
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparing a series of 8-quinolinolato complexes of aluminium, titanium and zinc as initiators for the ring-opening polymerization of rac-lactide†
Clare
Bakewell
a,
Giovanna
Fateh-Iravani
a,
Daniel W.
Beh
a,
Dominic
Myers
a,
Sittichoke
Tabthong
b,
Pimpa
Hormnirun
b,
Andrew J. P.
White
a,
Nicholas
Long
*a and
Charlotte K.
Williams
*a
aDepartment of Chemistry, Imperial College London, London SW7 2AZ, UK. E-mail: c.k.williams@imperial.ac.uk; n.long@imperial.ac.uk
bDepartment of Chemistry, Faculty of Science, Kasetsart University, Bangkok 10900, Thailand
First published on 6th March 2015
Abstract
The preparation and characterization of a series of 8-hydroxyquinoline ligands and their complexes with Ti(IV), Al(III) and Zn(II) centres is presented. The complexes are characterized using NMR spectroscopy, elemental analysis and, in some cases, by single crystal X-ray diffraction experiments. The complexes are compared as initiators for the ring-opening polymerization of racemic-lactide; all the complexes show moderate/good rates and high levels of polymerization control. In the case of the titanium or aluminium complexes, moderate iso-selectivity is observed (Pi = 0.75), whereas in the case of the zinc complexes, moderate hetero-selectivity is observed (Ps = 0.70).
Introduction
Lewis acidic metal alkoxide/amide complexes have become popular choices as the initiators in the ring-opening polymerization of lactones.1 This is relevant because ROP can be used to prepare bio-derived and/or bio-compatible polyesters, such as polylactide which are proposed as sustainable alternatives to common petrochemicals.2 Metal catalysed, or more precisely initiated, polymerizations are proposed to occur by a coordination-insertion mechanism whereby the Lewis acidic metal centre coordinates the lactide, activating it to attack by a metal bound alkoxide group. This attack leads to ring-opening and generation of a new metal alkoxide species. The selection of the initiator is important as it affects features such as the polymerization rate, the degree of polymerization control (end-groups/molecular weight, dispersity, facility to form block copolymers) and the stereocontrol. Initiators which are able to efficiently produce PLA, with stereocontrol are of interest as the different tacticities of PLA result in different performances, in particular in different thermal and mechanical properties.1a–d Well-defined, i.e. ligated, metal complexes are frequently targeted as catalysts; they are particularly attractive as the ligand–metal interactions moderate and control the catalysis. The application of earth-abundant metal centres is especially desirable as a means to reduce the cost and improve sustainability of the initiator selection. There is already a strong track record for use of some of the most earth-abundant metal centres including successful initiators of Al(III),3 Fe(III),4 Ca(II),5 Mg(II),5b,d,6 Na(I),7 K(I)7a,8 and Ti(IV).9 Despite these successes there is still a strong drive for new initiators particularly those able to exert high degrees of polymerization control, especially stereocontrol.Results and discussion
Our approach was to prepare complexes of earth abundant elements using a series of easily synthesised and moderated ancillary ligands. The use of 8-hydroxyquinoline ligands is attractive as they are either commercially available or easily synthesised and, from the point of view of catalysis, offer a large range of different sites for substitution, most notably at positions R1–3, which enable modifications of the steric and electronic features of the complexes.10 Some of us have previously reported Group 13 complexes of several 8-hydroxyquinoline ligands; these complexes are slow but iso-selective initiators in the polymerization of rac-lactide.10b It was discovered that modifications to the R1 and R3 substituents led to increased iso-selectivity and polymerization activity, respectively.10b Further, the (8-quinolinolato)gallium analogues were significantly faster initiators highlighting the importance of the metal centre in moderating catalysis.10a,b It was, therefore, of interest to explore a wider range of 8-hydroxyquinoline complexes, using Al(III), Ti(IV) and Zn(II), to explore the effects on polymerization catalysis.Pro-ligand syntheses
A series of 8-hydroxyquinoline pro-ligands were selected for the study; their structures are illustrated in Fig. 1. Pro-ligands A–D are either commercially available or were prepared by previously described literature procedures and all have methyl substituents at position R3 and a range of different halides/H at positions R1 and R2.10b Pro-ligand E was also prepared by a modified literature route (see ESI†) and differs from ligand D by having a larger phenyl substituent at position R3.11 Compounds F and G have phenyl and ethynyl ferrocene substituents at position R1, with the other substituents being the same as ligand A. They were targeted to investigate the influence of aromatic substituents at the position ortho- to the phenolate moiety. Compounds F and G were prepared from the mixed halide pro-ligand B, via sequences of protection of the phenol group; followed by cross-coupling reactions with the iodo-substituent (R1) using Suzuki (for F) or Sonagashira (for G) methods; followed by deprotection of the phenol which enabled isolations in good overall yields (57% for F and 68% for G). The new pro-ligands E–G were fully characterised by NMR spectroscopy, mass spectrometry and the stoichiometry was confirmed by elemental analysis. Further details of the ligand syntheses are available in the ESI (Schemes S1 and 2†).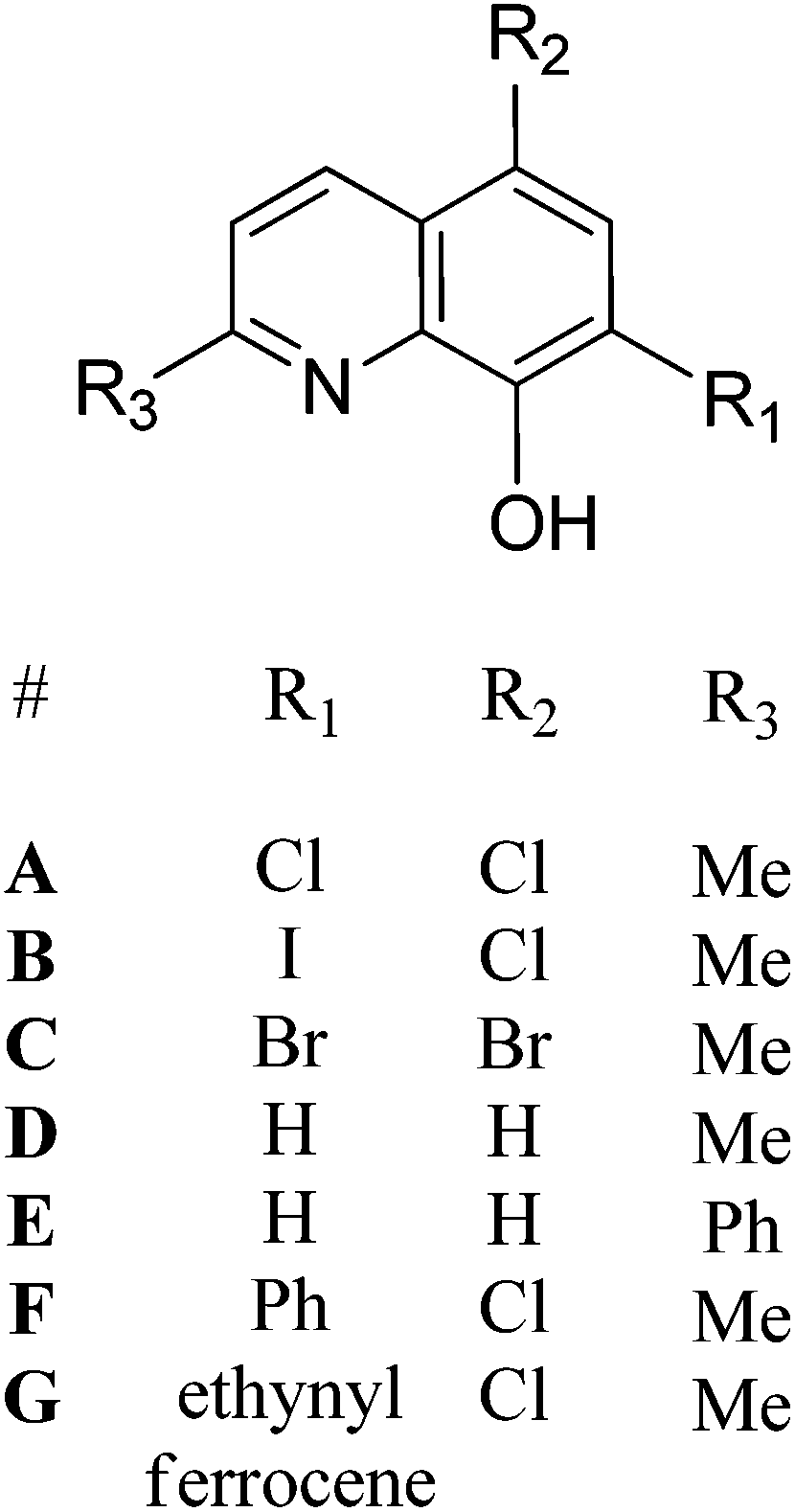 | ||
| Fig. 1 The structures of a series of 8-hydroxyquinoline compounds A–G. | ||
Complex syntheses
 | ||
| Fig. 2 General synthesis of initiators Al-E, F and G, numbering scheme included. Reagents and conditions: i. AlEt3, toluene, 298 K, 12 h, Al-E (71%), Al-F (34%), Al-G (63%). | ||
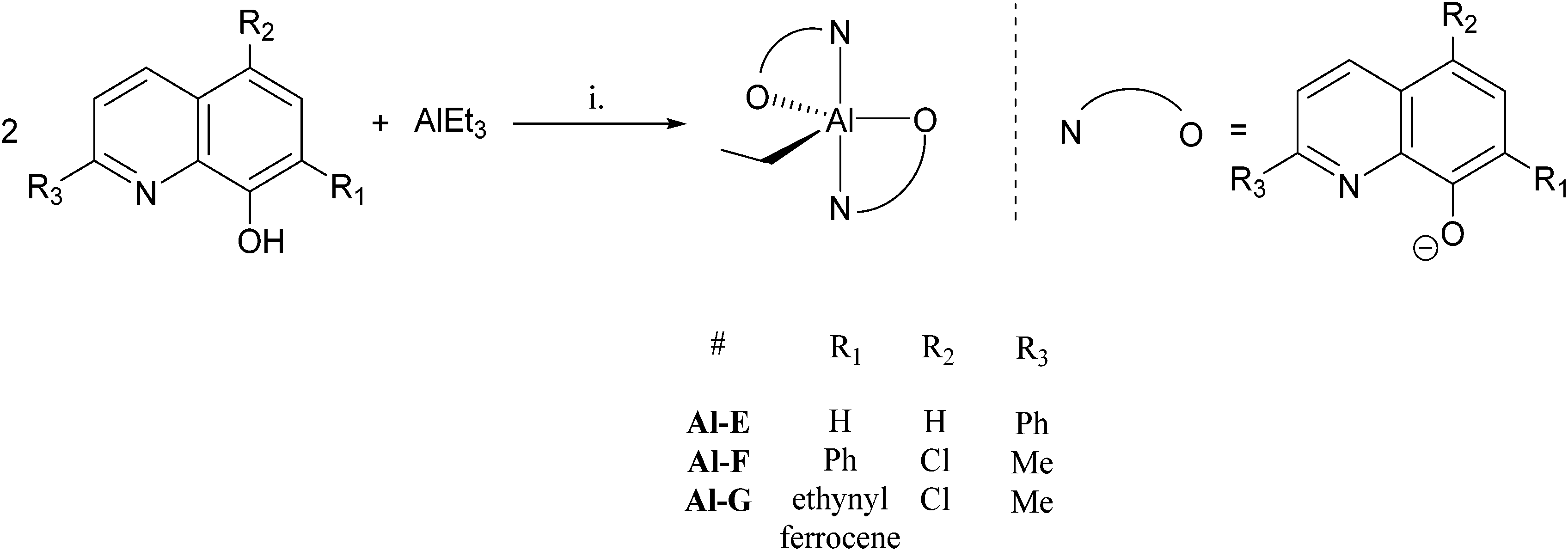
 | ||
| Fig. 3 General synthesis of initiators Ti-B, D and G, numbering scheme included. Reagents and conditions: i. Ti(OiPr)4, toluene, 298 K, 12 h, Ti-B (70%), Ti-D (43%), Ti-G (64%). | ||
The new compounds were characterised by NMR spectroscopy and the purity was confirmed by elemental analysis. The characteristic peaks of the iso-propoxy alkoxide groups resonate as a septet (4.8–5.2 ppm) and a doublet of doublets (1.2–1.4 ppm), and integrate at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with the quinolinate peaks confirming the proposed complex stoichiometry. Although X-ray crystal structures of the new titanium(IV) complexes were not obtained, the complexes are proposed to adopt distorted octahedral geometries with the N atoms, of the quinolinate ligands, being in a cis-disposition to one another and the O-atoms being in trans-positions (see Fig. 3). Such a geometry is different to that observed for the pentacoordinate Al(III) complexes, but is in line with other X-ray crystal structures reported for closely related bis(8-quinolinolato) bis(iso-propoxide) complexes of titanium(IV).12
:1 ratio with the quinolinate peaks confirming the proposed complex stoichiometry. Although X-ray crystal structures of the new titanium(IV) complexes were not obtained, the complexes are proposed to adopt distorted octahedral geometries with the N atoms, of the quinolinate ligands, being in a cis-disposition to one another and the O-atoms being in trans-positions (see Fig. 3). Such a geometry is different to that observed for the pentacoordinate Al(III) complexes, but is in line with other X-ray crystal structures reported for closely related bis(8-quinolinolato) bis(iso-propoxide) complexes of titanium(IV).12
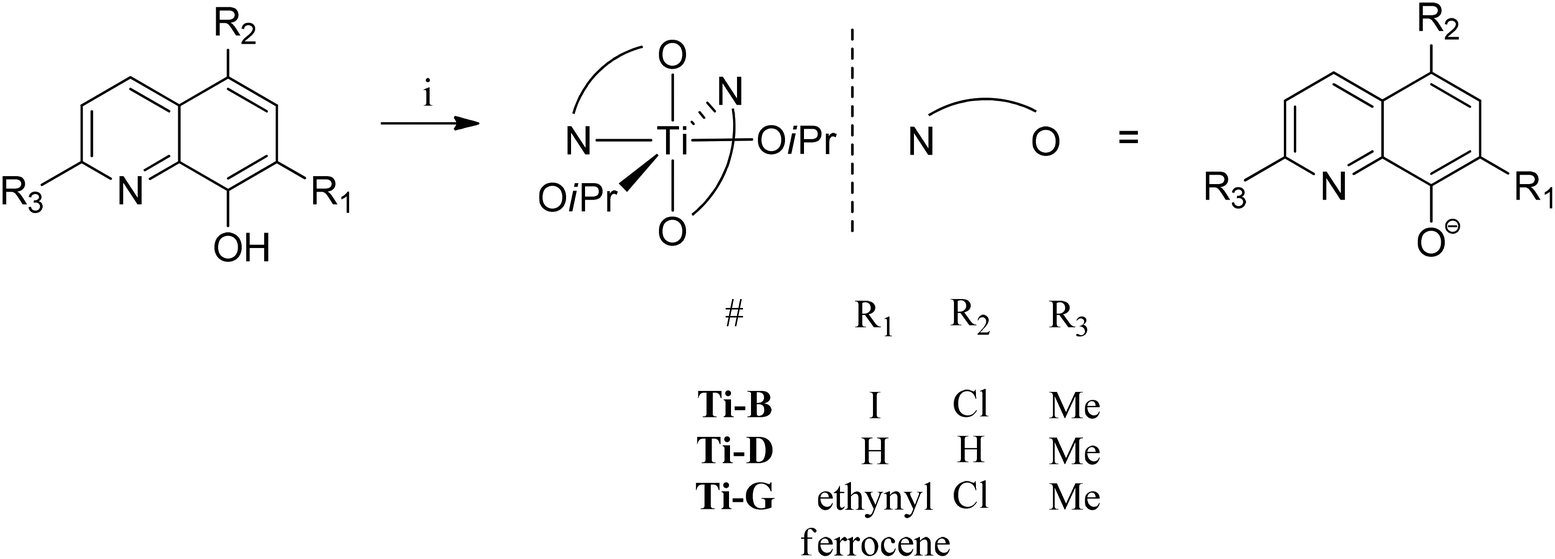
 | ||
| Fig. 4 General synthesis of initiators Zn-A, B, C, E and G, numbering scheme included. Reagents and conditions: i. ZnEt2, toluene, 298 K, 12 h, Zn-A (78%), Zn-B (70%), Zn-C (56%), Zn-D (67%). Zn-E (70%), Zn-G (60%). | ||
Single crystal X-ray diffraction experiments revealed that compound Zn-A exists as a dimer in the solid state, vide infra. On the basis of this finding, it is tentatively assumed that other complexes with sterically hindered substitutents at sites R1 and R2 are also dimeric in the solid state, i.e. complexes Zn-(A–C) and Zn-G. Consistent with this proposal is the finding that these complexes (Zn-(A–C), Zn-G) all showed well-defined 1H NMR spectra, when dissolved in THF-d8. In contrast, the 1H NMR spectra of compounds Zn-D and Zn-E (where R1 = R2 = H), in THF-d8 at 298 K, are broad and undefined, thus indicative of higher degrees of aggregation. The use of a stronger donor solvent, pyridine-d5, resulted in well-defined 1H NMR spectra being obtained, consistent with the pyridine coordinating to the zinc centre and favouring the formation of discrete mononuclear complexes. Indeed, there is already a good literature precedent for the formation of high order clusters/aggregates for unsubstituted (8-quinolinolato)zinc(tert-butyl) complexes.13 There is also a track record for pyridine coordinating to zinc complexes and disrupting aggregate structures.14 To further confirm the structures of Zn-D, a single crystal X-ray diffraction experiment (vide infra) showed the complex exhibited a trimeric structure in the solid state.
X-ray crystallography
Single crystals, suitable for X-ray diffraction experiments, were isolated for compounds Zn-A, Zn-D and Al-E from THF–hexane and toluene solutions, respectively. The crystallizations occurred at −18 °C for Zn-D and Al-E and at 25 °C for Zn-A. Illustrations of the structures are shown in Fig. 5–8 and Table 1 presents selected bond lengths and angles (for full data see the ESI†).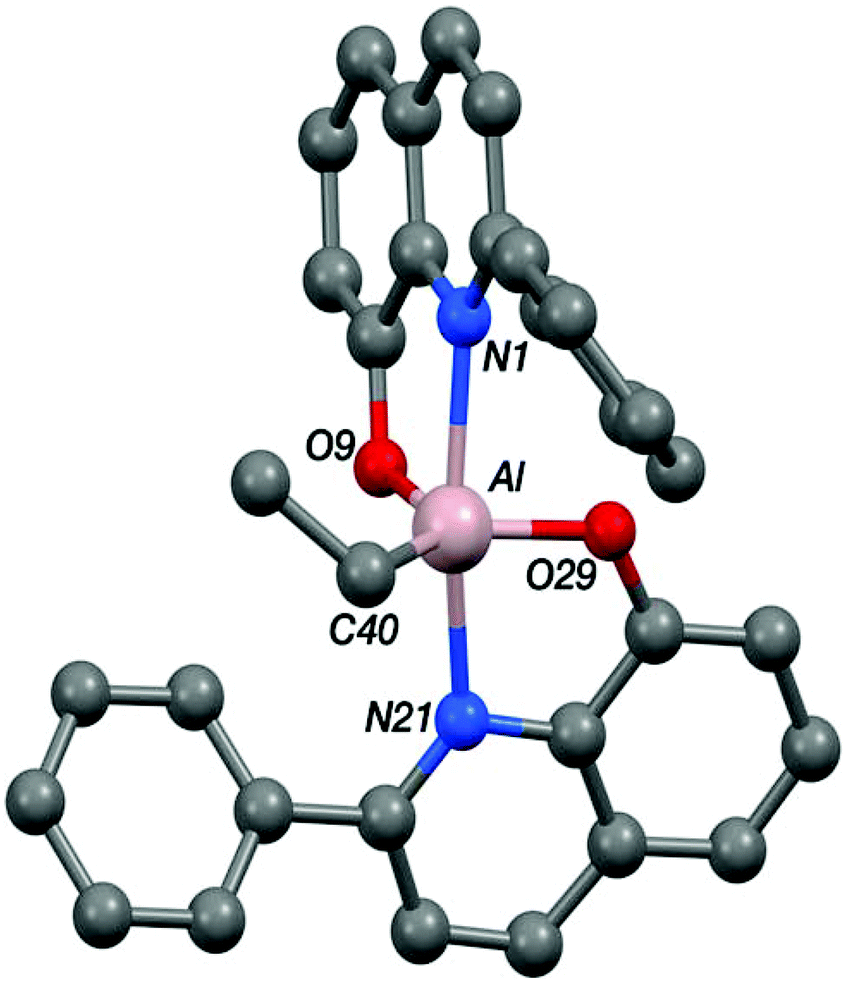 | ||
| Fig. 5 The crystal structure of Al-E. Selected bond lengths (Å) and angles (°); Al–N1 2.1892(10), Al–O9 1.7989(9), Al–N21 2.1527(10), Al–O29 1.8007(10), Al–C40 1.9746(13), N1–Al–O9 82.28(4), N1–Al–N21 165.14(4), N1–Al–O29 89.27(4), N1–Al–C40 94.77(5), O9–Al–N21 88.62(4), O9–Al–O29 109.02(5), O9–Al–C40 122.89(5), N21–Al–O29 82.59(4), N21–Al–C40 100.04(5), O29–Al–C40 128.02(5). | ||
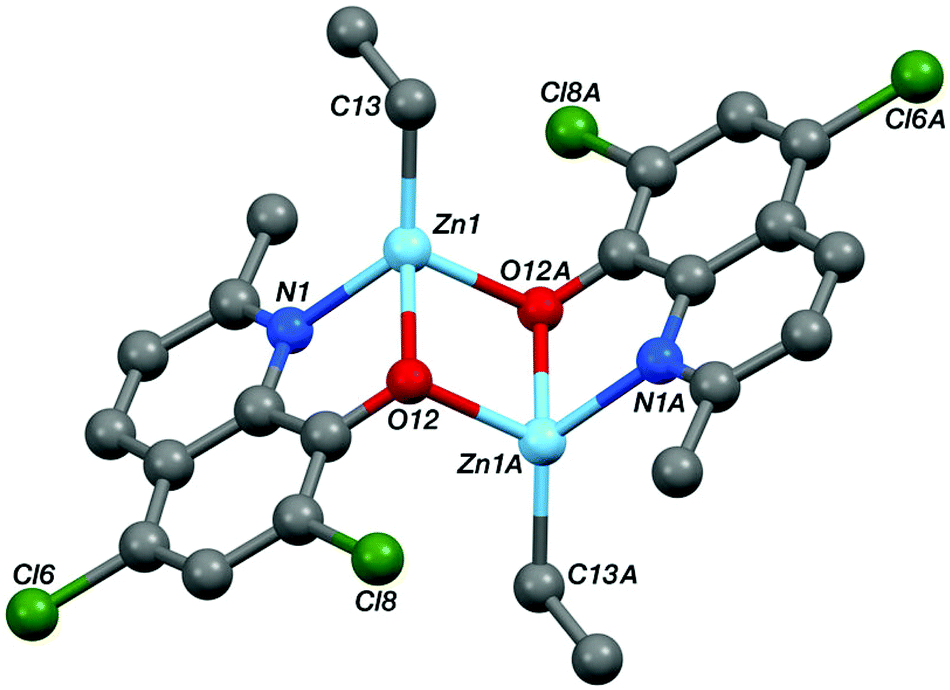 | ||
| Fig. 6 The crystal structure of the Ci-symmetric complex Zn-A. Selected bond lengths (Å) and angles (°); Zn1–N1 2.0866(17), Zn1–O12 2.0776(14), Zn1–C13 1.984(2), Zn1–O12A 2.0761(15), Zn1⋯Zn1A 3.09986, N1–Zn1–O12 80.54(6), N1–Zn1–C13 128.80(8), N1–Zn1–O12A 105.43(6), O12–Zn1–C13 129.36(8), O12–Zn1–O12A 83.46(6), C13–Zn1–O12A 117.05(8). | ||
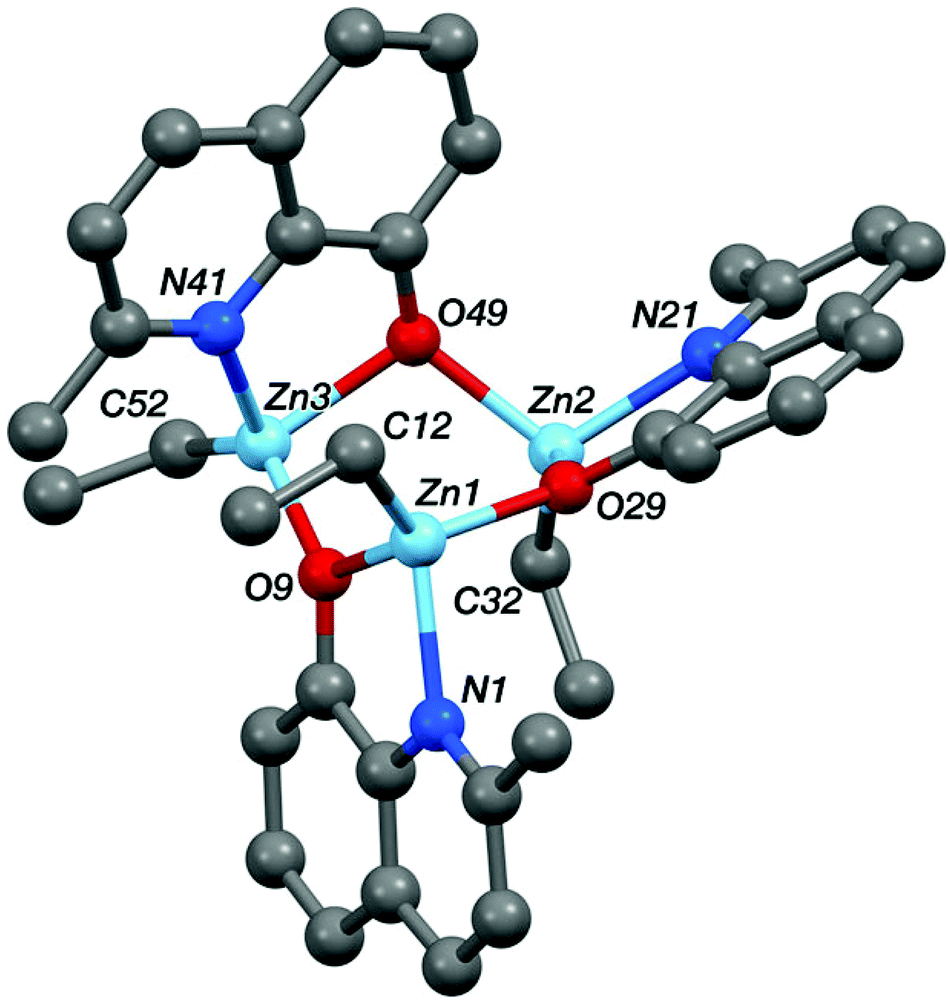 | ||
| Fig. 7 The crystal structure of Zn-D, see Table 1 for selected bond lengths and angles. | ||
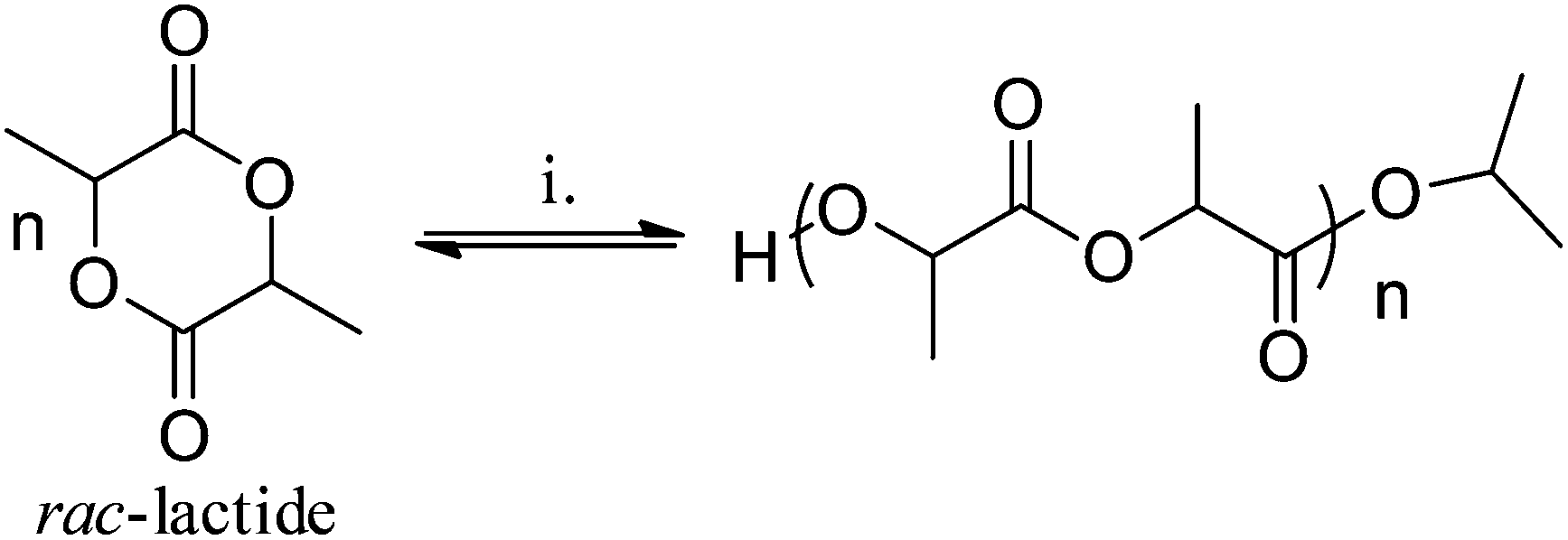 | ||
| Fig. 8 Illustrates the ring-opening polymerization of lactide to polylactide. Reagents and Conditions: Polymerization conditions: Al-(A–G): Toluene, 348 K, 1:1:100 [I]:[iPrOH]:[LA], 1 M [LA]. Ti-(B/D/G): Toluene, 348 K, 1:100 [I]:[LA], 1 M [LA]. Zn-(A–G): THF/CH2Cl2, 298 K, 1:1:100 [I]:[iPrOH]:[LA], 1 M [LA]. | ||
| Zn1–N1 | 2.083(2) | Zn2–N21 | 2.083(2) | Zn3–N41 | 2.097(2) |
| Zn1–O9 | 2.0514(17) | Zn2–O29 | 2.0487(16) | Zn3–O49 | 2.0537(16) |
| Zn1–C12 | 1.975(3) | Zn2–C32 | 1.977(3) | Zn3–C52 | 1.979(3) |
| Zn1–O29 | 2.0215(16) | Zn2–O49 | 2.0427(17) | Zn3–O9 | 2.0116(17) |
| N1–Zn1–O9 | 81.20(7) | N21–Zn2–O29 | 80.85(7) | N41–Zn3–O49 | 80.57(7) |
| N1–Zn1–C12 | 123.03(10) | N21–Zn2–C32 | 125.24(10) | N41–Zn3–C52 | 123.88(11) |
| N1–Zn1–O29 | 101.28(7) | N21–Zn2–O49 | 108.13(7) | N41–Zn3–O9 | 95.85(7) |
| O9–Zn1–C12 | 125.24(10) | O29–Zn2–C32 | 128.93(9) | O49–Zn3–C52 | 115.50(10) |
| O9–Zn1–O29 | 94.00(7) | O29–Zn2–O49 | 95.89(7) | O49–Zn3–O9 | 96.95(7) |
| C12–Zn1–O29 | 122.22(10) | C32–Zn2–O49 | 111.70(9) | C52–Zn3–O9 | 130.87(11) |
The structure of the aluminium complex Al-E shows a distorted trigonal bipyramidal coordination geometry (τ = 0.62) for the aluminium centre, with N1 and N21 occupying the axial sites (Fig. 5). Both of the C2NOZn chelate rings have envelope conformations; for the N1/O9 chelate ring the metal lies ca. 0.12 Å out of the C2NO plane (the atoms of which are coplanar to within ca. 0.01 Å), whilst for the N21/O29 case the aluminium lies ca. 0.18 Å out of the plane of the other four atoms (which are coplanar to better than 0.01 Å).
The structures of the two zinc complexes confirm the formation of aggregates in the solid state, presumably driven in part by the high stability of four coordinate, tetrahedral zinc centres. The crystal structure of Zn-A shows the complex to be a Ci-symmetric dimer with bridging phenoxide oxygen atoms (Fig. 6). The geometry at the zinc centre is noticeably distorted with the angles involving the ethyl ligand all being significantly increased from ideal, ranging between 117.05(8) and 129.36(8)°. The five-membered C2NOZn chelate ring has an envelope conformation, the zinc atom lying ca. 0.41 Å out of the plane of the other four atoms (which are coplanar to within ca. 0.01 Å). In contrast, the crystal structure of Zn-D shows a trimeric, cyclic structure based on three EtZn-D units (Fig. 7, Table 1). The Zn3O3 ring has a “two-up one-down” arrangement for the quinolinolate ligands; this ring has a twist-boat conformation with Zn3 and O49 lying ca. 1.42 and 1.85 Å, respectively, out of the [Zn1, Zn2, O9, O29] plane (which is coplanar to ca. 0.11 Å). All three zinc centres have distorted tetrahedral coordination geometries with angles in the ranges 81.20(7)–125.24(10)°, 80.85(7)–128.93(9)° and 80.57(7)–130.87(11)° at Zn1, Zn2 and Zn3 respectively; in each case the smallest angle is the bite of the N,O chelate ligand. Two of the three five-membered C2NOZn chelate rings are approximately flat (the Zn1 and Zn2 based rings are coplanar to within ca. 0.02 and 0.03 Å respectively), whilst the third has an envelope conformation with Zn3 lying ca. 0.11 Å out of the plane of the other four atoms which are coplanar to within ca. 0.01 Å.
It is notable that ligand G contains a redox-active ferrocene substituent and a number of catalysts containing such substituents have been shown to be capable of control/moderation of the polymerization properties by control of ferrocene redox chemistry.9g,h,15 Thus, it was relevant to investigate the redox chemistry of compounds Al-G, Ti-G and Zn-G. Cyclic voltammetry showed all three compounds to have reversible redox behaviour, however chemical oxidation proved problematic. Ethyl compounds Al-G and Zn-G showed evidence of alkyl abstraction using a range of different chemical oxidants including Ag+OTf−, Ag+BF4−, NO+BF4−, Fc+PF6− and Fc+BArF−, as signalled by the absence of the characteristic ethyl group signals in the 1H NMR spectra. Attempts to chemically oxidise compound Ti-G, a titanium bis(iso-propoxide) species, also failed to result in a paramagnetic Fe(III) species and showed poor stability of any chemically oxidised product formed. As such, compounds Al-G, Ti-G and Zn-G cannot be redox controlled, rather they are included in this study as initiators containing aromatic/sterically hindered substituents at position R1.
Ring-opening polymerization of rac-lactide
All the new compounds (Al, Zn and Ti) were tested as initiators for the ROP of rac-LA and for ease of comparison, the data for Al-(A–D) is also included (Fig. 8, Table 2).| Initiator (I) | Time (h) | Convsn.d (%) |
k
obs × 10−6 s−1e |
M
nf (g mol−1) |
M n, calc | PDIf |
P
ig |
|---|---|---|---|---|---|---|---|
|
a The results are reproduced from ref. 10b to enable comparisons between the initiators.
b Polymerization conditions: Toluene, 348 K, 1:1:100 [I]:[iPrOH]:[LA], 1 M [LA].
c Toluene, 348 K, 1:100 [I]:[LA], 1 M [LA].
d Determined by integration of the methine region of the 1H NMR spectrum (LA 4.98–5.04 ppm; PLA 5.08–5.22 ppm).
e Determined from the gradients of the plots of ln{[LA]0/[LA]t} versus time.
f Determined by GPC-SEC in THF, using a correction factor of 0.58.3a
g Determined by analysis of all the tetrad signals in the methine region of the homonuclear decoupled 1H NMR spectrum.
|
|||||||
| Al-A | 137 | 91 | 5.0 | 9900 | 13100 |
1.11 | 0.72 |
| Al-B | 169 | 80 | 2.5 | 7000 | 11500 |
1.04 | 0.75 |
| Al-C | 168 | 90 | 4.2 | 12400 |
12900 |
1.07 | 0.75 |
| Al-D | 169 | 94 | 4.3 | 9300 | 13500 |
1.19 | 0.62 |
| Al-E | 28 | 91 | 24 | 9700 | 13100 |
1.11 | 0.56 |
| Al-F | 165 | 93 | 4.3 | 10500 |
13400 |
1.13 | 0.75 |
| Al-G | 288 | 88 | 2.2 | 12700 |
12700 |
1.05 | 0.66 |
| Ti-B | 186 | 91 | 3.7 | 5400 | 6550 | 1.14 | 0.65 |
| Ti-D | 258 | 82 | 2.0 | 5600 | 5900 | 1.06 | 0.61 |
| Ti-G | 308 | 85 | 1.7 | 6600 | 6100 | 1.08 | 0.63 |
The polymerizations were conducted under a standard set of conditions; in toluene at 348 K for the aluminium and titanium complexes (note: an equivalent of iso-propyl alcohol was added to polymerizations using aluminium ethyl initiators) or in THF–methylene dichloride, at 298 K, with one equivalent of iso-propyl alcohol for the zinc initiators. All experiments were conducted at a standard concentration of rac-lactide (1 M) and using 10 mM concentration of initiator (i.e. 1:100 loading of initiator:lactide). In the case of the ethyl based initiators, i.e. all the Al and Zn complexes, an equivalent of iso-propyl alcohol was added. This alcohol reacts with the metal ethyl bond, in situ, forming an active metal iso-propoxide initiator. The polymerizations are all air and moisture sensitive and so were carried out in a nitrogen filled glovebox or on an argon Schlenk line. The polymerizations were monitored by taking aliquots at regular time intervals. The crude samples (aliquots) were then analysed using 1H NMR spectroscopy to determine the percentage monomer conversion. Size exclusion chromatography was used to determine the number-averaged molecular weight (Mn) and dispersity (PDI) for all samples. The tacticity of the resulting PLA was assessed by integration of the methyne region of the homonuclear decoupled NMR spectrum. The normalized tetrad integrals were compared with the expected probabilities determined by Bernoullian statistics.16
All the complexes were active initiators in the polymerization of rac-LA, the polymerization results are summarised in Tables 2 and 3.
| Ia | Solv. | Time (min) | Conv.b (%) |
k
obs × 10−4 s−1c |
M n, theo. |
M
n, NMRd (g mol−1) |
M
n, LSe (g mol−1) |
M
n, SECf (g mol−1) |
PDIe,f |
P
sg |
|---|---|---|---|---|---|---|---|---|---|---|
|
a Polymerization conditions: 298 K, 1:1:100 [I]:[iPrOH]:[LA], 1 M [LA].
b Determined by integration of the methine region of the 1H NMR spectrum (LA 4.98–5.04 ppm; PLA 5.08–5.22 ppm).
c Determined from the gradients of the plots of ln{[LA]0/[LA]t} versus time.
d Determined by integration of the hydroxyl chain-end versus the polymer methine protons.
e Determined by GPC in THF, using multiangle laser light scattering (GPC-MALLS).
f Determined by GPC in THF versus polystyrene standard and a correction factor on 0.58.
g Determined by analysis of all the tetrad signals in the methine region of the homonuclear decoupled 1H NMR spectrum.
|
||||||||||
| Zn-A | THF | 445 | 91 | 1.4 | 13100 |
12600 |
11000 |
7400 | 1.10 | 0.66 |
| Zn-A | DCM | 260 | 90 | 1.7 | 13000 |
8500 | 8900 | 5600 | 1.03 | 0.60 |
| Zn-B | THF | 240 | 90 | 2.1 | 13000 |
8750 | 8700 | — | 1.06 | 0.66 |
| Zn-B | DCM | 200 | 92 | 2.4 | 13250 |
8750 | 9000 | — | 1.03 | 0.60 |
| Zn-C | THF | 370 | 93 | 1.6 | 13400 |
8000 | 9400 | 6200 | 1.03 | 0.66 |
| Zn-C | DCM | 290 | 95 | 1.8 | 13700 |
9900 | 9100 | 6400 | 1.08 | 0.60 |
| Zn-D | THF | 560 | 85 | 0.8 | 12300 |
— | 8000 | 7000 | 1.07 | 0.67 |
| Zn-E | THF | 460 | 89 | 1.0 | 12800 |
6600 | 7500 | 5700 | 1.10 | 0.70 |
| Zn-G | THF | 480 | 93 | 0.9 | 13400 |
6700 | 10000 |
— | 1.09 | 0.65 |
The Al and Ti initiators showed similar performances, exhibiting slow rates compared to the very best catalysts for lactide polymerization but at values as expected for these metal centres. The polymerization kinetics were monitored for Al and Ti initiators, showing first order dependencies on lactide concentration in all cases; the pseudo first order rate constants, kobs, were obtained as the gradient of the linear fits to plots of ln([LA]0/[LA]t) versus time (Fig. 9, 10 and ref. 10b for the data for Al(A–D)). Of the Al compounds, Al-E (R3 = Ph) stands out as having a significantly faster rate (kobs = 24 × 10−6 s−1), it is also notable that a similarly higher rate was observed when R3 = tBu as previously reported by us (kobs = 58 × 10−6 s−1).10b It seems that the substitution at R3 position exerts more of an electronic influence on the aluminium centre, than substitution at sites R1 or R2. The other initiators Al-F and Al-G have comparable rates to the previously reported Al-(A–D).10b It is notable that compound Al-F has a short lag period at the start of the polymerization, likely owing to a relatively slow formation of the active aluminium alkoxide initiating species.
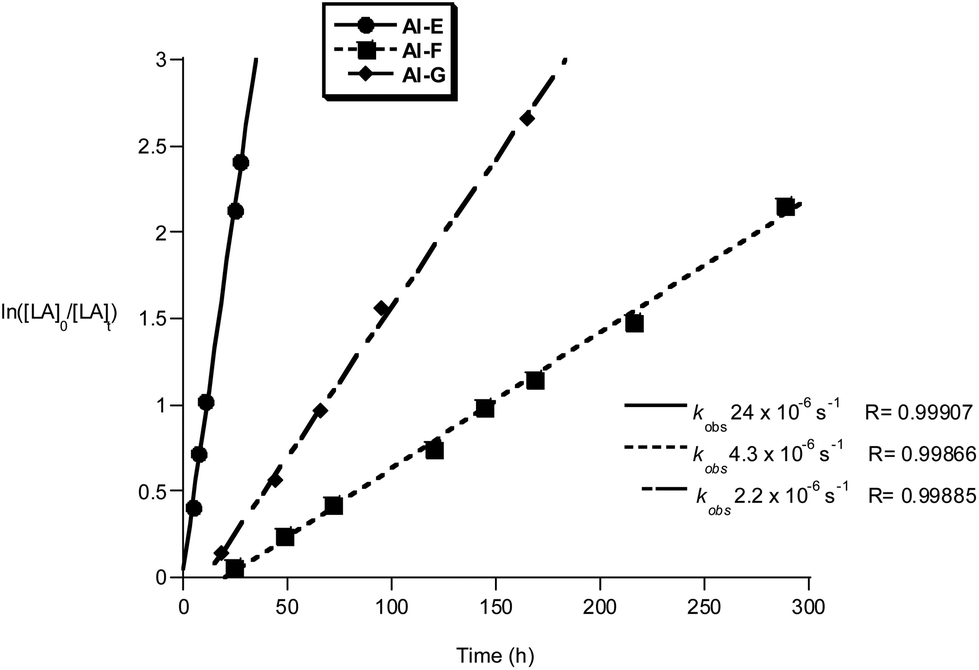 | ||
| Fig. 9 Plot of ln([LA]0/[LA]t) vs. time of initiator Al-E, F and G. Conditions: [LA]0 = 1 M, 1:1:100 [I]:[iPrOH]:[LA], toluene, 348 K. | ||
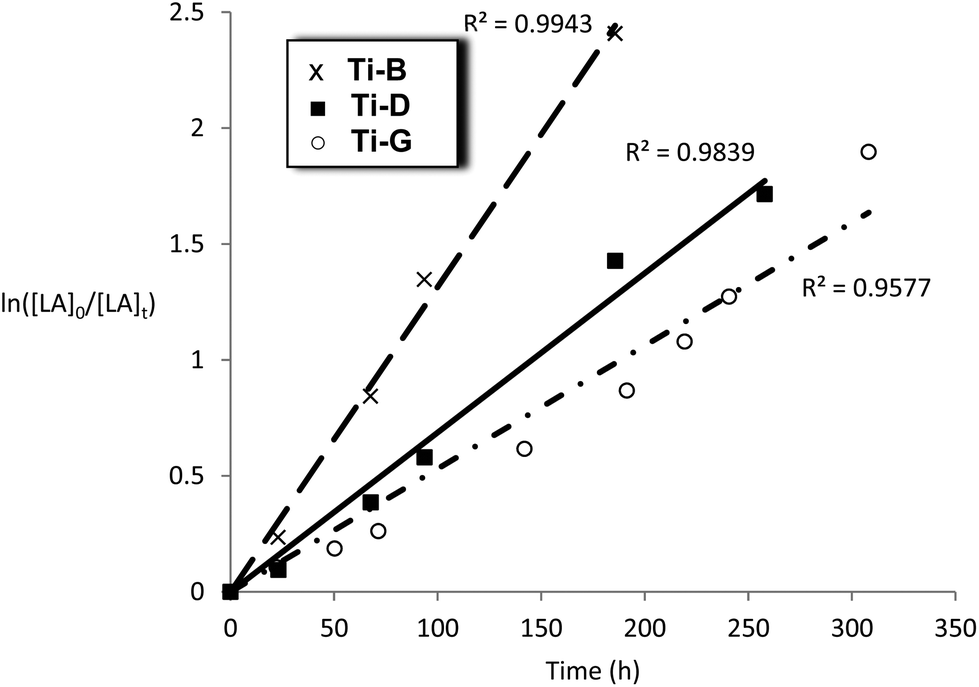 | ||
| Fig. 10 Plot of ln([LA]0/[LA]t) vs. time for initiators Ti-B, D and G Conditions: [LA]0 = 1 M, 1:100 [I]:[LA], toluene, 348 K. | ||
The titanium complexes, Ti-B, D and G were all slow initiators with comparable observed rate constants to the Al initiators (kobs = 1.7–3.7 × 10−6 s−1). It is interesting to note that the rate of polymerisation of Ti-B was faster than Ti-D, the opposite trend to that observed for the aluminium complexes bearing the same ligands. The slower rate of polymerization of compounds Ti-B and Ti-G, versusTi-D (where R1 = R2 = H), could have a steric origin note: limited electronic trends with variation of R1 and R2 could be identified in the series of related aluminium complexes. The data for the kinetics of Ti-G show a slight deviation from linearity (R2 = 0.9577) which is proposed to be due to the relatively long polymerization period and/or slower initiation.
Compounds Al-E, F and G exhibited a high degree of polymerization control, with all initiators showing a linear evolution of molecular weight with percentage conversion, Mn values being close to those predicted on the basis of the initiator concentration and dispersities are narrow throughout the course of the polymerizations (<1.11 in all cases). This level of polymerization control is comparable to the previously reported bis(8-quinolinolato)aluminium ethyl compounds and the bis(8-quinolinolato)gallium tert-butoxide compounds.10a,b The polymer end-groups were analysed using MALDI-ToF mass spectrometry, which showed that the major series were chain end-capped with iso-propyl ester groups (Fig. S18†). Compounds Ti-B, D and G also show a linear evolution of number averaged molecular weight (Mn) and narrow dispersities. The Mn values were consistent with two polymer chains growing from the two alkoxide initiating groups on the titanium catalysts (Table 2). This is rather different to the aluminium catalysts where a single polymer chain grows (for the single alkyl site). Thus, although equivalent rates are exhibited per equivalent of metal, the rate of the aluminium per active site is likely faster (approximately twice as fast).
The Al and Ti initiators all exert an isotactic bias during the polymerization of rac-LA. It is observed that the initiators with the most sterically hindered substituents at the R1 position result in higher degrees of iso-selectivity, i.e.Al-A, Al-B and Al-F. For the new initiators the best iso-selectivity is observed for Al-F (Pi = 0.75), with a phenyl substituent at R1. Attempts to prepare related ligands with more sterically hindered substituents at R1 were unsuccessful due to problems with ligand synthesis/purification. The analysis of the isotactic PLA produced by Al-F indicates that an enantiomorphic site control mechanism is dominant, with the relative integrals of stereoerror signals being: [sis]:[sii]:[iis]:[isi] = 1:1:1:2 (Fig. S19†). Compared to the Al analogues, the Ti complexes show lower iso-selectivities, with the maximum Pi = 0.65 for Ti-B (Fig. S20†). It should be noted that any degree of iso-selectivity for titanium initiators is rather unusual and this value may represent an interesting opportunity to prepare more selective titanium initiators in the future.17 In contrast, there have been several previous examples of hetero-selective titanium initiators and in such cases, the selectivity has been improved using heavier Group (IV) complexes, i.e. of Zr(IV) or Hf(IV).9d–f,i,j,18
Polymerizations were also conducted using the Zn initiators, either in methylene dichloride or THF solutions at 298 K (Table 3).
The polymerization kinetics were monitored for each initiator and show a first order dependence on lactide concentration in every case. The pseudo first-order rate constants, kobs, were determined when the polymerization was conducted in either THF (Fig. 11) or methylene dichloride (Fig. 12). In contrast to the Al initiators which require thermal activation, the Zn initiators are all active at 298 K. It may be that for the Al initiators, the higher temperatures are required to accelerate the formation of the active aluminium alkoxide species, whereas for the zinc initiators the reaction between alcohol and zinc–ethyl occurs without heating. Such a proposal is supported by the reduced bond dissociation energy of zinc–carbon bonds, Zn–C2H5 201 kJ mol−1, compared to aluminium–carbon bonds, Al–C 255 kJ mol−1.19
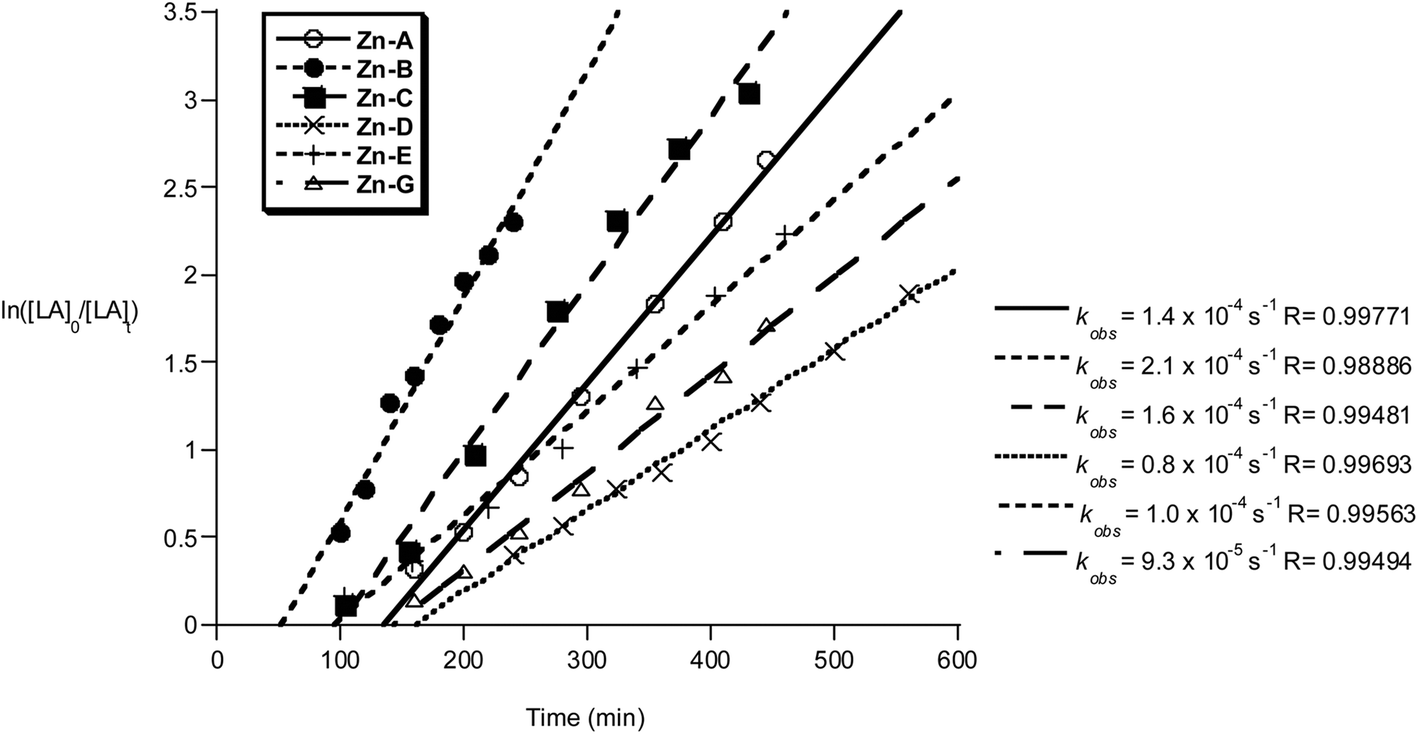 | ||
| Fig. 11 Plot of ln([LA]0/[LA]t) vs. time of initiator Zn-A–E and G. Conditions: [LA]0 = 1 M, 1:1:100 [I]:[iPrOH]:[LA], THF, 298 K. | ||
 | ||
| Fig. 12 Plot of ln([LA]0/[LA]t) vs. time of initiators Zn-A, B and C. Conditions: [LA]0 = 1 M, 1:1:100 [I]:[iPrOH]:[LA], CH2Cl2, 298 K. | ||
The polymerizations in THF exhibited a significant induction period (∼1–2 hours), after which the polymerizations progressed with good control and showed pseudo first-order kinetics. Interestingly, when methylene dichloride was employed as a solvent, the induction period was not observed. It is proposed that during the induction period, the active alkoxide initiator is forming and that THF coordination may slow the rate of Zn–C alcoholysis. During the propagation phases the rates of polymerization are not significantly influenced by the reaction solvent, with comparable values for kobs being obtained (Zn-A: kobs = 1.4 × 10−4 s−1 in THF and kobs = 1.7 × 10−4 s−1 in methylene chloride). The polymerization activity of compounds Zn-D, Zn-E and Zn-G in methylene dichloride were not monitored, due to the reduced solubility of the initiator in that solvent.
The zinc compounds were moderately fast initiators, reacting in the order Zn-B > Zn-C > Zn-A > Zn-E > Zn-G > Zn-D. Compounds Zn-A, Zn-B and Zn-C (where R1 = R2 = Cl, R1 = I R2 = Cl and R1 = R2 = Br, respectively) were the fastest, with a clear trend in rate with respect to the halide substituent I > Br > Cl and with the Lewis acidity of the active site. In contrast to the results using Al initiators, compound Zn-E (R3 = Ph), is slightly slower than the other Zn initiators. This suggests that different factors govern the activity of the two types of metal initiator. A comparison of the activity of these Zn initiators with other known literature systems reveals them to be of good rate (for Zn compounds). They show equivalent activity to Schiff base zinc complexes and zinc guanidinate complexes.20 They are faster than zinc ketoiminate compounds (1:100 I:[LA], 298 K, chloroform, 24 h, 100%), which also contain a quinolinolate ligand system.21 However, compared to the very best zinc catalysts, based on phenolate diamines/diimines, the activities of these Zn compounds are significantly lower.22
All the Zn initiators show a linear evolution of Mn with percentage conversion, and the dispersities are narrow throughout the polymerizations (<1.10 in all cases). The polymer molecular weights were compared using different size exclusion chromatographic methods, either as an absolute value using light scattering, or vs. polystyrene standards (with correction factors applied) or by 1H NMR analysis (by comparison of the signals for the main chain vs. the iso-propoxide end group). In all cases the values are slightly lower than expected. There is no significant initiation from the quinolinolate moieties on the ligand, as determined by 1H NMR and MALDI-ToF analysis of the PLA end-groups. The end group analysis using MALDI-ToF mass spectrometry, showed just one major series in which the chains were end-capped with iso-propyl ester groups and the peaks are separated by 144 amu, consistent with only limited inter-molecular transesterification occurring (Fig. S21†).
Compounds Zn-A–E and G polymerize rac-LA with a slight heterotactic bias, maximum Ps = 0.70 (Fig. S22†). The degree of stereocontrol does not change as the substituents at the R1 position are altered, this is in contrast to the ability to use this site to ‘tune’ the iso-selectivity of the Al initiators. Slightly increased hetero-selectivity was observed when the polymerizations are conducted in THF vs. methylene dichloride, Ps = 0.66 vs. 0.60 for both compounds Zn-A, B and C. This improvement in stereocontrol when THF is employed as a solvent has been observed for many other initiating systems.23 It has been postulated that the labile coordination of THF to the Lewis acidic metal centres facilitates the hetero-selectivity.23a,24
Conclusions
This study investigated the preparation of a series of new compounds of various 8-hydroxyquinoline ligands with earth-abundant metals, including Al(III), Ti(IV) and Zn(II). The pro-ligands contained different substituents at positions ortho- (R1) and para (R2) to the phenolate and ortho- (R3) to the N moieties. The coordination chemistry resulted in the formation of bis(8-quinolinolato)aluminium ethyl, bis(8-quinolinolato) bis(iso-propoxide) titanium(IV) and (8-quinolinolato)zinc(II) ethyl complexes which were characterized using spectroscopy, elemental analysis and, in some cases, using single crystal X-ray diffraction experiments. In the case of Al complexes, the coordination geometries were distorted trigonal bipyramidal, with the N-atoms occupying the axial sites. In the case of the zinc complexes, dimeric or higher order aggregates (trimer) were formed depending on the ligand substitution.All the new complexes were active initiators, in the cases of metal alkyl complexes in combination with exogenous alcohol, for lactide polymerization. The Al and Ti initiators showed similar rates which were typical for those particular metal centres. The Zn initiators, which operated under milder conditions, showed good rates which were significantly (qualitatively) faster than the Al/Ti analogues. In terms of the ligand substitution influences over the polymerization rates, the Al complexes showed significantly faster rates if the position ortho to the N atom, on the ligand, was substituted with a sterically hindered group. In contrast, for the Zn initiators no such effect was observable. The complexes all exerted high degrees of polymerization control, leading to PLA of predictable Mn with narrow dispersity (<1.11 in all cases). Furthermore, the Al and Ti initiators exhibited moderate iso-selectivities (Pi = 0.75) whilst the Zn initiators showed a moderate hetero-selectivities (Ps = 0.70).
This series of complexes demonstrates the potential and versatility of the 8-hydroxyquinoline ligand type; ligands which can be easily prepared and offer multiple sites for substitution. The experiments demonstrate the potential for good control, moderate rates and, in some cases, stereocontrol using this ligand class and earth-abundant metals. The influence of the metal coordination spheres over rate and stereochemistry differs from Al/Ti to Zn and this warrants further investigation in the future to help to understand the critical factors to prepare improved catalysts.
Experimental section
All reactions were conducted under an inert nitrogen atmosphere, using a nitrogen filled glovebox or standard Schlenk techniques. The pro-ligands A–D were prepared as previously described,10b whilst the experimental protocols for pro-ligands E–G are reported in the ESI.† All solvents and reagents were obtained from commercial sources. Triethyl aluminium and diethyl zinc were obtained from Strem and titanium(IV) tetrakis(iso-propoxide) was obtained from Sigma-Aldrich. Toluene and THF was distilled from sodium, de-gassed and stored under nitrogen. Methylene dichloride was distilled from CaH2. Iso-propyl alcohol was heated to reflux over CaH2, distilled onto fresh CaH2 and further refluxed, then distilled, degassed and stored under nitrogen. Benzene-d6 was distilled from sodium, THF-d8, toluene-d8 and CDCl3 were dried over CaH2, and all were then degassed and stored under nitrogen. rac-Lactide was obtained from Purac Plc. and was crystallised from dry toluene and sublimed at 323 K three times under vacuum.Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Av400 spectrometer operating at 400 MHz for 1H and 100 MHz for 13C{1H} spectra. Solvent peaks were used as internal references for 1H and 13C chemical shifts (ppm). Higher resolution 1H NMR and 1H{1H} NMR (homo-decoupled spectroscopy) experiments were performed on a Bruker Av500 spectrometer and also a DRX 400 spectrometer by Mr Peter Haycock. Spectra were processed and analyzed using Mestrenova software. MALDI-ToF mass spectra were performed on a Waters/Micromass MALDI micro MX, using potassium or sodium salts for ionization. Elemental analyses were determined by Mr Stephen Boyer at London Metropolitan University, Science Centre, 29 Hornsey Road, London N7 7DD. GPC-MALLS measurements were conducted on a Polymer Laboratories PL GPC-50 instrument at 35 °C, using two Polymer Laboratories Mixed D columns in series and THF as the eluent, at a flow rate of 1 mL min−1. The light scattering detector was a Dawn 8, Wyatt Technology, and data were analysed using Astra V version 5.3.4.18.
Aluminium complexes
Triethyl aluminium (53 mg, 0.46 mmol) in toluene (5 mL) was added, drop-wise with stirring, to a solution of the desired 8-hydroxyquinoline (0.93 mmol) in toluene (10 mL). The reaction was stirred for 12 h, after which time the solvent was removed in vacuo. The resulting solid was washed with hexane, filtered, and dried in vacuo to yield a yellow powder.![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C), 3JHH = 2.0 Hz), 4.27 (t, 4H, (Cp(CH)CC), 3JHH = 2.0 Hz), 4.19 (s, 10H, Cp(CH)), 3.34 (s, 6H, CH3), 0.67 (t, 3H, CH3, 3JHH = 8.0 Hz), 0.11 (dq, 2H, CH2, 3JHH = 8.0 Hz, 4JHH = 14.6 Hz); 13C{1H} NMR (100 MHz, toluene-d8) δ (ppm): 158.8 (CIV), 158.2 (CIV), 140.5 (CIV), 135.8 (CH), 131.0 (CH), 124.7 (CIV), 124.6 (CH), 116.2 (CIV), 109.1 (CIV), 93.7 (CC), 83.7 (CC), 71.7 (Cp(CH)CC), 70.3 (Cp(CH)), 69.8 (Cp(CH)CC), 66.8 (Cp(CIV)CC), 23.3 (CH3), 9.9 (CH2CH3), 1.4 (CH2CH3); Anal. Calc. (AlC46H35Cl2Fe2N2O2): C, 64.44; H, 4.11; N, 3.27 Found: C, 64.35; H, 4.05; N, 3.27.
C), 3JHH = 2.0 Hz), 4.27 (t, 4H, (Cp(CH)CC), 3JHH = 2.0 Hz), 4.19 (s, 10H, Cp(CH)), 3.34 (s, 6H, CH3), 0.67 (t, 3H, CH3, 3JHH = 8.0 Hz), 0.11 (dq, 2H, CH2, 3JHH = 8.0 Hz, 4JHH = 14.6 Hz); 13C{1H} NMR (100 MHz, toluene-d8) δ (ppm): 158.8 (CIV), 158.2 (CIV), 140.5 (CIV), 135.8 (CH), 131.0 (CH), 124.7 (CIV), 124.6 (CH), 116.2 (CIV), 109.1 (CIV), 93.7 (CC), 83.7 (CC), 71.7 (Cp(CH)CC), 70.3 (Cp(CH)), 69.8 (Cp(CH)CC), 66.8 (Cp(CIV)CC), 23.3 (CH3), 9.9 (CH2CH3), 1.4 (CH2CH3); Anal. Calc. (AlC46H35Cl2Fe2N2O2): C, 64.44; H, 4.11; N, 3.27 Found: C, 64.35; H, 4.05; N, 3.27.
Titanium complexes
Titanium(IV) tetrakis(iso-propoxide) (0.23 mL, 0.80 mmol) in toluene (15 mL) was added, drop-wise under nitrogen, to a solution of the 8-hydroxyquinoline (1.6 mmol) in toluene (15 mL), with stirring. The clear yellow solution was left to stir for 12 h at 298 K. The solvent was removed in vacuo and the product washed with hexane (10 mL) and isolated as a yellow solid.C), 80.1 (CC), 71.4 (CH), 71.3 (Cp(CH)), 70.1 (Cp(CH)), 68.9 (Cp(CH)), 66.0 (Cp(C)), 25.6 (CH3), 23.6 (CH3); Anal. Calc. (C50H44Cl2I2N2O4Ti): C, 62.08%; H, 4.58%; N, 2.90%. Found: C, 61.93%; H, 4.57%; N, 2.99%.
Zinc complexes
Diethyl zinc (0.27 g, 2.19 mmol) in toluene (5 mL) was added, dropwise with stirring, to a solution of 8-hydroxyquinoline (2.19 mmol) in toluene (15 mL). The solution was stirred for 12 h, after which time a yellow precipitate had formed. The precipitate was isolated by filtration, washed with hexane and dried in vacuo to yield a yellow solid.C), 3JHH = 1.6 Hz), 4.24 (s, 5H, (Cp(CH)), 4.21 (t, 2H, (Cp(CH)CC), 3JHH = 1.6 Hz), 2.83 (s, 3H, CH3), 1.33 (t, 3H, CH2CH3, 3JHH = 8.1 Hz), 0.44 (q, 2H, CH2CH3, 3JHH = 8.1 Hz); 13C{1H} NMR (125.3 MHz, THF-d8) δ (ppm): 165.5 (CIV), 157.2 (CIV), 141.5 (CIV), 137.1 (CH), 132.5 (CH), 126.2 (CIV), 124.0 (CH), 112.1 (CIV), 109.9 (CIV), 93.2 (CC), 85.2 (CC), 72.2 (Cp(CH)CC), 70.9 (Cp(CH)), 69.4 (Cp(CH)CC), 68.1 (Cp(CIV)CC), 24.8 (CH3), 13.6 (CH2CH3), −1.3 (CH2CH3); Anal. Calc. (ZnC24H20ClFeNO): C, 58.22%; H, 4.07%; N, 2.83% Found: C, 58.33%; H, 4.12%; N, 2.91%.
Acknowledgements
The research was supported by funding from the EPSRC (EP/K035274/1, EP/K014070/1). CB acknowledges an EPSRC Doctoral Prize Fellowship for financial support. DWB acknowledges the Imperial-UBC Summer Research Placement Exchange 2014 for financial support. Purac Plc. are thanked for the donation of rac-lactide.References
- (a) P. J. Dijkstra, H. Z. Du and J. Feijen, Polym. Chem., 2011, 2, 520–527 RSC; (b) J.-C. Buffet and J. Okuda, Polym. Chem., 2011, 2, 2758–2763 RSC; (c) N. Ajellal, J. F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC; (d) R. H. Platel, L. M. Hodgson and C. K. Williams, Polym. Rev., 2008, 48, 11–63 CrossRef CAS; (e) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC.
- M. J. L. Tschan, E. Brule, P. Haquette and C. M. Thomas, Polym. Chem., 2012, 3, 836–851 RSC.
- (a) A. Kowalski, A. Duda and S. Penczek, Macromolecules, 1998, 31, 2114 CrossRef CAS; (b) H. Du, A. H. Velders, P. J. Dijkstra, J. Sun, Z. Zhong, X. Chen and J. Feijen, Chem. – Eur. J., 2009, 15, 9836–9845 CrossRef CAS PubMed; (c) Z. Y. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed., 2002, 41, 4510–4513 CrossRef CAS; (d) M. Normand, V. Dorcet, E. Kirillov and J. F. Carpentier, Organometallics, 2013, 32, 1694–1709 CrossRef CAS; (e) M. Bouyahyi, T. Roisnel and J.-F. Carpentier, Organometallics, 2011, 31, 1458–1466 CrossRef; (f) M. Bouyahyi, T. Roisnel and J.-F. Carpentier, Organometallics, 2009, 29, 491–500 CrossRef; (g) M. Bouyahyi, E. Grunova, N. Marquet, E. Kirillov, C. M. Thomas, T. Roisnel and J. F. Carpentier, Organometallics, 2008, 27, 5815–5825 CrossRef CAS; (h) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316 CrossRef CAS PubMed; (i) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed; (j) D. J. Darensbourg and O. Karroonnirun, Organometallics, 2010, 29, 5627–5634 CrossRef CAS.
- (a) X. Y. Wang, K. R. Liao, D. P. Quan and Q. Wu, Macromolecules, 2005, 38, 4611–4617 CrossRef CAS; (b) B. J. O'Keefe, L. E. Breyfogle, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2002, 124, 4384–4393 CrossRef; (c) M. Stolt and A. Sodergard, Macromolecules, 1999, 32, 6412–6417 CrossRef CAS; (d) A. B. Biernesser, B. Li and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553–16560 CrossRef CAS PubMed.
- (a) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2004, 43, 6717–6725 CrossRef CAS PubMed; (b) M. H. Chisholm, J. Gallucci and K. Phomphrai, Chem. Commun., 2003, 48–49 RSC; (c) M. Lamberti, A. Botta and M. Mazzeo, Appl. Organomet. Chem., 2014, 28, 140–145 CrossRef CAS; (d) W. Yi and H. Ma, Inorg. Chem., 2013, 52, 11821–11835 CrossRef CAS PubMed; (e) B. Liu, T. Roisnel, L. Maron, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2013, 19, 3986–3994 CrossRef CAS PubMed; (f) M.-W. Hsiao and C.-C. Lin, Dalton Trans., 2013, 42, 2041–2051 RSC; (g) L. Clark, G. B. Deacon, C. M. Forsyth, P. C. Junk, P. Mountford, J. P. Townley and J. Wang, Dalton Trans., 2013, 42, 9294–9312 RSC; (h) B. Liu, T. Roisnel, J.-P. Guegan, J.-F. Carpentier and Y. Sarazin, Chem. – Eur. J., 2012, 18, 6289–6301 CrossRef CAS PubMed; (i) J. P. Davin, J.-C. Buffet, T. P. Spaniol and J. Okuda, Dalton Trans., 2012, 41, 12612–12618 RSC; (j) M. G. Cushion and P. Mountford, Chem. Commun., 2011, 47, 2276–2278 RSC; (k) J.-C. Buffet, J. P. Davin, T. P. Spaniol and J. Okuda, New J. Chem., 2011, 35, 2253–2257 RSC; (l) Z. Y. Zhong, M. J. K. Ankone, P. J. Dijkstra, C. Birg, M. Westerhausen and J. Feijen, Polym. Bull., 2001, 46, 51–57 CrossRef CAS.
- (a) M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2014, 6, 1892–1898 CrossRef CAS; (b) H. Wang, Y. Yang and H. Ma, Macromolecules, 2014, 47, 7750–7764 CrossRef CAS; (c) F. Drouin, T. J. J. Whitehorne and F. Schaper, Dalton Trans., 2011, 40, 1396–1400 RSC; (d) M. H. Chisholm, J. C. Gallucci and K. Phomphrai, Inorg. Chem., 2005, 44, 8004–8010 CrossRef CAS PubMed; (e) B. M. Chamberlain, M. Cheng, D. R. Moore, T. M. Ovitt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 3229–3238 CrossRef CAS PubMed; (f) M. H. Chisholm, J. Gallucci and K. Phomphrai, Inorg. Chem., 2002, 41, 2785–2794 CrossRef CAS PubMed; (g) V. Poirier, T. Roisnel, J.-F. Carpentier and Y. Sarazin, Dalton Trans., 2009, 9820–9827 RSC; (h) J. C. Wu, B. H. Huang, M. L. Hsueh, S. L. Lai and C. C. Lin, Polymer, 2005, 46, 9784–9792 CrossRef CAS; (i) W.-C. Hung and C.-C. Lin, Inorg. Chem., 2009, 48, 728–734 CrossRef CAS PubMed; (j) L. Wang and H. Ma, Macromolecules, 2010, 43, 6535–6537 CrossRef CAS; (k) T. J. J. Whitehorne, B. Vabre and F. Schaper, Dalton Trans., 2014, 43, 6339–6352 RSC; (l) C. Gallegos, V. Tabernero, F. M. Garcia-Valle, M. E. G. Mosquera, T. Cuenca and J. Cano, Organometallics, 2013, 32, 6624–6627 CrossRef CAS.
- (a) J. Zhang, J. Xiong, Y. Sun, N. Tang and J. Wu, Macromolecules, 2014, 47, 7789–7796 CrossRef CAS; (b) L. N. Saunders, L. N. Dawe and C. M. Kozak, J. Organomet. Chem., 2014, 749, 34–40 CrossRef CAS; (c) B. Calvo, M. G. Davidson and D. Garcia-Vivo, Inorg. Chem., 2011, 50, 3589–3595 CrossRef CAS PubMed; (d) Y. Huang, Y.-H. Tsai, W.-C. Hung, C.-S. Lin, W. Wang, J.-H. Huang, S. Dutta and C.-C. Lin, Inorg. Chem., 2010, 49, 9416–9425 CrossRef CAS PubMed.
- (a) Y. Huang, W. Wang, C.-C. Lin, M. P. Blake, L. Clark, A. D. Schwarz and P. Mountford, Dalton Trans., 2013, 42, 9313–9324 RSC; (b) Y. Lemmouchi, M. C. Perry, A. J. Amass, K. Chakraborty and E. Schacht, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 5348–5362 CrossRef CAS; (c) L. Sipos and M. Zsuga, J. Macromol. Sci., Part A: Pure Appl. Chem., 1997, 34, 1269–1284 CrossRef; (d) Z. Jedlinski, P. Kurcok and R. W. Lenz, J. Macromol. Sci., Part A: Pure Appl. Chem., 1995, 32, 797–810 CrossRef; (e) Z. Jedlinski, P. Kurcok, M. Kowalczuk, A. Matuszowicz, P. Dubois, R. Jerome and H. R. Kricheldorf, Macromolecules, 1995, 28, 7276–7280 CrossRef CAS; (f) H. R. Kricheldorf and C. Boettcher, Makromol. Chem., 1993, 73, 47–64 CrossRef CAS.
- (a) Y. Sarazin, R. H. Howard, D. L. Hughes, S. M. Humphrey and M. Bochmann, Dalton Trans., 2006, 340–350 RSC; (b) Z. R. Turner, J.-C. Buffet and D. O'Hare, Organometallics, 2014, 33, 3891–3903 CrossRef CAS; (c) C. J. Chuck, M. G. Davidson, G. G. du Sart, P. K. Ivanova-Mitseva, G. I. Kociok-Koehn and L. B. Manton, Inorg. Chem., 2013, 52, 10804–10811 CrossRef CAS PubMed; (d) A. Sauer, A. Kapelski, C. Fliedel, S. Dagorne, M. Kol and J. Okuda, Dalton Trans., 2013, 42, 9007–9023 RSC; (e) B. J. Jeffery, E. L. Whitelaw, D. Garcia-Vivo, J. A. Stewart, M. F. Mahon, M. G. Davidson and M. D. Jones, Chem. Commun., 2011, 47, 12328–12330 RSC; (f) J.-C. Buffet, A. N. Martin, M. Kol and J. Okuda, Polym. Chem., 2011, 2, 2378–2384 RSC; (g) C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410–7411 CrossRef CAS PubMed; (h) C. K. A. Gregson, I. J. Blackmore, V. C. Gibson, N. J. Long, E. L. Marshall and A. J. P. White, Dalton Trans., 2006, 3134–3140 RSC; (i) S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45, 4783–4790 CrossRef CAS PubMed; (j) A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn, M. F. Mahon, A. F. Johnson, P. Khunkamchoo, S. L. Roberts and S. S. F. Wong, Macromolecules, 2006, 39, 7250–7257 CrossRef CAS; (k) Y. Kim, G. K. Jnaneshwara and J. G. Verkade, Inorg. Chem., 2003, 42, 1437–1447 CrossRef CAS PubMed.
- (a) C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Inorg. Chem., 2013, 52, 12561–12567 CrossRef CAS PubMed; (b) C. Bakewell, R. H. Platel, S. K. Cary, S. M. Hubbard, J. M. Roaf, A. C. Levine, A. J. P. White, N. J. Long, M. Haaf and C. K. Williams, Organometallics, 2012, 31, 4729–4736 CrossRef CAS; (c) K. Sokołowski, I. Justyniak, W. Śliwiński, K. Sołtys, A. Tulewicz, A. Kornowicz, R. Moszyński, J. Lipkowski and J. Lewiński, Chem. – Eur. J., 2012, 18, 5637–5645 CrossRef PubMed; (d) W. Zhang, S. Liu, W. Yang, X. Hao, R. Glaser and W. H. Sun, Organometallics, 2012, 31, 8178–8188 CrossRef CAS; (e) B. Kamenar, C. K. Prout and J. D. Wright, J. Chem. Soc., 1965, 4851–4867 RSC; (f) M. Pasquali, P. Fiaschi, C. Floriani and P. F. Zanazzi, J. Chem. Soc., Chem. Commun., 1983, 613–614 RSC; (g) W.-H. Sun, M. Shen, W. Zhang, W. Huang, S. Liu and C. Redshaw, Dalton Trans., 2011, 40, 2645–2653 RSC.
- G. Delapierre, J. M. Brunel, T. Constantieux and G. Buono, Tetrahedron: Asymmetry, 2001, 12, 1345–1352 CrossRef CAS.
- (a) Y. Fazaeli, M. M. Amini and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, m1509–m1509 CAS; (b) M. Mirzaee and M. M. Amini, Appl. Organomet. Chem., 2005, 19, 339–342 CrossRef CAS; (c) Y. Fazaeli, E. Najafi, M. M. Amini and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m271–m271 CAS; (d) P. H. Bird, A. R. Fraser and C. F. Lau, Inorg. Chem., 1973, 12, 1322–1328 CrossRef CAS; (e) W. F. Zeng, Y. S. Chen, M. Y. Chiang, S. S. Chern and C. P. Cheng, Polyhedron, 2002, 21, 1081–1087 CrossRef CAS; (f) F. Grasset, J.-B. Cazaux, L. Magna, P. Braunstein and H. Oliver-Bourbigou, Dalton Trans., 2012, 41, 10396–10404 RSC.
- K. Sokołowski, I. Justyniak, W. Śliwiński, K. Sołtys, A. Tulewicz, A. Kornowicz, R. Moszyński, J. Lipkowski and J. Lewiński, Chem. – Eur. J., 2012, 18, 5637–5645 CrossRef PubMed.
- K. L. Orchard, J. E. Harris, A. J. P. White, M. S. P. Shaffer and C. K. Williams, Organometallics, 2011, 30, 2223–2229 CrossRef CAS.
- (a) X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed; (b) E. M. Broderick, P. S. Thuy-Boun, N. Guo, C. S. Vogel, J. Sutter, J. T. Miller, K. Meyer and P. L. Diaconescu, Inorg. Chem., 2011, 50, 2870–2877 CrossRef CAS PubMed; (c) E. M. Broderick, N. Guo, C. S. Vogel, C. L. Xu, J. Sutter, J. T. Miller, K. Meyer, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 9278–9281 CrossRef CAS PubMed.
- J. Coudane, C. Ustariz-Peyret, G. Schwach and M. Vert, J. Polym. Sci., Part A: Polym. Chem., 1997, 35, 1651–1658 CrossRef CAS.
- (a) E. L. Whitelaw, M. D. Jones and M. F. Mahon, Inorg. Chem., 2010, 49, 7176–7181 CrossRef CAS PubMed; (b) K.-C. Hsieh, W.-Y. Lee, L.-F. Hsueh, H. M. Lee and J.-H. Huang, Eur. J. Inorg. Chem., 2006, 2006, 2306–2312 CrossRef; (c) M. Hu, M. Wang, H. Zhu, L. Zhang, H. Zhang and L. Sun, Dalton Trans., 2010, 39, 4440–4446 RSC.
- (a) S. K. Russell, C. L. Gamble, K. J. Gibbins, K. C. S. Juhl, W. S. Mitchell, A. J. Tumas and G. E. Hofmeister, Macromolecules, 2005, 38, 10336–10340 CrossRef CAS; (b) A. J. Chmura, M. G. Davidson, C. J. Frankis, M. D. Jones and M. D. Lunn, Chem. Commun., 2008, 1293–1295 RSC; (c) A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS; (d) J.-C. Buffet and J. Okuda, Chem. Commun., 2011, 47, 4796–4798 RSC.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill Inc., 1999 Search PubMed.
- (a) M. H. Chisholm, J. C. Gallucci, H. Zhen and J. C. Huffman, Inorg. Chem., 2001, 40, 5051–5054 CrossRef CAS PubMed; (b) M. P. Coles and P. B. Hitchcock, Eur. J. Inorg. Chem., 2004, 2004, 2662–2672 CrossRef.
- C. C. Roberts, B. R. Barnett, D. B. Green and J. M. Fritsch, Organometallics, 2012, 31, 4133–4141 CrossRef CAS.
- (a) H. Y. Chen, H. Y. Tang and C. C. Lin, Macromolecules, 2006, 39, 3745–3752 CrossRef CAS; (b) C. K. Williams, L. E. Breyfogle, S. K. Choi, W. Nam, V. G. Young, M. A. Hillmyer and W. B. Tolman, J. Am. Chem. Soc., 2003, 125, 11350–11359 CrossRef CAS PubMed.
- (a) R. H. Platel, A. J. P. White and C. K. Williams, Chem. Commun., 2009, 4115–4117 RSC; (b) N. Ajellal, J. F. Carpentier, C. Guillaume, S. M. Guillaume, M. Helou, V. Poirier, Y. Sarazin and A. Trifonov, Dalton Trans., 2010, 39, 8363–8376 RSC; (c) M. Bouyahyi, N. Ajellal, E. Kirillov, C. M. Thomas and J. F. Carpentier, Chem. – Eur. J., 2011, 17, 1872–1883 CrossRef CAS PubMed; (d) K. Nie, L. Fang, Y. Yao, Y. Zhang, Q. Shen and Y. Wang, Inorg. Chem., 2012, 51, 11133–11143 CrossRef CAS PubMed; (e) H. Ma, T. P. Spaniol and J. Okuda, Angew. Chem., Int. Ed., 2006, 45, 7818–7821 CrossRef PubMed.
- M. H. Chisholm, K. Choojun, A. S. Chow and G. Fraenkel, Angew. Chem., Int. Ed., 2013, 52, 3264–3266 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1043547–1043549. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00192g |
| This journal is © The Royal Society of Chemistry 2015 |