
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Te–Te and Te–C bond cleavage reactions using a monovalent gallanediyl†
Chelladurai
Ganesamoorthy
,
Georg
Bendt
,
Dieter
Bläser
,
Christoph
Wölper
and
Stephan
Schulz
*
Institute of Inorganic Chemistry, University of Duisburg-Essen, 45117 Essen, Germany. E-mail: stephan.schulz@uni-due.de; Fax: +201 1833830; Tel: +201 1834635
First published on 5th February 2015
Abstract
LGa (L = [(2,6-i-Pr2-C6H3)NC(Me)]2CH) reacts with elemental tellurium with formation of the Te-bridged compound [LGa-μ-Te]21, whereas the reactions with Ph2Te2 and i-Pr2Te occurred with cleavage of the Te–Te and Te–C bond, respectively, and subsequent formation of LGa(TePh)22 and LGa(i-Pr)Tei-Pr 3. 1–3 were characterized by heteronuclear NMR (1H, 13C, 125Te) and IR spectroscopy and their solid state structures were determined by single crystal X-ray analyses.
Introduction
Univalent LGa containing the sterically crowded β-diketiminate ligand (L = [(2,6-i-Pr2-C6H3)NC(Me)]2CH)1 adopts a monomeric structure in the solid state and in solution. The gallium valence shell contains two bond pairs, a lone pair and an empty p-orbital. Therefore, LGa can react as electrophilic and nucleophilic reagent at the gallium atom. Computational calculations predicted that LGa is a good σ-donor but a poor π-acceptor due to the low energy and high s-character of the HOMO, and the large energy difference (95.3–110 kcal mol−1) between the HOMO and the rather diffuse acceptor 4p-orbital (LUMO+1).2–4 The σ-donor capacity of LGa was experimentally demonstrated for instance with the synthesis of the Lewis acid–base adduct LGa→B(C6F5)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5 and other p- or d-block metal complexes as well as with the synthesis of a large variety of (late) transition metal complexes.6 The latter were shown to be promising reagents for the activation of small molecules such as ethylene and have been used as precursors for the formation of heterometallic clusters,7,8 which in part can be described as molecular models for alloys.
5 and other p- or d-block metal complexes as well as with the synthesis of a large variety of (late) transition metal complexes.6 The latter were shown to be promising reagents for the activation of small molecules such as ethylene and have been used as precursors for the formation of heterometallic clusters,7,8 which in part can be described as molecular models for alloys.
While the coordination chemistry of LGa has been developed to a far greater extent compared to that of its lighter homologue LAl, its use in the transformation of unsaturated organic substrates has not,9–11 which most likely results from the weaker reducing properties of LGa. However, LGa was found to react with E–X bonds via insertion of the Ga(I) centre and subsequent formation of covalent Ga–E bonds.12 This reaction pathway was used for cluster formation reactions via oxidative insertion/reductive elimination processes. The synthesis of molecular gallium–tin intermetallic clusters upon reaction of LGa with SnCl213 as well as two galla-dibismuthenes containing covalent Ga–Bi single-bonds and Bi![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Bi double bonds, which were obtained from the reactions of LGa with Bi(OR)3 (R = O2SCF3, C6F5),14 represent remarkable experimental “snapshots” of these reactions. The cluster compounds can be considered as isolated reaction intermediates on the way to full reduction to tin metal and bismuth metal, respectively. The syntheses of these complexes demonstrate the promising potential of LGa to serve as selective reducing agent in the preparation of metalloid clusters and subvalent “metastable” compounds.
Bi double bonds, which were obtained from the reactions of LGa with Bi(OR)3 (R = O2SCF3, C6F5),14 represent remarkable experimental “snapshots” of these reactions. The cluster compounds can be considered as isolated reaction intermediates on the way to full reduction to tin metal and bismuth metal, respectively. The syntheses of these complexes demonstrate the promising potential of LGa to serve as selective reducing agent in the preparation of metalloid clusters and subvalent “metastable” compounds.
In addition to these interesting studies on the synthesis of intermetallic compounds, the capability of LGa in bond activation reactions was also studied. LGa was reacted with a large variety of compounds containing different element–element bonds including dihydrogen15 as well as electronically unsaturated molecules such as N2O, organic azides N3R and elemental sulphur. Many of these reactions were performed in order to synthesize compounds containing multiple bonds to gallium. However, reactions of LGa with N2O or S8 yielded the oxo- or sulfido bridged dimers [LGa-μ-E]2 (E = O, S),16 while its reaction with N3SiMe3 proceeded with formation of a cyclic gallium tetrazole and a gallium imide/azide compound. The most-likely formed reaction intermediate, a monomeric gallium imide LGaNR,17 was finally synthesized by reaction of LGa with the sterically encumbered azide Ar*N3 (Ar* = 2,6-Trip2-C6H3, Trip = 2,4,6-i-Pr3-C6H2) and structurally characterized by single crystal X-ray diffraction.18
Our long-term interest in the reactivity of low-valent organometallics of group 12 to 16 elements prompted us to start investigations on the general reactivity of LM (M = Al, Ga, In) toward group 15 compounds such as BiEt319 as well as tetraalkyldistibanes and dibismuthanes Et4E2 (E = Sb, Bi).20 These reactions were found to proceed with cleavage of the Bi–C as well as E–E bond and subsequent formation of LM(Et)BiEt2 and LM(EEt2)2, respectively, in which the Ga atom is oxidized from the formal oxidation state +I to +III. Moreover, oxidative addition reactions of monovalent Zn(I) compounds,21 germylenes and stannylenes22 as well as distibanes and dibismuthanes23 with dichalcogenanes were reported by us. In addition, insertion reactions of elemental chalcogens into metal–metal bonds were observed in reactions with tetraalkyldistibanes and -dibismuthanes.24,25 We herein report on the reactions of LGa with elemental tellurium as well as diphenylditellane Ph2Te2 and diisopropyltellane i-Pr2Te, which proceeded with Te–Te and Te–C bond cleavage and subsequent formation of compounds containing direct Ga–Te σ-bonds.
Results and discussion
Equimolar amounts of LGa were reacted with elemental tellurium, diphenylditellane Ph2Te2 and diisopropyltellane i-Pr2Te in toluene at ambient temperature, yielding the Te-bridged dimer [LGa-μ-Te]21 as well as LGa(TePh)22 and LGa(i-Pr)Tei-Pr 3, respectively (Scheme 1). 1–3 are moisture sensitive, yellow to pale yellow crystalline solids but moderately stable toward air. 1 is sparingly soluble in benzene, toluene and n-hexane whereas 2 and 3 are soluble in these solvents.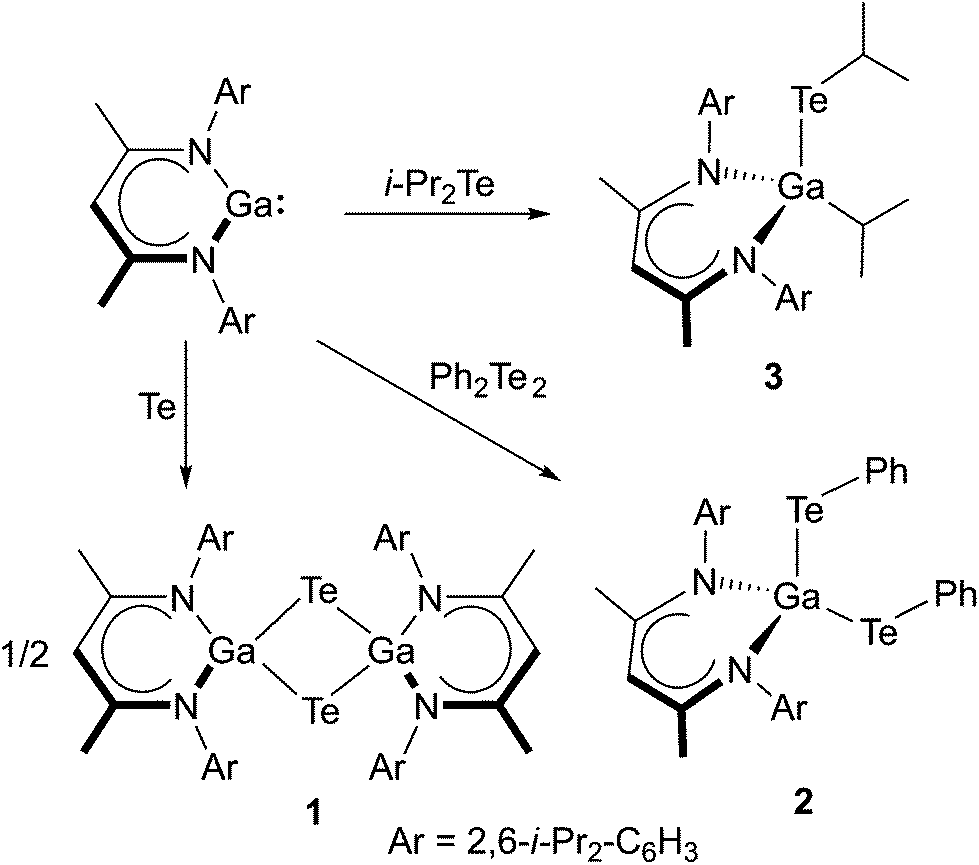 | ||
| Scheme 1 Synthesis of 1–3. | ||
The 1H and 13C NMR spectra of 1–3 show the characteristic resonances of the organic entities. 1H NMR spectral pattern of 1 and 2 are similar to those of LGa and C2v symmetric molecules related to the β-diketiminate ligands. They exhibit single resonances for the γ-CH (4.59 ppm, 1; 4.76 ppm, 2) and two methyl groups of the C3N2Ga rings (1.35 ppm 1; 1.50 ppm 2). The methyl protons of the isopropyl substituents and methine protons appear as two doublets (1.13 & 1.10 ppm 1; 1.48 & 1.08 ppm 2) and a septet (3.33 ppm 1; 3.62 ppm 2), respectively. Due to its low solubility, the 13C and 125Te NMR resonances of 1 were not intense enough. The 13C NMR spectrum of 2 shows 14 signals including the characteristic resonances due to the γ-CH (99.22 ppm) backbone carbon atom, both β-C atoms atoms of the C3N2Ga ring (170.63 ppm) and the methine (29.36 ppm) and methyl carbon atoms of isopropyl groups (27.01, 25.44 ppm). Compound 3 shows more distinct 1H and 13C NMR patterns than 1 and 2. Due to the presence of three different substituents at the Ga atom and a hindered rotation about the N–C bonds, the i-Pr groups in 3 are magnetically inequivalent, leading to six doublets (1.87, 1.61, 1.34, 1.31, 1.08, 0.67 ppm) in the 1H NMR spectrum. The six methine protons of the i-Pr groups appear as four septets (4.16, 3.82, 3.42, 1.04 ppm) with the integral ratios of 2:1:2:1, respectively. Integrals of 2 H belong to the i-Pr methine groups of the L ligand. The γ-CH and two methyl groups of the C3N2M ring are in the mirror plane and exhibit only single resonances at 4.67 and 1.51 ppm, respectively. The 13C{1H} NMR spectrum of 3 shows the expected 19 signals and some of them were tentatively assigned to the i-Pr carbons atoms in 3 in the Experimental section. Elemental analyses (C, H, N) of 1–3 confirm the structural compositions and their analytical pure nature. Furthermore, the NMR (1H, 13C, 125Te) and IR spectroscopic details are in accordance with the proposed formulations of 1–3.
1–3 are stable in solution and no reduction/decomposition occurs even at 90 °C in C6D6. Despite that the first Te–C bond cleavage of i-Pr2Te occurred smoothly at room temperature, the second Te–C bond couldn't be cleaved by reaction with an additional equivalent of LGa. In contrast, we successfully cleaved the Te–C and Te–Te bonds of Ph2Te2 upon reaction with an equimolar amount of the Lewis acid–base adduct LGa→B(C6F5)3 in C6D6, which yielded an yellow-orange solution at room temperature within three days. The 1H NMR spectrum of the reaction mixture shows the presence of four different types of γ-CH protons with different integral ratios (Fig. S11†) and the spectral comparison evidences the presence of traces of 1 (4.57, 3.39 ppm) and 2 (4.76, 3.62 ppm) along with two unknown compounds. Prolonged storage of the reaction mixture (10 days) at room temperature led to pale yellow crystals of 1. During this period the peaks corresponding to 2 gradually decreased. Unfortunately, our efforts to isolate the major component of the reaction mixture (see Fig. S12†) failed since the solution is highly sensitive and decomposes to oily substances. According to the 1H NMR pattern, the Ga atom in the major product has three different substituents.
Single crystals of 1 were grown separately in benzene and toluene solutions. 1a is the solvent-free compound (obtained from the 1:1 reaction of Ph2Te2 and LGa→B(C6F5)3) and crystallises in the monoclinic space group P21/n, while 1b is its toluene hemi-solvate (obtained from the 1:1 reaction of Te and LGa), which crystallises in the monoclinic space group C2/m. Single crystals of 2 were obtained from a freshly prepared n-hexane solution upon storage at room temperature, while single crystals of 3 were grown from saturated toluene solutions at 5 °C. 2 and 3 crystallise in the orthorhombic space groups Pbca (2) and Pnma (3). Fig. 1–3 show the solid state structures of 1a, 2 and 3 and the selected bond lengths and bond angles are given at the figure captions. Table 1 summarizes the crystal data and details of the structural determinations.
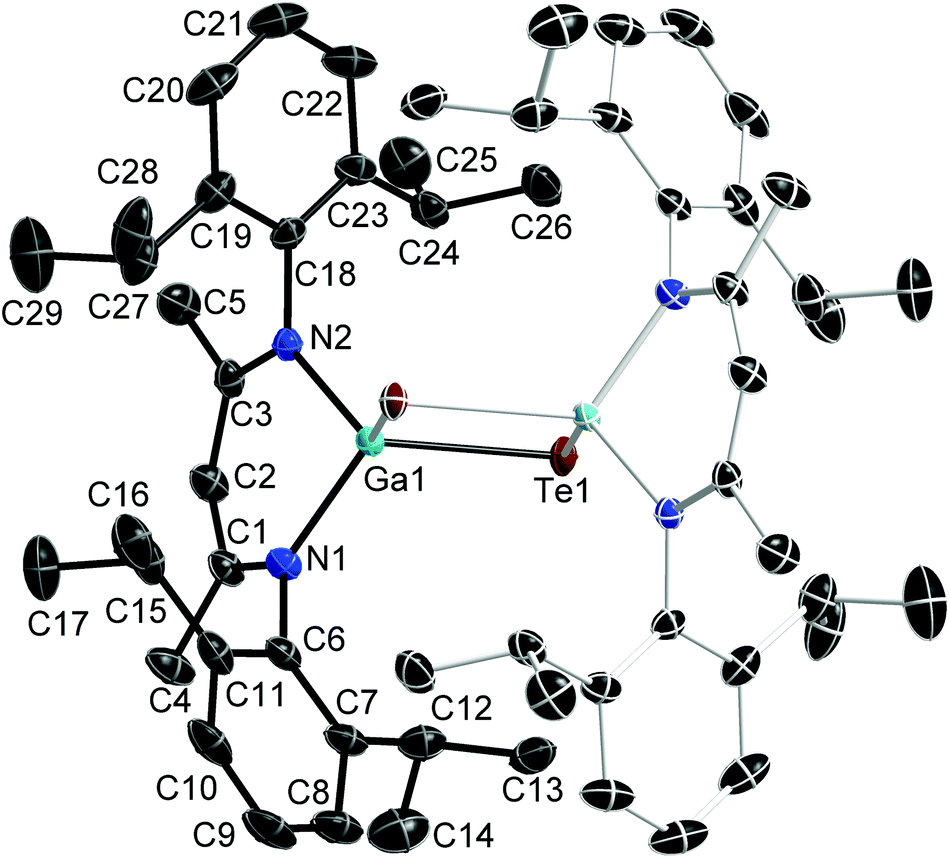 | ||
| Fig. 1 Solid state structure of 1a (thermal ellipsoids are shown at 50% probability levels); H atoms are omitted for clarity. Symmetry generated part in pale colours. Selected bond lengths and angles in Å and °: Te(1)–Ga(1)#1 2.5777(4), Te(1)–Ga(1) 2.5898(4), Ga(1)–N(2) 1.979(2), Ga(1)–N(1) 1.982(2), C(1)–C(2) 1.397(4), C(2)–C(3) 1.397(4), N(1)–C(1) 1.339(4), N(2)–C(3) 1.337(4), N(2)–Ga(1)–N(1) 94.92(10), N(2)–Ga(1)–Te(1)#1 118.56(7), N(1)–Ga(1)–Te(1)#1 115.72(7), N(2)–Ga(1)–Te(1) 114.87(7), N(1)–Ga(1)–Te(1) 115.13(7), Te(1)#1–Ga(1)–Te(1) 98.883(11), C(3)–C(2)–C(1) 128.4(3), N(1)–C(1)–C(2) 123.6(3), N(2)–C(3)–C(2) 124.1(3). | ||
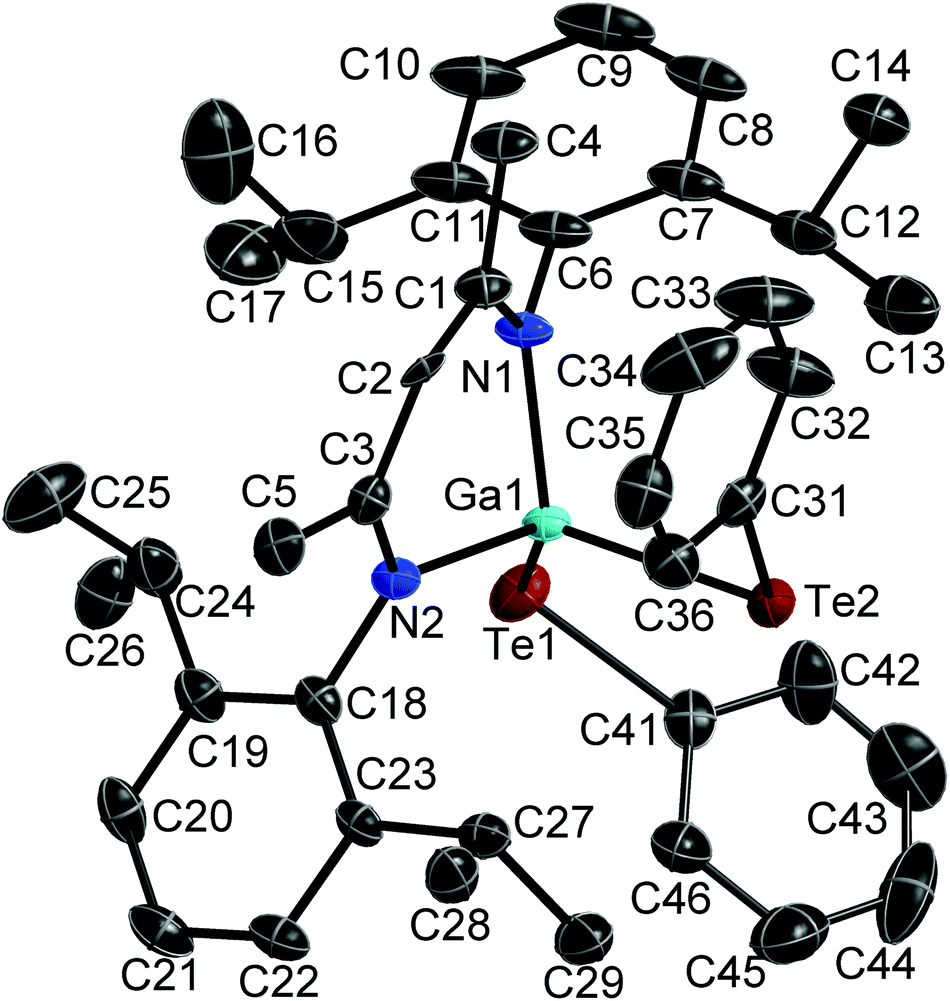 | ||
| Fig. 2 Solid state structure of 2 (thermal ellipsoids are shown at 50% probability levels); H atoms are omitted for clarity. Selected bond lengths and angles in Å and °: Ga(1)–N(1) 1.950(4), Ga(1)–N(2) 1.971(4), Ga(1)–Te(1) 2.5586(5), Ga(1)–Te(2) 2.6076(6), Te(1)–C(41) 2.114(5), Te(2)–C(31) 2.144(4), N(1)–C(1) 1.342(6), N(2)–C(3) 1.334(5), C(1)–C(2) 1.377(6), C(2)–C(3) 1.406(6), N(1)–Ga(1)–N(2) 97.45(15), N(1)–Ga(1)–Te(1) 110.65(11), N(2)–Ga(1)–Te(1) 114.06(11), N(1)–Ga(1)–Te(2) 110.98(12), N(2)–Ga(1)–Te(2) 109.00(11), Te(1)–Ga(1)–Te(2) 113.587(19), C(1)–C(2)–C(3) 130.5(4), N(1)–C(1)–C(2) 123.5(4), N(2)–C(3)–C(2) 122.7(4). | ||
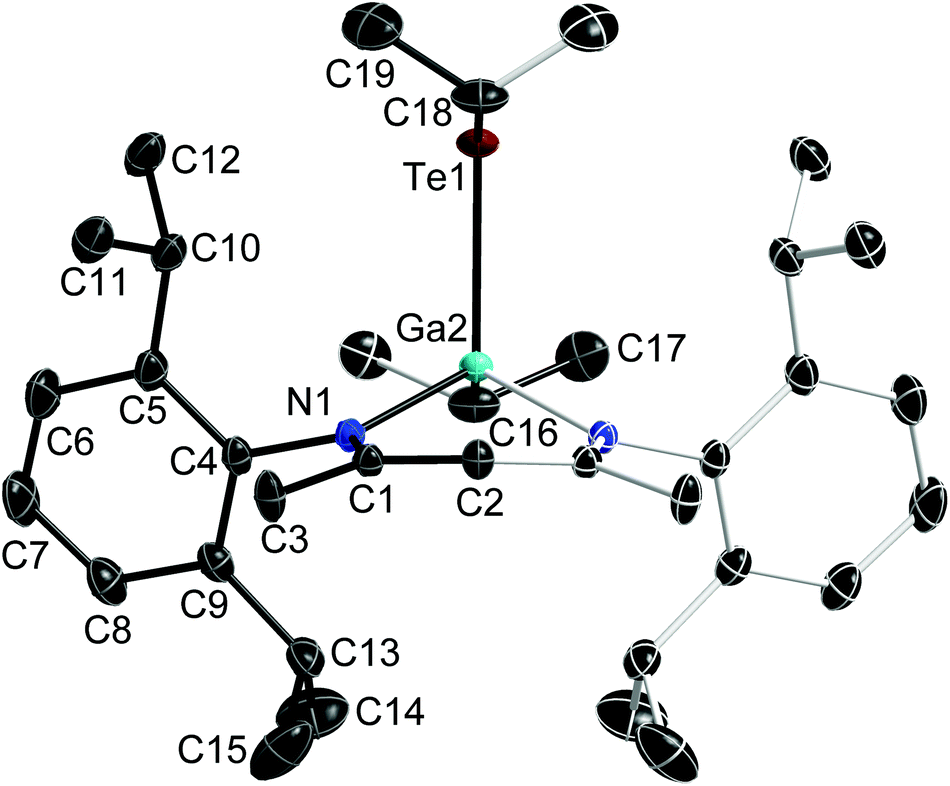 | ||
| Fig. 3 Solid state structure of 3 (thermal ellipsoids are shown at 50% probability levels); H atoms are omitted for clarity. Symmetry generated part in pale colours. Selected bond lengths and angles in Å and °: Te(1)–C(18) 2.1983(19), Te(1)–Ga(2) 2.5929(4), Ga(2)–N(1) 1.9739(10), Ga(2)–C(16) 1.9902(17), N(1)–C(1) 1.3321(13), C(1)–C(2) 1.4044(13), N(1)–Ga(2)–N(1)#1 95.66(5), N(1)–Ga(2)–C(16) 115.64(4), N(1)–Ga(2)–Te(1) 112.07(3), C(16)–Ga(2)–Te(1) 105.84(5), C(1)–C(2)–C(1)#1 128.03(14), N(1)–C(1)–C(2) 123.73(11). | ||
| 1a | 1b | 2 | 3 | |
|---|---|---|---|---|
| a R 1 = ∑(||Fo| − |Fc||)/∑|Fo| (for I > 2σ(I)). b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. c Goodness of fit = {∑[w(|Fo2| − |Fc2|)2]/(Nobservns − Nparams)}1/2. w−1 = σ2(Fo2) + (aP)2 + bP with P = [Fo2 + 2Fc2]/3, a and b are constants chosen by the program. | ||||
| Empirical formula | C58H82Ga2N4Te2 | C61.50H86Ga2N4Te2 | C41H51GaN2Te2 | C35H55GaN2Te |
| M | 1229.91 | 1275.98 | 896.75 | 701.13 |
| Crystal size [mm] | 0.280 × 0.220 × 0.180 | 0.220 × 0.180 × 0.130 | 0.415 × 0.326 × 0.198 | 0.200 × 0.190 × 0.100 |
| T [K] | 130(1) | 100(1) | 100(1) | 100(1) |
| Crystal system | Monoclinic | Monoclinic | Orthorhombic | Orthorhombic |
| Space group | P21/n | C2/m | Pbca | Pnma |
| a [Å] | 14.0688(10) | 18.6322(4) | 19.1512(8) | 16.888(3) |
| b [Å] | 13.8376(10) | 20.9742(5) | 19.7070(8) | 19.798(3) |
| c [Å] | 15.2807(11) | 16.4363(4) | 20.3343(8) | 10.2125(15) |
| α [°] | 90 | 90 | 90 | 90 |
| β [°] | 106.759(2) | 98.9820(10) | 90 | 90 |
| γ [°] | 90 | 90 | 90 | 90 |
| V [Å3] | 2848.5(4) | 6344.5(3) | 7674.4(5) | 3414.5(9) |
| Z | 2 | 4 | 8 | 4 |
| D calc [g cm−1] | 1.434 | 1.336 | 1.552 | 1.364 |
| μ (MoKα [mm−1]) | 1.988 | 1.788 | 2.237 | 1.668 |
| Transmissions | 0.75/0.50 | 0.75/0.68 | 0.75/0.52 | 0.75/0.63 |
| F(000) | 1248 | 2596 | 3568 | 1448 |
| Index ranges | −20 ≤ h ≤ 21 | −27 ≤ h ≤ 27 | −27 ≤ h ≤ 27 | −25 ≤ h ≤ 23 |
| −20 ≤ k ≤ 20 | −28 ≤ k ≤ 31 | −27 ≤ k ≤ 26 | −30 ≤ k ≤ 26 | |
| −21 ≤ l ≤ 22 | −24 ≤ l ≤ 24 | −26 ≤ l ≤ 28 | −15 ≤ l ≤ 15 | |
| θ max [°] | 32.618 | 32.155 | 30.255 | 33.202 |
| Reflections collected | 43835 |
86728 |
131905 |
77225 |
| Independent reflections | 10256 |
11354 |
11268 |
6666 |
| R int | 0.0375 | 0.0319 | 0.0446 | 0.0307 |
| Refined parameters | 298 | 342 | 415 | 193 |
| R 1 [I > 2σ(I)]a | 0.0440 | 0.0223 | 0.0469 | 0.0225 |
| wR2 [all data]b | 0.1300 | 0.0606 | 0.1249 | 0.0578 |
| GooFc | 1.081 | 1.072 | 1.127 | 1.046 |
| Δρfinal (max/min) [e Å−3] | 3.763/−1.417 | 0.986/−0.453 | 2.287/−1.586 | 0.677/−0.358 |
The Ga atoms in 1a, 1b, 2 and 3 each adopt slightly distorted tetrahedral coordination spheres.‡ The six-membered GaN2C3 rings show boat-type conformations, in which the Ga atoms are significantly out of-plane (deviation from best plane of the ligand's backbone: 0.521(3) Å 1a, 0.5853(19) and 0.560(2) Å 1b, 0.721(5) Å 2, 0.6309(18) Å 3) The average Ga–N bond length (1.981(2) Å 1a; 1.992(2) Å 1b; 1.961(4) Å 2; 1.9739(10) Å 3) and N–Ga–N bond angle (94.92(10)° 1a; 95.38(6)° 1b; 97.45(15)° 2; 95.66(5)° 3) as observed for 1, 2 and 3 are almost identical to that of LGa, for which an average Ga–N distance of 2.054(2) Å and a N–Ga–N bond angle of 87.56(6)° was reported.1 The Te–Ga–Te (98.88(2)° 1a; 100.39(1)° 1b; 113.59(2)° 2) and C–Ga–Te bond angles (105.84(5)° 3) are smaller compared to the N–Ga–N bond angles. The Ga–Te bond lengths (2.5777(4), 2.5898(4) Å 1a; 2.5809(2), 2.5909(2) Å 1b; 2.5586(5), 2.6076(6) Å 2; 2.5929(4) Å 3) are comparable to those reported for [K(OEt2)3][(PhTe)2Ga{[N(2,6-i-Pr2-C6H3)C(H)]2}] (2.5785(12), 2.6577(10) Å),26 the Te-bridged dimers {[Me3SiNC(Ph)C(SiMe3)2]Ga-μ-Te}2 (2.570 Å)27 and [i-Pr2PNP(i-Pr2)TeGa-μ-Te]2 (2.5783(7) Å) as well as the six-membered heterocycle [i-Pr2PNP(i-Pr2)TeGa-μ-Te]3 (2.582(2) Å).28 In addition, the mixed Te/carboxylate-bridged compound [R2Ga2(μ-Te)(μ-OOCCH3)2] (2.534 Å)29 as well as the base-stabilized compounds LGa(TePh)3 (L = NMe3, PCy3), which were obtained from the reaction of base-stabilized gallanes [LGaH3] with Ph2Te2 and which also contain fourfold-coordinated Ga atoms and twofold-coordinated Te atoms, show comparable Ga–Te bond distances of 2.580(1) Å and 2.597(2) Å, respectively.30 In contrast, the Ga–Te bonds in the [MeGa(TePh)3]+ monocation of [(Me3P)4Cu][MeGa(TePh)3] is slightly elongated (2.6406(3) Å).31 The same holds for the Ga–Te bonds in four structurally characterized Ga4Te4 heterocubanes, which range from 2.67 to 2.72 Å,32–35 as well as in dimeric compounds of the general type [R2Ga-μ-TeR]2, for which Ga–Te bond lengths in the range from 2.67 to 2.76 Å were reported.36–39 In contrast, slightly shorter Ga–Te bond lengths were reported for {[(Me3Si)2CH]2Ga-μ-Te} (2.5521(4) Å),40 [(Me3Si)2CH]2GaTeSi(SiMe3)3 (2.535(1) Å),41 whereas the Ga–Te bonds observed in Ga{TeSi(SiMe3)3}3 (av. 2.496(6) Å)42 as well as in Tp#GaTe (2.422(1)) (Tp# = tris(3,5-di-tert-butylpyrazolyl)hydroborato), to date the only compound containing a terminal Ga–Te double bond, are significantly shortened.43
Reactions of trivalent group 13 compounds (MR3, MH3; M = Al, Ga, In) with elemental chalcogens E or chalcogen sources such as R3PE (E = O, S, Se, Te) typically occur with insertion of the chalcogen atom into the M–C/H bond and subsequent formation of dimeric ([R2M-μ-ER]2) or tetrameric ([RME]4) compounds, whereas divalent (R4M2) as well as monovalent group 13 compounds RM (M = Al, Ga, In, Tl) react with insertion of the chalcogen atom into the M–M bond.44–49 In addition, E–Br bond cleavage reactions of PhEBr (E = Se, Te) upon treatment with the monovalent In(I) cluster [(Me3Si)3C]4In450 as well as of Te–Te bond cleavage reactions of Ph2Te2 by reaction with Ga(I)26 and In(I) compounds51 as well as with base-stabilized GaH330 were reported. However, to the best of our knowledge, the cleavage of the Te–C bond of diorganyltellanes upon reaction with monovalent group 13 compounds RM has never been observed before.
Conclusions
LGa was found to selectively insert into the Te–Te bond of diphenylditellane Ph2Te2 as well as one Te–C bond of diisopropyltellane i-Pr2Te, resulting in the formation of LGa(TePh)22 and LGa(i-Pr)Tei-Pr 3. Moreover, its reaction with elemental tellurium yielded the Te-bridged compound [LGa-μ-Te]21. We are currently investigating reactions of the somewhat stronger reducing agent LAl with various group 16 precursors. A previous report showed that LAl reacted with elemental sulfur with subsequent formation of [LAl(μ-S3)]2, containing an unusual Al2S6 ring.52Experimental
All manipulations were performed in an atmosphere of purified argon using standard Schlenk and glove-box techniques. Toluene and hexane were dried using a mBraun Solvent Purification System. THF was carefully dried over Na/K. Deuterated solvents were dried over activated molecular sieves (4 Å) and degassed prior to use. Anhydrous nature of the solvents was verified by Karl Fischer titration. The 1H (300 MHz), 13C{1H} (75.5 MHz) and 125Te NMR (95 MHz) (δ in ppm) spectra were recorded using a Bruker Avance DPX-300 spectrometer. The 1H and 13C{1H} spectra were referenced to internal C6D5H (1H: δ = 7.154; 13C: δ = 128.39) and C6D5CHD2 (1H: δ = 2.09; 13C: δ = 20.40). 125Te NMR spectra were referenced to Na2TeO3 in D2O. The microanalyses were performed at the elemental analysis laboratory of the University of Duisburg-Essen. IR spectra were measured with an ALPHA-T FT-IR spectrometer equipped with a single reflection ATR sampling module. Elemental tellurium and Ph2Te2 were commercially available and used as received, whereas i-Pr2Te was freshly prepared according to a slightly modified procedure described for the synthesis of Te(SiMe3)2.53 In necessary cases, the reaction mixtures were filtered using a oven dried Teflon cannula (3.5 mm D) in which one end of the tip was wrapped with Whatman® glass microfiber filters (CAT no. 1823-025) using Teflon tape.Single crystal X-ray diffraction
Crystallographic data of 1–3, which were collected on a Bruker D8 Kappa APEX2 diffractometer (MoKα radiation, λ = 0.71073 Å) at 130(1) K (1a), 100(1) K (1b), 100(1) K (2) and 100(1) K (3), are summarized in Table 1. The solid-state structures of 1–3 are shown in Fig. 1–3. The structures were solved by Direct Methods (SHELXS-97) and refined anisotropically by full-matrix least-squares on F2 (SHELXL-97).54,55 Absorption corrections were performed semi-empirically from equivalent reflections on basis of multi-scans (Bruker AXS APEX2). The toluene molecule of 1b is disordered via 2/m symmetry. Further solvent molecules that could not be modelled sufficiently were removed by a PLATON/SQUEEZE run.56 The crystal quality of 2 was rather low consequently the quantitative results of the model should be carefully assessed. The resolution of the data of 3 was high enough to show a mismatch of the calculated position of H2 and the residual electron density. Consequently H2 was refined freely with its displacement parameter constrained to be 1.2 times Ueq of the connected C atom. Other hydrogen atoms were refined using a riding model or rigid methyl groups.Acknowledgements
S.S. likes to thank the University of Duisburg-Essen for financial support.Notes and references
- N. J. Hardman, B. E. Eichler and P. P. Power, J. Chem. Soc., Chem. Commun., 2000, 1991–1992 RSC.
- N. J. Hardman and P. P. Power, ACS Symp. Ser., 2002, 822, 2–15 CrossRef CAS PubMed.
- C.-H. Chen, M.-L. Tsai and M.-D. Su, Organometallics, 2006, 25, 2766–2773 CrossRef CAS.
- M. S. Hill, P. B. Hitchcock and R. Pontavornpinyo, J. Chem. Soc., Dalton Trans., 2005, 273–277 RSC.
- N. J. Hardman, P. P. Power, J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, J. Chem. Soc., Chem. Commun., 2001, 1866–1867 RSC.
- C. Gemel, T. Steinke, M. Cokoja, A. Kempter and R. A. Fischer, Eur. J. Inorg. Chem., 2004, 4161–4176 CrossRef CAS.
- A. Kempter, C. Gemel, T. Cadenbach and R. A. Fischer, Organometallics, 2007, 26, 4257–4264 CrossRef CAS.
- A. Kempter, C. Gemel and R. A. Fischer, Chem. – Eur. J., 2007, 13, 2990–3000 CrossRef CAS PubMed.
- S. Schulz, Chem. – Eur. J., 2010, 16, 6416–6428 CrossRef CAS PubMed.
- R. J. Baker and C. Jones, Coord. Chem. Rev., 2005, 249, 1857–1869 CrossRef CAS PubMed.
- Y.-C. Tsai, Coord. Chem. Rev., 2012, 256, 722–758 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- G. Prabusankar, A. Kempter, C. Gemel, M.-K. Schröter and R. A. Fischer, Angew. Chem., Int. Ed., 2008, 47, 7234–7237 CrossRef CAS PubMed.
- G. Prabusankar, C. Gemel, P. Parameswaran, C. Flener, G. Frenking and R. A. Fischer, Angew. Chem., Int. Ed., 2009, 48, 5526–5529 CrossRef CAS PubMed.
- A. Seifert, D. Scheid, G. Linti and T. Zessin, Chem. – Eur. J., 2009, 15, 12114–12120 CrossRef CAS PubMed.
- N. J. Hardman and P. P. Power, Inorg. Chem., 2001, 40, 2474–2475 CrossRef CAS.
- N. J. Hardman and P. P. Power, J. Chem. Soc., Chem. Commun., 2001, 1184–1185 RSC.
- N. J. Hardman, C. Cui, H. W. Roesky, W. H. Fink and P. P. Power, Angew. Chem., Int. Ed., 2001, 40, 2172–2174 CrossRef CAS.
- C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, J. Chem. Soc., Chem. Commun., 2014, 50, 12382–12384 RSC.
- C. Ganesamoorthy, D. Bläser, C. Wölper and S. Schulz, Angew. Chem., Int. Ed., 2014, 53, 11587–11591 CrossRef CAS PubMed.
- S. Gondzik, S. Schulz, D. Bläser and C. Wölper, J. Chem. Soc., Chem. Commun., 2014, 50, 1189–1191 RSC.
- P. Steiniger, G. Bendt, D. Bläser, C. Wölper and S. Schulz, J. Chem. Soc., Chem. Commun., 2014, 50, 15461–15463 RSC.
- S. Heimann, A. Kuczkowski, D. Bläser, C. Wölper, R. Haak, G. Jansen and S. Schulz, Eur. J. Inorg. Chem., 2014, 4858–4864 CrossRef CAS.
- S. Heimann, S. Schulz, D. Bläser and C. Wölper, Eur. J. Inorg. Chem., 2013, 4909–4915 CAS.
- S. Heimann, D. Bläser, C. Wölper and S. Schulz, Organometallics, 2014, 33, 2295–2300 CrossRef CAS.
- R. J. Baker, C. Jones and M. Kloth, J. Chem. Soc., Dalton Trans., 2005, 2106–2110 RSC.
- K. S. Klimek, J. Prust, H. W. Roesky, M. Noltemeyer and H.-G. Schmidt, Organometallics, 2001, 20, 2047–2051 CrossRef CAS.
- M. C. Copsey and T. Chivers, J. Chem. Soc., Chem. Commun., 2005, 4938–4940 RSC.
- W. Uhl, L. Cuypers, T. Spies, F. Weller, B. Habrecht, M. Conrad, A. Greiner, M. Puchner and J. H. Wendorff, Z. Anorg. Allg. Chem., 2003, 629, 1124–1130 CrossRef CAS.
- W. J. Grigsby, C. L. Raston, V.-A. Tolhurst, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 1998, 2547–2556 RSC.
- O. Kluge and H. Krautscheid, Inorg. Chem., 2012, 51, 6655–6666 CrossRef CAS PubMed.
- S. Schulz, M. Andruh, T. Pape, T. Heinze, H. W. Roesky, L. Häming, A. Kuhn and R. Herbst-Irmer, Organometallics, 1994, 13, 4004–4007 CrossRef CAS.
- C. H. Harlan, E. G. Gillan, S. G. Bott and A. R. Barron, Organometallics, 1996, 15, 5479–5488 CrossRef CAS.
- U. App and K. Merzweiler, Z. Anorg. Allg. Chem., 1997, 623, 478–482 CrossRef CAS.
- W. Uhl, M. Benter, W. Saak and P. G. Jones, Z. Anorg. Allg. Chem., 1998, 624, 1622–1628 CrossRef CAS.
- M. A. Banks, O. T. Beachley Junior, H. J. Gysling and H. R. Luss, Organometallics, 1990, 9, 1979–1982 CrossRef CAS.
- C. Schnitter, A. Klemp, H. W. Roesky, H.-G. Schmidt, C. Ropken, R. Herbst-Irmer and M. Noltemeyer, Eur. J. Inorg. Chem., 1998, 2033–2039 CrossRef CAS.
- M. R. Kopp and B. Neumüller, Z. Anorg. Allg. Chem., 1997, 623, 796–804 CrossRef CAS.
- O. Kluge, S. Gerber and H. Krautscheid, Z. Anorg. Allg. Chem., 2011, 637, 1909–1921 CrossRef CAS.
- W. Uhl and U. Schütz, Organometallics, 1995, 14, 1073–1075 CrossRef CAS.
- W. Uhl, M. Layh, G. Becker, K. W. Klinkhammer and T. Hildenbrand, Chem. Ber., 1992, 125, 1547–1551 CrossRef CAS.
- S. P. Wuller, A. L. Seligson, G. P. Mitchell and J. Arnold, Inorg. Chem., 1995, 34, 4854–4861 CrossRef CAS.
- M. C. Kuchta and G. Parkin, Inorg. Chem., 1997, 36, 2492–2493 CrossRef CAS.
- C. J. Allan and C. L. B. Macdonald, in Comprehensive Inorganic Chemistry II, ed. J. Reedijk and K. Poeppelmeier, Elsevier, Oxford, 2013, vol. 1, pp. 485–566 Search PubMed.
- S. Schulz, in Topics in Organometallic Chemistry, Vol. 41 Modern Organoaluminum Reagents, ed. S. Woodward and S. Dagorne, Springer Verlag, 2013 Search PubMed.
- S. Aldridge, in The Group 13 Metals Aluminum, Gallium, Indium and Thallium, ed. S. Aldridge and A. J. Downs, Wiley, 2011 Search PubMed.
- S. Schulz, in Comprehensive Organometallic Chemistry III, ed. R. H. Crabtree and D. M. P. Mimgos, Compounds of Group 13–15, ed. C. E. Housecroft, Elsevier, 2007, vol. 3 Search PubMed.
- J. A. J. Pardoe and A. J. Downs, Chem. Rev., 2007, 107, 2–45 CrossRef CAS PubMed.
- G. He, O. Shynkaruk, M. W. Lui and E. Rivard, Chem. Rev., 2014, 114, 7815–7880 CrossRef CAS PubMed.
- W. Uhl, F. M. de Andrade, C. Peppe, J. Kösters and F. Rogel, J. Organomet. Chem., 2007, 692, 869–873 CrossRef CAS PubMed.
- C. Peppe, F. Molinos de Andrade and W. Uhl, J. Organomet. Chem., 2009, 694, 1918–1921 CrossRef CAS PubMed.
- Y. Peng, H. Fan, V. Jancik, H. W. Roesky and R. Herbst-Irmer, Angew. Chem., Int. Ed., 2004, 43, 6190–6192 CrossRef CAS PubMed.
- M. R. Detty and M. D. Seidler, J. Org. Chem., 1982, 47, 1354–1356 CrossRef CAS.
- SHELXS-97, G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, Universität Göttingen, 1997 CrossRef CAS PubMed See also: G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- PLATON/SQUEEZE, P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 194–201 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic data of 1a, 1b, 2, and 3 and spectroscopic details (1H, 13C, 125Te, IR). CCDC 1040132–1040135. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00172b |
| ‡ The crystallographic data of 1a, 1b, 2, and 3 (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication nos. CCDC-1040132 (1a), CCDC-1040134 (1b), CCDC-1040133 (2) and CCDC-1040135 (3). |
| This journal is © The Royal Society of Chemistry 2015 |