
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, structural studies and ligand influence on the stability of aryl-NHC stabilised trimethylaluminium complexes†
Melissa
Wu. M
a,
Arran M.
Gill
b,
Lu
Yunpeng
a,
Laura
Falivene
c,
Li
Yongxin
a,
Rakesh
Ganguly
a,
Luigi
Cavallo
cd and
Felipe
García
*a
aSchool of Physical and Mathematical Sciences, Division of Chemistry and Biological Chemistry, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: Fgarcia@ntu.edu.sg; Fax: (+65) 67911961
bChemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield Southampton, SO17 1BJ, UK
cKing Abdullah University of Science and Technology, Building 9, Level 4, Room #4358, Thuwal, 23955-6900, Saudi Arabia
dDipartimento di chimica, Univerità di Salerno, Via Ponte don Melillo, 84054, Fisciano, Italia
First published on 13th February 2015
Abstract
Treatment of a series of aromatic NHCs (IMes, SIMes, IPr and SIPr) with trimethylaluminium produced their corresponding Lewis acid–base adducts: IMes·AlMe3 (1), SIMes·AlMe3 (2), IPr·AlMe3 (3), and SIPr·AlMe3 (4). These complexes expand the few known examples of saturated NHC stabilised Group 13 complexes. Furthermore, compounds 1–4 show differential stability depending on the nature of the NHC ligand. Analyses of topographic steric maps and NHC %VBur were used to explain these differences. All the compounds have been fully characterised by multinuclear NMR spectroscopy, IR and single crystal X-ray analysis together with computational studies.
Introduction
Since the discovery of the first stable N-heterocyclic carbene (NHC) by Arduengo in 1991,1 these compounds have been extensively used as ligands in the chemistry of transition metals.2,3 Similarly to their phosphine counterparts, transition metal complexes containing various finely tuned NHC ligands have been used in a wide range of catalytic processes.2–4 As NHCs are highly nucleophilic Lewis bases, they have also been used to stabilise many Group 13 complexes.5,6 Our interest in NHC-Group 13 complexes arises from the discovery that their properties and reactivities have not been thoroughly studied. However, their potential has been demonstrated for a diverse range of applications; for example, sterically demanding NHC ligands have been used to synthesise neutral B–B double and triple bonded species,7 as well as stabilising a neutral aromatic Ga6 octahedron cluster.8 In addition, NHCs that do not form stable Lewis acid–base adducts, forming frustrated Lewis pair (FLP) systems, have shown interesting properties in the activation of small molecules.9 However, there is still much to be explored in terms of their properties and reactivity. The majority of NHC–aluminium complexes reported comprise hydride and halide groups (AlXnH3−n, n = 0, 1, 2).5 In contrast, there are only a few examples of aluminium alkyl complexes.6 In particular, in the case of the simplest alkyl substituent, trimethylaluminium, only five complexes have been fully characterised: IiPrMe (IiPrMe = 1,3-isopropyl-4,5-dimethyl-imidazol-2-ylidene, A);6h ItBu (ItBu = 1,3-di-tert-butylimidazol-2-ylidene, B);6d IMes (IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazol-2-ylidene, C);6e a bidentate amino ligand (D)6e and a chiral imidazolium sulfonate (E)6f (Fig. 1). Furthermore, to the best of our knowledge, there are only a few known examples of other saturated NHC stabilised Group 13 metals that have been fully characterised.5b Here, we report the synthesis, characterisation and theoretical studies of a series of NHC aluminium alkyl complexes.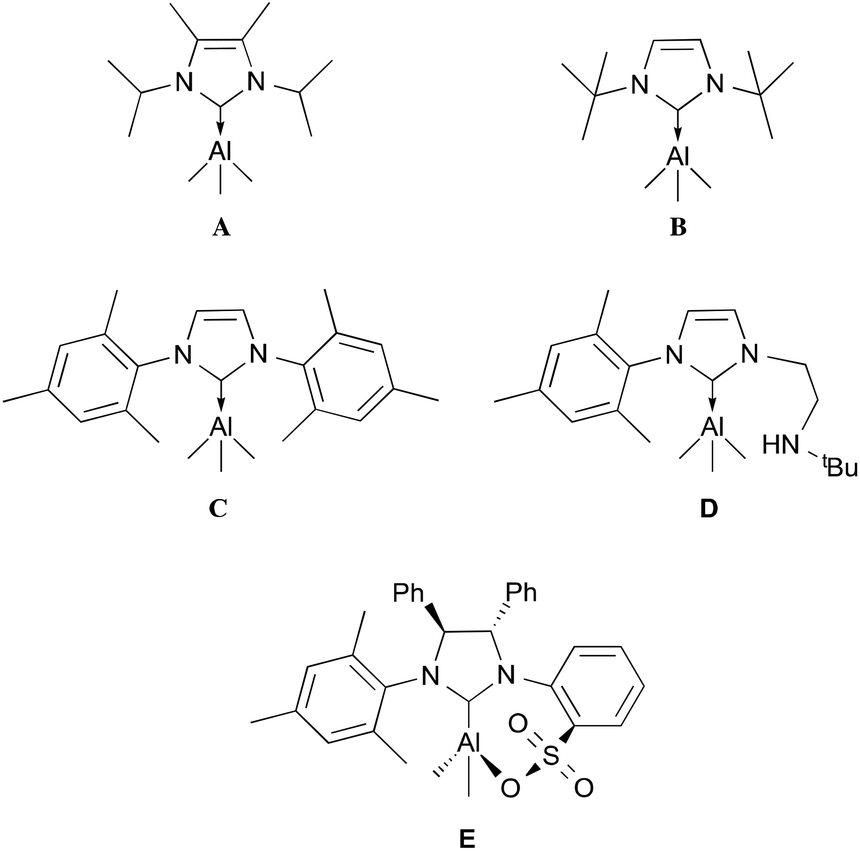
Results and discussion
Synthesis of complexes 1–4
The general synthetic route for the synthesis of target complexes, described in Scheme 1,6e,h involved the treatment of 1 equiv. of carbene (IMes, SIMes, IPr or SIPr) with trimethylaluminium (1 M in toluene), resulting in the isolation of their respective adducts: IMes·AlMe3 (1); SIMes·AlMe3 (2); IPr·AlMe3 (3) and SIPr·AlMe3 (4). Complex 1 was previously reported by Ong et al. using an analogous synthetic route,6e and has been included in this report to maintain the rigour of our studies (Fig. 2).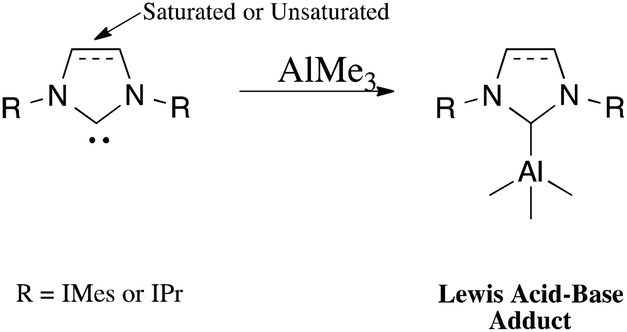 | ||
| Scheme 1 Synthetic strategy for the NHC adducts. | ||
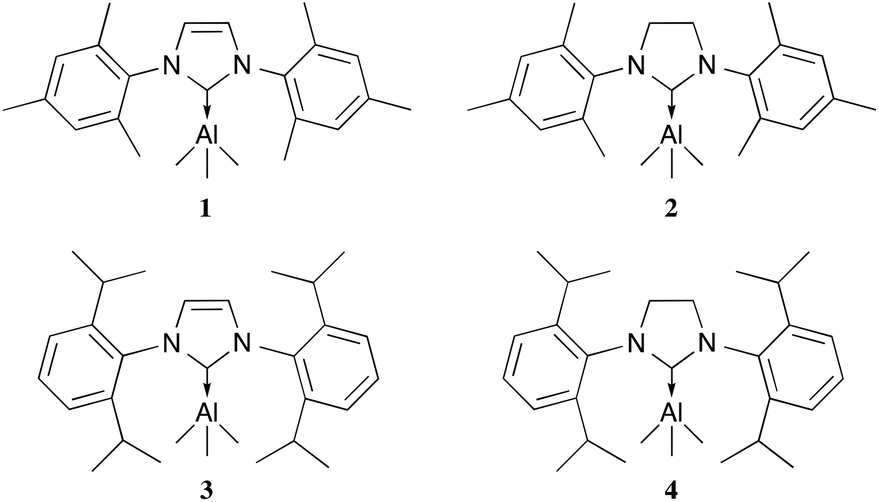 | ||
| Fig. 2 NHC trimethylaluminium complexes synthesised. IMes·AlMe3 (1), SIMes·AlMe3 (2), IPr·AlMe3 (3) and SIPr·AlMe3 (4). | ||
Compounds 1–4 are highly air- and moisture-sensitive; traces of decomposition were consistently observed during their characterisation, making their characterisation tedious. This was particularly pronounced in the case of complexes 3 and 4 where peaks corresponding to the imidazolylidenes were always present on the 1H and 13C NMR spectra. Moreover, this was also observed in the solid state, where argon-gas-stored samples of 3 and 4 slowly decomposed to imidazolylidene and imidazolinylidene respectively and other unidentified side-products at room temperature (see ESI†), whereas solids 1 and 2 can be stored over long periods of time without any observable decomposition.
Crystallographic studies of complexes 1–4
Single-crystal X-ray structures of complexes 1–4 are shown in Fig. 3–6. Complexes 2 and 3 crystallised out as two crystallographically independent but chemically equivalent molecules; hence only one molecule will be described herein (Table 1).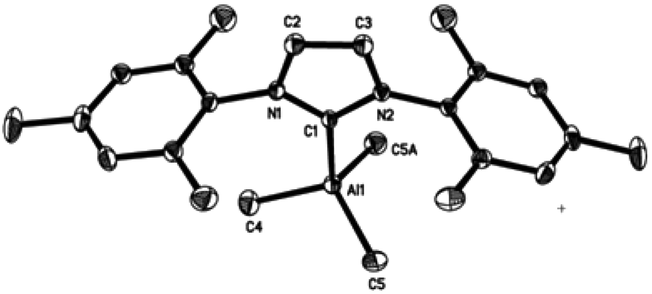 | ||
| Fig. 3 Molecular structure of IMes·AlMe3 (1). Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths [Å] and angles [°] for 1: Al(1)–C(1) 2.098(2), Al(1)–C(4) 1.978(2), Al(1)–C(5) 1.991(1), C(1)–N(1) 1.365(2), C(1)–N(2) 1.364(2), C(2)–N(1) 1.385(2), C(3)–N(2) 1.382(2), C(2)–C(3) 1.353(2), C(4)–Al(1)–C(5) 110.8(1), C(4)–Al(1)–C(1) 108.7(1), C(5)–Al(1)–C(5A) 114.4(1), C(5)–Al (1)–C(1) 105.8(1), N(1)–C(1)–N(2) 103.5. | ||
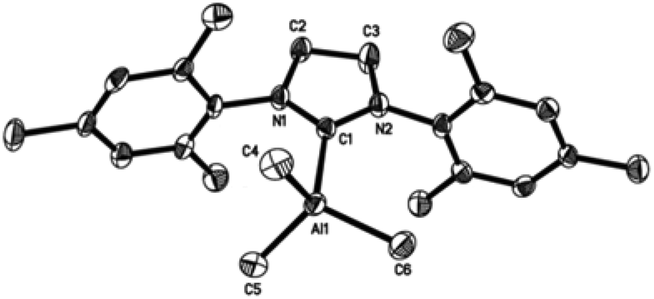 | ||
| Fig. 4 Molecular structure of SIMes·AlMe3 (2). Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and angles [°] for 2: Al(1)–C(1) 2.112(6), Al(1)–C(4), 1.984(6), Al(1)–C(5) 1.994(7), Al(1)–C(6) 1.983(6), C(1)–N(1) 1.341(7), C(1)–N(2) 1.343(7), C(2)–N(1) 1.477(7), C(3)–N(2) 1.474(7), C(2)–C(3) 1.534(8), C(4)–Al(1)–C(5) 110.7(3), C(4)–Al(1)–C(1) 105.7(3), C(4)–Al(1)–C(6) 112.3(3), C(5)–Al(1)–C(6) 111.8(3), C(5)–Al(1)–C(1) 107.6(3), C(6)–Al(1)–C(1) 108.4(2), N(1)–C(1)–N(2) 107.3(5). | ||
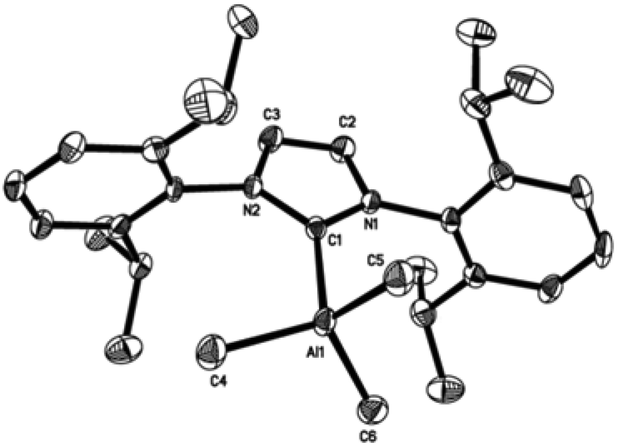 | ||
| Fig. 5 Molecular structure of IPr·AlMe3 (3). Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and angles [°] for 3: Al(1)–C(1) 2.103(3), Al(1)–C(4) 1.992(3), Al(1)–C(5) 1.994(3), Al(1)–C(6) 1.994(3), C(1)–N(1) 1.370(4), C(1)–N(2) 1.370(4), C(2)–N(1) 1.387(4), C(3)–N(2) 1.385(4), C(2)–C(3) 1.351(4), C(4)–Al(1)–C(5) 111.5(1), C(4)–Al(1)–C(1) 109.6(1), C(4)–Al(1)–C(6) 111.3(1), C(5)–Al(1)–C(6) 113.6(1), C(5)–Al(1)–C(1) 104.7(1), C(6)–Al(1)–C(1) 105.7(1), N(1)–C(1)–N(2) 103.1(2). | ||
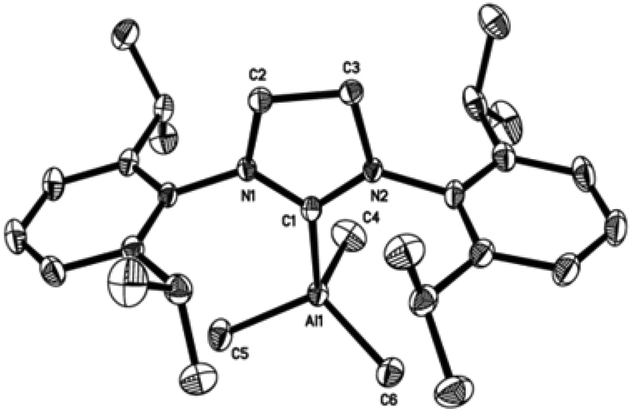 | ||
| Fig. 6 Molecular structure SIPr·AlMe3 (4). Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms were omitted for clarity. Selected bond lengths [Å] and angles [°] for 4: Al(1)–C(1) 2.127(2), Al(1)–C(4) 1.986(3), Al(1)–C(5) 1.992(2), Al(1)–C(6) 1.980(2), C(1)–N(1) 1.346(2), C(1)–N(2) 1.345(2), C(2)–N(1) 1.477(2), C(3)–N(2) 1.483(2), C(2)–C(3) 1.521(3), C(4)–Al(1)–C(5) 109.4(1), C(4)–Al(1)–C(1) 100.7(1), C(4)–Al(1)–C(6) 114.0(1), C(5)–Al(1)–C(6) 111.5(1), C(5)–Al(1)–C(1) 110.9(1), C(6)–Al(1)–C(1) 109.5(1), N(1)–C(1)–N(2) 107.1(1). | ||
Compounds 1–4 adopt a distorted tetrahedral geometry at the Al centre, with Al–Ccarbene bond lengths ranging from 2.098 to 2.127 Å, which are consistent with previously reported trimethylaluminium complexes (cf. 2.124(6) Å,6h 2.162(2) Å,6d 2.097(2) Å,6e 2.074(2) Å6e and 2.078(3) Å6f for A–E respectively). Interestingly, the Al–Ccarbene bond distance of SIPr (4) was similar to the less sterically bulky IiPrMe (A) (2.127(2) and 2.124(6) Å respectively). Moreover, Huynh et al. using an NHC-NMR spectroscopic probe reported that saturated NHC (sNHC) moieties are marginally more basic (i.e., stronger σ-donors) than their unsaturated (uNHC) counterparts (decreasing σ-donor strength SIPr ∼ SIMes > IPr > IMes). In our case the 1H NMR chemical shift of the methyl groups on the aluminium centre also supports Huynh's observations. In addition, a slight bond lengthening consistent with this property is expected for 2 and 4 with respect to 1 and 3 (containing sNHC and uNHC respectively).4g,10,11 However, clear bond lengthening is only observed between 3 and 4, since the difference between 1 and 2 could be attributed to statistical error range (3σ). Complex B (i.e., ItBu) has the longest reported Al–Ccarbene bond length reported to date, mainly due to additional steric hindrance introduced by the large tert-butyl groups (vide infra), 36.9%VBur, resulting in the complex being susceptible towards isomerization or decomposition depending on the experimental conditions (solvent dependent).6d
Spectroscopic studies of complexes 1–4
The 1H and 13C NMR spectra obtained for complexes 1–4 were consistent with the low temperature X-ray crystallographic analysis. The 1H and 13C NMR spectra for these compounds display singlets at δH −0.78 to −0.91 and at δC −7 ppm respectively. This is indicative of the presence of methyl groups on the aluminium centre. The IR spectra of these complexes show relatively strong stretching signals at around 620 cm−1, confirming the presence of these methyl groups.12 Moreover, the formation of the complex is further indicated by the upfield shifting of the Ccarbene signal that is consistent with a carbene–metal bond (Table 2).13The optimised geometrical parameters, bond lengths and angles for complexes 1–4 calculated using PBE0/6-311G(d,p) model chemistry are in good agreement with the experimental values obtained from the single-crystal X-ray diffraction studies. Furthermore, the calculated 1H and 13C NMR spectra using B972/6-311+G(2d,p) on the optimised geometries were consistent with the experimental data obtained, which provided further validation of the identity of the complexes synthesised (see ESI†).
Lewis acid–Lewis base properties
Comparison between NHCs and phosphines has been carried out to assess the relative donor abilities (Lewis basicity) of this important family of ligands. For this reason, NHC–Al complexes 1–4 were compared to selected phosphine-Al counterparts. Similarly to what Barron et al. reported with trimethylaluminium phosphine complexes,14 the lengths of the Al–C bonds increase (cf. 1.956 Å for AlMe3, 1.985 Å, 1.987 Å 1.993 Å and 1.986 Å for compounds 1–4 respectively) and the C–Al–C angles decrease (ca. 120° for AlMe3 and respective average angles 112.6°, 111.6°, 112.1°, 111.7° for 1–4) upon coordination to the NHC. Both changes indicate increased p-character in the Al–C bonds on changing from planar to tetrahedral geometries. The greater distortion from planarity observed for NHC complexes compared with their phosphine counterparts (see Table 3), indicates higher Lewis basicity of the former. This is further evidenced by the 1H NMR chemical shift of the methyl groups on the aluminium centre. Complexes 1–4 show signals at higher fields (δH −0.78 to −0.91) than previously reported basic trimethylaluminium phosphine complexes (cf. δH −0.02–0.41)15 indicative of a stronger donation from the NHC to the aluminium center (see ESI† and Table 3).| Complex | Al–Mea [Å] | C–Al–Ca [°] | 1H [AlCH3] (ppm) |
|---|---|---|---|
| a Average values were taken for both bond lengths and angles. | |||
| 1 | 1.985 | 112.6 | −0.78 |
| 2 | 1.987 | 111.6 | −0.86 |
| 3 | 1.993 | 112.1 | −0.86 |
| 4 | 1.986 | 111.7 | −0.91 |
AlMe3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16,17 16,17 |
1.956 | 123.2 | −0.35 |
| Me3P·AlMe314 |
1.973 | 117.1 | −0.41 |
| Ph3P·AlMe314,18 |
1.981 | 116.6 | −0.09 |
| (o-tolyl)3P·AlMe314 |
1.874 | 113.9 | −0.31 |
The Lewis acidity of trihalide and trihydride aluminium centres within NHC–aluminium complexes has been previously discussed in the literature.5a,d In the case of complexes 1–4, the trimethylaluminium moiety is found to be a poorer Lewis acid as compared to hydrides and halides. This was evident from the carbenic carbon to aluminium bond distances observed in the IMes (1) and IPr (3) complexes. The Lewis acidity trend, AlMe3 < AlH3 < AlX3, can be illustrated by Al–Ccarbene bond distances: 2.034(3) Å for IMes·AlH3;5t 2.017(2) Å for IMes·AlCl3;5h 2.056(2) Å for IPr·AlH3,5n and 2.031(2) Å for IPr·AlI3.5g The same tendency was also observed in the mixed alane gallane halide complexes.5d,f In the case of indium and thallium complexes, Jones et al. also observed the same Lewis acidic behaviour during the synthesis of bis-NHC (i.e., NHC–(CH2)2–NHC) group 13 complexes. Their studies showed monometallic pentacoordinate indium and thallium halide complexes containing chelating bis-NHC moieties, whereas hydride counterparts formed monodentate tetra-coordinate bimetallic species (i.e., R3E←NHC–(CH2)2–NHC→ER3) indicating the higher Lewis acidity of the former.5m,o,q Furthermore, the relative Lewis acidity can also be assessed using 13C NMR spectroscopy, despite the fact that many Al–Ccarbene signals have not been reported in the literature due to the quadrupolar nature of the aluminium metal centre to which they are attached. Nevertheless, the chemical shifts observed for complexes 1–4 show that trimethylaluminium is a poorer electron acceptor compared with AlH3 and AlX3 since the corresponding 13C NMR signals for Al–Ccarbene, shown in Table 2, were more downfield shifted with respect to hydride and halide counterparts (Al–Ccarbene signals at δC 174.3 for ItBu (B),6dδC 175.3 for IMes·AlH3,5t 153.9 for IMes·AlI3, and δC 153.3 for IPr·AlI35g).
Stability studies
Unstable NHC–AlMe3 complexes have previously been reported; for example, the tert-butyl NHC complex B isomerised to an ‘abnormal’ NHC–AlMe3 species in THF or toluene.6d We will use complex B as a benchmark throughout our comparative studies. Since the isomerization/decomposition of B was attributed to steric factors, and a standard parameter for quantifying the steric properties of NHCs is the percent buried volume, %VBur, this parameter was used to compare complexes 1–4 with other NHC·AlR3 species previously reported in the literature (Table 4).4b,c The %VBur for each complex was calculated with the Al–NHC bond distance fixed at the experimental value obtained by X-ray diffraction studies and also at 2.0 Å, in order to provide a point of comparison independent of the Al–NHC distances.| Entry | Complex | Al–Ccarbene [Å] | %VBur R = X-ray | %VBurR = 2.0 Å | E diss (kJ mol−1) |
|---|---|---|---|---|---|
| a Value obtained using DFT calculations with the PBE0/6-311G(d,p) basis set. | |||||
| 1 | IMes·AlMe3 (1) | 2.098(2) | 31.7 | 33.7 | 114.47 |
| 2 | SIMes·AlMe3 (2) | 2.112(6) | 32.0 | 34.1 | 104.76 |
| 3 | IPr·AlMe3 (3) | 2.103(3) | 34.2 | 36.2 | 97.14 |
| 4 | SIPr·AlMe3 (4) | 2.127(2) | 36.1 | 38.5 | 79.82 |
| 5 | IiPrMe (A) | 2.124(6) | 25.5 | 27.2 | 132.59 |
| 6 | ItBu·AlMe3 (B) | 2.162(2) | 34.3 | 36.9 | 59.33 |
| 7 | IMes·AlMe3 (C) | 2.097(2) | 31.8 | 33.6 | 114.47 |
| 8 | IMes·Al(C6F5)3 | 2.061(3) | 31.2 | 32.7 | 157.79 |
| 9 | IMe·Al(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CtBu)3 CtBu)3 |
2.051(2) | 25.3 | 25.9 | 161.73 |
| 10 | IPr·Al((CH2)3CH3)3 | 2.118(2) | 32.6 | 34.9 | 85.85 |
| 11a | SItBu·AlMe3 | 2.229 | 33.3 | 37.6 | 38.59 |
Calculations revealed that the buried volume of the new NHC complexes was 4 > 3 > 2 > 1. In order to provide a meaningful assessment of the steric influence of the NHC moiety on the overall stability of the NHC–AlMe3 complexes, the %VBur values of previously characterised counterparts were included. With this inclusion, the overall order is 4 > B > 3 > 2 > C ≈ 1 > A. It can be noted that complex B occupies a larger volume than that calculated for 1–2, and is comparable to that of 3 but is surprisingly lower than that of 4 (cf. 36.9% in B). Since the %VBur of compound 3 is larger than that of 1 and 2 and no decomposition was observed for either of the latter, the onset of decomposition may be attributed to the larger volume occupied by the isopropylphenyl groups as compared to the mesityl groups. The lower stability exhibited by the sterically encumbered complex B was previously rationalised by Dagorne et al. using the congested nature of the NHC present (36.9%VBur). Consequently, the %VBur calculated for 3 (36.2%, comparable to B) and for 4 (38.5%, greater than B) rationalises their lower stability (cf.1 and 2). To gain insight into the molecular level of the steric impact of the different NHCs, the topographic steric maps for compounds 1–4 and A–C were calculated (see ESI†). A comparative analysis of the topographic maps of complexes 2 and 4, chosen as representatives of a stable and of an unstable system, is reported in Fig. 7. The steric contour maps reveal that the distribution of the steric bulk of the ligand in 2 is quite symmetrical around the metal, with large grooves between the two mesityl rings. As expected, the enhanced steric hindrance in 4 is mainly localised around the bulkier ortho isopropyl groups, blocking the grooves between the two N-substituents. The difference in the nature of the distribution of the NHC ligands around the metal centre (similar maps are found for 1 and 3, see ESI†) can be related to the lower stability of 3 and 4 as compared to 1 and 2.
 | ||
| Fig. 7 Topographic steric maps of the SIMes and SIPr ligands in 2 and 4. The iso-contour curves of the steric maps are in Å. The maps have been obtained starting from the crystallographic data of the Al–NHC complexes (CIF), with the Al–Ccarbene distance fixed at 2.0 Å. The xz plane is the mean plane of the NHC ring, whereas the yz plane is the plane orthogonal to the mean plane of the NHC ring, and passing through the Ccarbene atom of the NHC ring. | ||
At this stage, it is also worth doing a comparative analysis of the topographic steric map of B, as the only reported unstable NHC–AlMe3 complex, with that of 4 (Fig. 8). The topographic steric map of complex B shows the two top quadrants being slightly more sterically hindered. However, this topographical asymmetry is lower compared to 4, where the distribution of the steric bulk is much more localised in the top left and top right quadrants. This difference is even more evident looking at the %VBur representative of each single quadrant, i.e. 39.6–40.2% for Bvs. 43.1–50.7% for 4. Once again, the greater localization of the ligand steric hindrance in one or two quadrants around the metal centre may be the reason for the lower stability of the complexes, in the case of 4 as compared to B.
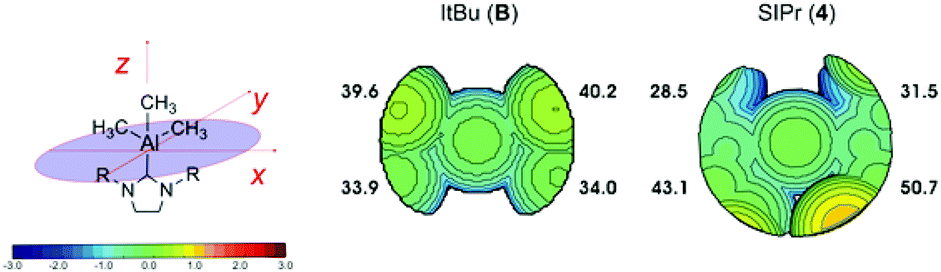 | ||
| Fig. 8 Topographic steric maps of the ItBu and SIPr ligands in B and 4. The iso-contour curves of the steric maps are in Å. The maps have been obtained starting from the crystallographic data of the Al–NHC complexes (CIF), with the Al–Ccarbene distance fixed at 2.0 Å. The xz plane is the mean plane of the NHC ring, whereas the yz plane is the plane orthogonal to the mean plane of the NHC ring, and passing through the Ccarbene atom of the NHC ring. | ||
In addition to the %VBur and topographic steric maps, bond dissociation energies were also evaluated to further rationalise the stability differences observed.4f DFT calculations show that the bond dissociation energy of complexes 1–4 decreases with increasing steric volume of the corresponding NHC: 1 > 2 > 3 > 4, which further corroborated the observation that complexes 1 and 2 were less susceptible to dissociation as compared to 3 and 4 (114.47 (1), 104.76 (2), 97.14 (3), and 79.82 (4) kJ mol−1 for 1–4 respectively). With the inclusion of the dissociation energy calculated for all NHC trimethylaluminium complexes, the order is as follows: B > 4 > 3 > 2 > 1 ≈ C > A (Table 5). It is worth noting that the %VBur calculated for complex 4 is higher than that calculated for B; however its Ediss is lower. This discrepancy may be explained by the differing electronic properties of the SIPr and ItBu NHCs moieties. On the one hand, going from the unsaturated (uNHC) to saturated (sNHC) NHCs contributes to an increased donor ability of the latter (sNHC > uNHC) (vide supra). On the other hand, the presence of withdrawing aryl substituents in the NHC leads to a decreased donor ability (alkyl-NHC > aryl-NHC). The opposite electronic effects present in both SIPr and ItBu (i.e., the donating effect of the sp3 backbone and withdrawing effects of the aryl groups in SIPr vs. the less donating sp2 backbone combined with more donating alkyl groups in ItBu) make the relative NHC→metal donation properties difficult to predict.10 However experimental evidence suggests that the SIPr N-heterocyclic carbene moiety present in 4 is a better donor ligand than ItBu since the 1H NMR chemical swift of the methyl group on 4 (δH −0.91) is more upfield than that found for B (δH −0.73). This is also supported by 11B NMR studies on NHC–BX3 species, where the chemical shift for the ItBu–BCl3 complex is more downfield than its IPr analogue.19 However, the overall stability of these complexes is a concomitant balance between the electronic and steric properties of the NHC moieties present.4f
| Complex | %VBur R = X-ray | %VBurR = 2.0 Å | E diss (kJ mol−1) | Overall stability |
|---|---|---|---|---|
| a Structure was optimised using PBE0/6-311G(d,p) model chemistry. | ||||
| SItBu·AlMe3a | 33.3 | 37.6 | 38.59 | Unstable |
| ItBu·AlMe3 (B) | 34.3 | 36.9 | 59.33 | |
| SIPr·AlMe3 (4) | 36.1 | 38.5 | 79.82 | |
| IPr·AlMe3 (3) | 34.2 | 36.2 | 97.14 | |
| SIMes·AlMe3 (2) | 32.0 | 34.1 | 104.76 | Stable |
| IMes·AlMe3 (1) | 31.7 | 33.7 | 114.47 | |
| IMes·AlMe3 (C) | 31.8 | 33.6 | 114.47 | |
| IiPrMe·AlMe3 (A) | 25.5 | 27.2 | 132.59 | |
A plot of the calculated %VBur (R = 2.0 Å) versus the calculated Ediss for all the crystallographically characterised structures is shown in Fig. 9. The linear correlation between the steric bulk of the NHC ligand and the dissociation energy of these complexes (R2 = 0.7057) shows that as the steric bulk increases, the dissociation energy decreases (see ESI†).
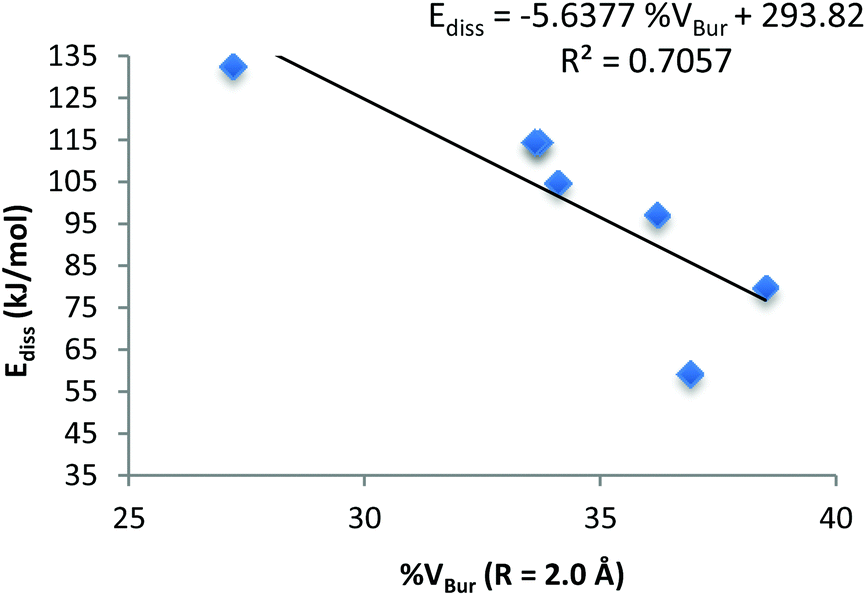 | ||
| Fig. 9 Plot of calculated %VBurvs. calculated Ediss for NHC trimethylaluminium complexes. | ||
On inspection of the calculated %VBur for all NHC·AlMe3 complexes, it is observed that all stable complexes fall within or below a calculated %VBur of 34%, whereas B, 3 and 4 have %VBur values exceeding 36%. Therefore, the difference in the %VBur observed between the stable and the unstable complexes is only 2–4% (Table 5). Despite the observed differences in %VBur between 1 and 4 being minor and concentrated in small areas (as indicated by the topographic maps) they exhibit profound effects on the stability and dissociation energies of these complexes (the asymmetry underlined by the maps adds value to this 2–4%).
To further test the proposed stability threshold of %VBur of 36% and in order to complete the series of trimethylaluminium complexes, we attempted to synthesise SItBu·AlMe3 (the saturated counterpart of B). Unfortunately, in all our synthetic attempts, only complex mixtures of products were obtained. The slurry formed in the reaction mixture was insoluble in most aprotic solvents (pentane, hexane, ether, THF, benzene, and toluene) which made the isolation of any viable product unsuccessful. To allow for comparison, the optimised geometry for SItBu·AlMe3 was calculated using DFT methods (see ESI†). The corresponding %VBur and the dissociation energy calculated are shown in Tables 4 and 5. From the theoretical values obtained and in comparison with the rest of the isolated NHC trimethylaluminium complexes, the %VBur for SItBu·AlMe3 falls within the range observed for the unstable complexes (37.6%), which may help explain our lack of success in its synthesis.
By-product obtained from SIPr·AlMe3 (4)
As discussed previously, compounds 3 and 4 were shown to be susceptible towards the formation of the imidazolylidenes and other unidentified decomposition products. Efforts were made to isolate and identify some of these side-products. Since the observed rate of decomposition was temperature dependent – and in order to accelerate this process – the reaction mixture, initially used to produce complex 4 (at RT), was refluxed overnight instead. Crystalline solids from this reaction proved to be remarkably air and moisture sensitive, and difficult to separate from the complex mixture of products obtained from the reaction. However, solid 5 was obtained when the reaction mixture was extracted in THF. Suitable single crystals for X-ray diffraction studies were grown in a THF–hexane mixture (Fig. 10).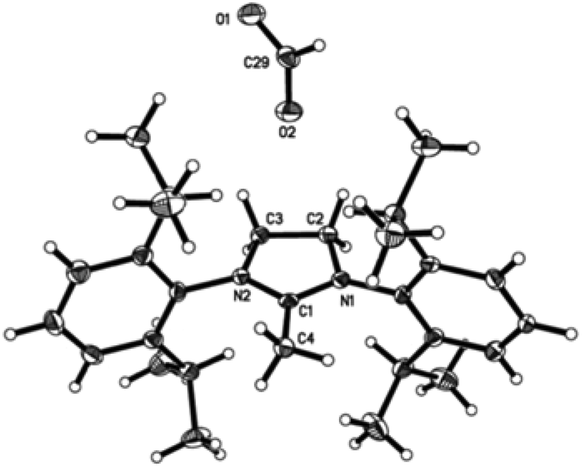 | ||
| Fig. 10 Molecular structure of complex 5 (1,3-bis(2,6-diisopropylphenyl)-2-methylimidazolium formate). Thermal ellipsoids are drawn at the 50% probability level. Hydrogen atoms, except for H(4) and H(29), are omitted for clarity. C(1)–N(1) 1.319(4), C(1)–N(2) 1.320(4), C(1)–C(4) 1.483(4), C(2)–N(1) 1.488(2), C(3)–N(2) 1.471(4), C(2)–C(3) 1.539(4), C(2)–H(2) 0.991, C(3)–H(3) 0.990, C(4)–H(4) 0.980, C(29)–O(1) 1.266(4), C(29)–O(2) 1.219(4), C(29)–H(29) 0.950, N(1)–C(1)–N(2) 111.9(3), N(1)–C(1)–C(4) 124.3(3), N(2)–C(1)–C(4) 123.7(3), N(1)–C(2)–C(3) 102.4(2), N(2)–C(3)–C(2) 102.8(2), C(1)–C(4)–H(4) 109.4, O(1)–C(29)–O(2) 127.1(3), O(1)–C(29)–H(29) 116.5, O(2)–C(29)–H(29) 116.4. | ||
Complex 5 crystallised out as a methylated imidazolium salt containing a formate counter ion and an acetic acid lattice molecule (1:1:1 ratio). Despite the extreme care taken to ensure inert atmosphere conditions, presumably trace impurities of water, oxygen or carbon dioxide were present in the reaction mixture. Therefore, in the presence of these impurities, the formation of compound 5 could be considered closely related to the reaction proposed by Rogers et al. that describes the generation of carboxylate zwitterion species with acetate ionic liquids due to their relative structural similarity.20 Further mechanistic studies are being conducted in order to rationalise the formation of 5.
Conclusions
In conclusion, the work presented here describes the synthesis and characterization of a series of new aromatic N-substituted NHC trimethylaluminium species. These complexes exhibit differing stabilities, which is attributed to differences in steric bulk of the NHCs used during their synthesis. Our studies demonstrate that the mesityl substituted NHC complexes (1 and 2) are more robust than their isopropylphenyl counterparts (3 and 4). In addition, comparison with previously characterised trimethylaluminium complexes showed that small variations (2–4%) in the steric bulk of the NHC substituent (%VBur) exert a profound effect on the overall stability of the complex formed. The results obtained indicate that all the reported stable NHC·AlMe3 complexes fall within or below a %VBur of 34%. The unstable nature of complexes with %VBur higher than 36% is illustrated by the new complexes 3 and 4 and the previously reported complex B. Mechanistic studies are currently underway to gain a better understanding of the reactivity of these trimethylaluminium complexes and to rationalise their decomposition pathways.Experimental section
General method
All manipulations were carried out using standard Schlenk and glove-box techniques under a dried argon atmosphere and with oven dried glassware. Toluene and ether were distilled over Na/benzophenone, degassed and purged with dry argon prior to use. Acetonitrile for high-resolution mass spectra (HRMS) was stirred over 4A molecular sieves and subsequently distilled under CaH2 prior to use. All solvents used after purification were stored under 4A molecular sieves. Deuterated C6D6 and THF-d8 were distilled over Na and stored under potassium mirror. Starting materials IMes, IPr, SIMes, and SIPr were obtained commercially from Strem and used as received. Solution of trimethylaluminum (1 M) in toluene was prepared from the neat compound purchased from Sigma-Aldrich.Instrumentation
1H, 13C NMR (400/100 MHz) spectra were recorded using a Bruker Avance DPX400 spectrometer with the 1H, 13C NMR chemical shifts internally referenced to the residual solvent peaks used. All NMR spectroscopic analyses were performed at room temperature (300 K). High-resolution mass spectra were obtained using a Water Q-Tof Premier, with ESI mode. Melting points were determined on an SRS-Optimelt MPA-100 apparatus using sealed glass capillaries under argon and were uncorrected. Infrared spectra were recorded as Nujol mulls using NaCl plates on a Shimadzu IR Prestige-21 FTIR spectrometer.Procedure for the synthesis of complexes 1–4
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 615 (ν Al–C stretch; m). HRMS: calcd for C24H33AlN2 [M + H]+: 377.25; found 377.25.
= 627 (ν Al–C stretch; m). HRMS: calcd for C24H35AlN2 [M + H]+: 379.27; found 379.27.
= 615 (ν Al–C stretch; m). HRMS: calcd for C30H45AlN2 [M + H]+: 461.35; found 461.35.
= 617 (ν Al–C stretch; m). HRMS: calcd for C30H47AlN2 [M + H]+: 463.36; found 463.36.
= 615 (ν Al–C stretch; m). HRMS: calcd for C24H33AlN2 [M + H]+: 377.25; found 377.25.
= 627 (ν Al–C stretch; m). HRMS: calcd for C24H35AlN2 [M + H]+: 379.27; found 379.27.
= 615 (ν Al–C stretch; m). HRMS: calcd for C30H45AlN2 [M + H]+: 461.35; found 461.35.
= 617 (ν Al–C stretch; m). HRMS: calcd for C30H47AlN2 [M + H]+: 463.36; found 463.36.
X-Ray crystallographic studies
Diffraction-quality crystals 1–4 were obtained in ether or toluene at room temperature or −25 °C, and 5 in a THF–hexane mixture at room temperature. The crystals were mounted onto quartz fibers, and the X-ray diffraction intensity data were collected at 103 K with a Bruker Kappa diffractometer equipped with a CCD detector, employing Mo Kα radiation (λ = 0.71073 Å), with the SMART suite of programs.21 All data were processed and corrected for Lorentz and polarization effects with SAINT and for absorption effects with SADABS.22 Structural solution and refinement were carried out with the SHELXTL suite of programs.23 The structures were solved by direct methods or Patterson maps to locate the heavy atoms, followed by difference maps for the light, non-hydrogen atoms. For 3 the isopropyl groups were disordered and these are modelled in two alternative sites and refined with appropriate restraints. All non-hydrogen atoms were refined with anisotropic thermal parameters.Computational details
Notes and references
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef CAS.
- L. Cavallo and C. S. J. Cazin, in N-heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Netherlands, 1996, vol. 32, p. 1 Search PubMed.
- (a) S. J. Hock, L. Schaper, W. A. Herrmann and F. E. Kühn, Chem. Soc. Rev., 2013, 42, 5073 RSC; (b) H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032 RSC; (c) A. T. Normand and K. J. Cavell, Eur. J. Inorg. Chem., 2008, 18, 2781 CrossRef; (d) I. J. B. Lin and C. S. Vasam, Coord. Chem. Rev., 2007, 251, 642 CrossRef CAS PubMed; (e) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239 CrossRef CAS PubMed; (f) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619 RSC; (g) M. C. Perry and K. Burgess, Tetrahedron: Asymmetry, 2003, 14, 951 CrossRef CAS; (h) W. G. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS.
- (a) L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705 CrossRef CAS PubMed; (b) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC; (c) A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 13, 1759 CrossRef; (d) S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874 CrossRef PubMed; (e) H. Jacobsen, A. Correa, C. Costabile and L. Cavallo, J. Organomet. Chem., 2006, 691, 4350 CrossRef CAS PubMed; (f) L. Cavallo, A. Correa, C. Costabile and H. Jacobsen, J. Organomet. Chem., 2005, 690, 5407 CrossRef CAS PubMed; (g) A. M. Magill, K. J. Cavell and B. F. Yates, J. Am. Chem. Soc., 2004, 126, 8717 CrossRef CAS PubMed.
- (a) C. Fliedel, G. Schnee, T. Avilés and S. Dagorne, Coord. Chem. Rev., 2014, 275, 63 CrossRef CAS PubMed; (b) P. Horeglad, G. Szczepaniak, M. Dranka and J. Zachara, Chem. Commun., 2012, 48, 1171 RSC; (c) S. Aldridge and A. J. Downs, in The Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities, ed. S. Aldridge and A. J. Downs, Wiley, UK, 2011, vol. 3 Search PubMed; (d) S. G. Alexander, M. L. Cole and C. M. Forsyth, Chem. – Eur. J., 2009, 15, 9201 CrossRef CAS PubMed; (e) S. G. Alexander, M. L. Cole, S. K. Furfari and M. Kloth, Dalton Trans., 2009, 2909 RSC; (f) M. L. Cole, S. K. Furfari and M. Kloth, J. Organomet. Chem., 2009, 694, 2934 CrossRef CAS PubMed; (g) R. S. Ghadwal, H. W. Roesky, R. Herbst-Irmer and P. G. Jones, Z. Anorg. Allg. Chem., 2009, 635, 431 CrossRef CAS; (h) B. Bantu, G. M. Pawar, K. Wurst, U. Decker, A. M. Schmidt and M. R. Buchmeiser, Eur. J. Inorg. Chem., 2009, 13, 1970 CrossRef; (i) N. Marion, E. C. Escudero-Adán, J. Benet-Buchholz, E. D. Stevens, L. Fensterbank, M. Malacria and S. P. Nolan, Organometallics, 2007, 26, 3256 CrossRef CAS; (j) J. H. Cotgreave, D. Colclough, G. Kociok-Köhn, G. Ruggiero, C. G. Frost and A. S. Weller, Dalton Trans., 2004, 1519 RSC; (k) A. Stasch, S. Singh, H. W. Roesky, M. Noltemeyer and H. G. Schmidt, Eur. J. Inorg. Chem., 2004, 20, 4052 CrossRef; (l) R. J. Baker and C. Jones, Appl. Organomet. Chem., 2003, 17, 807 CrossRef CAS; (m) R. J. Baker, M. L. Cole, C. Jones and M. F. Mahon, Dalton Trans., 2002, 9, 1992 RSC; (n) R. J. Baker, A. J. Davies, C. Jones and M. Kloth, J. Organomet. Chem., 2002, 656, 203 CrossRef CAS; (o) M. L. Cole, A. J. Davies and C. Jones, Dalton Trans., 2001, 17, 2451 RSC; (p) C. D. Abernethy, M. L. Cole and C. Jones, Organometallics, 2000, 19, 4852 CrossRef CAS; (q) M. D. Francis, D. E. Hibbs, M. B. Hursthouse, C. Jones and N. A. Smithies, Dalton Trans., 1998, 19, 3249 RSC; (r) D. E. Hibbs, M. B. Hursthouse, C. Jones and N. A. Smithies, Chem. Commun., 1998, 869 RSC; (s) S. J. Black, D. E. Hibbs, M. B. Hursthouse, C. Jones, K. M. A. Malik and N. A. Smithies, Dalton Trans., 1997, 4313 RSC; (t) A. J. Arduengo, H. V. R. Dias, J. C. Calabrese and F. Davidson, J. Am. Chem. Soc., 1992, 114, 9742 Search PubMed.
- (a) R. A. Musgrave, R. S. P. Turbervill, M. Irwin and J. M. Goicoechea, Angew. Chem., Int. Ed., 2012, 51, 10832 CrossRef CAS PubMed; (b) Y. Zhang, G. M. Miyake and E. Y.-X. Chen, Angew. Chem., Int. Ed., 2010, 49, 10158 CrossRef CAS PubMed; (c) A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Dalton Trans., 2010, 39, 9091 RSC; (d) A. L. Schmitt, G. Schnee, R. Welter and S. Dagorne, Chem. Commun., 2010, 46, 2480 RSC; (e) W. C. Shih, C. H. Wang, Y. T. Chang, G. P. A. Yap and T. G. Ong, Organometallics, 2009, 28, 1060 CrossRef CAS; (f) Y. Lee, B. Li and A. H. Hoveyda, J. Am. Chem. Soc., 2009, 131, 11625 CrossRef CAS PubMed; (g) M. Schiefer, N. D. Reddy, H.-J. Ahn, A. Stasch, H. W. Roesky, A. C. Schlicker, H.-G. Schmidt, M. Noltemeyer and D. Vidovic, Inorg. Chem., 2003, 42, 4970 CrossRef CAS PubMed; (h) X. W. Li, J. Su and G. H. Robinson, Chem. Commun., 1996, 2683 RSC.
- (a) Y. Wang, B. Quillian, P. Wei, C. S. Wannere, Y. Xie, R. B. King, H. F. Schaefer, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2007, 129, 12412 CrossRef CAS PubMed; (b) H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420 CrossRef CAS PubMed.
- B. Quillian, P. Wei, C. S. Wannere, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2009, 131, 3168 CrossRef CAS PubMed.
- (a) D. W. Stephan, Dalton Trans., 2009, 3129 RSC; (b) P. A. Chase and D. W. Stephan, Angew. Chem., Int. Ed., 2008, 47, 7433 CrossRef CAS PubMed; (c) D. Holschumacher, T. Bannenberg, C. G. Hrib, P. G. Jones and M. Tamm, Angew. Chem., Int. Ed., 2008, 47, 7428 CrossRef CAS PubMed.
- H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395 CrossRef CAS.
- S. LeuthäuBer, D. Schwarz and H. Plenio, Chem. – Eur. J., 2007, 13, 7195 CrossRef PubMed.
- G. Davidson, in Spectroscopic Properties of Inorganic and Organometallic Compounds, ed. G. Davidson and E. A. V. Ebsworth, Royal Society of Chemistry, UK, 1990, vol. 23, p. 227 Search PubMed.
- (a) X. Bantreil and S. P. Nolan, Nat. Protocols, 2011, 6, 69 CrossRef CAS PubMed; (b) A. J. Arduengo, R. Krafczyk and R. Schmutzler, Tetrahedron, 1999, 55, 14523 CrossRef CAS.
- D. A. Wierda and A. R. Barron, Polyhedron, 1989, 8, 831 CrossRef CAS.
- A. R. Barron, J. Chem. Soc., Dalton Trans., 1988, 3047 RSC.
- J. C. Huffman and W. E. Streib, J. Chem. Soc. D, 1971, 911 RSC.
- (a) M. Kessler, C. Knapp and A. Zogaj, Organometallics, 2011, 30, 3786 CrossRef CAS; (b) A. Kuczkowski, S. Schulz and M. Nieger, Eur. J. Inorg. Chem., 2001, 2605 CrossRef CAS; (c) D. C. Bradley, H. Chudzynska, M. M. Faktor, D. M. Frigo, M. B. Hursthouse, B. Hussain and L. M. Smith, Polyhedron, 1988, 14, 1289 CrossRef.
- V. V. Shatunov, A. A. Korlyukov, A. V. Lebedev, V. D. Sheludyakow, B. I. Kozyrkin and V. Y. Orlov, J. Organomet. Chem., 2011, 696, 2238 CrossRef CAS PubMed.
- S. Muthaiah, D. C. H. Do, R. Ganguly and D. Vidović, Organometallics, 2013, 32, 6718 CrossRef CAS.
- (a) G. Gurau, H. Rodríguez, S. P. Kelly, P. Janiczek, R. S. Kalb and R. D. Rogers, Chem. Commun., 2011, 50, 12024 CAS; (b) H. Rodríguez, G. Gurau, J. D. Holbrey and R. D. Rogers, Chem. Commun., 2011, 47, 3222 RSC.
- SMART version 5.628, Bruker AXS Inc., Madison, WI, USA, 2001 Search PubMed.
- G. M. Sheldrick, SADABS, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- SHELXTL version 5.1, Bruker AXS Inc., Madison, WI, USA, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and spectral data, and crystallographic data of 1–5. CCDC 1020090–1020094. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00079c |
| This journal is © The Royal Society of Chemistry 2015 |