nitrosyl and formate ligands (Fig. 7). The asymmetric unit of this minor isomer 6b(cis) contained two crystallographically independent tungsten species displaying intermolecular hydrogen bonding31 between the protic HN atom of one molecule and the Oformato of an adjacent molecule forming linear chains in the three dimensional lattice. It should be noted here that the HN atom and the formate group were found to be disposed anti in the solid state structure, which was somewhat surprising when cis addition of H2 to 3a,b is taken into consideration, followed by CO2 insertion into the M–H bond occurring with retention of the configuration at the metal centers (Scheme 2). One possible reason for the thermodynamically favored re-orientation of the HN atom by a prototopic rearrangement could be the adoption of an energetically preferred intermolecular hydrogen bonding network requiring anti disposition of the HN atom and the formate ligand in the crystal lattice.
 | ||
| Fig. 7 Molecular structure of 6b(cis). Thermal ellipsoids are drawn at the 50% probability level. All hydrogen atoms are omitted for clarity except H1. Selected bond distances [Å] and bond angles [°]: W1–N1 2.287(5), W1–C1 1.930(8), W1–N2 1.792(6), W1–O3 2.185(5), C1–O1 1.172(8), C2–O3 1.271(7), C2–O4 1.213(8), N1–W1–C1 91.6(3), N2–W1–O3 101.9(2), N1–W1–N2 178.3 (3) P1–W1–P2 158.32(7). | ||
In addition to these findings DFT calculations support the observation that the major trans NO/formate isomer 6b(trans) is thermodynamically more stable than the trans CO/formate isomer 6b(cis) by ΔE = −4.4 kcal mol−1 (see Scheme 2 and ESI†), which seems to match well reality. The fact that in the crystallization experiment of the 6b(cis,trans) mixture 6b(cis) was found to be less soluble than 6b(trans) can now be attributed to a secondary crystallization effect based on the mentioned hydrogen bonding system in the solid state causing lower solubility of the minor constituent in solution. In addition it should be noted that the proton at N1 and the formate ligand of the structure of 6b(cis) are in anti position, which somewhat contradicts the fact that in 5a,b(trans) and 5a,b(cis) they are expected to be syn due to the Z stereochemistry of the H2 addition and the expected retention of the configuration at the metal center associated with the CO2 insertion process. Therefore the proton at N1 is assumed to have undergone an inversion process when forming the crystals of 6b(cis), which seems likely on the basis of our DFT calculations, which revealed only minor energetic differences (approx. 2 kcal mol−1) between the syn and anti structures in favour of the anti arrangements.
Stoichiometric hydrogenation of carbon dioxide
The 6a,b(cis,trans) mixtures can be deprotonated at the HN atom employing 1 equiv. of Na[N(SiMe3)2] at room temperature with regeneration of 3a,b. Sodium formate was formed in addition in accord with eqn (1) closing thus a stoichiometric cycle of consecutive reaction steps of H2 and CO2 additions in a “pseudo-catalytic” reaction course. The permanent presence of a base seemed not to interfere with 6b(cis,trans) and to initiate side reactions, but the preferential addition of CO2 over the H2 addition to 3a,b seemed in the presence of a H2–CO2 mixture a great obstacle to establish a genuine catalytic reaction course. Therefore, if catalysis should be accomplishable, it could be envisaged at higher temperatures making the CO2 addition to some extent reversible enabling thus under catalytic conditions H2 before CO2 addition to 3a,b (Scheme 3).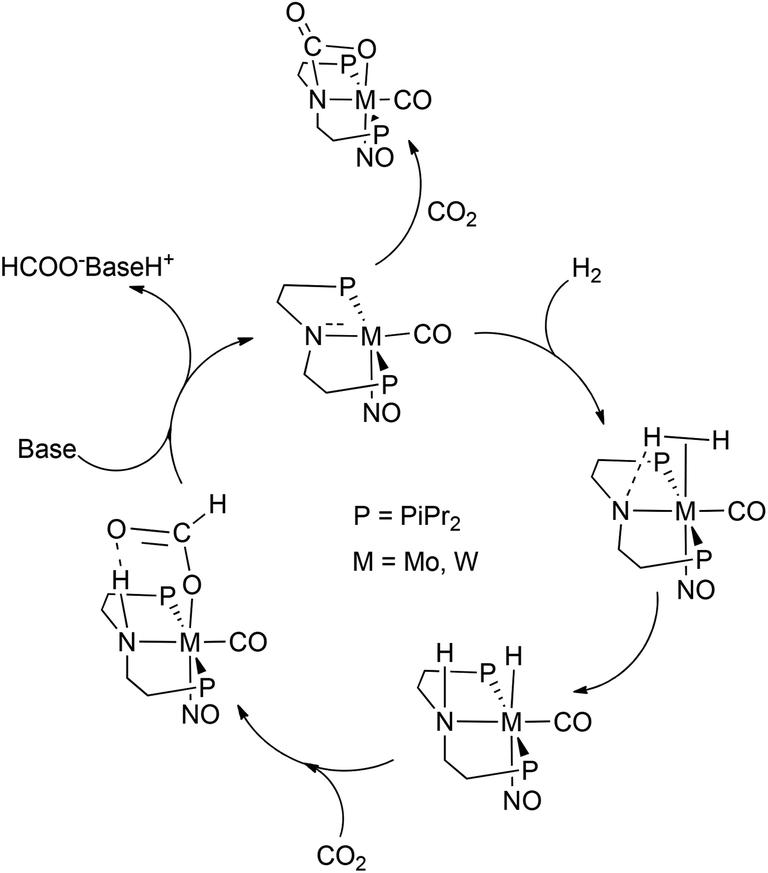 | ||
| Scheme 3 Schematic cycle for the CO2 hydrogenation using 3a,b. | ||
Being aware of this situation, we attempted catalysis and carried out at 140 °C 3 types of hydrogenation experiments of CO2 applying the simultaneous presence of H2 (70 bar) and CO2 (10 bar) and of catalytic amounts of 3a,b or of the 5b(cis,trans) mixture (5 mol% loading) in the presence of Na[N(SiMe3)2] as stoichiometric agent. After 15 h all the experiments revealed formation of HCOONa according to eqn (1), but the yields were low, only 4%, 2% and 3%, respectively, as examined by 1H NMR (DMF as internal standard) (Table 1). To improve the yield of the formate salt, we varied the type of the base using DBU, KOtBu or NEt3 in the presence of 3a as a catalyst with otherwise the same conditions as above. However, the yield of the formate salts [HCOO][DBUH] (3.5%), HCOOK (2.5%) or HCOOH–NEt3 (0.5%), did not increase (Table 1). Furthermore, the addition of B(C6F5)3 or the Et3SiH–B(C6F5)3 mixture as co-catalysts led in the presence of 3a and Na[N(SiMe3)2] even to a decrease in yields.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | Cat | Base | Time (h) | Yieldb (%) | |||
| a Unless and otherwise stated 10 bar CO2, 70 bar H2, THF as solvent, 140 °C temperature and 5 mol% catalyst with respect to base were used as reaction conditions. b Yield on the basis of 1H NMR integration using DMF as internal standard. c No catalyst was used. | |||||||
| 1 | 3a | Na[N(SiMe3)2] | 15 | 4 | |||
| 2 | 3b | Na[N(SiMe3)2] | 15 | 2 | |||
| 3 | 5b(cis,trans) | Na[N(SiMe3)2] | 15 | 3 | |||
| 4 | 3a | DBU | 15 | 3.5 | |||
| 5 | 3a | KOtBu | 15 | 2.5 | |||
| 6 | 3a | Et3N | 15 | 0.50 | |||
| 7c | — | Na[N(SiMe3)2] | 17 | 0.14 | |||
| 8 | 3a | TMP | 15 | — | |||
| 9 | 3a/BCF | Na[N(SiMe3)2] | 15 | 0.70 | |||
| 10 | 3a/Et3SiH/BCF | Na[N(SiMe3)2] | 22 | 0.30 | |||

At this stage it can be stated that CO2 hydrogenation is in principle possible, but for effective such catalyses further tuning efforts have to be taken into consideration, presumably by attempts to make the CO2 adducts of type 4 more labile. It should be mentioned at this point that since strong bases are known to react with CO2 particularly at high temperatures and pressures, a control experiment was also carried out in the absence of the metal catalyst applying only Na[N(SiMe3)2] as the catalyst and keeping all the other conditions the same. The yield of sodium formate was in this case only 0.14%, which is significantly lower than the yields obtained in the presence of 3a,b.
It deserves mentioning that after the CO2 hydrogenation experiments (applying especially the NaN(SiMe3)2 base), the reaction mixtures were found to contain a considerable amount of white precipitate in the THF reaction mixture. When THF was evaporated in vacuo and the residue was dissolved in D2O to quantify the yield of the formate salt by 1H NMR spectroscopy (DMF internal standard), the solution was still found to contain white particles even after addition of excess D2O. Therefore, formation of another species is anticipated to be produced in the hydrogenation of CO2 (note that the product sodium formate is expected to be soluble in D2O). The nature of this species could however not be unravelled, but in principle it should be added to the overall yield of these catalytic reactions.
Conclusions
In conclusion we have reported a new synthetic route for the preparation of isomeric diamagnetic M(NO)Cl3(PNHP) (M = Mo, 1a(syn,anti); W, 1b(syn,anti)) complexes and their reduction to rare paramagnetic species M(NO)Cl2(PNHP) {M = Mo, 2a(syn,anti); W, 2b(syn,anti)}. Furthermore, we have shown that the excessively reduced pentacoordinate amides Mo,W(0)(PNP)(NO)(CO) (3a,b; PNP = (iPr2PCH2CH2)2N)) can activate CO2 in a metal–ligand assisted way via 2e + 2e additions across the MN bonds forming the pseudo-octahedral 4a,b(trans) compounds. The CO2 approach occurs thermodynamically controlled from the CO ligand side. Kinetically there is practically no distinction between the sides of attack. The amine hydride species M(NO)(CO)H(PNHP) (M = Mo, 5a(cis,trans) W, 5b(cis,trans)) formed by H2 addition to 3a,b reacted with CO2 at room temperature to produce η1-formato complexes M(NO)(CO)(PNHP)(η1-OCHO) {M = Mo, 6a(cis,trans); W, 6b(cis,trans)}. Stoichiometric hydrogenations of CO2 to formate salts could thus be accomplished via elimination of the formate ligands of type 6 complexes induced through the presence of a strong base regenerating the amides 3a,b. Catalytic hydrogenation of CO2 was then also approached at somewhat elevated temperatures and pressures. However, the apparently too high stability of the CO2 addition products of type 4 are anticipated to largely block the catalytic reaction course and cause low yields. This conclusion rendered the idea that in these Mo and W systems catalysis could eventually be achieved by tuning the 4 type complexes for reversibility in CO2 additions. It is finally worth mentioning that this study demonstrated for the first time that reductions of CO2 are within reach utilizing middle, non-noble transition metal compounds as catalysts.
Experimental section
General procedures
All experiments were carried out under an atmosphere of nitrogen using either dry glove box or Schlenk techniques. Reagent grade solvents benzene, tetrahydrofuran, pentane, toluene, diethyl ether were dried with sodium benzophenone and distilled prior to use under N2 atmosphere. CH2Cl2 and Et3N were dried over calcium hydride and distilled. Deuterated solvents were dried with sodium benzophenone ketyl (THF-d8, toluene-d8 and C6D6) and calcium hydride (CD2Cl2) and distilled via freeze–pump–thaw cycle prior to use. The M(NO)Cl3(NCMe)2,29 M(NO)(CO)(PNP),23a {M = Mo, W; PNP = N(CH2CH2iPr2)2} complexes and HN(CH2CH2iPr2)230 were prepared according to literature procedures. KOtBu, Na[N(SiMe3)2] and DBU were purchased from commercially available sources and used without further purification. The NMR spectra were measured with a Varian Mercury 200 spectrometer (at 200.1 MHz for 1H, at 81.0 MHz for 31P), with Varian Gemini-300 instrument (1H at 300.1 MHz, 13C at 75.4 MHz), with Bruker-DRX 500 spectrometer (500.2 MHz for 1H, 202.5 MHz for 31P, 125.8 MHz for 13C) and Bruker-DRX 400 spectrometer (400.1 MHz for 1H, 162.0 MHz for 31P, 100.6 MHz for 13C). All chemical shifts for 1H and 13C{1H} are expressed in ppm relative to tetramethylsilane (TMS) and for 31P{1H} relative to 85% H3PO4 as an external standard reference. Signal patterns are as followed: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; v, virtual triplet. IR spectra were obtained either ATR or KBr methods using Bio-rad FTS-45 instrument. Elemental analyses were carried out at Anorganisch-Chemisches Institut of the University of Zurich.
Preparation of isomeric mixture of Mo(NO)Cl3(PNHP), 1a(syn,anti)
To a solution of Mo(NO)Cl3(NCMe)2 (0.200 g, 0.636 mmol) in 10 mL of THF, 0.16 g (196 mg, 0.64 mmol) (iPr2PCH2CH2)2NH ligand in 5 mL THF was added. The resulting mixture was kept stirring overnight to obtain a light green precipitate and the solution becomes dark green. Then the solvent was separated from the precipitate and the precipitate was washed with minimum amount of THF and extracted with dichloromethane and dried in vacuo to obtain the powdery isomeric mixture of 1a(syn,anti) in 55% yield.1H NMR (400 MHz, CD2Cl2): δ = 4.4 (broad singlet, NH, 1a(syn)), 3.63–3.5 (m, NCH2), 3.23–3.21 (m, NCH2), 3.1–3.02 (m, CH), 2.3–2.2 (m, –PCH2), 2.05 (m, PCH2), 1.52–1.39 (m, CH3, 1a(syn)), 1.34–1.30 (m, CH3, 1a(anti)). 13C{1H} NMR (100.6 MHz, CD2Cl2): δ = 55.9 (t, vJC–P = 3.6 Hz, NCH2, 1a(syn)), 29 (m, CH), 28 (m, PCH2), 21 (m, CH3), 20.4 (m, CH3). 31P{1H} NMR (400 MHz, CD2Cl2): 69.3 (s, iPr2P, 1a(syn)), 71 (s, 1a(anti)). IR (cm−1, ATR): 1653 (νNO), expected m/z: 538, observed m/z: 538.2. Anal. Calcd for C16H37Cl3MoN2OP2: C, 35.74; H, 6.94; N, 5.21. Found: C, 36.03; H, 6.39; N, 5.67.
Preparation of isomeric mixture of W(NO)Cl3(PNHP), 1b(syn,anti)
WCl3(NO)(NCMe)2 (200 mg, 0.5 mmol) was dissolved in 10 mL of THF. 0.160 g (0.52 mmol) of aminodiphosphine (iPr2PCH2CH2)2NH was added via a syringe. After a few minutes stirring a light red precipitate was observed and the reaction was allowed to stir overnight. Then the solution was removed partially under vacuum and the reddish residue was filtered off and washed with THF. The obtained residue was extracted with dichloromethane and the solvent was removed in vacuo to obtained the isomeric mixture of 1b(syn,anti) in 62% yield. 1H NMR (400 MHz, CD2Cl2): δ = 4.47 (broad singlet, NH, 1b(syn)), 5.5 (broad singlet, NH, 1b(anti)) 3.71–3.57 (m, NCH2, syn,anti), 3.19–3.05 (m, CH), 2.30–2.23 (m, –PCH2), 2.09–2.06 (m, PCH2), 1.47–1.19 (m, CH3). 13C{1H} NMR (100.6 MHz, CD2Cl2): δ = 55.9 (t, vJC–P = 5.9 Hz, NCH2, 1b(anti)), 55 (t, vJC–P = 4.8 Hz, NCH2, 1b(anti)), 27.8 (m, CH), 27.3 (m, –PCH2), 20 (m, CH3). 31P{1H} NMR (400 MHz, CD2Cl2): 56.8 [s, 1JP–W (d, satellite) = 184.8 Hz, 1b(syn)], 58.3 [s, 1JP–W (d, satellite) = 189.3 Hz, 1b(anti)]. IR (cm−1, ATR): 1596 (νNO), Anal. Calcd for C16H37Cl3N2OP2W: C, 30.72; H, 5.96; N, 4.48. Found: C, 30.72; H, 5.88; N, 4.55.Preparation of Mo(NO)Cl2(PNHP), 2a(syn,anti)
The isomeric mixture of 1a(syn,anti) (0.030 g, 0.056 mmol) was added to a stirred suspension of approximately 1% sodium amalgam (0.0013 g, 0.057 mmol) in 15 mL THF. Stirring was continued overnight at room temperature to ensure the completion of the reaction. The final supernatant green solution was filtered off from the mercury containing residue and evaporated to dryness. The resulting residue was washed with pentane and extracted with THF. Finally the isomeric mixture of 2a(syn,anti) was obtained as a green powder after removal of THF in vacuo. Yield 74%. IR (cm−1, ATR): 1595 (νNO), Anal. Calcd for C16H37Cl2MoN2OP2: C, 38.26; H, 7.42; N, 5.58. Found: C, 37.88; H, 7.12; N, 5.29.Preparation of W(NO)Cl2(PNHP), 2b(syn,anti)
0.075 g (0.12 mmol) of the isomeric mixture of 1b(syn,anti) was added to a stirred suspension of 1% sodium amalgam (0.003 g, 0.13 mmol) in 15 mL THF. Stirring was continued overnight at room temperature to ensure the completion of the reaction. The final supernatant solution was filtered off from the mercury containing residue and evaporated to dryness. The residue was washed with pentane and extracted with THF and later with toluene. Recrystallisation from cold toluene–pentane solution afforded green crystals of 2b(syn,anti). Yield 78%. IR (cm−1, ATR): 1547 (νNO). Anal. Calcd for W(NO)Cl2(PNP): C, 32.56; H, 6.32; N, 4.75. Found: C, 32.71; H, 6.22; N, 4.78.General procedure for the preparation of M(NO)(CO)(PNP)(OCO), (M = Mo, 4a(trans); W, 4b(trans))
M(NO)(CO)(PNP) (0.065 mmol, M = Mo, 3a; W, 3b) was dissolved in 0.5 mL THF-d8 in a J. Young NMR tube. The tube was taken out from the glove box and frozen with liquid nitrogen. The nitrogen atmosphere was removed via a freeze–pump–thaw cycle and the tube was filled with 2 bar of CO2 and sealed. Formation of 4a(trans) or 4b(trans) took place immediately within five minutes in quantitative yield.31P{1H} NMR data for the reaction of 3a with CO2 at 213 K: 31P{1H} NMR (121.48 MHz, 213 K, THF-d8): δ = 63.1 (s, formation of 4a(trans)), 62.7 (s, formation of 4a(cis)).
31P{1H} NMR data for the reaction of 3b with CO2 at 213 K: 31P{1H} NMR (121.48 MHz, 213 K, THF-d8): δ = 58.1 (s, formation of 4b(trans)), 57.9 (s, formation of 4b(cis)).
:1:1), the hydrogen pressure was slowly released and CO2 (2 bar) was purged into that mixture instantaneously. Immediate formation of 6a(cis) and 6a(trans) was observed along with 4a(trans) in a ratio of 1:1:1 as revealed by the 31P{1H} NMR.
Selected spectroscopic data: 1H NMR (400.1 MHz, THF-d8) data: δ = 8.5 (s, 1H, OCH, 6a(cis)), 8.1 (s, 1H, OCHO, 6a(trans)). 13C{1H} NMR (100.6 MHz, THF-d8): δ 248 (s, CO, 6a(trans)), 246.5 (s, CO, 6a(cis)), 170 (s, OCHO, 6a(cis)), 168.8 (s, OCHO, 6a(trans)), 156 (s, CO2, 4a(trans)). 31P{1H} NMR (161.97 MHz, THF-d8): 60.9 (s, 6a(trans)), 60.0 (s, 6b(cis)). Since the resulting product contains mixtures of 6a(cis), 6a(trans) and 4a(trans) which could not be separated, elemental analyses could not be provided.
:9 ratio. Suitable orange crystals were obtained for diffraction upon layering pentane to a concentrated THF solution of the product. Yield (by 31P{1H} NMR): 10% (6b(cis)), 90% (6b(trans)). 1H NMR (400.1 MHz, THF-d8): δ = 8.52 (s, 1H, OCH, 6b(trans)), 8.0 (s, 1H, OCH, 6b(cis)), 2.93 (m, 4H, –NCH2), 2.41–2.26 (m, 4H, –CH), 1.66–1.62 (m, 4H, CH2P), 1.35–1.25 (m, 24H, CH3). 13C{1H} NMR (100.6 MHz, THF-d8): δ 244.7 (s, CO), 164.3 (s, CO2H), 57.7 (s, NCH2), 24.5 (t, vJCP = 13.1 Hz, CH), 24.01 (t, vJCP = 9.5 Hz, CH), 19.08 (t, VJCP = 8.3 Hz, CH2), 17.2 (s, CH3), 14.80 (s, CH3). 31P{1H} NMR (161.97 MHz, THF-d8): 51.3 (s, 6b(trans)), 52.6 (s, 6b(cis)). IR (cm−1, KBr): 1578 (νNO), 1612 (νOCHO), 1863 (νCO). Anal. Calcd for C18H38N2O4P2W: C, 36.50; H, 6.47; N, 4.73. Found: C, 36.25; H, 6.41; N, 4.53.
General procedure for the reaction of the formate complexes (mixture of 6a(cis) and 6a(trans)) and (mixture of 6b(cis) and 6b(trans)) with Na[N(SiMe3)2]
The mixture of the formate complexes (0.03 mmol) of molybdenum (mixture of 6a(cis) and 6a(trans) along with 4b(trans)) and tungsten (mixture of 6b(cis) and 6b(trans)) was dissolved in THF in a J. Young NMR tube. Then 1 equiv. of Na[N(SiMe3)2] was added. The mixture was shaken vigorously. The 31P{1H} NMR spectra revealed immediate regeneration of 3a or 3b. Then the reaction mixture was dried in vacuo and extracted with pentane to remove the 3a or 3b. The remaining solid insoluble in pentane was dissolved in D2O. The 1H NMR spectra in D2O revealed the formation of HCOONa.X-ray diffraction analyses
The data collection and structure refinement data for 2a(anti), 2b(syn), 4b(trans) and 6b(cis) are presented in (Table 2). Single-crystal X-ray diffraction data were collected at 183(2) K on a Xcalibur diffractometer (Agilent Technologies, Ruby CCD detector) for all compounds using a single wavelength Enhance X-ray source with MoKα radiation (λ = 0.71073 Å).32a The selected suitable single crystals were mounted using polybutene oil on the top of a glass fiber fixed on a goniometer head and immediately transferred to the diffractometer. Pre-experiment, data collection, data reduction and analytical absorption corrections32b were performed with the program suite CrysAlisPro.32a The crystal structures were solved with SHELXS9732c using direct methods. The structure refinements were performed by full-matrix least-squares on F2 with SHELXL97.32c All programs used during the crystal structure determination process are included in the WINGX software.32d PLATON32e was used to check the result of the X-ray analyses and DIAMOND32f was used for the molecular graphics. CCDC 888516 (for 4b(trans)), 888517 (for 6b(cis)), 960495 (for 2a(anti)) and 960496 (for 2b(syn)) contain the supplementary crystallographic data (excluding structure factors) for this paper.| 2a(anti) | 2b(syn) | 4b(trans) | 6b(cis) | |
|---|---|---|---|---|
| a The unweighted R-factor is R1 = ∑(Fo − Fc)/∑Fo; I > 2σ(I) and the weighted R-factor is wR2 = {∑w(Fo2 − Fc2)2/∑w(Fo2)2}1/2. | ||||
| CCDC | 960495 | 960496 | 888516 | 888517 |
| Empirical formula | C16H37Cl2MoN2OP2 | C16H37Cl2N2OP2W | C18H36N2O4P2W | 8(C18H38N2O4P2W), C4H8O |
| Formula weight (g mol−1) | 502.26 | 590.16 | 590.28 | 4810.46 |
| Temperature (K) | 183(2) | 183(2) | 183(2) | 183(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system, space group | Orthorhombic, P212121 | Orthorhombic, P212121 | Monoclinic, P21/c | Tetragonal, I41/a |
| a (Å) | 7.3971(1) | 7.4129(1) | 7.8847(1) | 36.3651(6) |
| b (Å) | 13.2575(2) | 13.2003(2) | 26.3644(4) | 36.3651(6) |
| c (Å) | 23.8535(4) | 23.8800(5) | 11.5872(2) | 14.8550(3) |
| α (°) | 90 | 90 | 90 | 90 |
| β (°) | 90 | 90 | 106.979(2) | 90 |
| γ (°) | 90 | 90 | 90 | 90 |
| Volume (Å3) | 2339.24(6) | 2336.72(7) | 2303.70(7) | 19644.6(8) |
| Z, density (calcd) (Mg m−3) | 4, 1.426 | 4, 1.678 | 4, 1.702 | 4, 1.626 |
| Abs coefficient (mm−1) | 0.933 | 5.316 | 5.178 | 4.859 |
| F(000) | 1044 | 1172 | 1176 | 9632 |
| Crystal size (mm3) | 0.30 × 0.11 × 0.06 | 0.12 × 0.10 × 0.06 | 0.33 × 0.27 × 0.20 | 0.10 × 0.08 × 0.03 |
| θ range (°) | 2.88 to 32.58 | 2.88 to 28.28 | 2.70 to 28.28 | 2.44 to 25.68 |
| Reflections collected | 55639 |
16728 |
37089 |
27807 |
| Reflections unique | 8495/[Rint = 0.0485] | 5804/[Rint = 0.0372] | 5720/[Rint = 0.0263] | 9319/[Rint = 0.0800] |
| Completeness to θ (%) | 99.9 | 99.9 | 99.9 | 99.9 |
| Absorption correction | Analytical | Analytical | Analytical | Analytical |
| Max/min transmission | 0.951 and 0.799 | 0.765 and 0.644 | 0.432 and 0.275 | 0.995 and 0.987 |
| Data/restraints/parameters | 7529/0/225 | 5804/61/257 | 5574/0/252 | 5971/0/510 |
| Goodness-of-fit on F2 | 1.030 | 0.895 | 1.294 | 0.987 |
| Final R1 and wR2 indices [I > 2σ(I)] | 0.0351, 0.0852 | 0.0329, 0.0488 | 0.0203, 0.0400 | 0.0488, 0.0619 |
| R 1 and wR2 indices (all data) | 0.0420, 0.0872 | 0.0473, 0.0505 | 0.0212, 0.0403 | 0.0972, 0.0744 |
| Absolute structure parameter | −0.02(3) | −0.007(7) | ||
| Largest diff. peak and hole (e Å−3) | 1.107 and −0.613 | 2.044 and −1.158 | 0.657 and −1.231 | 1.241 and −0.845 |
Acknowledgements
Financial support from the Swiss National Science Foundation and the University of Zürich are gratefully acknowledged.Notes and references
- (a) Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010 Search PubMed; (b) M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- (a) T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed; (b) X. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27–59 CrossRef CAS.
- (a) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS; (b) P. G. Jessop, F. Joó and T. Chih-C, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS PubMed; (c) W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207–2221 CrossRef CAS.
- (a) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (b) M. D. Fryzuk and P. A. MacNeil, Organometallics, 1983, 2, 682–684 CrossRef CAS; (c) A. Friedrich, M. Drees, M. Käss, E. Herdtweck and S. Schneider, Inorg. Chem., 2010, 49, 5482–5494 CrossRef CAS PubMed; (d) C. M. Fafard, D. Adhikari, B. M. Foxman, D. J. Mindiola and O. V. Ozerov, J. Am. Chem. Soc., 2007, 129, 10318–10319 CrossRef CAS PubMed; (e) E. Khaskin, M. A. Iron, L. J. W. Shimon, J. Zhang and D. Milstein, J. Am. Chem. Soc., 2010, 132, 8542–8543 CrossRef CAS PubMed; (f) C. Gunanathan, B. Gnanaprakasam, M. A. Iron, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 14763–14765 CrossRef CAS PubMed; (g) L. Schwartsburd, M. A. Iron, L. Konstantinovski, E. Ben Ari and D. Milstein, Organometallics, 2011, 30, 2721–2729 CrossRef CAS; (h) S. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS PubMed; (i) E. Ben-Ari, G. Leitus, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2006, 128, 15390–15391 CrossRef CAS PubMed.
- (a) R. Noyori, M. Koizumi, D. Ishii and T. Ohkuma, Pure Appl. Chem., 2001, 73, 227–232 CrossRef CAS; (b) S. Clapham, A. Hadzovic and R. Morris, Coord. Chem. Rev., 2004, 248, 2201–2237 CrossRef CAS PubMed; (c) T. Ikariya and A. J. Blacker, Acc. Chem. Res., 2007, 40, 1300–1308 CrossRef CAS PubMed; (d) C. Gunanathan and D. Milstein, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (e) H. Grützmacher, Angew. Chem., Int. Ed., 2008, 47, 1814–1818 CrossRef PubMed; (f) Y. Blum, D. Czarkie, Y. Rahamim and Y. Shvo, Organometallics, 1985, 4, 1459–1461 CrossRef CAS.
- C. A. Huff, J. W. Kampf and M. S. Sanford, Organometallics, 2012, 31, 4643–4645 CrossRef CAS.
- (a) M. Vogt, M. Gargir, M. A. Iron, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2012, 18, 9194–9197 CrossRef CAS PubMed; (b) M. Vogt, A. Nerush, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. Sci., 2014, 5, 2043–2051 RSC.
- (a) C. M. Mömming, E. Otten, G. Kehr, R. Frçhlich, S. Grimme, D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2009, 48, 6643–6646 CrossRef PubMed; (b) A. E. Ashley, A. L. Thompson and D. O'Hare, Angew. Chem., Int. Ed., 2009, 48, 9839–9843 CrossRef CAS PubMed; (c) G. Ménard and D. W. Stephan, J. Am. Chem. Soc., 2010, 132, 1796–1797 CrossRef PubMed; (d) J. Boudreau, M. A. Courtemanche and F. G. Fontaine, Chem. Commun., 2011, 47, 11131–11133 RSC; (e) L. J. Hounjet, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 4714 CrossRef CAS PubMed; (f) I. Peuser, R. C. Neu, X. Zhao, M. Ulrich, B. Schirmer, J. A. Tannert, G. Kehr, R. Fröhlich, S. Grimme, G. Erker and D. W. Stephan, Chem. – Eur. J., 2011, 17, 9640 CrossRef CAS.
- Y. Jiang, O. Blacque, T. Fox and H. Berke, J. Am. Chem. Soc., 2013, 135, 7751–7760 CrossRef CAS PubMed.
- J. F. Hull1, Y. Himeda, W.-H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef PubMed.
- (a) C. Federsel, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 6254–6257 CrossRef CAS PubMed; (b) A. H. Chelsea and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef PubMed.
- (a) S. Wesselbaum, T. Vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS PubMed; (b) M. J. Sgro and D. W. Stephan, Angew. Chem., Int. Ed., 2012, 51, 11343–11345 CrossRef CAS PubMed.
- P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS.
- P. G. Jessop, Y. Hsiao, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 344–355 CrossRef CAS.
- (a) F. Joó, G. Laurenczy, L. Nádasdi and J. Elek, Chem. Commun., 1999, 971–972 RSC; (b) C. A. Huff and M. S. Sanford, ACS Catal., 2013, 3, 2412–2416 CrossRef CAS.
- E. Graf and W. Leitner, J. Chem. Soc., Chem. Commun., 1992, 623–624 RSC.
- F. Gassner and W. Leitner, J. Chem. Soc., Chem. Commun., 1993, 1465–1466 RSC.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702–712 CrossRef CAS.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS PubMed.
- (a) S. Enthaler, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3317–3321 CrossRef CAS PubMed; (b) S. Gaillard and J.-L. Renaud, ChemSusChem, 2008, 1, 505–509 CrossRef CAS PubMed; (c) A. Correa, O. G. Mancheño and C. Bolm, Chem. Soc. Rev., 2008, 37, 1108–1117 RSC; (d) C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 1–6 CrossRef; (e) R. Langer, Y. Diskin-Posner, G. Leitus, L. J. W. Shimon, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2011, 50, 9948–9952 CrossRef CAS PubMed.
- R. M. Bullock, Chem. – Eur. J., 2004, 10, 2366–2374 CrossRef CAS PubMed.
- (a) S. Chakraborty, O. Blacque, T. Fox and H. Berke, ACS Catal., 2013, 3, 2208–2217 CrossRef CAS; (b) E. Peterson, A. Y. Khalimon, R. Simionescu, L. G. Kuzmina, J. A. K. Howard and G. I. Nikonov, J. Am. Chem. Soc., 2009, 131, 908 CrossRef CAS PubMed; (c) V. K. Dioumaev and R. M. Bullock, Nature, 2000, 424, 530–532 CrossRef PubMed.
- (a) S. Chakraborty, O. Blacque, T. Fox and H. Berke, Chem. – Asian J., 2014, 9, 328–337 CrossRef CAS PubMed; (b) A. Dybov, O. Blacque and H. Berke, Eur. J. Inorg. Chem., 2011, 652–659 CrossRef CAS; (c) S. Chakraborty, O. Blacque, T. Fox and H. Berke, Chem. – Asian J., 2014, 9, 2896–2907 CrossRef CAS PubMed.
- S. Chakraborty, O. Blacque, T. Fox and H. Berke, Chem. – Eur. J., 2014, 20, 12641–12654 CrossRef CAS PubMed.
- S. Chakraborty and H. Berke, ACS Catal., 2014, 4, 2191–2194 CrossRef CAS.
- (a) P. J. Walsh, F. J. Hollander and R. G. Bergman, J. Am. Chem. Soc., 1988, 110, 8729–8731 CrossRef CAS; (b) P. J. Walsh, F. J. Hollander and R. G. Bergman, Organometallics, 1993, 12, 3105–3123 CrossRef; (c) J. L. Bennett and P. T. Wolczanski, J. Am. Chem. Soc., 1994, 116, 2179–2180 CrossRef CAS; (d) J. de With and A. D. Horton, Organometallics, 1993, 12, 1493–1496 CrossRef CAS.
- M. W. Bouwkamp, A. A. Batinas, P. T. Witte, T. Hubregtse, J. Dam, A. Meetsma, J. H. Teuben and B. Hessen, Organometallics, 2008, 27, 4071–4082 CrossRef CAS.
- (a) Z. Chen, H. W. Schmalle, T. Fox and H. Berke, Dalton Trans., 2005, 580–587 RSC; (b) J. Höck, H. Jacobsen, H. W. Schmalle, G. R. J. Artus, T. Fox, J. I. Amor, F. Bäth and H. Berke, Organometallics, 2001, 20, 1533–1544 CrossRef; (c) F. Furno, T. Fox, H. W. Schmalle and H. Berke, Organometallics, 2000, 19, 3620–3630 CrossRef CAS; (d) F. Liang, H. Jacobsen, H. W. Schmalle, T. Fox and H. Berke, Organometallics, 2000, 19, 1950–1962 CrossRef CAS; (e) Y. Zhao, H. W. Schmalle, T. Fox, O. Blacque and H. Berke, Dalton Trans., 2006, 73–85 RSC.
- L. Bencze and J. Kohan, Inorg. Chim. Acta, 1982, 65, L17–L19 CrossRef CAS.
- A. A. Danopoulos, A. R. Wills and P. G. Edwards, Polyhedron, 1990, 9, 2413–2418 CrossRef CAS.
- CCDC 888516 (for 4b(trans)) and 888517 (for 6b(cis)), 960495 (for 2a(anti)) 960496 for 2b(syn)) contain the supplementary crystallographic data (excluding structure factors) for this paper.
- (a) Agilent Technologies (formerly Oxford Diffraction), Yarnton, England, 2011 Search PubMed; (b) R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1995, 51, 887–897 CrossRef; (c) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (d) L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef CAS; (e) A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS; (f) K. Brandenburg, DIAMOND, Crystal Impact GbR, Bonn, Germany, 2007 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 888517, 888516, 960496 and 960495. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00278h |
| This journal is © The Royal Society of Chemistry 2015 |