
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMagnesium amino-bis(phenolato) complexes for the ring-opening polymerization of rac-lactide†
Katalin
Devaine-Pressing
a,
Joshua H.
Lehr
a,
Michelle E.
Pratt
a,
Louise N.
Dawe‡
ab,
Amy A.
Sarjeant
c and
Christopher M.
Kozak
*a
aDepartment of Chemistry, Memorial University of Newfoundland, St. John's, Newfoundland, Canada A1B 3X7. E-mail: ckozak@mun.ca; Tel: +1-709-864-8082
bC-CART X-ray Diffraction Laboratory, Memorial University of Newfoundland, St. John's, Newfoundland, Canada
cDepartment of Chemistry, Northwestern University, 2145 Sheridan Rd, Evanston, IL 60208, USA
First published on 17th March 2015
Abstract
Magnesium compounds of tetradentate amino-bis(phenolato) ligands, Mg[L1] (1) and Mg[L2] (2) (where [L1] = 2-pyridyl-N,N-bis(2-methylene-4-methoxy-6-tert-butylphenolato), and [L2] = dimethylaminoethylamino-N,N-bis(2-methylene-4-methyl-6-tert-butylphenolato)) were prepared. The proligands, H2[L1] and H2[L2] were reacted with di(n-butyl)magnesium in toluene to give the desired compounds in high yields. Compounds 1 and 2 exhibit dimeric structures in solutions of non-coordinating solvents as observed by NMR spectroscopy and in the solid state as shown by the single crystal X-ray structure of 2. These compounds exhibit good activity for rac-lactide polymerization in solution and in molten lactide.
Introduction
Polylactide continues to be at the forefront of biodegradable, biosourced and biocompatible polymer synthesis. It is central to the development of sustainable polymers, an area of critical importance for the design of new materials that reduce environmental and health impacts resulting from their manufacture, use, and their end-of-life properties.1–4 The array of different metal complexes studied for the ring-opening polymerization (ROP) of lactide has been described in several reviews5–8 and examples of metal-containing initiators vary considerably in terms of the metal centre and ligands employed. Complexes of amino-bis(phenolates) exhibit interesting structural and electronic properties, as well as catalytic behaviour; the variety of metal complexes with this ligand class has also been reviewed.9 Derivatives of this ligand class have been used in main group and d-block metal chemistry, including lithium,10–18 magnesium,19–25 calcium,26 rare-earths,27,28 zinc,20,29–37 aluminum,7,38,39 zirconium40,41 and titanium.42 Many of these complexes have been reported to be excellent initiators for the ROP of cyclic esters such as lactide and ε-caprolactone. For the ROP of cyclic esters, earth abundant alkali and alkaline earth metal compounds have become particularly important in the search for efficient, stable, inexpensive and non-toxic catalysts. We have previously reported the structures and catalytic lactide ROP activity of lithium and potassium amino-bis(phenolato) compounds.43,44 Magnesium compounds also have great potential as ROP catalysts for the preparation of PLA particularly for biomedical applications, as Mg is a biocompatible and non-toxic metal and thus the residue of these metals in the resulting polylactide is not of great concern.45,46 Furthermore, magnesium compounds have been shown to elicit high reaction rates for copolymerization of CO2 and epoxides.47Magnesium complexes reported by Ma and co-workers exhibited some of the highest activities in ROP of cyclic esters by this metal.48,49 The high activity for the ROP of rac-lactide may be due to the monomeric nature of these magnesium bis(silyl)amido complexes supported by tetradentate monophenolate ligands and the presence of a reactive monodentate amido ligand. A TOF of 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 560 h−1 was achieved when 10000 equiv. of rac-lactide was converted to polylactide in 15 minutes at 25 °C in the absence of any co-catalyst. One of these complexes showed very high activity even under melt conditions at 110 °C giving 72% conversion with a TOF of 86880 h−1. Magnesium compounds supported by tridentate monophenolate Schiff-base ligands and benzyl alkoxide also proved active for L-lactide polymerization in CH2Cl2 solutions.21 In both these cases, it is likely the monodentate amido or alkoxide ligand that serves as the initiation site.
560 h−1 was achieved when 10000 equiv. of rac-lactide was converted to polylactide in 15 minutes at 25 °C in the absence of any co-catalyst. One of these complexes showed very high activity even under melt conditions at 110 °C giving 72% conversion with a TOF of 86880 h−1. Magnesium compounds supported by tridentate monophenolate Schiff-base ligands and benzyl alkoxide also proved active for L-lactide polymerization in CH2Cl2 solutions.21 In both these cases, it is likely the monodentate amido or alkoxide ligand that serves as the initiation site.
In light of these high activities, we investigated the activity of magnesium amino-bis(phenolato) complexes towards ROP of rac-lactide under melt conditions and in solution. Magnesium complexes of aminophenolate ligands have previously been reported by others but their activity for ROP of lactides was not tested or they showed very low or no activity.50–53 In this report, we present the synthesis and characterization of two new magnesium amine-bis(phenolate) complexes that show good lactide ROP activity in both melt and solution conditions.
Results and discussion
Synthesis and characterization of proligands and magnesium complexes
Tetradentate tripodal amine-bis(phenol) proligands (Scheme 1) were synthesized via a modified Mannich condensation reaction in water.54 The substituted phenols contained t-butyl groups in the ortho positions and methoxy (H2[L1]) or methyl groups (H2[L2]) para to the hydroxyl group. The proligands possessed pyridyl (H2[L1]) or dimethylaminoethyl (H2[L2]) groups as the neutral pendant N-donors. The synthesis of H2[L1] using a four step procedure55 and without the addition of any further solvents56 was previously reported. The synthesis of H2[L2] in methanol was reported by Bochmann and co-workers.53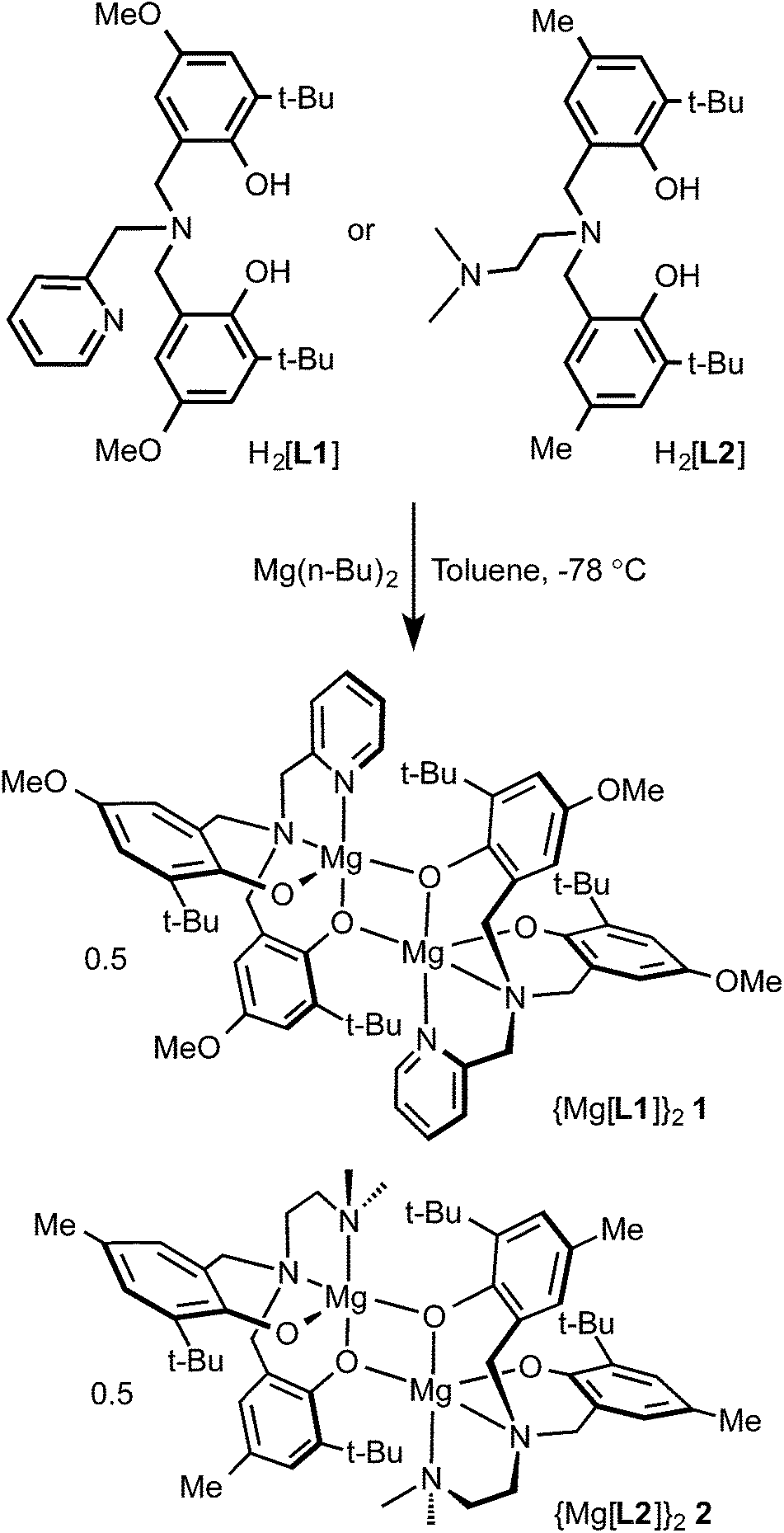 | ||
| Scheme 1 Synthesis of complexes 1 and 2. | ||
The corresponding ligand was reacted with one equivalent of di(n-butyl)magnesium in toluene at −78 °C to afford complexes 1 and 2 (Scheme 1). The NMR spectra of complexes 1 and 2 in non-coordinating solvents revealed that the compounds exist as dimers in solution. The 1H-NMR spectrum of 2 in C6D6 showed two t-butyl environments implying inequivalent phenolates. This was further supported by the existence of four aromatic proton resonances arising from the phenolate rings occupying inequivalent coordination sites at the magnesium centres. The methylene resonances exist as four doublets. Two doublets are expected for a monometallic complex given the diastereotopic environments resulting from coordination to the metal centre. The existence of the four doublets arises from asymmetric phenolate sites where one phenolate of each ligand occupies a bridging position between two magnesium ions. The solid-state structure of compound 2 confirmed the proposed connectivity (see below). In coordinating solvents, such as pyridine or DMSO, the NMR spectra are consistent with monometallic compounds. The 1H NMR spectra of 1 in C6D6 or toluene-d8 showed the presence of several isomers suggesting the existence of dimeric species but also of more than one structural isomer (e.g. syn and anti oriented pendent donors) present in solution. Similar isomerism has been observed in zirconium amine-tris(phenolates).57
MALDI-TOF mass spectrometry (see Fig. S1 and S2 in ESI†) showed complexes 1 and 2 exhibit dimeric structures in the gas phase as well as in solution and the solid state. Peaks at m/z 1028.50 (rel. intensity 100%) and m/z 514.26 (58%) corresponding to the dimer and monomer fragment ions, respectively, were observed for complex 1. For complex 2, the peak assigned to the dimer at m/z 924.52 was very weak (3%) and the peak at m/z 462.25 (100%) corresponds to the monomer.
Colourless crystals of 2 suitable for single crystal X-ray diffraction were obtained from saturated toluene solutions at −35 °C. The solid-state molecular structure with selected bond lengths and angles is shown in Fig. 1. Crystallographic and structure refinement data can be found in Table S1 in ESI.† The dimeric structure with chemically distinct phenolate groups is consistent with the NMR studies discussed above. Each magnesium centre is five-coordinate and bound to two nitrogen donors and three oxygen donors. Four of the coordination sites are occupied by the chelating diamine-bis(phenolate) ligand, while the fifth coordination site is occupied by a bridging phenolate oxygen. The non-planar Mg2O2 core exhibits a O(2)–Mg(1)–O(2)*–Mg(1)* torsion angle of 32.69(4)°. The magnesium complexes that show polymerization activity are believed to initiate cyclic ester ring-opening polymerizations through a coordination insertion mechanism;6,45 therefore, the creation of a vacant site by breaking the Mg2O2 ring (in the presence of a polar monomer or a coordinating solvent, for example) may induce activity in these typically inactive bimetallic complexes. NMR studies of monomer formation in the presence of benzyl alcohol (a lactide ROP cocatalyst) are discussed below. The Mg(1)–O(1) bond (1.950(2) Å) in 2 is considerably shorter than the Mg(1)–O(2) bond (2.049(2) Å), because of the bridging bonding mode of the O(2) atom. The bond lengths for Mg(1)–N(1) and Mg(1)–N(2) were very similar at 2.243(3) Å and 2.265(3) Å, respectively, and are within the range observed in similar complexes.50–52
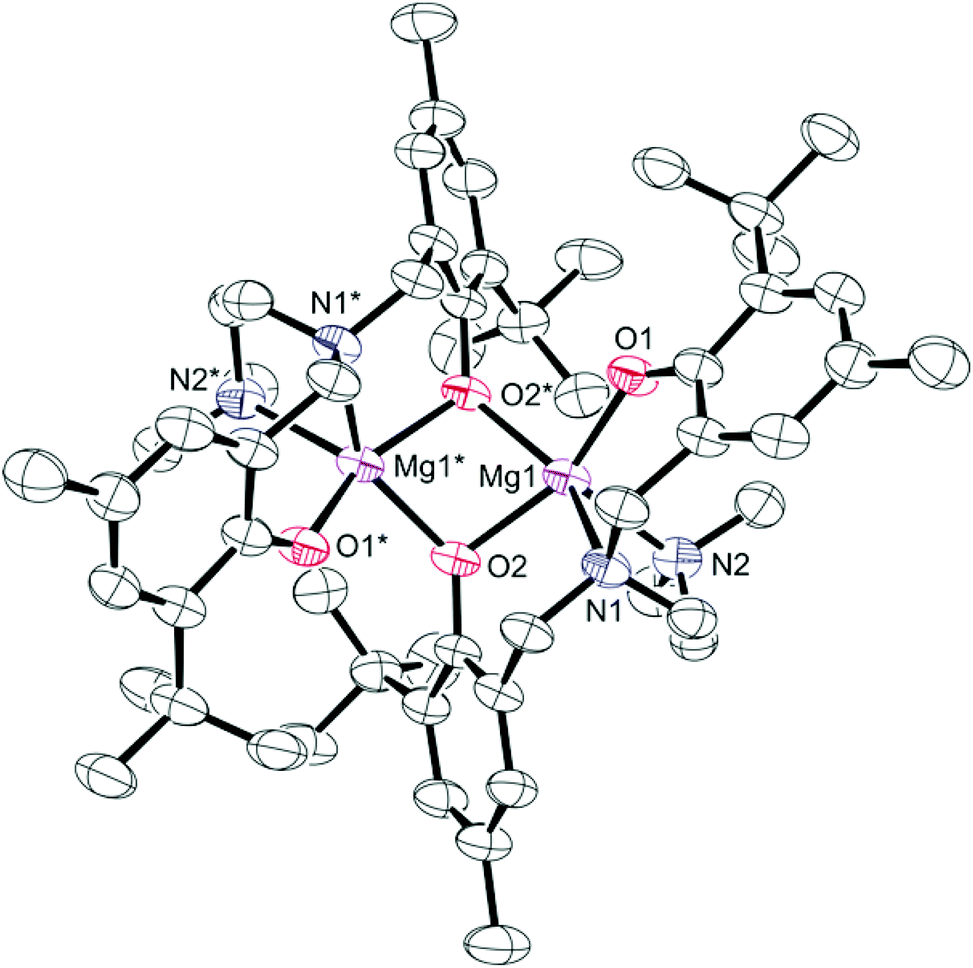 | ||
| Fig. 1 Partially labelled molecular structure (ORTEP) of the dimer of complex 2. Thermal ellipsoids are drawn at 50% probability and H atoms are excluded for clarity. Selected bond lengths (Å): Mg(1)–O(1), 1.950(2); Mg(1)–O(2), 2.049(2); Mg(1)–O(2)*, 2.049(2); Mg(1)–N(1), 2.243(3); Mg(1)–N(2), 2.265(3). Bond angles (°): O(1)–Mg(1)–O(2), 153.48(10); O(1)–Mg(1)–N(1), 88.25(9); O(2)–Mg(1)–O(2)*, 74.53(9); Mg(1)–O(2)–Mg(1)*, 95.71(8), O(2)*–Mg(1)–N(1), 140.38(9); O(2)–Mg(1)–N(2), 102.07(9); O(1)–Mg(1)–O(2)*, 91.64(9). *Symmetry operations used to generate equivalent atoms: 1 − x, y, ½ − z. | ||
A brief comparison of the structural properties of magnesium amine-bis(phenolate) complexes follows. The trigonality index,58τ, for 2 is 0.27 at both magnesium centres, which more closely approaches a distorted square pyramidal (τ = 0) rather than a trigonal bipyramidal geometry (τ = 1). The related magnesium complex reported by Jerzykiewicz and co-workers differs from 2 in that it contains one methyl substituent at the para position of the phenolate ring and no substituents at the ortho position resulting in a much less sterically encumbered metal site.50 This is manifested in a rhombic Mg2O2 core that is nearly perfectly planar, possessing a sum of angles of 359.9°. The τ values for this compound are 0.35 and 0.61 for Mg(1) and Mg(2), respectively, indicating geometries around the metal centres that are intermediate to square pyramidal and trigonal bipyramidal.
A similar complex was reported by Sobota and co-workers possessing the tripodal amine-bis(phenolate) ligand, N,N-bis(3,5-di-tert-butylbenzyl-2-hydroxy)tetrahydrofurfurylamine.52 A dimeric complex was observed in C6D6 solutions and in the solid state, where it displayed a distorted rhombic Mg2O2 core whose sum of angles was 346.8°. The structure exhibited τ values for the two pentacoordinate magnesium centres that were almost identical at 0.41 and 0.39, intermediate to trigonal bipyramidal and square pyramidal arrangements, similar to complex 2. This complex, however, also proved to be inactive in ROP of L-lactide at 25 °C in toluene.
A related phenolate-bridged magnesium dimer possessing N,N′-bis(2-hydroxido-3,5-di-tert-butyl)-N,N′-dimethylethane-1,2-diamine (salan) ligands was reported by Davidson, O'Hara and co-workers.51 As in the compound reported by Jerzykiewicz, each magnesium ion in this structure possesses different coordination environments with Mg(1) and Mg(2) centres having τ values of 0.32 and 0.25, respectively. A Mg2O2 core is also present in this structure with a sum of angles of 357.6°. 13C- and 1H-NMR spectra for this compound in aromatic solvents suggested a dimeric structure in solution, similar to complex 2. Two distinct phenolate environments and complicated methylene resonances indicate the dimer remains intact in non-coordinating aromatic solvents. This complex showed no activity in ring opening polymerization of lactide even at 110 °C in solution, which was attributed to the bulkiness of the ligand and the persistence of the dimeric nature of the complex in non-coordinating, aromatic solvents.
Magnesium N,N-bis(3,5,-di-tert-butylbenzyl-2-hydroxy)dimethylethylenediamine was prepared by Bochmann and co-workers using magnesium bis[bis(trimethylsilyl)amide] (instead of the more commonly used di(n-butyl)magnesium) to give a complex that, although not structurally authenticated, is expected to be similar to those discussed above.53 This complex showed poor activity (only 5% conversion) in the ROP of ε-caprolactone (CL) in toluene at 60 °C for a [CL]:[Mg] ratio of 200:1.
Although the solution-state ROP experiments of the previously reported magnesium-bis(phenolate) complexes were not encouraging, we believed that under melt conditions compounds 1 and 2 may exhibit activity. Indeed, rac-lactide can be polymerized by 1 and 2 in the bulk at temperatures above 125 °C and, to our surprise, in toluene solutions at 90 °C.
Ring opening polymerization in the melt
Melt phase ROP has attracted much interest recently as it offers a viable technique for industry.59 There are examples for lactide ROP in the melt catalysed by a variety of metal complexes, such as sulphonamide-supported aluminum complexes,60 Ni(II) and Ni(II)–Sm(III) salen complexes,61 aluminum-salen62 and salen-like63 complexes, zirconium and hafnium amine tris(phenolate) alkoxides,64 but solvent-free lactide ROP with an earth-abundant, non-toxic metal initiator at low catalyst loading still remains a challenge. Magnesium complexes 1 and 2 were assessed for their capabilities toward the ROP of rac-lactide under industrially relevant melt (solvent-free) conditions above 125 °C. The polymerization results are summarized in Table 1. Polymerization occurred with or without a co-initiator (benzyl alcohol, BnOH). In general, the polymer molecular weights observed by gel permeation chromatography were lower than expected, likely due to mass transfer issues in the melt. Also, lower molecular weight polymers were obtained when BnOH was used, but the dispersity values were lower indicating a more controlled polymerization. Higher molecular weight and monodisperse polymers were obtained when catalyst loading is decreased. Interestingly, the physical appearance of the polymers was found to depend on whether BnOH was used (which gave thin films) or not (which gave crystalline white solids). The mechanical properties of the polymers are discussed below. Complex 1 showed slightly better conversions than complex 2, so 1 was chosen to carry out further reactions.| Entrya | Complex | Time (min) | T (°C) | [Mg]:[rac-LA]:[BnOH] |
Conv.b (%) | Activityc (kg mol−1 h−1) |
M
nd (calc'd) |
M
ne (GPC) |
Đ (Mw/Mn) |
|---|---|---|---|---|---|---|---|---|---|
| a All polymerization reactions were carried out in neat rac-lactide (0.5–0.6 g). b Conversion determined by 1H-NMR. c Calculated as mass of lactide converted in grams/(mol of Mg × time in h). d Calculated from ([LA]/[Mg]) × % conv. × 144.13 g mol−1. e Molecular weights (g mol−1) of entries 1 to 8 determined by gel permeation chromatography (GPC) in CHCl3 calibrated against polystyrene standards using the Mark–Houwink correction of 0.58.65 GPC of entries 9 to 15 were conducted in THF by triple detection. See Experimental for full details. f ND = Not Determined, yield too low. | |||||||||
| 1 | 1 | 100 | 125 | 1:100:0 |
99 | 8.56 | 14400 |
20300 |
2.74 |
| 2 | 1 | 100 | 125 | 1:100:1 |
99 | 8.56 | 14400 |
11300 |
1.77 |
| 3 | 1 | 100 | 125 | 1:500:0 |
99 | 42.8 | 72000 |
36200 |
3.01 |
| 4 | 1 | 100 | 125 | 1:500:1 |
98 | 42.4 | 70600 |
9700 | 2.01 |
| 5 | 2 | 100 | 125 | 1:100:0 |
100 | 8.65 | 14400 |
16500 |
2.40 |
| 6 | 2 | 100 | 125 | 1:100:1 |
82 | 6.23 | 14400 |
7200 | 1.54 |
| 7 | 2 | 100 | 125 | 1:500:0 |
72 | 31.1 | 51800 |
30600 |
2.72 |
| 8 | 2 | 100 | 125 | 1:500:1 |
98 | 42.4 | 70600 |
2400 | 1.25 |
| 9 | 1 | 100 | 125 | 1:1000:0 |
93 | 80.4 | 133900 |
44900 |
1.25 |
| 10 | 1 | 100 | 125 | 1:1000:1 |
93 | 80.4 | 133900 |
42900 |
1.24 |
| 11 | 1 | 180 | 125 | 1:1000:0 |
93 | 44.7 | 133900 |
74300 |
1.05 |
| 12 | 1 | 180 | 125 | 1:1000:1 |
89 | 42.8 | 128200 |
50100 |
1.24 |
| 13 | 1 | 100 | 150 | 1:1000:0 |
92 | 79.6 | 132500 |
35200 |
1.22 |
| 14 | 1 | 100 | 150 | 1:1000:1 |
96 | 83.0 | 138200 |
70290 |
1.08 |
| 15 | 1 | 180 | 150 | 1:2500:0 |
16 | 19.2 | 57600 |
NDf | ND |
When the catalyst loading was lowered to 0.1 mol% (entries 9–14), lactide conversion remained high translating to an increase in activity with good control as shown by the narrow dispersities of the polymers obtained. Polymerization remained controlled even in the absence of BnOH co-initiator. Highest molecular weights were obtained when the polymerization was run for 180 min (entry 11) or when the temperature was raised to 150 °C (entry 14). Activity was still observed upon further decreasing the catalyst loading to 0.04 mol% at this temperature, but with low conversion (16%) (entry 15).
Kinetic studies performed with 1 under conditions given in Table 1, entry 3 revealed that the polymerization reached completion in 60 minutes (see Table 2). Molecular weights increase with increasing time and conversion and the dispersity values are generally low (1.14–1.40). The difference in conversion between entry 5 and 6 is not significant, but the difference in molecular weight is in accordance with the phenomenon observed earlier in Table 1, entry 11 that it is favourable to leave the polymerization to occur for longer time to achieve higher molecular weight polymers.
| Entrya | Time (min) | Conv.b (%) |
M
nc (calcd) |
M
nd (GPC) |
Đ (Mw/Mn) |
|---|---|---|---|---|---|
|
a All polymerization reactions were carried out in neat rac-lactide (0.5 g), 125 °C with [1]:[LA] = 1:500.
b Determined by 1H-NMR.
c Calculated from ([LA]/[Mg]) × % conv. × 144.13 g mol−1.
d Molecular weights (g mol−1) determined by triple detection gel permeation chromatography (GPC) in tetrahydrofuran using a dn/dc value of 0.049 mL g−1.
|
|||||
| 1 | 10 | 7 | 5000 | 5400 | 1.40 |
| 2 | 20 | 36 | 25900 |
16600 |
1.22 |
| 3 | 30 | 78 | 56000 |
18100 |
1.34 |
| 4 | 40 | 91 | 65500 |
25100 |
1.37 |
| 5 | 50 | 93 | 67000 |
31400 |
1.32 |
| 6 | 60 | 96 | 69100 |
55100 |
1.14 |
The conversion vs. time plot (Fig. S3 in ESI†) shows that polymerization starts once a homogenous melt is achieved (after 5 minutes at 125 °C), the rate of conversion remains almost linear for the next 10 min and then accelerates for 5 min before returning to a constant rate. The discontinuity in conversion rate implies an induction period, which could be a result of dimer dissociation in order to form the active species. Solution polymerization kinetics studies (see below) are consistent with this theory. The MALDI-TOF mass spectra of the polymers obtained in the melt in the absence of BnOH showed the presence of both cyclic poly(rac-lactide) and linear chains terminated with –OH and carboxylic acid end groups (Fig. S4 and S5 in ESI†).
Mechanical properties of the polymers obtained under melt conditions
Polymerizations were performed on a larger scale using 1.0 g (instead of 0.5 g) of rac-lactide according to the conditions in entries 3 and 4 of Table 1 to investigate the correlation between conversion and isolated yield. The scaled-up reactions improved the recovery of the polylactide after washing as determined by 1H NMR. The physical appearance of the polymers obtained with and without added benzyl alcohol is significantly different as a result of the difference in polymer molecular weights obtained (Fig. S6 in ESI†). Without BnOH, the polymer is obtained as a thin film whereas in the presence of alcohol a crystalline polymer results. Differential Scanning Calorimetry (DSC) and Thermogravimetric Analysis (TGA) were performed on these samples and revealed very similar thermal properties. The midpoint of the glass transition temperature taken from the third heating curve was 52.59 °C for the polymer obtained from entry 3 and 50.86 °C for entry 4, which is expected given the polymer's much lower observed Mn. TGA conducted with a ramp rate of 10 °C min−1 showed the decomposition temperature of the polymer obtained in the absence of BnOH was 354.5 °C and slightly higher (356.7 °C) for the polymer obtained in presence of BnOH. Slower rates of 5 °C min−1 resulted in slightly lower decomposition temperatures for both the polymers obtained with BnOH (onset at 340 °C, end at 375 °C) and without BnOH (onset at 344 °C, end at 367 °C, see Fig. S7 and S8 in ESI†). These decomposition temperatures are higher than expected,66 but they are similar to carboxylic acid terminated branched polymers reported by S. H. Lee et al.67 It should be noted that the polymers used for thermal analysis were highly purified by precipitation, centrifugation, decanting of solvents, washing with cold methanol and drying by vacuum oven overnight at 40 °C.Regarding polymer tacticity, the solvent-free (melt) ROP of rac-lactide using 1 gives an atactic microstructure of poly(rac-lactide). This is not surprising given the achiral ligands used in compounds 1 and 2 and the poor polymer Mn control. A representative 1H{1H}-NMR spectrum of the methine resonances of poly(rac-lactide) is given in Fig. S9 in ESI.† Highly isotactic polylactide stereopolymers can be obtained solvent-free at 130 °C using salen aluminum alkoxides at a monomer/initiator molar ratio of 200, but the reaction rates were very slow (95% conversion after 2 days).68
Ring opening polymerization in solution
Polymerization at room temperature did not occur in either CH2Cl2 or THF with complexes 1 and 2. However, upon heating to 90 °C with 1 equivalent of isopropyl alcohol (iPrOH) as co-initiator complex 2 exhibited fast propagation in toluene achieving 98% conversion in 12 minutes (Table 3, entry 1 and Fig. 2). Use of iPrOH accelerates the polymerization activity compared to using 2 alone, but gives lower than expected polymer molecular weights likely resulting from transesterification. Increasing the amount of iPrOH as co-initiator to 10 equivalents with respect to 2 resulted in faster reaction rate but much lower molecular weight and narrower dispersity (entry 2). The plot of conversion vs. time (Fig. 2) indicates an induction period of approximately 7 minutes before the onset of the expected conversion curve. This non-linearity is apparent when attempting to model the data using a first order kinetics plot over the whole reaction time (Fig. S10 in ESI†). These observations can be explained if one considers the dimeric form of 2 in toluene as being inactive to ROP of rac-lactide. Hence, activation of the complex is achieved in the presence of co-initiator, possibly via formation of monomeric alcohol adducts. Once an active species is obtained, ROP proceeds quickly at 90 °C, thus explaining the rapid increase in conversion after 7 minutes and effectively complete consumption of monomer after 12 minutes. This is consistent with the activity vs. t plot (Fig. S11 in ESI†), which shows the highest activity was observed between 9 and 15 min. NMR spectra following the reaction profile are given in Fig. S12 in ESI.†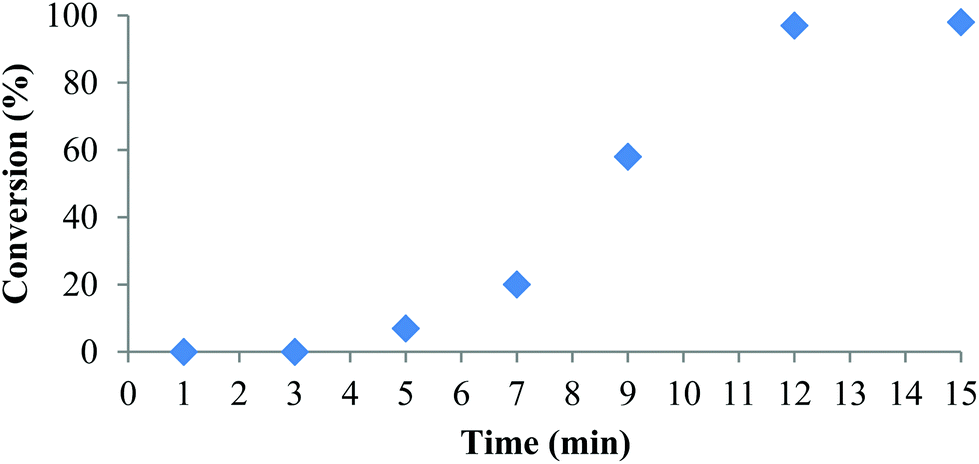 | ||
| Fig. 2 Conversion (%) vs. time for the ROP of rac-LA initiated by 2 under the conditions in Table 3, entry 1. | ||
| Entrya | [Mg]:[LA]:[ROH] |
Time (min) | Conv.b (%) |
M
nc (calcd) |
M
nd (GPC) |
Đ (Mw/Mn) |
|---|---|---|---|---|---|---|
| a All polymerization reactions were carried out in toluene (10 mL) at 90 °C. b Determined by 1H-NMR. c Calculated from ([LA]/[Mg]/[ROH]) × % conv. × 144.13 g mol−1. d Molecular weights (g mol−1) determined by gel permeation chromatography (GPC) in CHCl3 calibrated against polystyrene standards using the Mark–Houwink correction of 0.58.65 | ||||||
| 1 | 1:100:1 (iPrOH) |
30 | 99 | 14300 |
5700 | 1.70 |
| 2 | 1:100:10 (iPrOH) |
3 | 99 | 1400 | 480 | 1.42 |
| 3 | 1:100:0 |
210 | 100 | 14400 |
46000 |
1.26 |
| 4 | 1:100:1 (BnOH) |
12 | 98 | 14100 |
13900 |
1.07 |
| 5 | 1:100:2 (BnOH) |
7 | 97 | 7200 | 7100 | 1.02 |
The influence of the co-initiator on polymerizations in toluene was investigated. The polymerization without a co-initiator (Table 3, entry 3) proceeded very slowly and any conversion of lactide was only observed after 30 min in toluene at 90 °C, which is consistent with a slow initiation period. In the presence of BnOH, however, the reaction proceeded very quickly and achieved 98% conversion within 6 minutes (Table 3, entry 4). The polymerization demonstrates first order reaction profiles with respect to [LA] in either the presence or absence of BnOH (Fig. 3). The observed rate constant, kobs, in the presence of BnOH was 0.828 min−1 whereas in the absence of BnOH was 9.87 × 10−2 min−1. When the concentration of BnOH was doubled (Table 3, entry 5), the molecular weight of the polymer decreased by half, which suggests a well-behaved immortal ROP with rapid, reversible chain transfer between growing PLA fragments and dormant hydroxyl-terminated polylactide chains.69,70
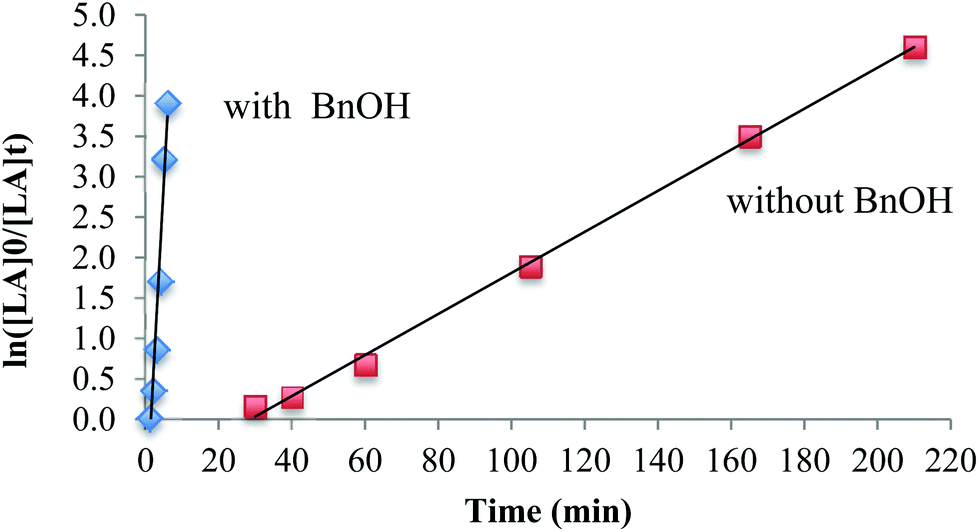 | ||
| Fig. 3 Plot of ln[LA]0/[LA]tvs. t, [LA]0/[Mg]0 = 100, in toluene at 90 °C according to the conditions in Table 3, entries 3 and 4. | ||
Mechanistic proposal
Without addition of exogenous alcohol, the ROP of LA is slower in solution than when the alcohol co-initiator is present. Initiation by nucleophilic attack of the phenolate ligand on the monomer has been observed in related lithium-phenolate complexes.10,44,69 In melt polymerization, however, there is no significant loss of control of the polymerization when conducted in the absence of alcohol co-initiator. The observation in the MALDI-TOF mass spectrum of cyclic polylactide alongside acyclic polymer possessing carboxylic acid and hydroxyl end groups (resulting from hydrolytic quenching of polymerization with acidified methanol) is consistent with phenolate-initiated ROP (Scheme 2).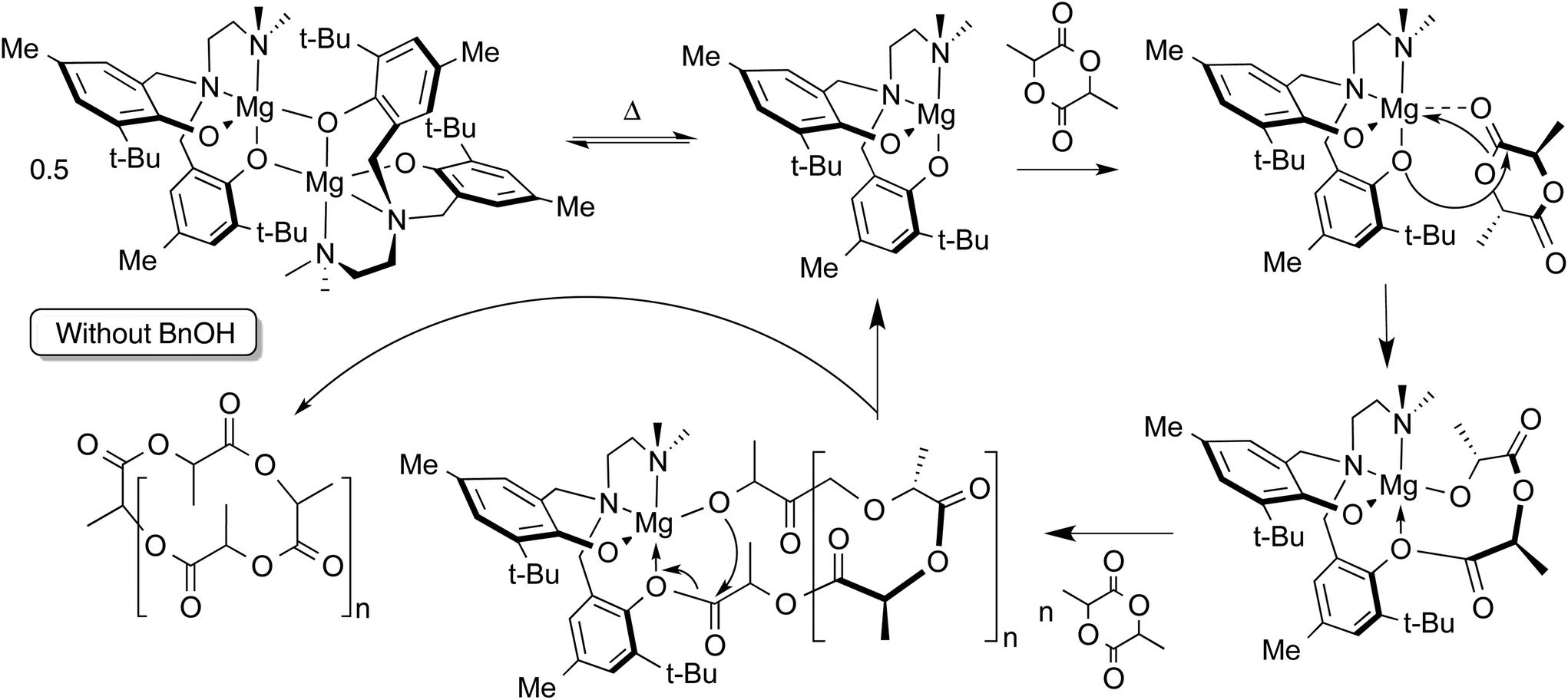 | ||
| Scheme 2 Proposed mechanism for ROP of LA by 2 without added alcohol co-initiator. Generation of cyclic polymer is shown, but hydroxyl or carboxylic acid end groups may be obtained by protonolysis. | ||
The mechanism of polymerization and the role of the alcohol co-initiator require closer examination, particularly since the majority of binary systems (employing a Lewis acidic metal site and an added alcohol as initiators) hypothesize coordination–insertion mechanisms. Carpentier, Sarazin and co-workers suggest that activated monomer mechanisms should not be excluded and have performed elegant studies of alkali aminoether-phenolate complexes that provide strong evidence that this mechanism at play under living or immortal ROP conditions of L-LA.69
Preliminary stoichiometric studies by 1H NMR spectroscopy of reactions involving complex 2, rac-LA and BnOH were conducted in toluene-d8 at 363 K, i.e. the conditions employed in solution ROP catalysis shown in Table 3. The reaction of a 1:1 ratio (per Mg centre) of 2 and BnOH was followed by 1H NMR at 363 K. After 10 minutes at this temperature, in addition to those of complex 2 alone, new resonances were observed consistent with monomeric Mg compounds (see Fig. S14 ESI† for 1H NMR spectra). It is not unequivocal whether the BnOH is coordinated to the metal centre since the benzyl methylene peak is slightly broadened at this temperature, possibly from an exchange process as suggested by the continued presence of dimer in the spectrum. Also, a very small peak at 9.19 ppm could be observed upon expansion of the region of the spectrum, suggesting that the presence of protonated ligand, H2[L2] is negligible.
The reaction of a 1:1:1 mixture of rac-LA, 2 and BnOH was monitored under the same conditions. Within 10 minutes at 363 K the product of lactide ring-opening, benzyl-2-((2-hydroxypropanoyl)oxy)propanoate or its deprotonated alkoxide form, was observed. It is possible that the ring-opened lactide remains coordinated to the Mg site, since a phenolic –OH resonance is observable at 9.22 ppm indicating protonation of a phenolate oxygen. The proposed mechanism is shown in Scheme 3. Based on these observations, an activated monomer mechanism may be occurring, but further investigation is needed.
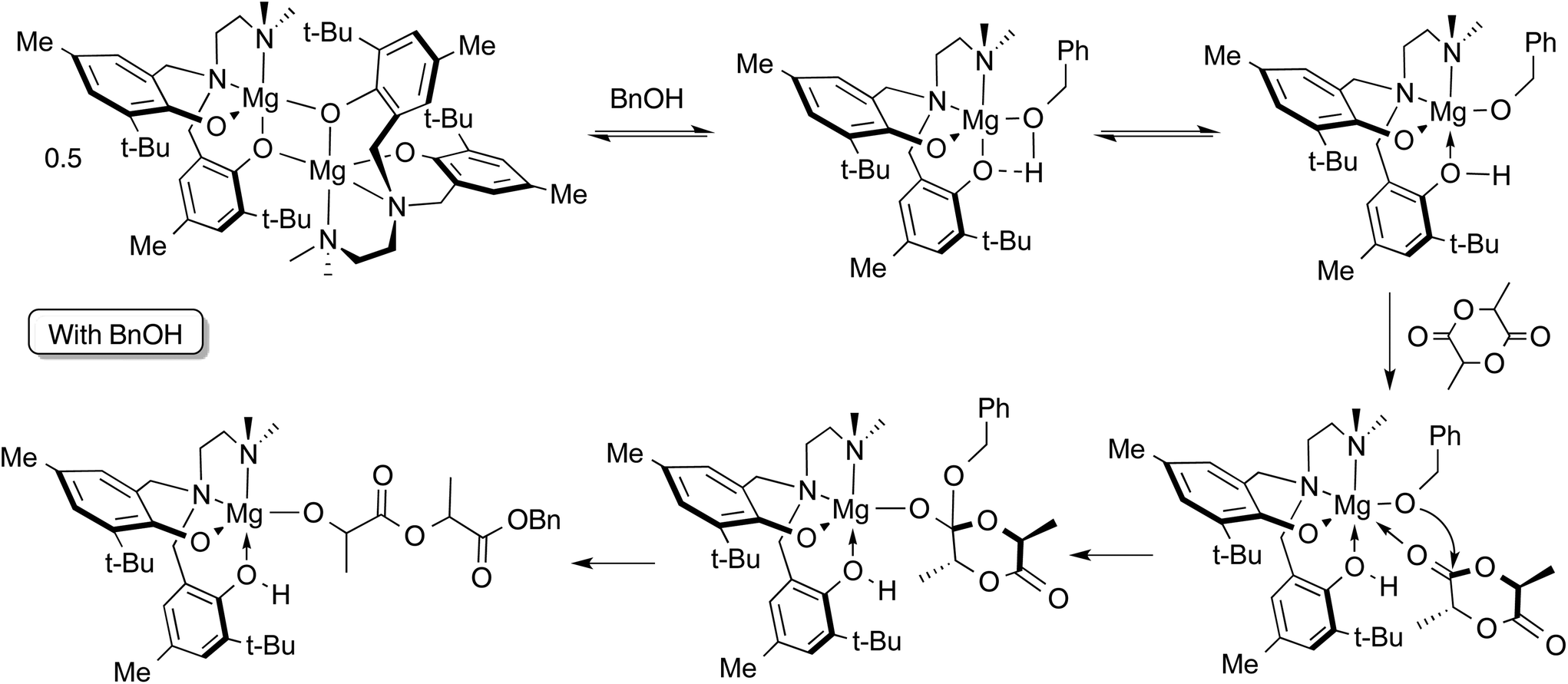 | ||
| Scheme 3 Proposed mechanism for ROP of LA by 2 with benzyl alcohol co-initiator. | ||
Conclusions
Magnesium amine-bis(phenolate) compounds 1 and 2 showed catalytic activity for the ROP of rac-lactide both in toluene solutions and solvent-free (melt) to give atactic poly(rac-lactide). Kinetic studies showed that conversion of rac-lactide is essentially complete within 60 minutes in the melt at 125 °C, although the resulting polymers exhibit broad dispersities. Solution polymerization studies in toluene at 90 °C indicate long initiation periods in the absence of an alcohol co-initiator, but rapid initiation when a suitable co-initiator is used. Complex 2 proved to be an efficient initiator in solution at 90 °C with a [LA]:[Mg] ratio of 100:1 regardless of whether a co-initiator was used or not. Without a co-initiator an initiation period of ∼30 min was observed, after which time the propagation showed a slow first order reaction profile with respect to rac-lactide concentration and effectively complete conversion within 210 minutes. The polymerization was accelerated significantly by the addition of a co-initiator, either iPrOH or BnOH. The highest activity was observed for [LA]:[Mg]:[BnOH] ratios of 100:1:1 at 90 °C in toluene giving 177.3 kg of rac-PLA per mol Mg per h (kobs = 1.38 × 10−2 s−1). Preliminary mechanistic studies support an activated monomer pathway, but more detailed studies are ongoing.
Experimental
General experimental conditions
Unless otherwise stated, all manipulations were performed under an atmosphere of dry, oxygen-free nitrogen by means of standard Schlenk techniques or using an MBraun Labmaster DP glove box. Anhydrous THF was distilled from sodium benzophenone ketyl under nitrogen. Isopropyl alcohol was dried over calcium oxide, then distilled under nitrogen. Toluene was purified by an MBraun Manual Solvent Purification System. Reagents were purchased either from Aldrich or Alfa Aesar and used without further purification. Rac-lactide was purchased from Aldrich or Alfa Aesar and dried over sodium sulphate in THF, recrystallized and stored under an inert atmosphere prior to use. Benzyl alcohol was purchased from Alfa Aesar and dried over activated 4 Å molecular sieves, distilled under reduced pressure and stored under nitrogen in an ampule prior to use.Instrumentation
MALDI-TOF MS was performed using an Applied Biosystems 4800 MALDI TOF/TOF Analyzer equipped with a reflectron, delayed ion extraction and high performance nitrogen laser (200 Hz operating at 355 nm). Samples were prepared in the glove box and sealed under nitrogen in a Ziploc© bag for transport to the instrument. Anthracene was used as the matrix for compounds 1 and 2 and 2,5-dihydroxybenzoic acid (DHBA) was used as the matrix for the polymers. The matrix was dissolved in THF at a concentration of 10 mg mL−1. Polymer was dissolved in THF at approximately 1 mg mL−1. The matrix and polymer solutions were mixed together at ratios of 5 to 1, 4 to 1 or 3 to 1; 1 μL of this was spotted on the MALDI plate and left to dry. Images of mass spectra were prepared using mMass™ software (http://www.mmass.org).GPC analysis was performed either in CHCl3 on a Viscotek VE 2001 GPCMax at 35 °C equipped with a Viscotek VE 3580 Refractive Index Detector or in THF at 25 °C on a Wyatt Triple Detection (triple angle light scattering, viscometry and refractive index) system with Agilent 2600 series sample and solvent handling. The Viscotek system used two Phenogel 5μ Linear Mixed Bed 300 × 4.60 mm columns whereas the Wyatt system used two Phenogel 103 Å 300 × 4.60 mm columns. Samples were prepared at a concentration of 2 mg mL−1 in CHCl3 and left to equilibrate for ∼2 h. The samples were filtered through syringe filters before analysis and eluted with HPLC grade solvents at flow rates of 0.30 mL min−1 with 100 μL injection volumes. For conventional calibration, six polystyrene standards (Viscotek) were used in making the calibration curve, bracketing molecular ranges from 1050 to 400000 Da. A correction factor of 0.58 was used to calculate the Mn value.65
NMR spectra were recorded at 300 MHz for 1H and 75.5 MHz for 13C or at 500 MHz for 1H NMR kinetic studies. CDCl3, C6D6, DMSO-d6 were purchased from Cambridge Isotope Laboratories and pyridine-d5 from Aldrich. All deuterated solvents except for DMSO-d6 used in the analysis of the magnesium complexes were dried over calcium hydride (CDCl3 and pyridine-d5) or sodium/potassium alloy (C6D6), vacuum transferred and stored under nitrogen in ampules fitted with Teflon valves. Elemental analyses were performed at Guelph Chemical Laboratories, Guelph, Ontario, Canada.
Thermogravimetric analysis (TGA) was performed with a TA Instruments Q500. Samples (4–15 mg) were loaded onto a platinum pan and subjected to a dynamic high-resolution scan, with an initial heating rate of 5 or 10 °C min−1. Each sample was heated from room temperature to 600 °C. Glass transition (Tg) temperatures were measured using a Mettler Toledo DSC 1 STARe System equipped with a Julabo FT 100 immersion cooling system, using R1150 refrigerant in an EtOH bath with a working range of −100 to +20 °C. Samples (∼5 mg) were weighed into 40 μL aluminum pans and subjected to three heating cycles. The first heating cycle consisted of heating from 0 to 100 °C at a rate of 10 °C min−1, held for 2 min at 100 °C and then cooled back to 0 °C at 10 °C min−1. The sample was held at this temperature for 2 min and subjected to a two heating cycles from 0 to 190 °C at a rate of 10 °C min−1.
Synthesis of compounds
1: 2.46 g (5.00 mmol) of H2[L1] was dissolved in 100 mL of toluene and cooled to −78 °C. To this solution was added 5.0 mmol of MgBu2 (5.0 mL of a 1.0 M solution in heptane) via syringe. The solution was warmed to room temperature and stirred overnight. Volatiles were removed in vacuo to yield a pale yellow powder. Yield = 2.42 g (94%). Anal. calc'd for C30H38MgN2O4: C 69.97, H 7.44, N 5.44. Found: C 69.26, H 7.17, N 5.13. The lower than expected %C is attributed to a 5–10% contamination by MgO or Mg(OH)2 (e.g. anal. calc'd for C30H38MgN2O4(MgO2H2)0.05: C 69.58, H 7.42, N 5.41. 1H NMR (300 MHz, 298 K, DMSO-d6, δ): 8.49 (d, 3JHH = 4.43 Hz, Py: NCH, 1H); 7.68 (td, 3JHH = 7.62 Hz, 4JHH = 1.51 Hz, Py: NCHCH, 1H); 7.21 (dd, coupling constants could not be determined because of the overlapping residual toluene peaks, Py: NCHCHCH, 1H); 7.06 (d, 3JHH = 7.8 Hz, Py: NCCH, 1H); 6.44 (d, 4JHH = 3.23 Hz, Ar: CHCCH2N, 2H); 6.38 (d, 4JHH = 3.23 Hz, Ar: CHCCMe3, 2H); 3.76 (d, 3JHH = 11.65 Hz, NCH2Ar, 2H); 3.58 (s, NCH2Py, 2H); 3.55 (s. OCH3, 6H); 3.15 (d, 3JHH = 11.72 Hz, NCH2Ar, 2H); 1.29 (s, C(CH3)3, 18H). 13C{1H} NMR (75.5 MHz, 298 K, DMSO-d6, δ): 160.71 (COMg); 158.04 (COMe); 148.60 (Ar: CCH2N); 145.35 (Py: NCCH); 138.46 (Py: NC); 136.66 (Py: NCCHCH); 130.38 (Py: NCHCH); 122.69 (Py: NCH); 122.03 (Ar: CCMe3); 113.46 (Ar: CHCCH2N); 112.94 (Ar: CHCCMe3); 60.68 (CH2Py); 56.10 (CH2Ar); 55.69 (OMe); 34.54 (CMe3); 29.34 (CMe3). MS (MALDI-TOF) m/z (%, ion): 514.26 (58, [M]+); 1028.50 (100, [M]2+).
2: 2.21 g (5.0 mmol) of H2[L2] was dissolved in 70 mL of toluene and cooled to −78 °C. To this solution was added 5.0 mmol of MgBu2 (5.0 mL of a 1.0 M solution in heptane) via syringe, forming a bright yellow solution. The solution was warmed to room temperature and stirred overnight, upon which a large amount of white precipitate formed within a colourless solution. Volatiles were removed in vacuo, yielding an analytically pure white powder. Yield = 2.29 g (99%). Recrystallization from toluene at −35 °C afforded colourless crystals. Anal. calc'd for C34H54MgN2O2: C 72.64, H 9.14, N 6.05. Found: C 72.38, H 9.09, N 6.22. 1H NMR (300 MHz, 298 K, C6D6, δ): 7.29 (m, Ar, 4H); 6.80 (d, 4JHH = 2.35 Hz, Ar, 2H); 6.69 (d, 4JHH = 2.30, Ar, 2H); 4.84 (d, 2JHH = 12.01 Hz, Ar–CH(H)–N, 2H); 4.63 (d, 2JHH = 12.41 Hz, Ar–CH(H)–N, 2H); 2.97 (d, 2JHH = 12.38 Hz, Ar–CH(H)–N, 2H); 2.63 (d, 2JHH = 12.06 Hz, Ar–CH(H)–N, 2H); 2.00–2.70 (br, CH2NMe2, 4H); 2.35 (s, NMe2, 6H); 2.31 (s, NMe2, 6H); 1.05–1.70 (br, CH2CH2NMe2, 4H); 1.69 (s, CMe3, 18H); 1.65 (s, ArMe, 6H); 1.59 (s, ArMe, 6H); 1.29 (s, CMe3, 18H). 13C{1H} NMR (75.5 MHz, 298 K, C6D6, δ): 164.59 (C–O); 160.09 (C–O); 137.80, 136.95, 130.13, 129.61, 128.92, 128.06, 128.74, 125.55, 123.01, 119.89 (All Ar); 65.49 (ArCH2); 64.48 (ArCH2); 60.54 (CH2CH2NMe2); 48.69 (CH2NMe2); 47.55 (NMe2); 47.42 (NMe2); 35.12 (CMe3); 34.77 (CMe3); 32.82 (CMe3); 30.34 (CMe3); 20.98 (ArMe); 20.90 (ArMe). 1H NMR (300 MHz, 298 K, pyridine-d5, δ): 7.35 (d, 4JHH = 2.45 Hz, Ar, 2H); 6.92 (d, 4JHH = 2.51 Hz, Ar, 2H); 4.04 (d, 2JHH = 12.49, Ar–CH(H)–N, 2H); 3.11 (d, 2JHH = 12.51 Hz, Ar–CH(H)–N, 2H); 2.75 (br, CH2NMe2, 2H); 2.40 (s, NMe2, 6H); 2.23 (br, CH2CH2NMe2, 2H); 1.97 (s, ArMe, 6H); 1.75 (s, CMe3, 18H). 13C{1H} NMR (75.5 MHz, 298 K, pyridine-d5, δ): 167.80 (C–O); 138.71, 132.41, 130.65, 130.08, 126.64 (All Ar); 66.88 (ArCH2); 61.54 (CH2CH2NMe2); 51.27 (CH2NMe2); 49.51 (NMe2); 37.24 (CMe3); 32.43 (CMe3); 23.05 (CMe3). MS (MALDI-TOF) m/z (%, ion): 462.25 (100, [M]+); 924.52 (3, [M]2+).
Polymerization procedure
:1 [rac-lactide]:[1] ratio was carried out with 500 mg (3.47 mmol) rac-lactide and 3.57 mg (6.93 × 10−3 mmol) of complex 1 weighed into a 10 mL scintillation vial equipped with a small stir bar in the glove box. The closed vial was taken out of the glove box and placed into an aluminum block vial holder, which was pre-heated on a hotplate to 125 °C. A typical polymerization was run for 100 minutes. The vial was then placed in an ice bath to halt the reaction and solidify the polymer. The solids were dissolved in dichloromethane or chloroform and the polymer was precipitated with acidified methanol. Centrifugation was applied where needed for better separation of the solids. Solvents were decanted and the white solids (either crystalline or thin film-like) were dried in vacuo followed by drying in a vacuum oven at 40 °C overnight.
Acknowledgements
Financial support was provided by the Canada Foundation for Innovation (CFI), Newfoundland and Labrador Research Development Corporation, the Natural Sciences and Engineering Research Council (NSERC) of Canada and Memorial University of Newfoundland. J. H. L. and M. E. P. thank NSERC for Undergraduate Student Research Awards.References
- R. T. Martin, L. P. Camargo and S. A. Miller, Green Chem., 2014, 16, 1768–1773 RSC.
- J. M. Becker and A. P. Dove, in Green Polymerization Methods: Renewable Starting Materials, Catalysis and Waste Reduction, eds. R. T. Mathers and M. A. R. Meier, Wiley-VCH, Weinheim, 1st edn, 2011, pp. 201–220 Search PubMed.
- R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS.
- O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- C. A. Wheaton, P. G. Hayes and B. J. Ireland, Dalton Trans., 2009, 4832–4846 RSC.
- M. H. Chisholm and Z. P. Zhou, J. Mater. Chem., 2004, 14, 3081–3092 RSC.
- B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC.
- O. Wichmann, R. Sillanpää and A. Lehtonen, Coord. Chem. Rev., 2012, 256, 371–392 CrossRef CAS PubMed.
- N. Ikpo, C. Hoffmann, L. N. Dawe and F. M. Kerton, Dalton Trans., 2012, 41, 6651–6660 RSC.
- Y. Huang, Y.-H. Tsai, W.-C. Hung, C.-S. Lin, W. Wang, J.-H. Huang, S. Dutta and C.-C. Lin, Inorg. Chem., 2010, 49, 9416–9425 CrossRef CAS PubMed.
- L. Wang, X. B. Pan, L. H. Yao, N. Tang and J. C. Wu, Eur. J. Inorg. Chem., 2011, 632–636 CrossRef.
- C.-A. Huang, C.-L. Ho and C.-T. Chen, Dalton Trans., 2008, 3502–3510 RSC.
- C. A. Huang and C. T. Chen, Dalton Trans., 2007, 5561–5566 RSC.
- F. M. Kerton, C. M. Kozak, K. Lüttgen, C. E. Willans, R. J. Webster and A. C. Whitwood, Inorg. Chim. Acta, 2006, 359, 2819–2825 CrossRef CAS PubMed.
- B.-T. Ko and C.-C. Lin, J. Am. Chem. Soc., 2001, 123, 7973–7977 CrossRef CAS PubMed.
- W. Clegg, M. G. Davidson, D. V. Graham, G. Griffen, M. D. Jones, A. R. Kennedy, C. T. O'Hara, L. Russo and C. M. Thomson, Dalton Trans., 2008, 1295–1301 RSC.
- Z. Janas, T. Nerkowski, E. Kober, L. B. Jerzykiewicz and T. Lis, Dalton Trans., 2012, 41, 442–447 RSC.
- L. E. Breyfogle, C. K. Williams, V. G. Young, M. A. Hillmyer and W. B. Tolman, Dalton Trans., 2006, 928–936 RSC.
- J. Ejfler, S. Szafert, K. Mierzwicki, L. B. Jerzykiewicz and P. Sobota, Dalton Trans., 2008, 6556–6562 RSC.
- W. C. Hung and C. C. Lin, Inorg. Chem., 2009, 48, 728–734 CrossRef CAS PubMed.
- E. L. Marshall, V. C. Gibson and H. S. Rzepa, J. Am. Chem. Soc., 2005, 127, 6048–6051 CrossRef CAS PubMed.
- Y. Sarazin, V. Poirier, T. Roisnel and J.-F. Carpentier, Eur. J. Inorg. Chem., 2010, 3423–3428 CrossRef CAS.
- A. D. Schofield, M. L. Barros, M. G. Cushion, A. D. Schwarz and P. Mountford, Dalton Trans., 2009, 85–96 RSC.
- X. Zhang, T. J. Emge and K. C. Hultzsch, Organometallics, 2010, 29, 5871–5877 CrossRef CAS.
- D. J. Darensbourg, W. Choi, O. Karroonnirun and N. Bhuvanesh, Macromolecules, 2008, 41, 3493–3502 CrossRef CAS.
- H. E. Dyer, S. Huijser, A. D. Schwarz, C. Wang, R. Duchateau and P. Mountford, Dalton Trans., 2008, 32–35 RSC.
- A. Amgoune, C. M. Thomas and J.-F. Carpentier, Macromol. Rapid Commun., 2007, 28, 693–697 CrossRef CAS.
- M. H. Chisholm, J. C. Gallucci, H. H. Zhen and J. C. Huffman, Inorg. Chem., 2001, 40, 5051–5054 CrossRef CAS.
- C. K. Williams, N. R. Brooks, M. A. Hillmyer and W. B. Tolman, Chem. Commun., 2002, 2132–2133 RSC.
- C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed.
- D. J. Doyle, V. C. Gibson and A. J. P. White, Dalton Trans., 2007, 358–363 RSC.
- J. D. Farwell, P. B. Hitchcock, M. F. Lappert, G. A. Luinstra, A. V. Protchenko and X.-H. Wei, J. Organomet. Chem., 2008, 693, 1861–1869 CrossRef CAS PubMed.
- G. Labourdette, D. J. Lee, B. O. Patrick, M. B. Ezhova and P. Mehrkhodavandi, Organometallics, 2009, 28, 1309–1319 CrossRef CAS.
- L. Wang and H. Ma, Dalton Trans., 2010, 39, 7897–7910 RSC.
- N. Ikpo, L. N. Saunders, J. L. Walsh, J. M. B. Smith, L. N. Dawe and F. M. Kerton, Eur. J. Inorg. Chem., 2011, 5347–5359 CrossRef CAS.
- V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2011, 40, 523–534 RSC.
- J. Lewinski, P. Horeglad, M. Dranka and I. Justyniak, Inorg. Chem., 2004, 43, 5789–5791 CrossRef CAS PubMed.
- J.-T. Issenhuth, J. Pluvinage, R. Welter, S. Bellemin-Laponnaz and S. Dagorne, Eur. J. Inorg. Chem., 2009, 4701–4709 CrossRef CAS.
- S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783–4790 CrossRef CAS PubMed.
- S. Groysman, E. Sergeeva, I. Goldberg and M. Kol, Inorg. Chem., 2005, 44, 8188–8190 CrossRef CAS PubMed.
- L. M. Broomfield, Y. Sarazin, J. A. Wright, D. L. Hughes, W. Clegg, R. W. Harrington and M. Bochmann, J. Organomet. Chem., 2007, 692, 4603–4611 CrossRef CAS PubMed.
- L. N. Saunders, L. N. Dawe and C. M. Kozak, J. Organomet. Chem., 2014, 749, 34–40 CrossRef CAS PubMed.
- R. K. Dean, A. M. Reckling, H. Chen, L. N. Dawe, C. M. Schneider and C. M. Kozak, Dalton Trans., 2013, 3504–3530 RSC.
- R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS.
- J. H. Khan, F. Schue and G. A. George, Polym. Int., 2009, 58, 296–301 CrossRef CAS.
- M. R. Kember and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 15676–15679 CrossRef CAS PubMed.
- L. Wang and H. Ma, Macromolecules, 2010, 43, 6535–6537 CrossRef CAS.
- S. Song, H. Ma and Y. Yang, Dalton Trans., 2013, 42, 14200–14211 RSC.
- E. Kober, Z. Janas, T. Nerkowski and L. B. Jerzykiewicz, Dalton Trans., 2013, 42, 10847–10854 RSC.
- M. G. Davidson, M. D. Jones, D. Meng and C. T. O'Hara, Main Group Chem., 2006, 5, 3–12 CrossRef CAS.
- J. Ejfler, K. Krauzy-Dziedzic, S. Szafert, L. B. Jerzykiewicz and P. Sobota, Eur. J. Inorg. Chem., 2010, 3602–3609 CrossRef CAS.
- Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC.
- F. M. Kerton, S. Holloway, A. Power, R. G. Soper, K. Sheridan, J. M. Lynam, A. C. Whitwood and C. E. Willans, Can. J. Chem., 2008, 86, 435–443 CrossRef CAS.
- S. Menage, G. Gellon, J.-L. Pierre, D. Zurita and E. Saint-Aman, Bull. Soc. Chim. Fr., 1997, 134, 785–791 CAS.
- S. Heidari, E. Safaei, A. Wojtczak, P. Cotic and A. Kozakiewicz, Polyhedron, 2013, 55, 109–116 CrossRef CAS PubMed.
- T. R. Forder, M. F. Mahon, M. G. Davidson, T. Woodman and M. D. Jones, Dalton Trans., 2014, 43, 12095–12099 RSC.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
- A. C. Silvino, D. Bonilha de Abreu Talina Martins, A. da Costa Rodrigues and M. L. Dias, J. Polym. Environ., 2013, 21, 1002–1008 CrossRef CAS PubMed.
- A. D. Schwarz, Z. Chu and P. Mountford, Organometallics, 2010, 29, 1246–1260 CrossRef CAS.
- G. Xiao, B. Yan, R. Ma, W. J. Jin, X. Q. Lu, L. Q. Ding, C. Zeng, L. L. Chen and F. Bao, Polym. Chem., 2011, 2, 659–664 RSC.
- C. Paetz and R. Hagen, Chem. Ing. Tech., 2014, 86, 519–523 CrossRef CAS.
- M. Bouyahyi, E. Grunova, N. Marquet, E. Kirillov, C. M. Thomas, T. Roisnel and J.-F. Carpentier, Organometallics, 2008, 27, 5815–5825 CrossRef CAS.
- A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295 RSC.
- J. Baran, A. Duda, A. Kowalski, R. Szymanski and S. Penczek, Macromol. Rapid Commun., 1997, 18, 325–333 CrossRef CAS.
- K. Jamshidi, S. H. Hyon and Y. Ikada, Polymer, 1988, 29, 2229–2234 CrossRef CAS.
- S.-H. Lee, S. Hyun Kim, Y.-K. Han and Y. H. Kim, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 973–985 CrossRef CAS.
- Z. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS.
- Y. Sarazin, S.-C. Rosca, D.-A. Rosca, V. Dorcet, J.-F. Carpentier, F. M. Kerton and C. M. Kozak, Dalton Trans., 2013, 9361–9375 Search PubMed.
- N. Ajellal, J.-F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic experimental and spectroscopic data (PDF). CCDC 1043113. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00236b |
| ‡ Current Address: Department of Chemistry and Biochemistry, Wilfrid Laurier University, Waterloo, Ontario, Canada |
| This journal is © The Royal Society of Chemistry 2015 |