
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ring-opening polymerization of cyclohexene oxide using aluminum amine–phenolate complexes†‡
Hart
Plommer
,
Immanuel
Reim
and
Francesca M.
Kerton
 *
*
Department of Chemistry, Memorial University of Newfoundland, St. John's, NL A1B 3X7, Canada. E-mail: fkerton@mun.ca
First published on 11th February 2015
Abstract
Remarkably active (down to 0.001% Al) catalysts for ring-opening polymerization of cyclohexene oxide under neat reaction conditions are reported. High molecular weight polymers with uniform dispersity are produced. Kinetic data from NMR studies and MALDI-TOF MS data of the polymers provide some mechanistic insight.
Aluminum is the most abundant of all metals in the earth's crust and its complexes can be used as Lewis acid catalysts or as co-catalysts for a range of reactions.1 In particular, they have found broad applicability as homogeneous catalysts in polymerization processes.2 Recently, significant attention has been given to their use in ring-opening polymerization (ROP) reactions of lactide (LA) and ε-caprolactone (CL) that yield biodegradable polyesters of controlled molecular weight and near uniform dispersity.3 They are also being widely explored as catalysts for reactions of carbon dioxide with epoxides that yield either polycarbonates or cyclic carbonate products.4 Aluminum species can also be active for polymerizations of propylene oxide (PO),5 cyclohexene oxide (CHO),3b,4h,6 and CHO-anhydride7 and PO-LA copolymerizations.8 The areas of polyether synthesis and stereoselective polymerization/copolymerizations of epoxides have recently been reviewed and provide good overviews of the concepts and possible mechanistic considerations in these reactions.9
We recently showed that chloro–aluminum complexes of N-piperazinyl and N-morpholinyl aminephenolate ligands, including 2 herein, were active in ROP of CL to yield polymers with the general formula (Cl{CL}nOH) i.e. containing chloride end groups.10 We proposed that these reactions proceeded in a similar manner to ROP of trimethylene carbonate using aluminum-salen chloride catalysts,11 which is to say that reactions proceed via insertion into the Al–Cl bond. We also showed that one of the N-morpholinyl aminephenolate complexes was catalytically active in CO2/CHO copolymerization to yield copolymers with a mix of ether and carbonate linkages (i.e. polyether-carbonates).10 Because of the relatively large number of ether linkages in these polymers, we decided to investigate the first step in this process in more detail, namely, the reaction of CHO with aluminum chloride complexes.
Complexes 1 and 2 are easily prepared in near quantitative yields via alkane elimination reactions of diethylaluminum chloride with two equivalents of the corresponding protio ligand (Scheme 1). Attempts to investigate the interaction of one equivalent of CHO with the aluminum complexes on an NMR scale in a range of solvents were unsuccessful due to the fluxional nature of the complexes. Therefore, larger amounts of CHO were added to samples and it was discovered that polyether forms.
 | ||
| Scheme 1 | ||
ROP of CHO was carried out using complexes 1 and 2 (Table 1) in the absence of solvent at room temperature. In most cases, polymerization occurred rapidly to yield a viscous monomer–polymer mixture that could no longer be stirred magnetically. For 2 however, the polymerizations were much slower, leading to less viscous solutions even after extended reaction times. This indicates that the nature of the outersphere heteroatom in the N-containing heterocycle has a significant effect on these reactions. Differences in reactivity between the N-piperazinyl and N-morpholinyl containing catalysts were also seen in ROP of CL and CO2/CHO copolymerizations.10
| Entry | Complex | [CHO]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[Al] :[Al] |
t/min | Conv./% (yield/%) | M n | Đ | TOF/min−1 |
|---|---|---|---|---|---|---|---|
| a Reactions were performed neat at room temperature in the absence of solvent unless otherwise indicated in an inert atmosphere workstation. Conversions determined by 1H NMR spectroscopy. Isolated yields indicated in parentheses. b Determined (in kg mol−1) by GPC equipped with a multi-angle light scattering detector. c Reaction performed in the presence of 160 μL toluene. Note, 7% conversion was obtained after 18 h in 5 mL toluene. | |||||||
| 1 | 1 | 100000:1 |
60 | 15 (13) | 500 | 1.17 | 250 |
| 2 | 1 | 1000:1 |
1 | 58 (53) | 180 | 1.20 | 580 |
| 3 | 2 | 1000:1 |
50 | 15 (6) | — | — | 3 |
| 4 | 2 | 200:1 |
30 | 16 (3) | 76.2 | 1.15 | 1 |
| 5 | 3 | 10000:1 |
1260 | <1% | — | — | — |
| 6 | 3 | 1000:1 |
45 | 29 | 262 | 1.36 | 6 |
| 7 | 3 | 500:1 |
25 | 49 | 255 | 1.21 | 10 |
| 8 | 3 | 200:1 |
2 | 56 | 125 | 1.12 | 56 |
| 9c | 3 | 500:1 |
40 | 66 | 110 | 1.08 | 8 |
In an effort to determine the essential features of active complexes, 3 was prepared (Scheme 2). Amine-bis(phenolate) aluminum-alkyl and -alkoxide complexes similar to 3 have been prepared previously and used for ROP of lactide.123 showed good activity in ROP of CHO and using small amounts of toluene a higher conversion and less disperse polymer could be obtained compared to the neat reactions (entries 9 and 7). In contrast to 1, catalyst loadings could not be reduced past 0.1 mol%.
 | ||
| Scheme 2 | ||
The activities of these aluminum amine–phenolate complexes are clearly dependent on the nature of the ligand. Since the maximum conversion of CHO that can be attained during neat polymerization depends on the molecular weight of PCHO produced and the ability of CHO to solvate the catalyst and polymer, turnover frequencies (TOFs) are the most practical way to compare the catalysts. The conversion of 1000 equiv. of CHO was much quicker for 1 than 2 and 3 (Table 1, entries 2, 3 and 6) and TOF values are an order of magnitude higher for 1. The observed reactivity trend for complexes 1–3 is in the order 1 ≫ 3 > 2. This implies that, although chloro complexes of both monoanionic and dianionic aminephenolate ligands give rise to active species, there is a more complex relationship between ligand structure and reactivity at play that warrants further investigation. Control polymerizations were performed using two simple chloro–aluminum reagents under similar reaction conditions to those employed with 1–3. AlCl3 (0.5 mol%) or Et2AlCl (0.25 mol%) gave TOFs (min−1) of 4 and 160, respectively, and a maximum Mn of 32000 Da with relatively narrow dispersities (∼1.3). These are significantly lower activities and molecular weights than achieved using 1 at similar loadings.
To avoid gelation and potentially increase conversions, some solution polymerizations with 1 were attempted using CH2Cl2, toluene or THF as reaction media. Reaction rates were significantly reduced and for a 200:1 ratio of CHO: 1 in 5 mL solvent conversions of 28% (CH2Cl2), 18% (toluene) and <1% (THF) were obtained after 20 h. The latter may be due to THF preferentially coordinating to the Al centre over CHO. Darensbourg and Chung have recently reinvestigated the basicities of a range of ethers and esters,13 and this statement is in agreement with the reported pKb values. A difference in reactivity for ROP reactions in CH2Cl2 and toluene was also observed by Martínez et al.6 in their study of multinuclear Al complexes for CHO polymerizations, e.g. 96% yield in CH2Cl2 and 75% yield in toluene. They found that the addition of benzenesulfonyl chloride to toluene improved activities, reaching similar conversions to those performed in CH2Cl2 with no chloride additive. This suggests that the chloride group is important in these ROP reactions.
Of note is the very low loadings of 1 that could be used, 0.001 mol% relative to CHO (Table 1, entry 1), giving low conversion (15%) of CHO but yielding a polymer with very high Mn (500000 Da) and uniform dispersity. The only catalysts reported to date for CHO polymerization that can produce such high molecular weight polymers (albeit at longer reaction times) are group 4 benzotriazole–phenolate complexes, which could achieve Mn values of up to 980000 Da.14 Other systems reported that give PCHO with high molecular weights include the rare earth catalysts of Cui and co-workers15 that attain Mn up to 147000 Da with a dispersity of 2.38, as well as the Zn and Mg catalysts prepared by Bochmann and co-workers16 that achieve up to 380000 Da with a dispersity of 3.8. It should be noted that previous authors invariably report Mn determined via GPC calibrated with polystyrene standards, and therefore some care must be taken in making direct comparisons with our results based on multi-angle light scattering.
The polymerization of various other epoxides (PO, styrene oxide, limonene oxide, and epichlorohydrin) was attempted using 1–3 (Table S1‡). In all cases, including microwave heated reactions conducted at 60 °C with propylene oxide and at 130 °C with limonene oxide, no polymer was detected by 1H NMR. Catalysts for ROP of CHO containing a range of other metals such as iron,17 zinc, magnesium,16 uranium,18 titanium, zirconium, and halfnium14 are able to polymerize a number of different epoxides (e.g. PO, epichlorohydrin). Therefore, it was somewhat surprising that 1 could not polymerize any of the other monomers studied. It is well known that the ring-opening reactions of CHO are more facile than PO. It has been suggested that this is due to the release of extra strain energy upon polymerizing the bicyclic CHO monomer,19 but more recently others have shown that the strain energies of CHO and PO are essentially the same.20 This is also reflected by nearly identical enthalpies of polymerization: −96.7 kJ mol−1 for CHO21 and −94.5 kJ mol−1 for PO (calculated using thermodynamic data in the NIST chemistry webbook). Therefore, differences in reactivity must be due to kinetic effects and thus determination of reaction pathways computationally would aid in discerning the differences observed experimentally. One possible reason for the differences in epoxide reactivity is steric i.e. the CHO monomer is slimmer, due to its bicyclic nature, than the other monomers studied,18a and this means it can coordinate to the metal centre more readily prior to ring-opening by a nucleophile.
MALDI-TOF mass spectra were obtained on the polymers in order to identify end-groups, as no phenolate or other end groups were seen in their 1H NMR spectra. It has been reported that laser desorption techniques induce significant fragmentation of polyethers and consequently only small oligomers are seen when using this technique,8 but these methods have been useful in identifying chloride end groups in polyethers and copolymers. The MALDI-TOF spectrum of the PCHO formed using 3 (Table 1, entry 9), Fig. S1,‡ is described here. The spectrum shows two prominent series of peak distributions with a mass difference between them of 36 Da, which corresponds to the mass of HCl. Successive peak distributions of either series are separated by 98 Da which equals the mass of the CHO monomer. These data are consistent with the formation of sodium adducts of both cyclic PCHO ({CHO}n·Na+) [e.g. n = 8, m/z 807.58 (expt), m/z 807.52 (calcd)] and PCHO chains, the latter being capped with hydroxyl and chloride end groups (Cl{CHO}nH·Na+) [e.g. n = 8, m/z 843.55 (expt), m/z 843.50 (calcd)]. Since HCl was unable to catalyze these reactions under identical conditions, the identification of a chloride end group confirms the role of the chloride ligand in ring-opening of the epoxide. In previous research, P(CL) polymers containing chloride end groups could be made with complex 2.10
The polymers obtained with 1–3 were analyzed by DSC. Glass transition temperatures obtained were 68–69 °C, in agreement with values found for PCHO by Kim et al.,22 who polymerized CHO to give PCHO with Mn in the range of 12400 to 24900. No exotherm corresponding to a crystallization process could be observed on cooling, indicating that these polymers are amorphous. The stereochemistry of the PCHO was analyzed via1H and 13C NMR spectroscopy (Fig. S2 and S3‡). No stereocontrol was achieved in these polymerizations, as three peaks at δ 3.53, 3.44, and 3.37 in the 1H NMR spectra were observed corresponding to the methine protons are characteristic of syndiotactic (rr), heterotactic (rm and mr), and isotactic (mm) triads. The atactic nature of the polymer was also confirmed by the presence of three broad peaks at δ 78.8, 77.7, and 75.6 (methine carbons) in the 13C NMR spectra. The stereochemistry is in accordance with the finding of others using similar achiral Al catalysts.4h,6
Kinetic studies on the ROP of CHO using 2 were carried out under neat conditions using a co-axial NMR tube containing CDCl3 for locking. The slower reactivity of this catalyst meant it was a practical choice for NMR monitoring. Rates of stirring could not be readily controlled within the spectrometer and may have led to inhomogeneity that produced scattering and curvature in the data for two samples. These studies show that the polymerization is first-order in [Al], indicated by the linear relationship between kobs and [2] (Fig. 1). The slope of this graph gives a propagation rate of 0.0568 M−1 min−1 and the intercept is close to the origin suggesting that minimal catalyst deactivation occurred through the presence of impurities. More studies are needed to determine whether the reactions proceed via a monometallic (intramolecular) or bimetallic (intermolecular) mechanism. Namely, whether the chloride initiator originates on the same Al centre as the coordinated CHO or on a neighbouring complex. For recently reported ROP of epoxides using [UO2Cl2(THF)3] as a catalyst,18b an intermolecular process is favoured and we think that a similar mechanism is at work in our catalyst system.
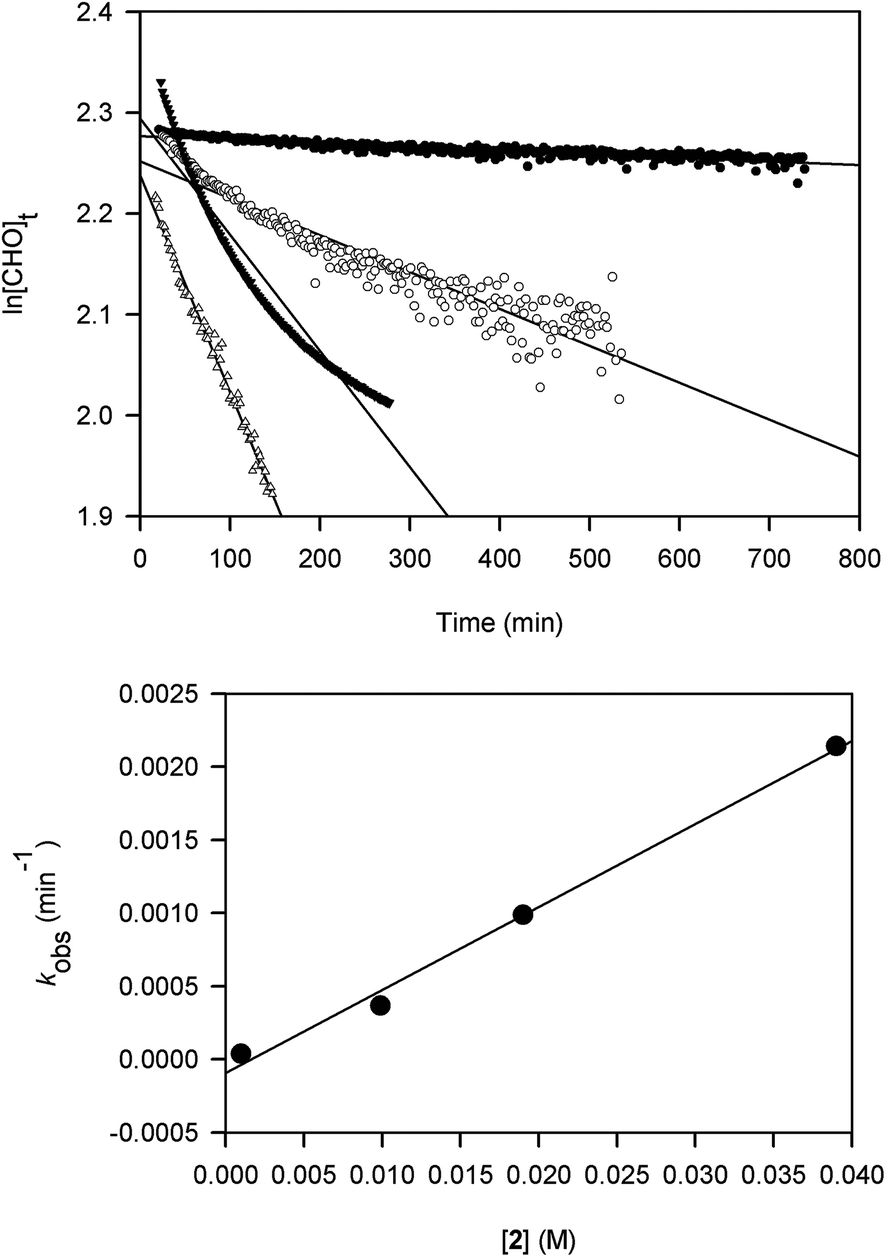 | ||
| Fig. 1 Plots of ln[CHO]t against time at catalyst loadings 0.01 (●), 0.1 (○), 0.2 (▼) and 0.4 (△) mol% using 2 (top) and kobs (min−1) against [2] (M) (bottom). | ||
In summary, we have shown that amine–phenolate complexes of aluminum containing ancillary chloride ligands are active catalysts for ROP of CHO under neat conditions. In some cases, very low catalyst loadings (0.001%) can be used and high molecular weight amorphous polymers (Mn up to 500000) are obtained. The reactions are generally inhibited in the presence of solvents, especially THF, and are first order in [Al]. The reactions are initiated by nucleophilic attack of the coordinated epoxide by a chloride anion, as evidenced by MALDI-TOF analysis of the resulting polymers. Further studies are needed to unequivocally determine whether the reaction proceeds via a monometallic or bimetallic process, and to determine the extent of reactivity across a wider range of aluminum amine–phenolate species.
We thank NSERC, Memorial University, Canada Foundation for Innovation, and RDC-Newfoundland for funding. Immanuel Reim thanks DAAD Rise for a scholarship.
Notes and references
- (a) M. R. Mason, Aluminum: Organometallic Chemistry, in Encyclopedia of Inorganic Chemistry, John Wiley & Sons, Ltd, 2006 Search PubMed; (b) J. Lewinski and A. E. H. Wheatley, Top. Organomet. Chem., 2013, 41, 1–58 CrossRef CAS.
- S. Dagorne and C. Fliedel, Top. Organomet. Chem., 2013, 41, 125–171 CrossRef CAS.
- (a) M. H. Chisholm, J. C. Gallucci, K. T. Quisenberry and Z. Zhou, Inorg. Chem., 2008, 47, 2613–2624 CrossRef CAS PubMed; (b) B. Liu, H. Li, C.-S. Ha, I. Kim and W. Yan, Macromol. Res., 2008, 16, 441–445 CrossRef CAS; (c) H. Du, A. H. Velders, P. J. Dijkstra, J. Sun, Z. Zhong, X. Chen and J. Feijen, Chem. – Eur. J., 2009, 15, 9836–9845 CrossRef CAS PubMed; (d) K. Phomphrai, P. Chumsaeng, P. Sangtrirutnugul, P. Kongsaeree and M. Pohmakotr, Dalton Trans., 2010, 39, 1865–1871 RSC; (e) A. D. Schwarz, Z. Chu and P. Mountford, Organometallics, 2010, 29, 1246–1260 CrossRef CAS; (f) M.-H. Thibault and F.-G. Fontaine, Dalton Trans., 2010, 39, 5688–5697 RSC; (g) C. Bakewell, R. H. Platel, S. K. Cary, S. M. Hubbard, J. M. Roaf, A. C. Levine, A. J. P. White, N. J. Long, M. Haaf and C. K. Williams, Organometallics, 2012, 31, 4729–4736 CrossRef CAS; (h) M. Bouyahyi, T. Roisnel and J.-F. Carpentier, Organometallics, 2012, 31, 1458–1466 CrossRef CAS; (i) M. Lamberti, I. D'Auria, M. Mazzeo, S. Milione, V. Bertolasi and D. Pappalardo, Organometallics, 2012, 31, 5551–5560 CrossRef CAS; (j) W. Alkarekshi, A. P. Armitage, O. Boyron, C. J. Davies, M. Govere, A. Gregory, K. Singh and G. A. Solan, Organometallics, 2013, 32, 249–259 CrossRef CAS; (k) X. Liu, C. Jian, D. Yu, J. Zhang, N. Tang, C. Wang and J. Wu, Inorg. Chem. Commun., 2013, 36, 206–211 CrossRef CAS PubMed; (l) L. Postigo, M. d. C. Maestre, M. E. G. Mosquera, T. Cuenca and G. Jimenez, Organometallics, 2013, 32, 2618–2624 CrossRef CAS; (m) K. Bakthavachalam, A. Rajagopal and N. Dastagiri Reddy, Dalton Trans., 2014, 43, 14816–14823 RSC; (n) M. J. Go, S. H. Kim, Y. Y. Kang, H.-R. Park, Y. Kim and J. Lee, Inorg. Chem. Commun., 2014, 44, 139–142 CrossRef CAS PubMed; (o) S.-Y. Hsu, C.-H. Hu, C.-Y. Tu, C.-H. Lin, R.-Y. Chen, A. Datta and J.-H. Huang, Eur. J. Inorg. Chem., 2014, 2014, 1965–1973 CrossRef CAS; (p) C. Romain, C. Fliedel, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2014, 33, 5730–5739 CrossRef CAS; (q) H. Kampova, E. Riemlova, J. Klikarova, V. Pejchal, J. Merna, P. Vlasak, P. Svec, Z. Ruzickova and A. Ruzicka, J. Organomet. Chem., 2015, 778, 35–41 CrossRef CAS PubMed.
- (a) W. Kuran, T. Listos, M. Abramczyk and A. Dawidek, J. Macromol. Sci., Part A: Pure Appl. Chem., 1998, 35, 427–437 CrossRef; (b) J. H. Jung, M. Ree and T. Chang, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3329–3336 CrossRef CAS; (c) T. Sârbu and E. J. Beckman, Macromolecules, 1999, 32, 6904–6912 CrossRef; (d) M. H. Chisholm and Z. Zhou, J. Am. Chem. Soc., 2004, 126, 11030–11039 CrossRef CAS PubMed; (e) H. Sugimoto, H. Ohtsuka and S. Inoue, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4172–4186 CrossRef CAS; (f) T. A. Zevaco, A. Janssen, J. Sypien and E. Dinjus, Green Chem., 2005, 7, 659–666 RSC; (g) J. Melendez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323–3326 CrossRef CAS; (h) T. A. Zevaco, J. K. Sypien, A. Janssen, O. Walter and E. Dinjus, J. Organomet. Chem., 2007, 692, 1963–1973 CrossRef CAS PubMed; (i) M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946–2948 CrossRef CAS PubMed; (j) W. Clegg, R. W. Harrington, M. North and R. Pasquale, Chem. – Eur. J., 2010, 16, 6828–6843 CrossRef CAS PubMed; (k) C. Chatterjee and M. H. Chisholm, Inorg. Chem., 2011, 50, 4481–4492 CrossRef CAS PubMed; (l) K. Nishioka, H. Goto and H. Sugimoto, Macromolecules, 2012, 45, 8172–8192 CrossRef CAS; (m) M. North, P. Villuendas and C. Young, Tetrahedron Lett., 2012, 53, 2736–2740 CrossRef CAS PubMed; (n) D. Tian, B. Liu, Q. Gan, H. Li and D. J. Darensbourg, ACS Catal., 2012, 2, 2029–2035 CrossRef CAS; (o) M. A. Fuchs, C. Altesleben, T. A. Zevaco and E. Dinjus, Eur. J. Inorg. Chem., 2013, 2013, 4541–4545 CrossRef CAS; (p) N. Ikpo, J. C. Flogeras and F. M. Kerton, Dalton Trans., 2013, 42, 8998–9006 RSC; (q) C. J. Whiteoak, N. Kielland, V. Laserna, E. C. Escudero-Adan, E. Martin and A. W. Kleij, J. Am. Chem. Soc., 2013, 135, 1228–1231 CrossRef CAS PubMed; (r) J. A. Castro-Osma, C. Alonso-Moreno, A. Lara-Sánchez, J. Martínez, M. North and A. Otero, Catal. Sci. Technol., 2014, 4, 1674 RSC; (s) S. H. Kim, D. Ahn, M. J. Go, M. H. Park, M. Kim, J. Lee and Y. Kim, Organometallics, 2014, 33, 2770–2775 CrossRef CAS; (t) X. Sheng, Y. Wang, Y. Qin, X. Wang and F. Wang, RSC Adv., 2014, 4, 54043–54050 RSC; (u) C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gomez, E. Martin, E. C. Escudero-Adan, C. Bo and A. W. Kleij, Chem. – Eur. J., 2014, 20, 2264–2275 CrossRef CAS PubMed.
- (a) M. Akatsuka, T. Aida and S. Inoue, Macromolecules, 1994, 27, 2820–2825 CrossRef CAS; (b) M. H. Chisholm and D. Navarro-Llobet, Macromolecules, 2002, 35, 2389–2392 CrossRef CAS; (c) W. Braune and J. Okuda, Angew. Chem., Int. Ed., 2003, 42, 64–68 CrossRef CAS; (d) L. Tang, E. P. Wasserman, D. R. Neithamer, R. D. Krystosek, Y. Cheng, P. C. Price, Y. He and T. J. Emge, Macromolecules, 2008, 41, 7306–7315 CrossRef CAS; (e) J.-T. Issenhuth, J. Pluvinage, R. Welter, S. Bellemin-Laponnaz and S. Dagorne, Eur. J. Inorg. Chem., 2009, 2009, 4701–4709 CrossRef; (f) S. Dagorne, M. Bouyahyi, J. Vergnaud and J.-F. Carpentier, Organometallics, 2010, 29, 1865–1868 CrossRef CAS; (g) N. Nakata, Y. Saito and A. Ishii, Organometallics, 2014, 33, 1840–1844 CrossRef CAS.
- G. Martínez, S. Pedrosa, V. Tabernero, M. E. G. Mosquera and T. Cuenca, Organometallics, 2008, 27, 2300–2305 CrossRef.
- E. H. Nejad, C. G. W. van Melis, T. J. Vermeer, C. E. Koning and R. Duchateau, Macromolecules, 2012, 45, 1770–1776 CrossRef CAS.
- M. H. Chisholm, D. Navarro-Llobet and W. J. Simonsick Jr., Macromolecules, 2001, 34, 8851–8857 CrossRef CAS.
- (a) A.-L. Brocas, C. Mantzaridis, D. Tunc and S. Carlotti, Prog. Polym. Sci., 2013, 38, 845–873 CrossRef CAS PubMed; (b) M. I. Childers, J. M. Longo, N. J. Van Zee, A. M. LaPointe and G. W. Coates, Chem. Rev., 2014, 114, 8129–8152 CrossRef CAS PubMed.
- N. Ikpo, S. M. Barbon, M. W. Drover, L. N. Dawe and F. M. Kerton, Organometallics, 2012, 31, 8145–8158 CrossRef CAS.
- D. J. Darensbourg, P. Ganguly and D. Billodeaux, Macromolecules, 2005, 38, 5406–5410 CrossRef CAS.
- (a) C.-T. Chen, C.-A. Huang and B.-H. Huang, Dalton Trans., 2003, 3799–3803 RSC; (b) Z. Tang and V. C. Gibson, Eur. Polym. J., 2006, 43, 150–155 CrossRef PubMed; (c) E. D. Cross, G. K. Tennekone, A. Decken and M. P. Shaver, Green Mater., 2013, 1, 79–86 CrossRef CAS.
- D. J. Darensbourg and W.-C. Chung, Polyhedron, 2013, 58, 139–143 CrossRef CAS PubMed.
- S. Pappuru, E. R. Chokkapu, D. Chakraborty and V. Ramkumar, Dalton Trans., 2013, 42, 16412–16427 RSC.
- D. Cui, M. Nishiura and Z. Hou, Macromolecules, 2005, 38, 4089–4095 CrossRef CAS.
- Y. Sarazin, M. Schormann and M. Bochmann, Organometallics, 2004, 23, 3296–3302 CrossRef CAS.
- E. Ertürk, M. A. Tezeren, T. Tilki, T. Erdogan and A. C. Gören, Polym. Int., 2012, 61, 795–799 CrossRef.
- (a) R. J. Baker and A. Walshe, Chem. Commun., 2012, 48, 985–987 RSC; (b) J. Fang, A. Walshe, L. Maron and R. J. Baker, Inorg. Chem., 2012, 51, 9132–9140 CrossRef CAS PubMed.
- Z. Liu, M. Torrent and K. Morokuma, Organometallics, 2002, 21, 1056–1071 CrossRef CAS.
- M. W. Lehenmeier, C. Bruckmeier, S. Klaus, J. E. Dengler, P. Deglmann, A.-K. Ott and B. Rieger, Chem. – Eur. J., 2011, 17, 8858 CrossRef CAS PubMed.
- F. Andruzzi, L. Shaofeng, G. Pilcher and F. Heatley, Makromol. Chem., 1987, 188, 2643–2650 CrossRef CAS.
- B. Y. Liu, H. Q. Li, C. S. Ha, I. Kim and W. D. Yan, Macromol. Res., 2008, 16, 441–445 CrossRef CAS.
Footnotes |
| † For themed issue on Earth Abundant Element Compounds in Homogeneous Catalysis. |
| ‡ Electronic supplementary information (ESI) available: Experimental details, MALDI-TOF mass spectrum and NMR spectra of PCHO, conversion vs. time plot, table of attempted ROP of other epoxides. See DOI: 10.1039/c5dt00220f |
| This journal is © The Royal Society of Chemistry 2015 |