DOI:
10.1039/C5DT00248F
(Paper)
Dalton Trans., 2015,
44, 7032-7040
Pentaborate(1−) salts templated by substituted pyrrolidinium cations: synthesis, structural characterization, and modelling of solid-state H-bond interactions by DFT calculations†
Received
19th January 2015
, Accepted 9th March 2015
First published on 10th March 2015
Abstract
The synthesis and characterization of a series of pentaborate(1−) salts of substituted pyrrolidinium cations [C4H8NH2][B5O6(OH)4] (1), [C4H8NMe2][B5O6(OH)4] (2) [C4H8NMeH][B5O6(OH)4] (3), [(2-CH2OH)C4H7NH2][B5O6(OH)4] (4) is reported. All compounds were characterized by single-crystal XRD studies with 3 (1/2CH3COCH3) and 4 (1/2H2O) solvated. TGA/DSC analysis of the pentaborates 1–4 showed that they thermally decomposed in air at 800 °C to 2.5 B2O3, in a 2 step process involving dehydration (<250 °C) and oxidative decomposition (250–600 °C). BET analysis of materials derived thermally from the pentaborates 1 and 2 had internal porosities of <1 m2 g−1, indicating they were non-porous. All compounds show extensive supramolecular H-bonded anionic lattices. H-bond interactions are described in detail and motifs found in these and in other pentaborate structures have been examined and modelled by DFT calculations. These calculations confirm that H-bonds interactions in pentaborates are moderately strong (ca. −10 to −21 kJ mol−1) and are likely to dominate the energetics of their templated syntheses.
1. Introduction
Many organic bases react with B(OH)3 in aqueous solution to yield pentaborate(1−) salts, [NMC][B5O6(OH)4], in which the protonated organic base is a non-metal cation (NMC).1 On rare occasions salts containing three,2 four,3 seven,4 eight,5 nine,6 fourteen7 and fifteen8 B atoms have been obtained. Variations arise since B(OH)3 in basic aqueous solution forms a dynamic combinatorial library9 (DCL) of polyborate anions whose concentrations are pH and boron concentration dependent.10 However, in mildly basic solutions it is estimated that <5% of the total boron is in the form of the [B5O6(OH)4]− anion, with [B3O3(OH)4]− and [B4O5(OH)4]2− the dominant species.11 We have recently performed DFT calculations (gas-phase)12 on the relative stabilities of the polyborate anions and concluded that in isolation the order of stability follows monoborate(1−) > triborate(1−) > pentaborate(1−) > triborate(2−) > tetraborate(2−). However, contrary to this order of stability, pentaborate(1−) salts are readily crystallized from aqueous solutions. The cations in these polyborate salts are structure directing and actively template the architecture of the NMC polyborate salts. The cations can influence the structures by their size, charge, and in some cases by their ability to form strong H-bond interactions. H-bonds are ranked high for intermolecular interaction energies in crystal engineering.13 H-bond interactions between hydrated polyborate anions are ubiquitous14 in polyborate salts, although cation–anion interactions will also play a significant role in the solid-state energetics. In this manuscript we prepare four (substituted) pyrrolidinium cation pentaborate salts, and confirm the structures by X-ray crystallography. We also examine their solid-state H-bond interactions and calculate (DFT) energies of the anion–anion interactions found in these structures. For completeness, we also calculate H-bond energies for anion–anion H-bonding motifs found in other pentaborate structures, and propose an explanation as to why pentaborate salts are so readily formed.
2. Results and discussion
2.1. Synthesis and characterization
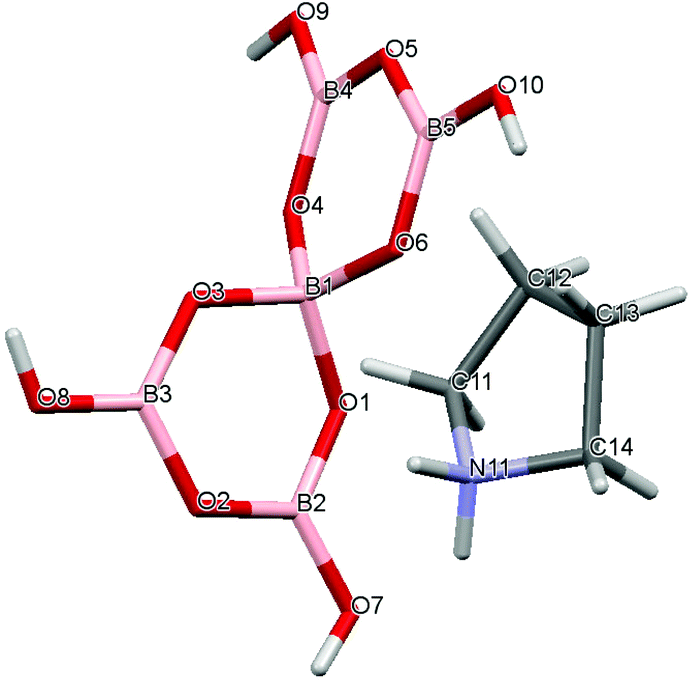
The pyrrolidinium pentaborate salts were all prepared in high yields in MeOH–H2O solution from the reaction of the free base (1, 3, 4) or the quaternary amine hydroxide salt (2) with B(OH)3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 molar ratio (eqn (1)–(4)). The structures of the organic cations and the pentaborate(1−) anions found in compounds 1–4 are shown in Fig. 1.
:5 molar ratio (eqn (1)–(4)). The structures of the organic cations and the pentaborate(1−) anions found in compounds 1–4 are shown in Fig. 1.| | | cyclo-C4H8NH + 5B(OH)3 → [C4H8NH2][B5O6(OH)4] (1) + 5H2O | (1) |
| | | [cyclo-C4H8NMe2][OH] + 5B(OH)3 → [cyclo-C4H8NMe2][B5O6(OH)4] (2) + 6H2O | (2) |
| | | cyclo-C4H8NMe + 5B(OH)3 → [C4H8NMeH][B5O6(OH)4] (3) + 5H2O | (3) |
| | | 2-HOCH2-cyclo-C4H7NH + 5B(OH)3 → [2-HOCH2-cyclo-C4H7NH2][B5O6(OH)4] (4) + 5H2O | (4) |
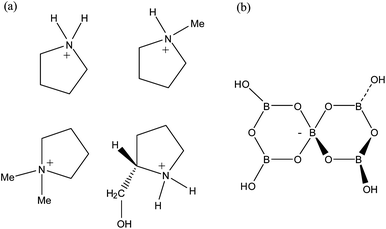 |
| | Fig. 1 Diagrams of the (a) pyrrolidinium cations and (b) the pentaborate(1−) anion, [B5O6(OH)4]−, as found in [C4H8NH2][B5O6(OH)4] (1), [C4H8NMe2][B5O6(OH)4] (2), [C4H8NMeH][B5O6(OH)4] (3) and [(2-CH2OH)C4H7NH2][B5O6(OH)4] (4). | |
Salts 1–4 were characterized by elemental composition, spectroscopy (NMR and IR), and thermal analysis. These data indicated that they were pentaborates and their structures were confirmed by single-crystal XRD studies. Spectroscopic measurements of 1–4 were in accord with previously reported non-metal cation pentaborates salts. 11B NMR spectra of moderately concentrated aqueous solutions (D2O) of these salts displayed the 3 characteristic signals at ∼18, 13 and 1 ppm which are assigned to B(OH)3/[OH], [B3O3(OH)4]− and the 4-coordinate centre of [B5O6(OH)4]−, respectively.15 These species arise due to the complex borate equilibria present in aqueous solution.10,1611B NMR spectra obtained under very dilute conditions can give some diagnostic information. Under these conditions, the formation of polyborate species is suppressed, and a single peak is observed due to equilibrium monoborate (B(OH)3/[B(OH)4]−) species, and the observed chemical shift is dependent upon the relative proportions of Btrig and Btet in solution.17 Thus, the pentaborate(1−) anion should show, at ‘infinite’ dilution, one peak at 16.1 ppm, and the pentaborate salts 1–4 all give a signal at this chemical shift when in dilute solution. The total B/charge ratio can be calculated from an observed chemical shift for dilute solutions (see experimental). This chemical shift value is not often noted but may have utility in helping to formulate products of unknown composition. 1H and 13C spectra (in D2O) were fully consistent with those expected for pyrrolidinium cations, with the NH (1, 3, 4), OH (4) and BOH protons overlapping as represented by a broad signal at ∼4.7 ppm due to rapid exchange. IR spectra of 1–4 clearly all show the diagnostic band of pentaborate salts at ∼925 cm−1.18 The recrystallized sample of 3 from acetone–H2O afforded a solvated species 3·1/2CH3COCH3 (confirmed by XRD, see below) with consistent analytical and spectroscopic data. Crystallisation of 4 from H2O–EtOH gave the solvated 4·1/2H2O, again confirmed by XRD.
2.2. Thermal properties
The thermal properties of the non-metal polyborate salts 1–4 were examined by TGA (in air) and DSC analysis. Previous studies on non-metal pentaborate salts has shown that they usually thermally dehydrate at temperatures up to 250 °C (via an endothermic process) to afford anhydrous non-metal cation pentaborate salts.1,19 At higher temperatures (up to 800 °C) in air exothermic processes occur (consistent with oxidation of the cation) and leaving B2O3 as a glassy residual solid, via an expanded intumesced material.5,20 B2O3 is also observed as the final product if the DSC thermolysis is recorded in an inert (Ar/N2) atmosphere.1,21 Compounds 1–4 all followed this expected path of decomposition, with observed weight losses for the dehydration, and residual masses of B2O3 after oxidation being consistent with calculated values (see experimental section). This is illustrated for 1 in eqn (5) and (6).| | | 1 → [C4H8NH2][B5O8] + 2H2O | (5) |
| | | [C4H8NH2][B5O8] + excess O2 → 2.5B2O3 + volatile oxidation products | (6) |
Samples of 1 and 2 were each separately calcined in air at 250 °C, 500 °C and 750 °C for 24 h in order to obtained significant quantities of the ‘anhydrous’, ‘intumesced’, and ‘residual’ materials. BET analysis of the 6 calcined materials showed that they were all essentially non-porous with porosities of <1.0 m2 g−1. These data are in agreement with BET analysis of thermal materials derived from other NMC pentaborates.20
2.3. Crystallographic studies on NMC pentaborate salts 1, 2, 3·1/2CH3COCH3, and 4·1/2H2O
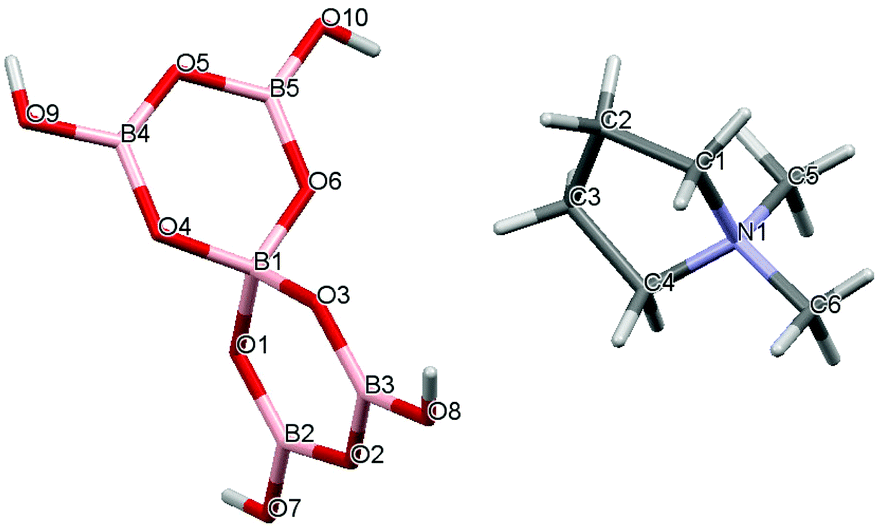
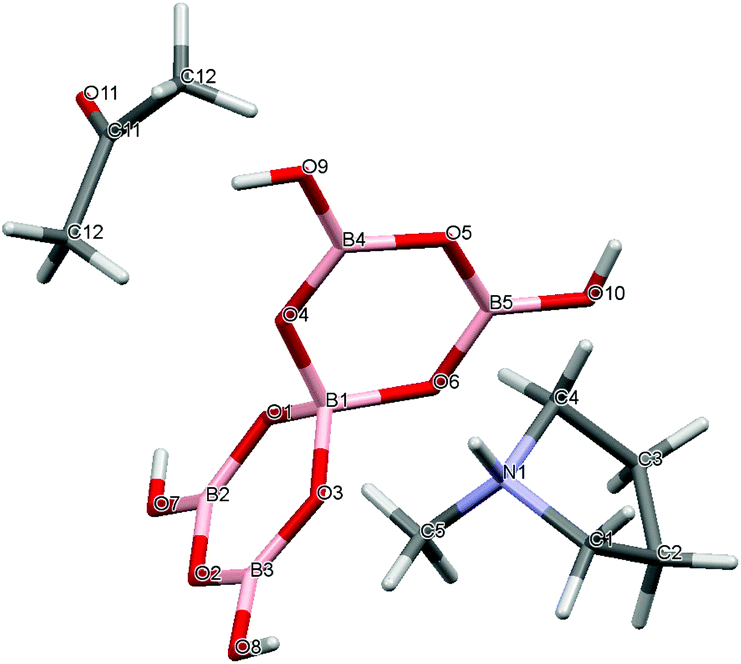
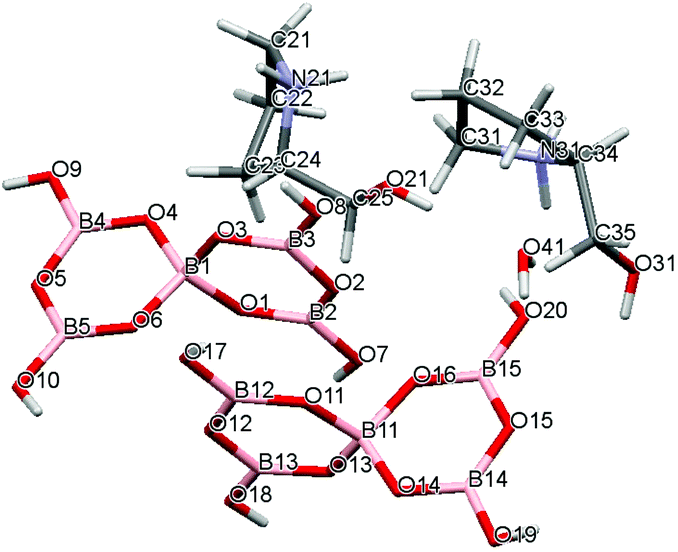
Crystal data for compounds 1, 2, 3·1/2CH3COCH3 and 4·1/2H2O are given in Table 1. These four structures are free from disorder and are characterized by having discrete (substituted) pyrrolidinium cations and pentaborate anions. Diagrams of the cations and anions present, and their associated numbering schemes are shown in Fig. 2–5 respectively. Compound 4·1/2H2O has two independent cations and anions per unit cell. The bond lengths and internuclear angles observed within the pentaborate anions’ boroxyl (B3O3) rings of 1–4 are within the ranges observed for previously reported [NMC][B5O6(OH)4] structures.1,5,15,19–22 The bond lengths and internuclear angles are also within ranges found in related boroxole (B3O3) structures which also contain both 4-coordinate and 3-coordinate B centres bound to O.23
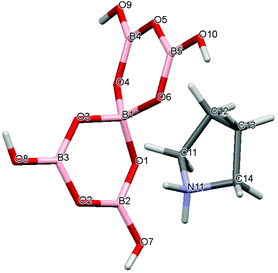 |
| | Fig. 2 Drawing of the structure of [C4H8NH2][B5O6(OH)4] (1), showing the atomic numbering scheme. | |
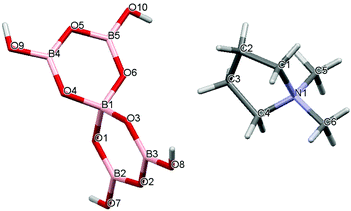 |
| | Fig. 3 Drawing of the structure of [C4H8NMe2][B5O6(OH)4] (2) showing the atomic numbering scheme. | |
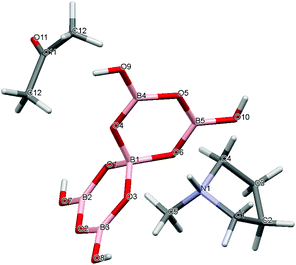 |
| | Fig. 4 Drawing of the structure of [C4H8NMeH][B5O6(OH)4]·1/2C3H6O (3·1/2CH3COCH3), showing the atomic numbering scheme. | |
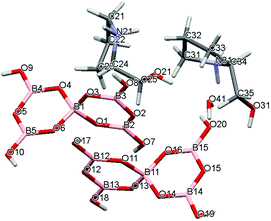 |
| | Fig. 5 Drawing of the structure of [2-HOCH2C4H7NHMe][B5O6(OH)4]·1/2H2O (4·1/2H2O) showing the atomic numbering scheme. | |
Table 1 Crystal data and structural refinement data for 1, 2, 3·1/2CH3COCH3 and 4·1/2H2O
| Crystal |
1
|
2
|
3·1/2CH3COCH3 |
4·1/2H2O |
| Empirical formula |
C4H14B5NO10 |
C6H18B5NO10 |
C6.5H19B5NO10.5 |
C5H17B5NO11.5 |
| Formula wt/g mol−1 |
290.21 |
318.26 |
333.28 |
329.24 |
| Crystal system, space group |
Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Triclinic, P |
Monoclinic, C2/c |
Triclinic, P1 |
|
a/Å |
8.8681(5) |
9.166(4) |
14.496(3) |
9.1164(5) |
|
b/Å |
8.8820(6) |
9.380(5) |
11.640(2) |
9.1691(5) |
|
c/Å |
9.6340(6) |
9.883(4) |
18.255(4) |
9.5532(7) |
|
α/° |
77.006(5) |
64.88(2) |
90 |
75.798(5) |
|
β/° |
75.896(5) |
75.49(3) |
107.200(4) |
69.608(4) |
|
γ/° |
64.320(5) |
84.73(4) |
90 |
82.611(5) |
| Vol/Å3 |
657.07(8) |
744.7(6) |
2942.5(10) |
724.81(8) |
|
Z, calc density (Mg m−3) |
2, 1.467 |
2, 1.445 |
8, 1.505 |
2, 1.509 |
| Abs coeff (mm−1) |
0.132 |
0.125 |
0.131 |
0.136 |
|
F(000) |
300 |
348 |
1392 |
342 |
| Crystal |
Colourless block |
Colourless plate |
Colourless block |
Colourless prism |
| Crystal dimensions/mm3 |
0.32 × 0.22 × 0.10 |
0.23 × 0.10 × 0.03 |
0.18 × 0.18 × 0.09 |
0.14 × 0.11 × 0.07 |
|
θ range (°) |
3.06–27.47 |
3.08–27.47 |
2.94–27.48 |
2.73–27.49 |
| No. of reflections collected |
7117 |
9366 |
13841 |
11353 |
|
R
int
|
0.0176 |
0.0428 |
0.0177 |
0.0239 |
| No. of data/restraints/parameters |
2998/0/185 |
3355/0/205 |
3340/0/215 |
5960/3/419 |
| Final R indices [F2 > 2σ(F2)]: R1, wR2 |
0.0383, 0.0981 |
0.0524, 0.1473 |
0.0320, 0.0870 |
0.0349, 0.0950 |
|
R indices (all data): R1, wR2 |
0.0609, 0.1301 |
0.0595, 0.1549 |
0.0337, 0.0884 |
0.0360, 0.0960 |
| Largest diff. peak and hole/e Å−3 |
0.782, −0.326 |
0.362, −0.314 |
0.241, −0.259 |
0.438, −0.238 |
Structures 1–4 all possess giant H-bond anionic lattices, with cations (and co-crystallized species) situated within ‘cavities’ of the lattice. It is informative to compare the structures of 1 and 2. The unsubstituted cation (in 1) is smaller and able to partake in H-bonding interactions whereas the dimethylated cation (in 2) is larger and is unable to partake in H-bond interactions. Despite these differences, 1 and 2 both crystallize in the same space group with triclinic unit cells, and have very similar supramolecular giant structures. Appropriately, the unit cell of 2 is expanded by 13.3% to accommodate the larger dimethylated cation. The anion–anion H-bond interactions in both of these structures may be described5,20,24 as ‘brickwall’ with each pentaborate part of a C(8) chain (involving a β acceptor site) and 3 reciprocal pair R22(8) interactions (involving α acceptor sites). The unsubstituted pyrrolidinium cation in 1 is involved in H-bonding to both an α (O1) and a β (O8) pentaborate acceptor site. Details of these H-bond interactions are given in Table 2. The inferences from this are that whilst additional H-bond interactions in 1 may further stabilize its solid-state structure, the brickwall structure is sufficiently flexible to accommodate larger cations, and that the pentaborate–pentaborate H-bond interactions dominate the energetics. These anion–anion H-bond interactions (and others commonly encountered in pentaborate structures) are discussed in section 2.4 in a computational study.
Table 2 H-bond interactions in 1, 2, 3·1/2CH3COCH3, 4·1/2H2O. H–O and N–H distances at 0.84 Å 0.99 Å, respectively, and ′ indicates the H-bond acceptor site is on a neighbouring polyborate anion. Data are arranged d(H⋯O) Å, d(D⋯A) Å and angle DHA (°)
|
1
|
O7H7⋯O9′ (β), 1.93, 2.7708 (18), 173.8; O8H8⋯O3′ (α), 1.86, 2.6939 (17), 176.5; O9H9⋯O4′ (α), 1.85, 2.6755 (17), 168.6; O10H10⋯O6′ (α), 1.87, 2.7081 (17), 171.2. N11H11⋯O8 (β), 2.05, 2.924 (2), 145.9; N11H11⋯O1 (α), 1.84, 2.801 (2), 162.1. |
| |
|
2
|
O7H7⋯O1′ (α), 1.86, 2.6933 (18), 172.5; O8H8⋯O3′ (α), 1.87, 2.702 (2), 171.4; O9H9⋯O8′ (β), 1.94, 2.746 (2), 159.6; O9H9⋯O6′ (α), 1.93, 2.763 (2), 170.7. |
| |
|
3
|
O7H7⋯O3′ (α), 1.88, 2.7199 (10), 173.4; O8H8⋯O1′ (α), 1.95, 2.7912 (10), 174.4; O9H9⋯O4′ (α), 1.96, 2.7975 (11), 172.4; O10H10⋯O8′ (β), 2.04, 2.8275 (11), 155.7; N1H1⋯O6 (α), 1.81, 2.7968 (11), 170.3. |
| |
|
4
|
O7H7⋯O11′ (α), 1.86, 2.688 (2), 171.1; O8H8⋯O13′ (α), 1.88, 2.712 (2), 169.2; O9H9⋯O7′ (β), 1.91, 2.701 (2), 156.4; O10H10⋯O16′ (α), 1.88, 2.717 (2), 175.0; O17H17⋯O1′ (α), 1.84, 2.680 (2), 172.2; O18H18⋯O3′ (α), 1.86, 2.698 (2), 175.0; O19H19⋯O17′ (β), 1.96, 2.736 (2), 153.4; O20H20⋯O6′ (α), 1.91, 2.750 (2), 174.1;N21H21A⋯O31, 1.97, 2.853 (2), 154.7; N21H21B⋯O12 (γ), 2.11, 3.018 (3), 152.3; O21H21⋯O41, 1.97, 2.807 (3), 179.5; N31H31C⋯O14 (α), 1.83, 2.791 (3), 162.7; N31H31D⋯O41, 2.18, 3.018 (3), 140.8; O31H31⋯O4 (α), 1.872.704 (2), 170.0; O41H41A⋯O9 (β), 1.92, 2.748 (2), 159.0; O41H41B⋯O20 (α), 2.11, 2.940 (3), 158.8. |
Compound 3 also has a brickwall structure with αααβ pentaborate acceptor sites, and R22(8) and C(8) chains. There is an additional cation–anion (NH⋯O) H-bond interaction to the α-site (O6), and the co-crystallized acetone molecule simply fills space within the lattice and is not involved in H-bonding. Taking into account different Z numbers the volume of the comparable unit of 3 is only 1.3% smaller than in 2.
The structure of 4 is closely related to the ‘brickwall’ structure with each pentaborate forming a C(8) chain (involving a β acceptor site) and 3 reciprocal pair R22(8) interactions (involving α acceptor sites). The two independent cations each interact via H-bonds to an O site on one or other of the two independent pentaborate anions, at O9 (β) or O12 (γ) positions. The co-crystallized H2O molecule also forms additional donor H bonds to β-sites of two pentaborates (O9 and O20) and is an H-bond acceptor from the hydroxy group of one cation (O21H) and the NH group (N31H) of the other cation. The volume of the unit cell is only 2.7% smaller than that of 2.
2.4. DFT calculations on solid-state H-bonding motifs observed in pentaborate salts
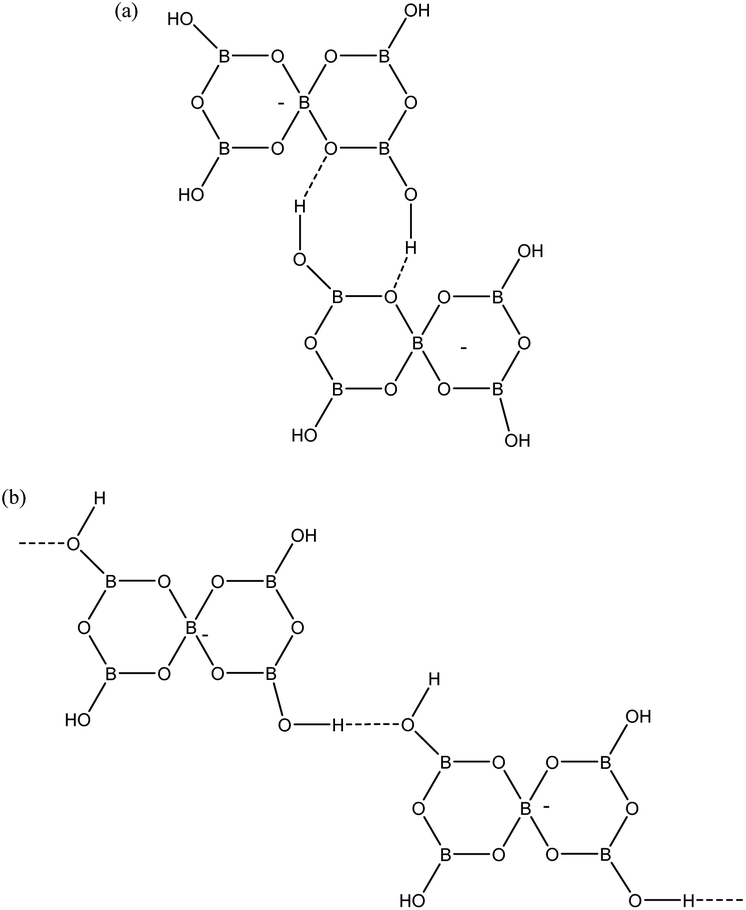
Given that pentaborate(1−) salts are most commonly crystallized from the DCL of polyborate anions available in aqueous solution, and that anion–anion H-bond interactions are found in all pentaborate structures, the energetics of these interactions have been examined computationally. Our QTAIM studies on gas-phase polyborate anions12 noted that H atoms are at a minimum energy when in the plane of a boroxole ring and that the pentaborate(1−) anion has 4 low energy rotamers which vary in energy by 22 kJ mol−1; the lowest energy rotamer having all four H atoms directed inwards towards α-O atoms (no bond critical points) and coplanar with the boroxole rings. This rotamer has only been observed once in the solid-state for [1,2,3-Me3C3N2H2][B5O6(OH)4], which has significantly non-planar boroxole rings.25 The rotamer which is most commonly observed has one H-atom pointing away from the 4-coordinate B centre towards the γ-O atoms (coplanar with the boroxole rings and no bond critical point) and 3 H atoms pointing inwards. This rotamer is 4 kJ mol−1 higher energy12 and is found as a basis for interanionic interactions in 1–4 and in most other reported pentaborate structures. The anion–anion H-bond interactions found in 1–4 are illustrated in Fig. 6. Each pentaborate is involved with three R22(8) interactions involving reciprocal-α sites and one C(8) interaction to a β site.24 The ‘outward’ pointing H-atom is involved in this chain interaction.
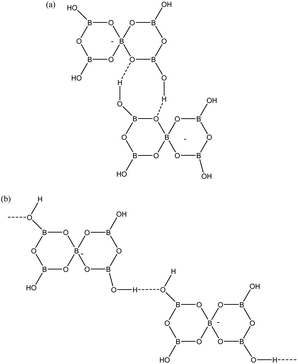 |
| | Fig. 6 H-bond interactions as found in 1–4. (a) R22(8) reciprocal-α, (b) C(8) β-chain. | |
The gas-phase 3 ‘inward’/1 ‘outward’ rotamer (iiio) was used as a starting geometry for DFT calculations involving pairing of anions in the geometries appropriate for the R22(8) and C(8) interactions. Initially, attempts to pair the anions resulted in endothermic rather than exothermic interactions, presumably a result of unfavorable coulombic forces. We attempted to solve this issue by protonating the pentaborate anions on γ-O atoms on the boroxole rings not involved in H-bonding. The interactions now became exothermic but minimised structures were considered unrepresentative since they contained boroxole rings which were distorted away from their idealised planar conformations. An alternative procedure, which we believe was successful, involved using ‘solvated’ rather than ‘gas-phase’ DFT energies in the calculations. Fang and co-workers26 have recently calculated solvated energies of the pentaborate anion (but did not specify the rotamer) at a lower computational level. Our data for the solvated iiio isomer was similar to their calculated value but not directly comparable since different basis sets were used. The solvated rotamers were dimerized and exothermic energies were computed, without boroxole distortions. The data for these two interactions are given in Table 3. The R22(8) α-reciprocal dimer is considerably more favoured per H-bond (−21 kJ mol−1) than the β-chain (−16 kJ mol−1). Durka et al. have calculated H-bond energies for boronic acid dimers, which also contains a R22(8) ring, and have reported an energy of −23.7 kJ mol−1.27 Our calculated structural data for the R22(8) system for D⋯A, angle OHO, H⋯O and H–O are 2.78 Å, 178.1°, 1.77 Å and 0.98 Å and these agree well with Durka's values (2.73 Å, 176.8°, 1.73 Å, 0.99 Å) which were computed at a lower level. It is difficult to compare the calculated values with those observed by X-ray crystallographically since the O–H distance in structures 1–4 was crystallographically fixed at 0.84 Å. Despite this, the calculated data does agree (with the exception for the OHO angle which is 1.6° larger than the range) within the observed ranges for the structural data available for 1–4 (Table 2). However, this OHO angle is within the range of structures published elsewhere.20 This leads us to conclude that our approach is valid and that the reciprocal-α H-bonds in these systems are relatively strong, and strongly influence the structure.
Table 3 DFT calculated energies for H-bond motifs commonly found in solid-state structures containing pentaborate(1−) anions. Relative energy is calculated energy of dimer – (2 × energy of iiio monomer)
| Species |
Abs energy (103 kJ mol−1) |
Rel. energy (kJ mol−1) |
H-bond energy (kJ mol−1) |
| [B5O6(OH)4]− (iiio) |
−2310.943 |
0 |
|
| R22(8) (α) |
−4621.927 |
−42 |
−21 |
| R22(12) (β) |
−4621.905 |
−19 |
−10 |
| R22(8) (γ) |
−4621.917 |
−32 |
−16 |
| C(8) (β-chain) |
−4621.901 |
−16 |
−16 |
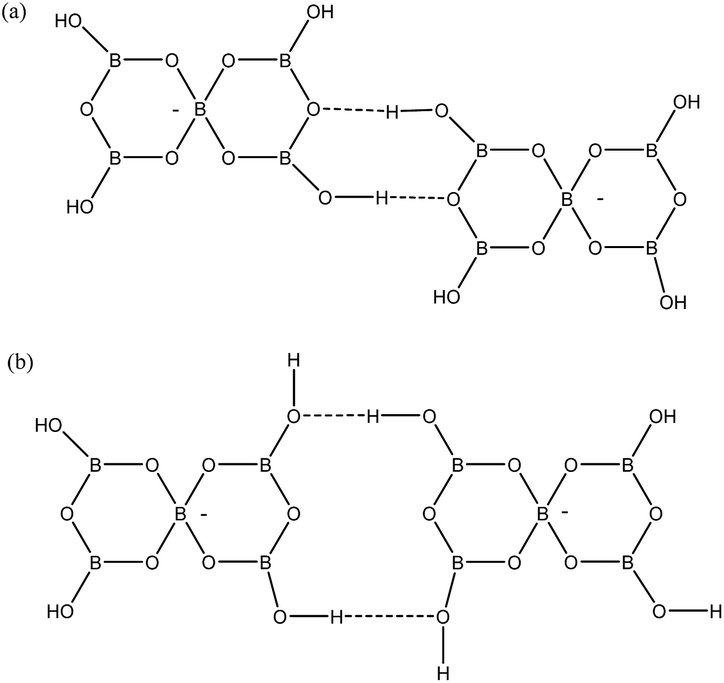
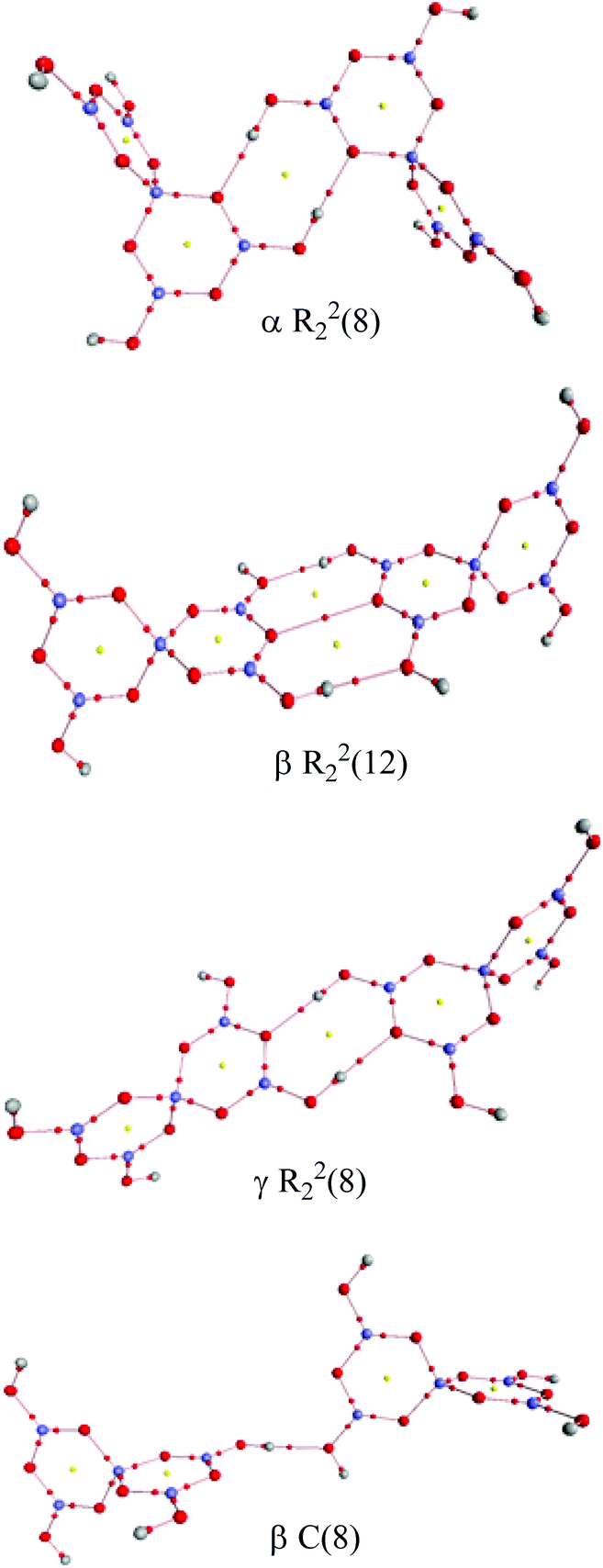
Two other H-bond motifs which have been less frequently observed in pentaborate structures are R22(8) reciprocal-γ interactions5,18,20 and R22(12) reciprocal-β interactions25,28 (Fig. 7). For completeness these were calculated by the same methods and their data are included in Table 3. The H-bond strengths for the R22(8) reciprocal-γ interaction is comparable to that of a C(8) β-chain, and is favoured over that of the R22(12) reciprocal-β interaction. QTAIM calculations (Fig. 8 and ESI†) on all H-bonded systems show that the H-bonds have bond critical points, with the energies of the H-bonds mirroring the electron density (ρb) at their bond critical points. There is also a red-shift in calculated O–H (donor) stretching vibrational frequencies of up to 450 cm−1, which correlates with the relative energies of the H-bonds. The calculated R22(12) reciprocal-β interaction has a close O⋯O contact (3.04 Å) which is similar to that observed in [2-iPrN2C3H4][B5O6(OH)4] (2.98 Å)25 and QTAIM analysis indicates that in addition to the two H-bonds, there exists a further bond critical point between these two O atoms. ρb for these H-bonds are the lowest of the 4 calculated H-bond interactions and this is in agreement with less favourable H-bond energies.
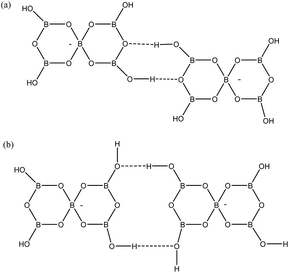 |
| | Fig. 7 H-bond interactions observed ion pentaborate structures (a) R22(8) reciprocal-γ (b) R22(12) reciprocal-β. | |
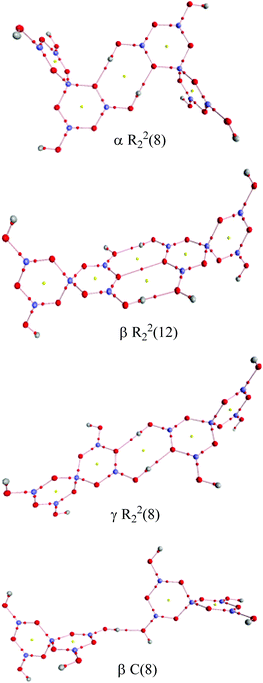 |
| | Fig. 8 QTAIM analysis of the H-bond interactions between pairs of pentaborate anions. Bond critical points (small red spheres) and ring critical points (small yellow spheres) are shown. | |
3. Conclusion
As noted in sections 1 and 2.3 the majority of polyborate salts are pentaborates and the majority of pentaborate salts crystallize with either a ‘brickwall’ or a ‘herringbone’ giant H-bonded lattice with cations in the cavities. Both of these common structural types contain αααβ acceptor sites H-bond interactions in the guise of three energetically favourable R22(8) reciprocal-α interactions and one C(8) β-chain. The ability for pentaborate anions to form strong 3D networks is an important driving force behind the facile syntheses of these salts. As shown in section 2.3 there is sufficient flexibility in the lattice to accommodate (within limits) cations of various sizes. Cation–anion interactions (as observed in 1, 3 and 4) can further stabilize the structure but do not necessarily outweigh the anion–anion contributions, which primarily arise through the reciprocal-α H-bonds. We surmise that given the energetically favoured pentaborate lattice, polyborates other than pentaborates would only be formed when the lattice cannot be stretched to accommodate the cations, and/or when there is sufficient cation–anion interactions to dominate the energetics.
4. Experimental
4.1. General
All chemicals were obtained commercially from Sigma Aldrich (UK) or Lancaster Synthesis (UK) and were used as supplied. N,N-Dimethyl pyrrolidinium iodide was prepared from N-methylpyrrolidine by use of MeI following standard procedures. NMR spectra were recorded at room temperature (298 K) on a Bruker Ultrashield™ Plus 400, using TopSpin™ 3.2 software package; spectra were further analysed using MestReNova v6.0.2–5475. 11B, 13C and 1H NMR spectra were obtained at 400 MHz (1H), 128 MHz (11B), 100 MHz (13C), with samples dissolved in D2O. Fourier transform Infrared spectra (FTIR) were obtained as KBr pellets on a Perkin-Elmer 100 FTIR spectrometer over 450–4000 cm−1. TGA and DSC analysis was performed between 25–100 °C (in air) on an SDT Q600 V4.1 Build 59 instrument using Al2O3 crucibles, with a ramp temperature rate of 2 °C min−1. Powder X-ray diffraction (p-XRD) was carried out on a Phillips X-Pert 3040/60 XRD diffractometer, with spectra obtained using the Phillips X'Pert Data Collector software package. X-ray crystallography was carried out at the EPSRC National Crystallography service at the University of Southampton. BET multipoint analyses were performed on a Micromeritics Gemini III 2375 instrument. CHN analysis was carried out at OEA laboratories Ltd in Callington, Cornwall. The chemical shift (δcalc) for equilibrium ratio of Btrig and Btet at infinite dilution was calculated from δcalc = δ[B(OH)4] + {[(Btet + Btrig) − Btet]/[Btet + Btrig]}·[δ(B(OH)3) − δ(B(OH)4)−] where δ(B(OH)3) and δ[B(OH)4]− are +19.48 and +2.48 ppm, respectively. The total B/charge ratio (B/1) was calculated from B/1 = −17.0/(δobs −19.48), i.e., δobs of 16.1 ppm gives a ratio of 5.02/1.
4.2. Preparation pyrrolidinium pentaborate(1−) salts (1–4)
Compounds 1, 3 and 4 were prepared by a general procedure as described below for 1.
[C4H8NH2][B5O6(OH)4] (1).
B(OH)3, (5.01 g, 81.0 mmol), was dissolved in 1:1 MeOH–H2O (100 ml). cyclo-C4H8NH2 (1.15 g, 16.2 mmol) was added with stirring. The solvent was removed under reduced pressure resulting in the formation of the product as a white solid, which was oven-dried at 60 °C for 24 hours (4.683 g, 99% yield). Recrystallisation from water yielded colourless crystals suitable for single-crystal XRD. NMR: δH (ppm): 1.95–1.99 (4H, m, CH2, 3J = 6.8 Hz), 3.24–3.27 (4H, m, CH2, 3J = 6.8 Hz), 4.79 (HOD, OH and NH rapidly exchanging in the D2O). δC (ppm): 23.58 (CH2), 45.44 (CH2N). δB (ppm): 1.1, 13.0, 18.1. IR (KBr) (νmax/cm−1): 3378, 2360, 1425, 1320, 1185 (m), 1120 (m), 1017 (m), 923 (vs), 777 (s), 697 (s), 485 (m). p-XRD: d-spacing/Å (% rel. int.): 5.66 (100.00), 4.39 (80.35), 6.78 (72.38), 3.39 (68.51), 4.33 (49.10), 9.38 (46.36). TGA: Loss of H2O: 12.3% (12.4% calc.); oxidation of cation: 26.8% (28.9% calc.); residual B2O3: 60.5% (60.0% calc.). Elemental Anal. Calc. (%) for 1, C4H14NO10B5; C, 16.55; H, 4.86; N, 4.82. Found (%): C, 16.78; H, 4.87; N, 4.92.
[C4H8NMeH][B5O6(OH)4]·1.5H2O (3·1.5H2O).
Yield 4.853 g from 5.01 g B(OH)3, (98%). NMR: δH (ppm): 2.08 (4H, s, CH2), 2.90 (3H, s, CH3), 3.05 (s, 2H, CH2), 3.61 (2H, s, CH2), 4.79 (s, HOD, OH and NH rapidly exchanging in the D2O). δC (ppm): 22.80 (CH2), 40.55 (CH3), 55.72 (CH2N). δB (ppm): 1.2, 13.2, 18.8. IR (KBr) (νmax/cm−1): 3381, 2775, 2360, 1426, 1182 (m), 1087 (m), 1026 (m), 922 (vs), 778 (s), 699 (s), 480 (m). p-XRD: d-spacing/Å (% rel. int.): 3.54 (100.00), 5.09 (97.66), 6.32 (82.79), 4.05 (56.23), 6.00 (47.83), 8.36 (36.23). TGA: Loss of interstitial H2O: 8.2% (8.2% calc.); loss of H2O: 18.6% (19.0% calc.); oxidation of cation: 26.3% (28.3% calc.); residual B2O3: 56.7% (52.5% calc.). Elemental Anal. Calc. (%) for 2·1.5H2O, C5H19NB5O11.5; C, 18.13; H, 5.78; N, 4.22. Found (%): C, 18.35; H, 5.19; N, 4.32. Recrystallisation of 3·1.5H2O from water–acetone yielded a few colourless crystals of [C5H12N][B5O6(OH)4]·1/2CH3COCH3 (3·1/2CH3COCH3), suitable for single-crystal XRD. Elemental Anal. Calc. (%) for 3·1/2CH3COCH3, C6.5H19NB5O10.5; C, 23.42; H, 5.75; N, 4.20. Found (%): C, 23.42; H, 5.75; N, 4.23.
(2-CH2OH)C4H7NH2][B5O6(OH)4] (4).
Yield 3.193 g from 3.092 g B(OH)3, (98%). NMR: δH (ppm): 1.73–1.78 (1H, m, CH), 1.99–2.19 (3H, m, CH & CH2), 3.32–3.36 (2H, t, CH2N, 3J = 7.2 Hz), 3.67–3.78 (1H, m, CH), 3.67–3.80 (2H, m, CH2OH), 3.86–3.90 (1H, dd, CHN, J = 3.6 Hz & 12 Hz), 4.79 (DOH, NH and OH rapidly exchanging in D2O). δC (ppm): 23.28 (CH2), 25.72 (CH2N), 45.44 (CH2OH), 60.24 (CH2), 61.15 (CHN). δB (ppm): 1.1, 13.1, 18.0. IR (KBr) (νmax/cm−1): 3308 (br), 1621 (m), 1423 (s), 1183, 1149, 1027 (m), 923 (s), 827, 774 (s), 704 (s). p-XRD: d spacing/Å (% rel. int):): 4.77 (100.00); 3.54 (96.43); 3.71 (85.64); 5.17 (67.94); 3.66 (67.63); 4.40 (55.49). TGA: Loss of interstitial H2O: 2.8% (2.7% calc.); loss of H2O 11.9% (10.9% calc.); residual B2O3: 53.8% (52.9% calc.). Recrystallisation from water–ethanol yielded a few single-crystal XRD quality colourless crystals of 4·1/2H2O. Elemental Anal. Calc (%) for 4·1/2H2O, C5H17NB5O10.5: C, 18.24; H, 5.20; N, 4.26. Found (%): C, 18.05; H, 5.05; N, 4.14.
[C4H8NMe2][B5O6(OH)4]·1/2H2O (2·1/2H2O).
N,N-Dimethylpyrrolidinium iodide (3.68 g, 16.2 mmol) was dissolved in deionised water (50 ml), to which an excess of Dowex™ Monosphere™ 550A ion exchange resin (OH− form) was added. The solution was stirred for 24 h, the resin removed by filtration, and MeOH (50 ml) was added to the filtrate. B(OH)3, (5.01 g, 81.0 mmol), was added, with stirring, and the solution warmed gently. The solvent was removed under pressure after 45 minutes, resulting in the formation of a cream solid, 2·1/2H2O which was oven-dried at 60 °C for 24 hours (5.066 g, 99% yield).
NMR: δH (ppm): 2.22–2.23 (4H, m, CH2), 3.13 (6H, s, CH3), 3.49–3.52 (4H, m, CH2), 4.79 (HOD, s, OH and NH rapidly exchanging in the D2O). δC (ppm): 21.58 (CH2), 51.61 (CH3), 65.77 (CH2N). δB (ppm): 1.1, 13.2, 18.4. IR (KBr) (νmax/cm−1): 3419, 3254, 2352, 1415, 1309 (m), 1148, 1093 (m), 1018 (m), 913 (vs), 772 (s), 724 (m), 708 (s), 478 (m), 464 p-XRD: d-spacing/Å (% rel. int.): 3.60 (100.00), 4.77 (91.42), 5.34 (90.14), 7.20 (67.08), 3.66 (55.14), 3.81 (47.33). TGA: Loss of interstitial H2O: 2.7% (2.7% calc.); loss of H2O: 13.8% (13.8% calc.); oxidation of cation: 29.6% (30.6% calc.); residual B2O3: 53.3% (53.2% calc.). Elemental Anal. Calc. (%) for 2·1/2H2O, C6H19NB5O10.5; C, 22.01; H, 5.85; N, 4.28. Found (%): C, 22.41; H, 5.67; N, 4.28. Recrystallisation of 2·1/2H2O from water–acetone yielded a few single-crystal XRD quality colourless crystals of 2. Elemental Anal. Calc. (%) for 2, C6H18NB5O10; C, 22.64; H, 5.70; N, 4.40. Found (%): C, 22.63; H, 5.74; N, 4.23.
4.3. Thermolysis experiments on 1 and 2 at 250 °C, 500 °C and 750 °C
Compounds of 1 and 2 (1–3 g, per experiment) were subjected to the following thermal treatments at 250 °C, 500 °C and 750 °C (detailed for 250 °C) and BET analyses were performed on the thermally produced materials. Samples of each were placed in open top Vitreosil (SiO2) crucibles and positioned within the furnace (air atmosphere). The furnace temperature was set to increase from room temperature to 250 °C at a ramp rate of 10 °C min−1. After reaching 250 °C, the samples were held at a constant temperature for 24 hours, before being allowed to cool to room temperature. Samples obtained from thermolysis at 500 °C had intumesced, and increased their volume ∼3 fold; the samples at 750 °C yielded glassy black solids. Samples were then removed from the furnace, ground using a mortar and pestle, then used for BET analysis.
1. 250 °C: 1.1709 g obtained from 1.3686 g. 14.45% weight loss (−2.3 H2O per unit formula). BET: surface area 0.3875 m2 g−1. 500 °C: 1.0642 g obtained from 1.7843 g (40.36% loss). BET: surface area 0.4842 m2 g−1. 750 °C: 1.5189 g (B2O3) obtained from 2.5741 g (40.9% loss). BET: surface area 0.7157 m2 g−1.
2. 250 °C: 1.1430 g obtained from 1.3024 g. 12.24% weight loss (−2.2 H2O per unit formula). BET: surface area 0.7517 m2 g−1. 500 °C: 0.8426 g obtained from 1.5223 g (44.65% loss). BET: surface area 0.5591 m2 g−1. 750 °C: 1.7625 g (B2O3) obtained from 3.2365 g (45.54% loss). BET: surface area 0.6605 m2 g−1.
4.4. Computational studies
Density Functional Theory (DFT) calculations were performed using Gaussian09 at the B3LYP/6-311++G(d,p) level of theory and analysed using GaussView 5.0 and WebMO visualization packages.29 Implicit water (ε = 78.3553) solvation was performed using the Polarizable Continuum Model (PCM) Self-Consistent Reaction Field (SCRF) approach.30 QTAIM (Quantum Theory of Atoms in Molecules) analyses were performed using AIM2000.31 Data and diagrams are supplied as ESI.†
4.5. X-ray crystallography
Suitable crystals were selected and data collected following a standard method.32 For compound 1 on a Rigaku SPIDER RAPID diffractometer at 120 K with an image plate detector. For compounds 2–4 on a Rigaku AFC12 goniometer at 100 K equipped with an enhanced sensitivity (HG) Saturn724+ detector mounted at the window of an FR-E-Superbright molybdenum anode generator with VHF Varimax optics (70 mm focus). Cell determination and data collection, data reduction, cell refinement and absorption correction were carried out using CrystalClear,33 structure solution and refinement using SHELX programs.34
Acknowledgements
We thank the EPSRC for use of the NCS X-ray crystallographic service (Southampton). We also thank Dr Simon Curling (Bangor) for BET analysis.
Notes and references
- C. C. Freyhardt, M. Wiebcke, J. Felsche and G. Engelhardt, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 18, 161–175 CrossRef CAS; M. Wiebcke, C. C. Freyhardt, J. Felsche and G. Englehardt, Z. Naturforsch., B: Chem. Sci., 1993, 48, 978–985 CrossRef.
- D. M. Schubert, M. Z. Visi and C. B. Knobler, Inorg. Chem., 2008, 47, 2017–2023 CrossRef CAS PubMed; M. A. Beckett, P. N. Horton, M. B. Hursthouse and J. L. Timmis, RSC Adv., 2013, 3, 15185–15191 RSC.
- G. M. Yang, Y. Q. Sun and G. Y. Yang, J. Solid State Chem., 2004, 177, 4648–4654 CrossRef CAS PubMed; T. J. R. Weakley, Acta Crystallogr., 1985, 41, 377–379 Search PubMed; M. A. Beckett, P. N. Horton, S. J. Coles and D. W. Martin, Inorg. Chem., 2011, 50, 12215–12218 CrossRef PubMed.
- C. Y. Pan, G. M. Wang, S. Y. Zheng and G. Y. Yang, Z. Anorg. Allg. Chem., 2007, 633, 336–340 CrossRef CAS; M. A. Beckett, P. N. Horton, M. B. Hursthouse, J. L. Timmis and K. S. Varma, Dalton Trans., 2012, 41, 4396–4403 RSC; D. M. Schubert, M. Z. Visi, S. Khan and C. B. Knobler, Inorg. Chem., 2008, 47, 4740–4745 CrossRef PubMed.
- M. Z. Visi, C. B. Knobler, J. J. Owen, M. I. Khan and D. M. Schubert, Cryst. Growth Des., 2006, 6, 538–545 CAS.
- D. M. Schubert, M. Z. Visi and C. B. Knobler, Inorg. Chem., 2000, 39, 2250–2251 CrossRef CAS; D. M. Schubert, R. A. Smith and M. Z. Visi, Glass Technol., 2003, 44, 63–70 Search PubMed.
- Z. H. Liu, L. Q. Li and W. J. Zhang, Inorg. Chem., 2006, 45, 1430–1432 CrossRef CAS PubMed.
- S. Merlino and F. Sartori, Science, 1971, 171, 377–379 CAS.
- P. T. Corbett, J. Leclaire, L. Vial, K. R. West, J.-L. Wietor, J. K. M. Sanders and S. Otto, Chem. Rev., 2006, 106, 3652–3711 CrossRef CAS PubMed.
- J. B. Farmer, Adv. Inorg. Chem., 1982, 25, 187–237 CAS.
- J. M. Simon and R. A. Smith, Glass Technol., 2000, 41, 169–173 Search PubMed; J. L. Anderson, E. M. Eyring and M. P. Whittaker, J. Phys. Chem., 1964, 68, 1128–1132 CrossRef CAS; D. M. Schubert, Struct. Bonding, 2003, 105, 1–40 CrossRef.
- M. A. Beckett, R. A. Davies and C. D. Thomas, Comput. Theor. Chem., 2014, 1044, 74–79 CrossRef CAS PubMed.
- J. D. Dunitz and A. Gavezzotti, Cryst. Growth Des., 2012, 12, 5873–5877 CAS.
- For early reviews: G. Heller, Top. Curr. Chem., 1986, 131, 39–98 CrossRef CAS; C. L. Christ and J. R. Clark, Phys. Chem. Miner., 1977, 2, 59–87 CrossRef.
- M. A. Beckett, C. C. Bland, P. N. Horton, M. B. Hursthouse and K. S. Varma, Polyhedron, 2007, 692, 2832–2838 CAS.
- C. G. Salentine, Inorg. Chem., 1983, 22, 3920–3924 CrossRef CAS.
- H. D. Smith Jr. and R. J. Wiersema, Inorg. Chem., 1972, 11, 1152–1154 CrossRef.
- J. Li, S. Xia and S. Goa, Spectrochim. Acta, Part A, 1995, 51, 519–532 CrossRef CAS; M. A. Beckett, P. N. Horton, S. J. Coles, D. A. Kose and A.-M. Kreuziger, Polyhedron, 2012, 38, 157–16119 CrossRef PubMed.
- M. A. Beckett, P. N. Horton, M. B. Hursthouse, J. L. Timmis and K. S. Varma, Collect. Czech. Chem. Commun., 2010, 75, 971–980 CrossRef CAS.
- M. A. Beckett, P. N. Horton, M. B. Hursthouse, D. A. Knox and J. L. Timmis, Dalton Trans., 2010, 39, 3944–3951 RSC.
- G. M. Wang, Y. Q. Sun and G. Y. Yang, J. Solid State Chem., 2006, 179, 1545–1553 CrossRef CAS PubMed; G. M. Wang, Y. Q. Sun and G. Y. Yang, J. Solid State Chem., 2005, 178, 729–735 CrossRef PubMed.
- H. X. Liu, Y. X. Liang and X. Jiang, J. Solid State Chem., 2008, 181, 3243–3247 CrossRef CAS PubMed; Z. H. Liu, J. J. Zheng and W. J. Zhang, Inorg. Chim. Acta, 2006, 359, 519–524 CrossRef PubMed; R. A. Baber, J. P. H. Charmant, N. C. Norman, G. A. Orpen and J. Rossi, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o1086–o1088 Search PubMed.
- M. A. Beckett, C. C. Bland, P. N. Horton, M. B. Hursthouse and K. S. Varma, Inorg. Chem., 2007, 46, 3801–3803 CrossRef CAS PubMed; M. A. Beckett, D. E. Hibbs, M. B. Hursthouse, P. Pwen, K. M. A. Malik and K. S. Varma, Main Group Chem., 1998, 2, 251–258 CrossRef; M. A. Beckett, S. J. Coles, M. E. Light, L. Fischer, B. M. Stiefvater-Thomas and K. S. Varma, Polyhedron, 2006, 25, 1011–1016 CrossRef PubMed.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- M. A. Beckett, P. N. Horton, M. B. Hursthouse and J. L. Timmis, Polyhedron, 2014, 77, 96–102 CrossRef CAS PubMed.
- Y. Zhou, C. Fang, Y. Fang and F. Zhu, Spectrochim. Acta, Part A, 2011, 83, 82–87 CrossRef CAS PubMed.
- K. Durka, K. N. Jarzembska, R. Kaminski, S. Lulinski, J. Serwatowski and K. Wozniak, Cryst. Growth Des., 2012, 12, 3720–3734 CAS.
- Z.-H. Liu, J.-J. Zhang and W.-J. Zhang, Inorg. Chim. Acta, 2006, 359, 519–524 CrossRef CAS PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. Tomasi, B. Mennucci and E. Cancès, J. Mol. Struct. (THEOCHEM), 1999, 464, 211–226 CrossRef CAS; J. L. Pascual-Ahuir, E. Silla and I. Tuñón, J. Comput. Chem., 1994, 15, 1127–1138 CrossRef; G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed; J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef PubMed.
- F. Biegler-Konig and J. Schonbohm, J. Comput. Chem., 2002, 23, 1489–1494 CrossRef CAS PubMed.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
-
Rigaku, CrystalClear- SM Expert 2.0 r1 or 3.1 b18 or 3.1 b27, 2013 Search PubMed.
- G. M. Shedrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1043674–1043677. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00248f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
R. Andrew
Davies
a,
Peter N.
Horton
b and
Charlotte L.
Jones
a
b,
R. Andrew
Davies
a,
Peter N.
Horton
b and
Charlotte L.
Jones
a