
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An anionic phosphenium complex as an ambident nucleophile†
B.
Stadelmann
a,
J.
Bender
a,
D.
Förster
a,
W.
Frey
b,
M.
Nieger
c and
D.
Gudat
*a
aInstitut für Anorganische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70550 Stuttgart, Germany. E-mail: gudat@iac.uni-stuttgart.de; Fax: +49 711 685 64241; Tel: +49 711 685 64186
bInstitut für Organische Chemie, Universität Stuttgart, Pfaffenwaldring 55, 70550 Stuttgart, Germany
cLaboratory of Inorganic Chemistry, Dept. of Chemistry, University of Helsinki, P.O. Box 55, 00014 University of Helsinki, Finland
First published on 10th February 2015
Abstract
A unique anionic phosphenium complex was prepared from reaction of an N-heterocyclic chlorophosphine with Collman's reagent or K[HFe(CO)4]/NaH and characterized by spectral and XRD data. The complex behaves as an ambident nucleophile. Reactions with acetic acid, ClSnPh3, and a further equivalent of an N-heterocyclic chlorophosphine proceed via electrophilic functionalization at the metal site to yield appropriate mono- or bis-phosphenium complexes. Reaction with MeI at −70 °C produces a P-alkylation product as the first spectroscopically detectable intermediate, which decays at a higher temperature to give a mixture of free P-methylated N-heterocyclic phosphine and its Fe(CO)4 complex. The different reaction products were characterized by spectral and XRD data. Computational studies indicate that the NHP units in all complexes display π-acceptor behaviour but show no disposition to adopt phosphide-like character or formally oxidize the metal centre.
Introduction
Cyclic diaminophosphenium ions (Ia, Scheme 1) are isosteric analogues of N-heterocyclic carbenes (NHCs, Ib) and, like NHCs, form a large variety of transition metal complexes.1 However, whereas NHCs are nucleophiles and act mainly as strong σ-donor ligands, N-heterocyclic phosphenium ions (NHPs) are ambiphiles that combine a strong (π-)acceptor power with limited (σ-)donor ability, and interact with electron poor metal fragments to form a main dative M → L π-bond and a weaker dative L → M σ-bond.1,2 This bonding situation compares to that in metal carbonyls or Fischer-type (electrophilic) carbene complexes,1e,2 and the M–P-bonds exhibit accordingly distinct double bond character (cf. IIa, Scheme 1). In complexes with electron rich and weak Lewis acidic metal fragments, NHPs can also act as electrophiles3 (“Z-type ligands”4) that have been considered to bind essentially via a dative M → P bond (cf. IIb, Scheme 1).5 It has recently been pointed out that the bonding in complexes of this type may be portrayed as the interaction of an anionic (“X-type”) phosphido ligand with a formally oxidized metal centre, and that the dualism of both descriptions together with the pyramidalization at the phosphorus atom and M–P bond lengthening upon transition from IIa to IIb allows one to draw parallels to the “non-innocent” behaviour of metal nitrosyls.6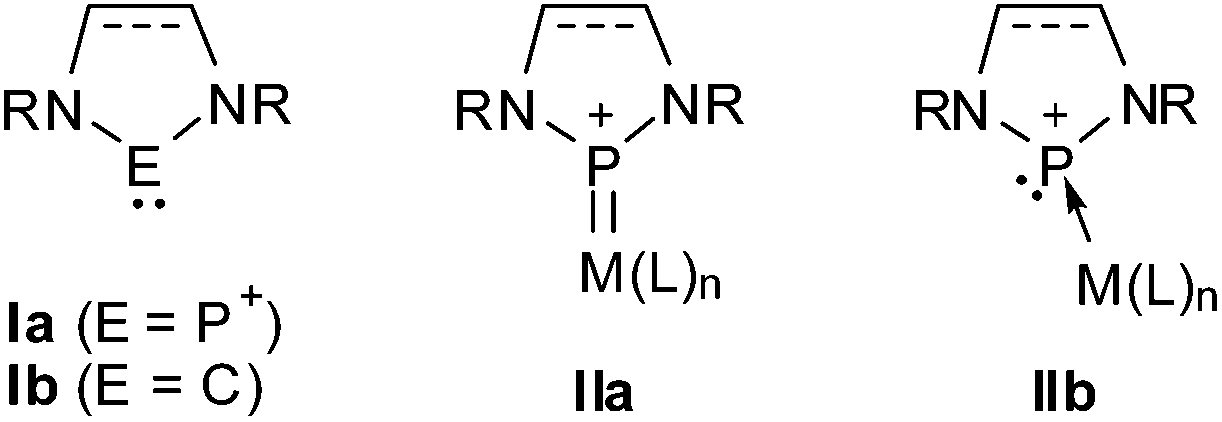 | ||
Scheme 1 Generic molecular structures of NHCs (Ib), NHPs (Ia), and NHP complexes (IIa, b). The symbol  indicates that the heterocycle contains either a single or, alternatively, a double bond. indicates that the heterocycle contains either a single or, alternatively, a double bond. | ||
The unique disposition of NHPs to interact with low valent metal centres has repeatedly been used to stabilise anionic metal fragments [M(L)nX]− (containing usually a zero-valent metal atom and an anionic ligand X),2b,3,7,8 so that to date both cationic ([(NHP)M(L)n]+) and neutral NHP complexes ([(NHP)M(L)nX]) are known.1–8 Although the chemistry of the latter has not been widely explored, they are expected to display a similar distributed nucleophilicity as neutral complexes of acyclic aminophosphenium ions which can be attacked by electrophiles at the metal atom, the phosphorus atom, or the MP bond, respectively.9
In the light of these findings, complexes [(NHP)M(L)n]− with an overall negative charge and CC-unsaturated NHP ligands (1,3,2-diazaphospholenium ions) make an interesting synthetic target. These specimens should be accessible from suitable phosphorus electrophiles and metallate dianions featuring a metal atom in the negative formal oxidation state. Coordinated diazaphospholenium ions, apart from providing tunable steric protection, exhibit low tendency toward “non-innocent” behaviour,7b and should thus provide electronic stabilization without entirely removing the electron excess at the metal atom. The formed complexes might then still behave as metal-centred nucleophiles, a class of compounds that has lately gained attention for uses in catalysis.10
Following these lines, we report here on the synthesis of a NHP carbonyl ferrate, which is a first anion analogue of cationic and neutral phosphenium complexes, and on initial reactivity studies, which confirm that this species behaves in fact as a metal-centred nucleophile but retains also some ambident character.
Results and discussion
By analogy to the known synthesis of neutral phosphenium tricarbonylcobaltates,5 2-chloro-1,3,2-diazaphospholene 1 reacted with Collman's reagent Na2[Fe(CO)4] via salt elimination and decarbonylation to afford the anionic NHP complex [(NHPDipp)Fe(CO)3]− (2; NHPDipp = 1,3-di-(2,6-di-isopropylphenyl-1,3,2-diazaphospholenium, Scheme 2).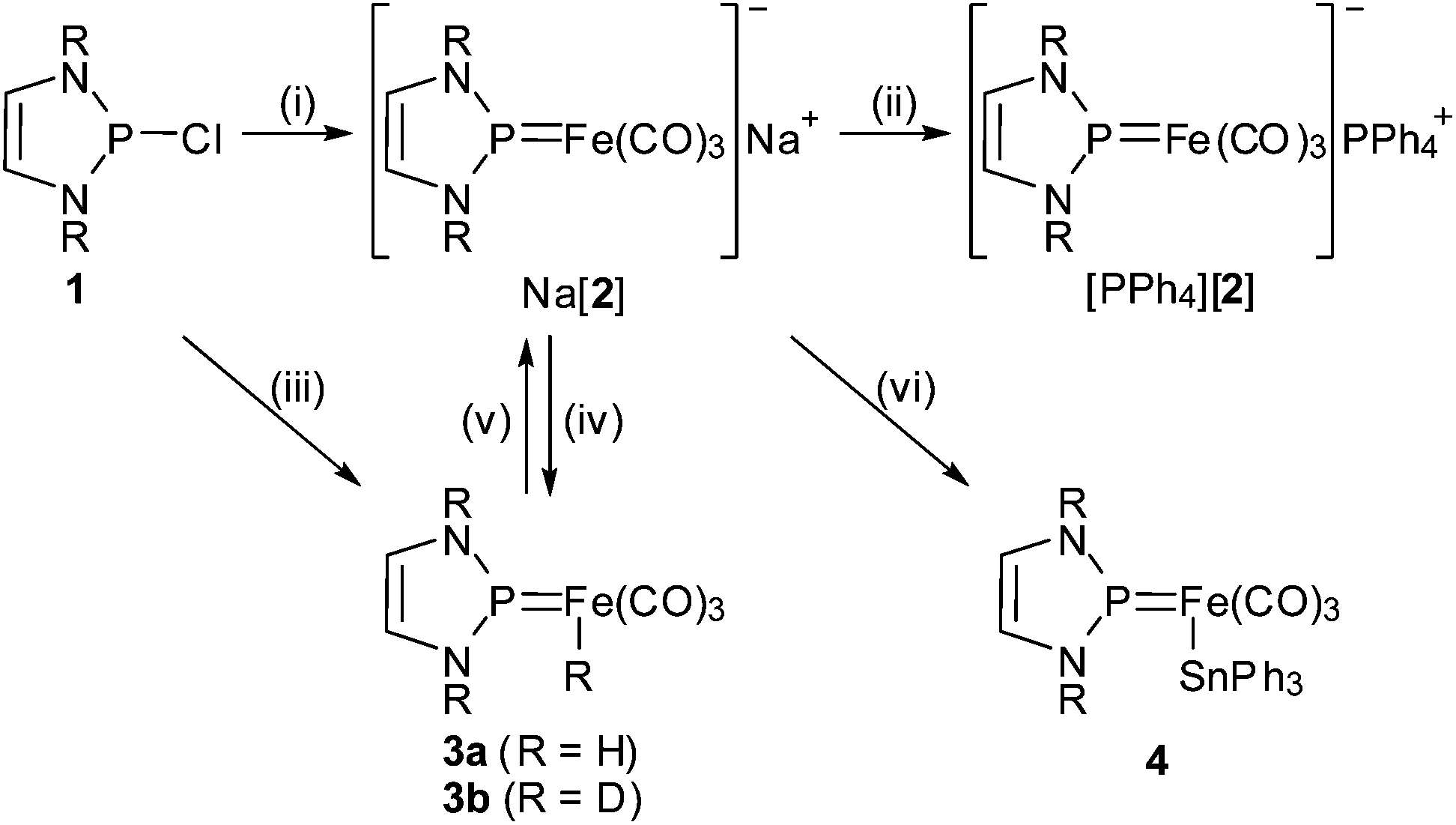 | ||
| Scheme 2 R = Dipp; conditions: (i) 1 equiv. Na2[Fe(CO)4], THF, −78 °C, 2 h; (ii) 1 equiv. Ph4PCl, THF; (iii) 1 equiv. K[HFe(CO)4], −78 °C → r.t., 2 h; (iv) CH3CO2H/CH3CO2D, r.t.; (v) 1 equiv. NaH, 0 °C, 12 h; (vi) 1 equiv. Ph3SnCl, r.t., 5 min. | ||
A crude product of composition Na[2] was isolated as a brown-red, highly air and moisture sensitive solid after separation of NaCl and evaporation of the solvent. Crystalline [PPh4][2] was isolated after anion exchange with [PPh4]Cl. Both products were readily identified by spectral data and a single-crystal XRD study of [PPh4][2] (Fig. 1). The orthorhombic crystals (space group Pna21, no. 33) contain two crystallographically independent pairs of isolated Ph4P+ and [(NHPDipp)Fe(CO)3]− ions with similar metric parameters. The distorted tetrahedral and trigonal planar (sum of bond angles 360.0(5)°) coordination at the iron and phosphorus atoms and the bond lengths in the planar NHP-rings of the anion (see Fig. 1) are similar as in the isosteric complexes [(NHPR)Co(CO)3]5 (R = Mes, tBu). The Fe–P bonds (1.989(2) Å) are shorter than in iron complexes with diaminophosphenium (Fe–P 2.018(2)–2.15 Å11) or -phosphine ligands (Fe–P > 2.23 Å,126 (vide infra): 2.212(1) Å), and Fe–C distances (1.750(6)–1.768(5) Å) match those in tetra-carbonyl ferrates (1.730(4)–1.762(5) Å13) and Tl[Fe(CO)3NO] (1.800(8) Å14). Noteworthily, spectroscopic features of 2 include a marked deshielding of the 31P NMR signal (which is typical of most aminophosphenium complexes),1,6 and a large counter ion dependence of 31P chemical shifts (δ31P = 211.1 for Na[2] vs. 197.9 for [PPh4][2]) and νCO normal modes (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 1912, 1834, 1767 cm−1 for Na[2] vs. = 1894, 1813, 1798 cm−1 for [PPh4][2]) which reflects presumably differences in the ion pairing behaviour.15 The νCO frequencies in both species are lower than in anionic tricarbonyl iron complexes such as [(Ge8)Fe(CO)3]3− or [(C8H8)Fe(CO)3]− where the negative charge seems mainly ligand centred,16 but do not come close to the values observed for gaseous or matrix isolated [Fe(CO)3]− (νCO = 1780–1795 cm−1 (ref. 17)) and dianions like [Fe(CO)4]2− (νCO = 1920, 1830, 1780, 1730 cm−1 (ref. 18)) or [Fe(CO)3(PR3)]2− (νCO = 1817, 1803, 1710, 1663 cm−1 for R = OMe; 1789, 1692, 1637 cm−1 for R = Ph19). Although the unavailability of exact force constants prohibits a quantitative comparison, these trends confirm, in line with the results of a previous study on cationic NHP complexes,7a the picture of the cationic NHP unit as a strong π-acidic ligand which outperforms neutral acceptors like CO or phosphites.
= 1912, 1834, 1767 cm−1 for Na[2] vs. = 1894, 1813, 1798 cm−1 for [PPh4][2]) which reflects presumably differences in the ion pairing behaviour.15 The νCO frequencies in both species are lower than in anionic tricarbonyl iron complexes such as [(Ge8)Fe(CO)3]3− or [(C8H8)Fe(CO)3]− where the negative charge seems mainly ligand centred,16 but do not come close to the values observed for gaseous or matrix isolated [Fe(CO)3]− (νCO = 1780–1795 cm−1 (ref. 17)) and dianions like [Fe(CO)4]2− (νCO = 1920, 1830, 1780, 1730 cm−1 (ref. 18)) or [Fe(CO)3(PR3)]2− (νCO = 1817, 1803, 1710, 1663 cm−1 for R = OMe; 1789, 1692, 1637 cm−1 for R = Ph19). Although the unavailability of exact force constants prohibits a quantitative comparison, these trends confirm, in line with the results of a previous study on cationic NHP complexes,7a the picture of the cationic NHP unit as a strong π-acidic ligand which outperforms neutral acceptors like CO or phosphites.
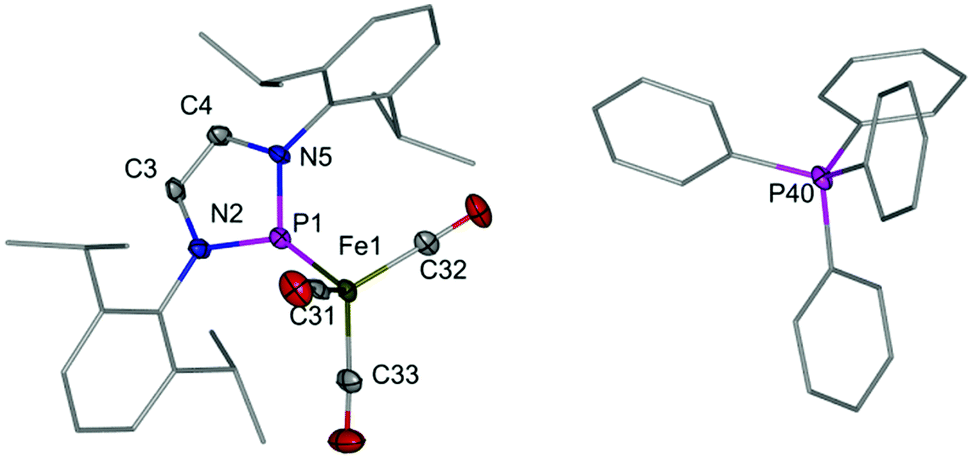 | ||
| Fig. 1 Representation of the molecular structure of one crystallographically independent ion pair in [PPh4][2]. H-atoms were omitted for clarity. Selected bond lengths [Å] (numbers in brackets refer to the second crystallographically independent unit): Fe1–C31 1.762(5) [1.768(5)], Fe1–C32 1.768(5) [1.750(6)], Fe1–C33 1.758(6) [1.754(5)], Fe1–P1 1.989(1) [1.989(2)], P1–N5 1.689(4) [1.703(4)], P1–N2 1.712(4) [1.710(4)], N2–C3 1.380(6) [1.399(6)], C3–C4 1.337(6) [1.333(7)], C4–N5 1.408(6) [1.416(6)]. | ||
All in all, the structural and spectral features of 2 match those of Fischer-carbene analogue phosphenium complexes2,5,7 and indicate that the NHP unit acts as an ambiphilic σ-donor/π-acceptor rather than the pure Z-type ligand. In order to validate this hypothesis by DFT calculations (B3LYP/def2-tzvp with inclusion of solvent effects using a polarizable continuum model; see Experimental for details), we performed a geometrical optimization and ensuing NBO calculation20 on the model anion [(NHPMe)Fe(CO)3]− (2′). The computed molecular structure replicates all important structural features of 2, including in particular the planar coordination at phosphorus and short P–Fe bond length (2.006 Å). Inspection of KS orbitals (see ESI†) and Wiberg Bond Indexes (WBI) confirms that the Fe–P bond receives a strong covalent contribution from a combination of dative σ- and π-bonding interactions, and has thus notable double bond character (WBI 1.41 vs. 1.35 for the Fe–C bonds). The overall natural charge of the coordinated NHP unit (+0.86) is lower than in the free ligand, indicating that the NHP plays a part in delocalizing the negative charge on the carbonyl ferrate moiety but retains its electrophilic character. The large opposite charges on the iron (−2.41) and phosphorus atoms (+1.52) suggest that the metal–ligand bond also has a strong electrostatic component. The population analyses indicate that, regardless of any superficial analogies, the behaviour of the NHP moiety in 2/2′ differs markedly from that of the strongly reduced nitrosyl unit21 in [Fe(CO)3(NO)]−.
Considering that such a charge distribution should support a behaviour as an iron-centred nucleophile, we studied reactions of Na[2] with various electrophiles. Treatment with acetic acid gave iron hydrido complex 3a (Scheme 1) which was identified by the observation of mutually coupled (2JPH = 88 Hz) NMR signals for a metal bound H-atom (δ1H = −8.88) and the phosphorus atom of a phosphenium ligand (δ31P = 225.0). The reaction of Na[2] with deuterated acetic acid gave appropriately 3b. Complex 3a is also directly accessible from 1 and K[HFe(CO)4]22 which reacts below ambient temperature under metathesis and decarbonylation (Scheme 2). This conversion contrasts known reactions of chlorophosphines, which proceed via transfer of the hydride to the phosphorus atom and yield phosphine complexes.23 A single-crystal XRD study revealed that the iron atom in 3a exhibits trigonal bipyramidal coordination and the H- and NHP-ligands occupy axial and equatorial positions, respectively (Fig. 2).
 | ||
| Fig. 2 Representation of the molecular structure of 3a in the crystal. H-atoms (with the exception of H1) were omitted for clarity. Selected bond lengths [Å]: Fe1–C1A 1.765(3), Fe1–C3C 1.789(3), Fe1–C2B 1.797(3), Fe1–P1 2.011(1), Fe1–H1 1.48(2), P1–N2 1.673(2), P1–N5 1.676(2), N2–C3 1.406(3), C3–C4 1.330(4), C4–N5 1.386(3). | ||
The protonation of 2 to 3a can be reversed by the action of a strong base (NaH), and combining the reaction of K[HFe(CO)4] and 1 with a subsequent deprotonation offers thus alternative access to anion 2. Monitoring the deprotonation of 3a by 31P NMR allowed us to establish that the reaction is slow and 2 and 3a undergo no dynamic exchange on the NMR time scale, indicating that the kinetic acidity of 3a is unusually low.
Reaction of Na[2] with ClSnPh3, which is a softer electrophile than H+, proceeds likewise under attack at the metal to afford complex 4 (Scheme 2) which was identified by spectral data and a single-crystal XRD study (see ESI†). The iron atom adopts trigonal-bipyramidal coordination like in 3a, but the Ph3Sn and NHP ligands occupy now both axial positions, presumably due to steric reasons.
Electrophilic attack at iron is also observed upon treatment of Na[2] with one more equivalent of 1. The reaction proceeds with spontaneous decarbonylation to yield bis-NHP complex 5 (Scheme 3) which is a rare example of a complex featuring two terminal phosphenium ligands coordinated to the same metal atom.24 A single-crystal XRD study (Fig. 3) reveals distorted tetrahedral coordination at iron, whereas the Fe–C and Fe–P bond lengths and the trigonal planar coordination at the phosphorus atoms are similar as in 2–4.
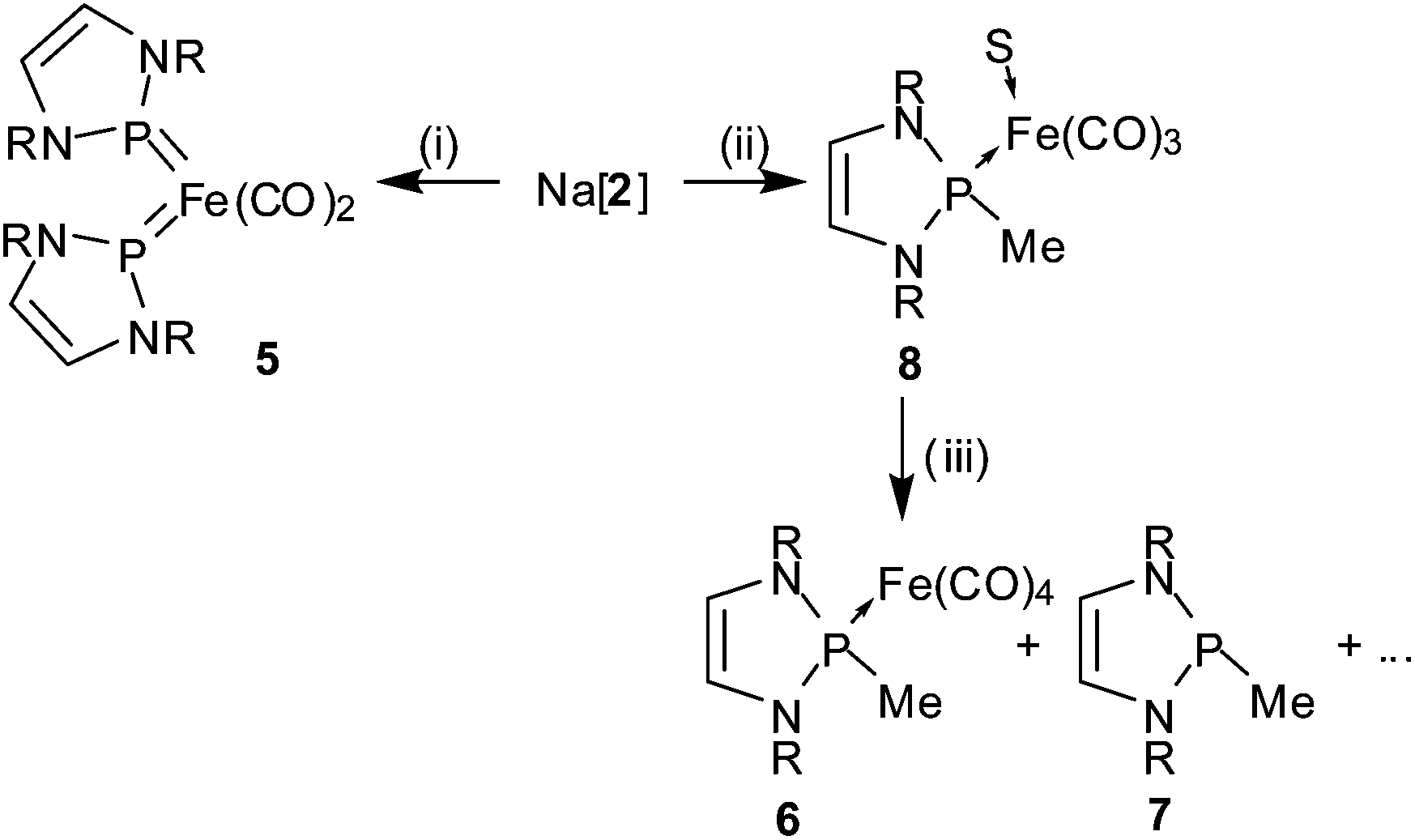 | ||
| Scheme 3 R = Dipp; S = nothing or solvent (THF); (i) 1 equiv. 1, THF, −78 °C, 2 h; (ii) 1 equiv. MeI, THF, −70 °C, r.t.; (iii) −70 °C → r.t. | ||
 | ||
| Fig. 3 Representation of the molecular structure of 5 in the crystal. H atoms were omitted for clarity. Selected bond lengths (Å) and angles (°): Fe1–C1A 1.766(3), Fe1–C2B 1.772(3), Fe1–P1 2.009(1), Fe1–P2 2.016(1), P1–N2 1.696(2), P1–N5 1.702(2), P2–N30 1.700(2), P2–N33 1.704(2), P1–Fe1–P2 126.1(1), other Fe-centred angles 103.4(1)–108.0(1). | ||
Computational studies on model complexes 3a′/5′ (with NMe instead of NDipp residues) imply a slight weakening of Fe–P bonds (Fe–P 2.047/2.017 Å; WBI 1.20/1.33) compared to 2′, which is mainly because of the M → L π-bonding and creates an enlarged partial charge (1.01/1.19) on the NHP unit. These findings confirm the role of the phosphenium moieties as Fischer-carbene analogue ligands which exert their stabilizing influence by a combination of ambiphilic σ-donor/π-acceptor and electrostatic interactions. A marked π-acceptor character of the NHP unit is once more highlighted by the large blue shift of the νCO modes of 3a (2014, 1960, 1933 cm−1) and 4 (2064, 1978, 1909 cm−1) in comparison with analogous phosphite complexes like [HFe(CO)3{P(OMe)3}]− (a1- and e-modes at 1941 and 1841 cm−1 (ref. 15b)) and [Ph3SnFe(CO)3{P(OMe)3}]− (a1- and e-modes at 1925 and 1820 cm−1 (ref. 19)), respectively. On the other hand, the observation of higher νCO frequencies in [Fe(CO)2(NO)2]25 (2092, 2046 cm−1) suggests that the π-acceptor capability of the NHP unit remains inferior to that of a nitrosyl.
In contrast to the cases described so far, the reaction of Na[2] with MeI (Scheme 3) affords no metal-functionalized product but a mixture of a P-alkylated phosphine complex 7 and its iron complex 6 which was isolated in modest yield and characterised by a single-crystal XRD study. The identity of both species was further established in situ from multinuclear NMR studies and verified by their independent synthesis (see Experimental). The observed product distribution implies that elemental iron or some iron-containing compounds are formed as by-products, even if none of these was directly identified.
The reaction leading to 6 and 7 seems appealing as it may at first glance be considered to involve nucleophilic reactivity of an electrophilic phosphorus atom. However, in view of a previous report by the group of Nakazawa on the assembly of a related P-alkylated ligand through migratory insertion of a phosphenium unit into a metal-alkyl bond,26 it is entirely possible that the initial electrophilic functionalization occurs at the iron site and the final products are formed in subsequent rearrangement steps.
In order to cast further light on these mechanistic aspects, we followed the reaction of Na[2] with MeI at low temperature by VT NMR spectroscopy. The 1H and 31P NMR spectra recorded immediately after mixing both reactants at −70 °C disclosed that under these conditions a previously unobserved species 8 (Scheme 3) had formed as a major product (approx. 90% of all phosphorus-containing species by integration of the 31P NMR spectrum) besides minor amounts of 6 and 7 (<10%) and trace quantities of unidentified by-products. The composition of the reaction mixture did not change perceptibly as long as the temperature was kept below −50 °C. At this point, the onset of a slow and irreversible conversion of 8 into a mixture of 6 and 7 was noted. This process accelerated upon warming, and went eventually to completion after the sample had reached ambient temperature. Further spectroscopically observable intermediates were not detected. Since the initial product distribution varied somewhat (in a non-reproducible manner) between repeated experiments, we assume that 8 is the primary reaction product, and the initially present amounts of 6 and 7 are formed as a consequence of insufficient control of local temperature or concentration during the mixing of reactants.
The constitutional assignment of the intermediate 8 was feasible from the analysis of a set of one- and two-dimensional NMR spectra recorded at low temperature. The finding that the signals of the extra CH3 group (δ1H = 2.02, δ13C = 26.7) do not show the negative chemical shifts which are typical of Fe–CH3 units,26 but come close to those of the P-bound substituents in 6 (δ1H = 1.96, δ13C = 29.5), gives a first indication that the methyl is bound, like in 6 and 7, to the phosphorus rather than the iron atom. This assignment is further supported by (i) the presence of two distinguishable anisochronic environments for the iPr-groups in the NDipp-substituents, which implies that the coordination at phosphorus is pyramidal or tetrahedral (as in 6, 7) rather than planar (as in 2–5), and (ii) the observation of 1H-NOESY cross peaks which connect the CH3-signal with both the signal of the CH-atoms in the diazaphospholene ring and the signals of one anisochronic iPr-group (the one on the same side of the five-membered ring; see ESI†). On this basis, we formulate the product as a complex in which the iron atom is bound to three carbonyls and the neutral phosphine 7, and the remaining coordination site remains empty or is occupied by a solvent molecule (8, Scheme 3). Starting from this assignment, the conversion of 8 into a mixture of 6 and 7 is explained as the consequence of ligand extrusion/redistribution reactions which are common for carbonyl complexes that are coordinatively unsaturated or carry weakly bound solvent ligands.
Considering that the VT NMR studies provided no evidence for the formation of a metal functionalized species as a precursor to 8, we consider it likely that this species may indeed constitute the initial reaction product. Even if this interpretation seems at first glance counterintuitive, it is backed by the finding that the HOMO−1 of 2′ (which arises from mixing a filled metal-centred orbital with a σ*(PN) ligand orbital) contains a perceptible P-centred contribution, and the anion retains thus a certain degree of ambident character. It is currently under investigation if the attack of carbon-based electrophiles can be directed by steric effects.
Conclusions
In conclusion, we describe the successful synthesis of a NHP carbonyl ferrate which complements known cationic and neutral phosphenium complexes by a first anionic derivative. Structural and computational studies indicate that the ligand retains its electrophilic nature and shows no disposition to adopt phosphide-like character or formally oxidize the metal centre. Accordingly, the NHP ferrate reacts mainly as a metal centred nucleophile, but still retains some of the ambident character that is otherwise typical of neutral metallophosphenium complexes.Experimental
General conditions
All manipulations were carried out under dry argon. Solvents were dried by standard procedures. NMR spectra were recorded on Bruker Avance AV 400 or AV 250 instruments (1H: 400.1/250.0 MHz, 13C: 100.5/62.9 MHz, 31P: 161.9/101.2 MHz); chemical shifts were referenced to ext. TMS (1H, 13C), 85% H3PO4 (Ξ = 40.480747 MHz, 31P). FT-IR spectra were recorded on a Thermo Scientific Nicolet iS5 instrument equipped with an iD5 ATR accessory. Elemental analyses were carried out using an Elementar Micro Cube. Melting points were determined with a Büchi B-545 melting point apparatus in sealed capillaries. Collman's reagent was prepared as described elsewhere.27Syntheses
Na[2]: (a) A solution of Na2[Fe(CO)4] (390 mg, 1.12 mmol) in THF (20 ml) was cooled in a dry ice/acetone bath to −78 °C. A solution of 1 (500 mg, 1.12 mmol) in THF (5 ml) was added dropwise. The solution was allowed to warm to r.t., stirred for 2 h, and filtered. The filtrate was concentrated to a volume of approx. 5 ml. Addition of hexane (15 ml) produced a brown-red precipitate which was filtered off, washed with additional hexane, and dried under vacuum to give 312 mg (48%) of a brown-red, air and moisture sensitive solid, m.p. 306 °C (dec.).(b) Solid NaH (8.7 mg, 0.37 mmol) was added to a solution of complex 3 (200 mg, 365 μmol) in THF (10 ml THF). The mixture was stirred overnight. The solution was filtered to remove undissolved NaH. The solvent was removed under reduced pressure. The remaining solid was washed with hexane and dried under vacuum to give 141 mg (67%) of a brown-red, air and moisture sensitive solid, m.p. 306 °C (dec.).
31P{1H} NMR (d8-THF): δ = 211.1 (s). 1H NMR (d8-THF) δ = 7.21 (m, 6 H, C6H3), 6.48 (d, 2 H, ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH, 3JPH = 3.8 Hz), 3.48 (sept, 4 H, CH(CH3)2, 3JHH = 6.9 Hz), 1.37 (d, 12 H, CH(CH3)2, 3JHH = 6.9 Hz), 1.21 (d, 12 H, CH(CH3)2, 3JHH = 6.9 Hz). 13C{1H} NMR (d8-THF) δ = 229.2 (d, 2JPC = 10.8 Hz, CO), 148.1 (s, p-C), 137.6 (d, 2JPC = 6.9 Hz, i-C), 127.8 (d, 3JPC = 3.9 Hz, o-C), 123.4 (s, m-C), 122.3 (d, 2JPC = 4.6 Hz, CH), 28.9 (s, C(CH3)2), 23.5 (s, CH3), 23.3 (s, CH3). IR: = 1912, 1834, 1767 cm−1 (νCO). 1H and 13C NMR spectra disclosed the presence of varying amounts of THF (presumably for the solvation of Na+) which precluded obtaining a consistent elemental analysis.
CH, 3JPH = 3.8 Hz), 3.48 (sept, 4 H, CH(CH3)2, 3JHH = 6.9 Hz), 1.37 (d, 12 H, CH(CH3)2, 3JHH = 6.9 Hz), 1.21 (d, 12 H, CH(CH3)2, 3JHH = 6.9 Hz). 13C{1H} NMR (d8-THF) δ = 229.2 (d, 2JPC = 10.8 Hz, CO), 148.1 (s, p-C), 137.6 (d, 2JPC = 6.9 Hz, i-C), 127.8 (d, 3JPC = 3.9 Hz, o-C), 123.4 (s, m-C), 122.3 (d, 2JPC = 4.6 Hz, CH), 28.9 (s, C(CH3)2), 23.5 (s, CH3), 23.3 (s, CH3). IR: = 1912, 1834, 1767 cm−1 (νCO). 1H and 13C NMR spectra disclosed the presence of varying amounts of THF (presumably for the solvation of Na+) which precluded obtaining a consistent elemental analysis.
[PPh4][2]: A solution of Na[2] was prepared as described above from 3 (200 mg, 365 μmol) and NaH (8.7 mg, 0.37 mmol). Solid [Ph4P]Cl (137 mg, 365 μmol) was added with stirring. The resulting mixture was filtered to remove precipitated NaCl. The filtrate was evaporated to dryness and the residue was dissolved in a minimum amount of THF. Storing the solution at 4 °C gave 230 mg (71%) of orange crystals, m.p. >350 °C (dec.).
31P{1H} NMR (d8-THF): δ = 197.9 (s, P–Fe), 22.7 (s, PPh4). 1H NMR (d8-THF) δ = 7.73 (m, 4 H, C6H5), 7.60 (m, 16 H, C6H5), 7.01 (m, 6 H, C6H3), 6.19 (d, 2 H, CH, 3JPH = 3.2 Hz), 3.41 (sept, 4 H, CH(CH3)2, 3JHH = 6.9 Hz), 1.19 (d, 12 H, CH(CH3)2, 3JHH = 6.9 Hz), 1.04 (d, 12 H, CH(CH3)2, 3JHH = 6.9 Hz). 13C{1H} NMR (d8-THF) δ = 229.2 (d, 2JPC = 13.2 Hz, CO), 147.4 (s, o-C), 137.6 (d, 2JPC = 6.4 Hz, i-C), 134.8 (d, 4JPC = 2.95 Hz, p-C(PPh4)), 134.3 (d, 2JPC = 10.5 Hz, o-C(PPh4)), 130.0 (d, 3JPC = 12.9 Hz, m-C(PPh4)), 126.6 (s, p-C), 122.5 (s, m-C), 120.6 (d, 2JPC = 4.6 Hz, CH), 28.0 (s, CH(CH3)2), 23.8 (s, CH3), 22.8 (s, CH3). IR: = 1894, 1813, 1798 cm−1 (νCO). The high sensitivity of the product precluded to obtain a meaningful elemental analysis.
3a, b: (a) K[HFe(CO)4] (234 mg, 1.12 mmol) and THF (3 ml) were introduced into a Schlenk flask and stirred for 10 min at r.t. The flask was then cooled to −78 °C in a dry ice-acetone bath, and a solution of 1 (500 mg, 1.12 mmol) in THF (5 ml) was added dropwise. The solution was allowed to warm to r.t. and stirred for another 2 h. The solvent was removed under reduced pressure. The remaining black solid was extracted with hexane. The mixture was filtered and evaporated to dryness. The residue was dissolved in a minimum amount of hexane. The resulting solution was stored at 4 °C to give 573 mg (93%) of 3a as yellow crystals, m.p. >350 °C (dec.).
(b) A solution of Na[2] (5 mg) in THF-d8 was treated with a drop of glacial acetic acid or acetic acid-d4. The formed products (3a, b) were identified by their NMR data.
3a: 31P{1H} NMR (C6D6): δ = 225.0 (s). 1H NMR (C6D6): δ = 7.18 (m, 6 H, C6H3), 6.13 (d, 2 H, 3JPH = 6.7 Hz, CH), 3.07 (sept, 4 H, 3JHH = 6.9 Hz, CH(CH3)2), 1.36 (d, 12 H, 3JHH = 6.9 Hz, CH(CH3)2), 1.10 (d, 12 H, 3JHH = 6.9 Hz, CH(CH3)2)), −8.88 (d, 1 H, 2JPH = 88.1 Hz, FeH). 13C{1H} NMR (C6D6): δ = 214.30 (d, 2JPC = 13.6 Hz, CO), 147.0 (d, 2JPC = 3.0 Hz, o-C), 132.9 (d, 3JPC = 5.9 Hz, i-C), 130.2 (d, 5JPC = 1.4 Hz, p-C), 124.4 (d, 4JPC = 1.0 Hz, m-C), 123.6 (d, 2JPC = 1.7 Hz, CH), 28.9 (s, C(CH3)2), 24.4 (s, CH3), 23.0 (s, CH3). IR: = 2014, 1960, 1933 cm−1 (νCO). Elemental analysis: calcd for C29H37FeN2O3P (548.19): C 63.51, H 6.80, N 5.11; found C 63.06, H 6.86, N 5.09.
3b: 31P{1H} NMR (d8-THF): δ = 228.1(t, 2JPD = 13.5 Hz). 1H NMR (d8-THF): δ = 7.36 (m, 2 H, C6H3), 7.24 (m, 4 H, C6H3), 6.88 (d, 2 H, 3JPH = 6.8 Hz, CH), 2.89 (sept, 4 H, CH(CH3)23JHH = 6.8 Hz), 1.19 (d, 12 H, CH(CH3)2, 3JHH = 6.8 Hz), 1.11 (d, 12 H, CH(CH3)2), 3JHH = 6.8 Hz).
4: A solution of Na[2] was prepared by dissolving complex 3a (30 mg, 54 μmol) in THF-d8 and deprotonating with NaH (1.3 mg, 54 μmol). After quantitative conversion had been established by NMR spectroscopy, triphenyltin chloride (20 mg, 54 μmol) was added. After 5 min, the reaction was complete, and precipitated solids were removed via decantation. After the evaporation of d8-THF, the product was isolated as red crystals, yield 47 mg (93%), m.p. >350 °C (dec.).
31P{1H} NMR (d8-THF): δ = 251.2 (s, 2J119/117SnP = 220/211 Hz). 1H NMR (d8-THF): δ = 7.45–7.05 (m, 23 H, C6H3, C6H5 and CH), 3.13 (sept, 4 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.24 (d, 6 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.23(d, 6H, 3JHH = 6.7 Hz, CH(CH3)2). 13C{1H} NMR (d8-THF): δ = 214.7 (d, CO, 2JPC = 8.8 Hz), 147.0 (d, 2JPC = 3.4 Hz, i-C), 145.2 (d, 3JPC = 4.1 Hz, o-C), 137.0 (s, o-C), 133.4 (d, 3JPC = 5.4 Hz, o-C), 130.5 (d, 4JPC = 1.3 Hz, m-C), 127.9 (d, 5JPC = 1.7 Hz, p-C), 127.69 (s, m-C), 127.68 (s, p-C), 124.6 (s, i-C), 29.4 (s, C(CH3)2), 24.8 (s, CH3), 22.8 (s, CH3). IR: = 2064, 1978, 1909 cm−1 (νCO). Analytical data were found to show a strong dependence on the combustion conditions which implied that even at elevated temperatures no complete combustion could be achieved. As several attempts to establish optimum conditions failed, no meaningful elemental analysis was obtained. The homogeneity of the product was established using spectral data (see ESI†).
5: (a) A solution of Na2[Fe(CO)4] (390 mg, 1.12 mmol) in THF (20 ml) was cooled in a dry ice/acetone bath to −78 °C. A solution of 1 (500 mg, 1.12 mmol) in THF (5 ml) was added dropwise with stirring. The solution was allowed to warm to r.t., stirred for 2 h, and filtered. The solution was again cooled to −78 °C, and another equivalent of 1 (500 mg, 1.12 mmol) was added. Stirring was continued for 2 h during which the mixture was allowed to warm to r.t. Solids were then filtered off, and the solvent was removed under reduced pressure. The remaining dark solid was treated with hexane, the mixture filtered, and the filtrate evaporated to dryness. The residue was dissolved in a minimum amount of hexane and stored at 4 °C, to give 570 mg (55%) of red crystals, m.p. >350 °C (dec.).
(b) Alternatively, 5 was directly accessible from reaction of Collman's reagent with 2 equiv. of 1: Na2[Fe(CO)4] (390 mg, 1.12 mmol) and THF (20 ml) were introduced into a Schlenk flask and stirred for 10 min at r.t. The flask was then cooled to −78 °C in a dry ice-acetone bath, and a solution of 1 (1.00 g, 2.25 mmol) in THF (10 ml) was added dropwise. The mixture was allowed to warm to r.t. and stirred for another 2 h. The solvent was removed under reduced pressure and the remaining dark solid was treated with hexane. The mixture was filtered and the filtrate was evaporated to dryness. The residue was dissolved in a minimum amount of hexane and stored at 4 °C, to give 747 mg (72%) of red crystals, m.p. >350 °C (dec.).
31P{1H} NMR (C6D6): δ = 217.7 (s). 1H NMR (C6D6): δ = 7.19 (m, 2 H, C6H3), 7.09 (m, 4 H, C6H3), 6.09 (s, 2 H, CH), 3.25 (sept, 4 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.23 (d, 12 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.12 (d, 12 H, 3JHH = 6.8 Hz, CH(CH3)2). 13C{1H} NMR (C6D6): δ = 222.4 (s, CO), 147.7 (s, o-C), 136.6 (s, i-C), 129.5 (s, p-C), 124.8 (s, CH), 124.6 (s, m-C), 29.2 (s, C(CH3)2), 25.6(s, CH3), 24.0 (s, CH3). IR: = 1930, 1883 cm−1 (νCO). Analytical data were found to show a strong dependence on the combustion conditions which implied that even at elevated temperatures no complete combustion could be achieved. As several attempts to establish optimum conditions failed, no meaningful elemental analysis was obtained. The homogeneity of the product was established using spectral data (see ESI†).
(b) A solution of Na[2] (30 mg, 53 μmol) in d8-THF (0.4 ml) was placed in an NMR tube equipped with a Teflon-coated septum and cooled to −70 °C. A slight excess of MeI (0.10 ml of a 0.54 M stock solution containing 7.5 mg/54 μmol of reagent) was added via a syringe. Recording of multinuclear (1H, 13C, 31P) NMR spectra at −70 °C immediately after mixing revealed the presence of a major product identified as complex 8 (approx. 90% of phosphorus-containing species by integration of the 31P NMR signals) besides minor amounts of 6 and 7 (<10%, presumably formed as a consequence of insufficient control of local temperature or concentration during the mixing process) and trace quantities of unidentified by-products. Monitoring the further progress of the reaction during slow warming to r.t. allowed us to establish that 8 decays to yield a mixture of 6 and 7. No further intermediates were spectroscopically observable during this process.
6: 31P{1H} NMR (C6D6): δ = 169.4 (s). 1H NMR (C6D6): δ = 7.07 (m, 6 H, C6H3), 5.76 (d, 2 H, 3JPH = 11.0 Hz, CH), 3.75 (sept, 2 H, 3JHH = 6.9 Hz, CH(CH3)2), 3.41 (sept, 2 H, 3JHH = 7.0 Hz, CH(CH3)2), 1.96 (d, 3 H, 2JPH = 5.2 Hz, PCH3), 1.48 (d, 6 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.25 (d, 6 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.16 (d, 6 H, 3JHH = 6.7 Hz, CH(CH3)2), 1.08 (d, 6 H, 3JHH = 6.7 Hz, CH(CH3)2). 13C{1H} NMR (C6D6): δ = 212.1 (d, 2JPC = 16.9 Hz, CO), 148.5 (d, 3JPC = 1.9 Hz, o-C), 134.2 (d, 2JPC = 4.6 Hz, i-C), 128.0 (d, 5JPC = 1.1 Hz, p-C), 123.0 (s, m-C), 119.9 (d, 2JPC = 2.7 Hz, CH), 29.5 (s, PCH3), 28.2 (s, C(CH3)2), 27.6 (s, C(CH3)2), 26.2 (s, CH3), 24.7 (s, CH3), 21.6 (s, CH3), 21.1 (s, CH3). IR: = 2039, 1963, 1917 cm−1 (νCO).
8: 31P{1H} NMR (d8-THF, 203 K): δ = 191.3 (s). 1H NMR (d8-THF, 253 K): δ = 7.22–7.10 (m, 6 H, C6H3), 5.97 (d, 2 H, 3JPH = 7.6 Hz, CH), 3.78 (sept, 2 H, 3JHH = 6.6 Hz, CH(CH3)2), 3.65 (sept, 2 H, 3JHH = 6.6 Hz, CH(CH3)2), 2.02 (d, 3 H, 2JPH = 3.6 Hz, PCH3), 1.43 (d, 6 H, 3JHH = 6.6 Hz, CH(CH3)2), 1.34 (d, 6 H, 3JHH = 6.6 Hz, CH(CH3)2), 1.20 (d, 6 H, 3JHH = 6.7 Hz, CH(CH3)2), 1.13 (d, 6 H, 3JHH = 6.7 Hz, CH(CH3)2). 13C{1H} NMR (d8-THF, 203 K): δ = 217.7 (d, 2JPC = 27.7 Hz, CO), 149.3 (s, o-C), 147.9 (s, o-C), 138.9 (d, 2JPC = 5.4 Hz, i-C), 128.0 (s, p-C), 123.9 (s, m-C), 123.0 (s, m-C), 120.2 (s, CH), 28.9 (s, C(CH3)2), 28.6 (s, C(CH3)2), 27.6 (s, CH3), 26.7 (d, 1JPC = 26 Hz, PCH3), 25.0 (s, CH3), 22.4 (s, 2 CH3).
7: 31P{1H} NMR (C6D6): δ = 111.9 (s). 1H NMR (C6D6): δ = 7.2–7.1 (m, 6 H, C6H3), 5.74 (d, 2 H, 3JPH = 2.0 Hz, CH), 3.83 (dsept, 2 H, 3JHH = 6.8 Hz, 5JPH = 3.8 Hz, CH(CH3)2), 3.65 (sept, 2 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.43 (d, 6 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.28 (d, 6 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.18 (d, 6 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.12 (d, 6 H, 3JHH = 6.8 Hz, CH(CH3)2), 1.04 (d, 3 H, 1JPH = 8.5 Hz, PCH3). 13C{1H} NMR (C6D6): δ = 148.9 (d, 3JPC = 2.1 Hz, o-C), 148.4 (s, o-C), 138.4 (d, 2JPC = 12.6 Hz, i-C), 127.7 (d, 4JPC = 1.7 Hz, m-C), 124.9 (s, p-C), 124.7 (d, 4JPC = 1.3 Hz, m-C), 119.3 (d, 2JPC = 6.5 Hz, CH), 29.1 (d, 4JPC = 6.1 Hz, CH(CH3)2), 28.7 (s, CH(CH3)2), 25.9 (s, CH3), 25.4 (s, CH3), 25.1 (s, CH3), 24.2 (d, 5JPC = 2.7 Hz, CH3), 19.8 (d, 1JPC = 48.2 Hz, CH3).
Crystal structure determinations
Diffraction studies were carried out using a Bruker diffractometer equipped with a Kappa APEX II Duo CCD-detector and a KRYO-FLEX cooling device with Mo-Kα radiation (λ = 0.71073 Å; complexes [PPh4][2], 3a, 5) or Cu-Kα radiation (λ = 1.54178 Å; complexes 4, 6) at T = 100 K. The structures were solved by direct methods (SHELXS-9728) and refined with a full-matrix least-squares scheme on F2 (SHELXL-2014 and SHELXL-9728). Semi-empirical absorption corrections were applied for all structures. Non-hydrogen atoms were refined anisotropically, and H atoms with a riding model, on F2. The hydrogen atom attached to the iron atom in 3a was located and refined freely using isotropic thermal displacement parameters. Details of the crystal structure determinations are listed in Table 1.| [PPh4][2] | 3a | 4 | 5 | 6 | |
|---|---|---|---|---|---|
| CCDC | 1032393 | 1032390 | 1032394 | 1032391 | 1032392 |
| Formula | C29H36FeN2O3P – C24H20P – 1.5THF | C29H37FeN2O3P | C47H51FeN2O3PSn | C54H72FeN4O2P2 | C31H39FeN2O4P |
| Formula weight | 994.94 | 548.43 | 897.40 | 926.95 | 590.46 |
| Crystal size/mm | 0.28 × 0.22 × 0.04 | 0.32 × 0.22 × 0.18 | 0.15 × 0.09 × 0.06 | 0.27 × 0.18 × 0.12 | 0.14 × 0.11 × 0.03 |
| T/K | 100(2) | 100(2) | 100(2) | 100(2) | 100(2) |
| Crystal system | Orthorhombic | Monoclinic | Triclinic | Monoclinic | Orthorhombic |
| Space group | Pna21 | P21/n |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/n | Pnma |
| a/Å | 19.0086 (16) | 10.9098(8) | 11.5843(6) | 13.5063(5) | 19.7315(14) |
| b/Å | 12.8494 (10) | 15.3843(11) | 12.2340(6) | 21.9821(8) | 20.5098(16) |
| c/Å | 43.849 (3) | 17.7380(11) | 17.8887(11) | 18.0479(5) | 7.4442(6) |
| α (°) | 90 | 90 | 92.606 (3) | 90 | 90 |
| β (°) | 90 | 99.998(2) | 103.175 (3) | 107.775 (1) | 90 |
| γ (°) | 90 | 90 | 116.117 (2) | 90 | 90 |
| V/Å3 | 10710.2 (14) | 2931.9 (4) | 2185.4 (2) | 5102.6 (3) | 3012.6 (4) |
| Density (Mg m−3) | 1.234 | 1.242 | 1.364 | 1.207 | 1.302 |
| F(000) | 4224 | 1160 | 924 | 1984 | 1248 |
| Z | 8 | 4 | 2 | 4 | 4 |
| Abs. coeff. μ/mm−1 | 0.390 | 0.599 | 7.870 | 0.401 | 4.814 |
| Absorption correction | Semi-empirical | Semi-empirical | Semi-empirical | Semi-empirical | Semi-empirical |
| Data collected | 56![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 223 223 |
21309 |
39721 |
32673 |
16472 |
| Unique data | 18565 (Rint = 0.100) |
6011 (Rint = 0.077) | 7394 (Rint = 0.053) | 8997 (Rint = 0.048) | 2677 (Rint = 0.038) |
| Observed data with I > 2σ(I) | 13658 |
3685 | 6119 | 6259 | 2441 |
| Restraints | 179 | 1 | 277 | 0 | 0 |
| Variables | 1231 | 329 | 464 | 568 | 198 |
| R 1 (I > 2σ(I)) | 0.060 | 0.047 | 0.040 | 0.038 | 0.029 |
| wR2 | 0.118 | 0.097 | 0.092 | 0.080 | 0.081 |
| GOF | 1.04 | 0.99 | 1.03 | 0.99 | 1.06 |
| Max. diff. dens./e Å−3 | 0.715 | 0.440 | 0.825 | 0.285 | 0.396 |
| Min. diff. dens./e Å−3 | −0.476 | −0.290 | −1.314 | −0.313 | −0.388 |
[PPh4][2] is a racemic twin (x = 0.473(15)), and an extinction correction was applied. Furthermore, one solvent molecule (THF) is disordered, and the high Uij-values in another one indicate a slight disorder, which was not resolved. 4 shows high Uij-values for the Fe(CO)3 moiety as well as for one 2,6-diisopropylphenyl substituent indicating a disorder. The disorder was resolved and modelled for the Fe(CO)3 moiety, the 2,6-diisopropylphenyl substituent and one iPr-group of the other 2,6-diisopropylphenyl substituent. Disordered C- and O-atoms in [PPh4][2] and 4 were refined isotropically, and the Fe-atoms were refined anisotropically (see the cif-files for further details).
Computational studies
Computational studies were performed with the Gaussian0329 suite of programs using def2-tzvp basis sets30 (obtained from the basis set exchange home page31,32) on model compounds 2′, [Li(OMe2)3][2′], 3′ and 5′ featuring NMe instead of the larger NDipp substituents. Solvent effects were included by using a PCM model as implemented in the Gaussian package (SCRF = THF). Molecular structures were first energy optimized without symmetry constraints. Calculation of harmonic vibrational frequencies was performed in order to ensure that all stationary points located were of local minima on the energy hypersurface (in case of [2′][Li(OMe2)3], the molecular structure had to be re-optimized using a smaller def2-svp basis set for the [Li(OMe2)3]+ cation, and the frequency calculation was performed at the same level. The structural features of both models showed no significant deviations). The NBO analysis33 was performed with the NBO 3.1 program as implemented in the Gaussian package. MOLDEN34 was used for the visualization of Kohn–Sham orbitals.Acknowledgements
Financial support by Deutsche Forschungsgemeinschaft (grant GU 415/15-1) is gratefully acknowledged.Notes and references
- Reviews: (a) A. H. Cowley and R. A. Kemp, Chem. Rev., 1985, 85, 367 CrossRef CAS; (b) M. Sanchez, M. R. Mazières, L. Lamandé and R. Wolf, in Multiple Bonds and Low Coordination Chemistry in Phosphorus Chemistry, ed. M. Regitz and O. J. Scherer, Thieme, Stuttgart, 1990, 129ff Search PubMed; (c) D. Gudat, Coord. Chem. Rev., 1997, 163, 71 CrossRef CAS; (d) H. Nakazawa, Adv. Organomet. Chem., 2004, 50, 107 CrossRef; (e) L. Rosenberg, Coord. Chem. Rev., 2012, 256, 606 CrossRef CAS PubMed.
- (a) M. B. Abrams, B. L. Scott and R. T. Baker, Organometallics, 2000, 19, 4944 CrossRef CAS; (b) C. A. Caputo, M. C. Jennings, H. M. Tuononen and N. D. Jones, Organometallics, 2009, 28, 990 CrossRef CAS.
- L. Hutchins, E. Duesler and R. Paine, Organometallics, 1982, 1, 1254 CrossRef CAS.
- M. L. H. Green, J. Organomet. Chem., 1995, 200, 127 CrossRef.
- S. Burck, J. Daniels, T. Gans-Eichler, D. Gudat, K. Nättinen and M. Nieger, Z. Anorg. Allg. Chem., 2005, 631, 1403 CrossRef CAS.
- B. Pan, Z. Xu, M. W. Bezpalko, B. M. Foxman and C. M. Thomas, Inorg. Chem., 2012, 51, 4170 CrossRef CAS PubMed.
- (a) D. Gudat, A. Haghverdi and M. Nieger, J. Organomet. Chem., 2001, 617, 383 CrossRef; (b) D. Förster, J. Nickolaus, M. Nieger, Z. Benkö, A. W. Ehlers and D. Gudat, Inorg. Chem., 2013, 52, 7699 CrossRef PubMed.
- (a) L. D. Hutchins, R. T. Paine and C. F. Campagna, J. Am. Chem. Soc., 1980, 102, 4521 CrossRef CAS; (b) L. D. Hutchins, H.-U. Reisacher, G. L. Wood, E. N. Duesler and R. T. Paine, J. Organomet. Chem., 1987, 335, 229 CrossRef CAS; (c) C. A. Caputo, A. L. Brazeau, Z. Hynes, J. T. Price, H. M. Tuononen and N. D. Jones, Organometallics, 2009, 28, 5261 CrossRef CAS.
- (a) H.-U. Reisacher, E. N. Duesler and R. T. Paine, J. Organomet. Chem., 1998, 564, 13 CrossRef CAS; (b) H.-U. Reisacher, E. N. Duesler and R. T. Paine, J. Organomet. Chem., 1997, 539, 37 CrossRef CAS; (c) H.-U. Reisacher, W. F. McNamara, E. N. Duesler and R. T. Paine, Organometallics, 1997, 16, 449 CrossRef CAS; (d) H.-U. Reisacher, E. N. Duesler and R. T. Paine, Chem. Ber., 1996, 129, 279 CrossRef CAS; (e) W. E. McNamara, E. N. Duesler, R. T. Paine, J. V. Ortiz, P. Koelle and H. Nöth, Organometallics, 1986, 5, 380 CrossRef CAS.
- S. Magens and B. Plietker, J. Org. Chem., 2010, 75, 3715 CrossRef CAS PubMed.
- (a) A. H. Cowley, E. A. Kemp, E. A. V. Ebsworth, D. W. H. Rankin and M. D. Walkinshaw, J. Organomet. Chem., 1984, 265, C19 CrossRef CAS; (b) H. Nakazawa, Y. Yamaguchi, K. Kawamura and K. Miyoshi, Organometallics, 1997, 16, 4626 CrossRef CAS.
- J.-J. Brunet, R. Chauvin, O. Diallo, B. Donnadieu, J. Jaffart and D. Neibecker, J. Organomet. Chem., 1998, 570, 195 CrossRef CAS.
- (a) H. B. Chin and R. Bau, J. Am. Chem. Soc., 1976, 98, 2434 CrossRef CAS; (b) R. G. Teller, R. G. Finke, J. P. Collman, H. B. Chin and R. Bau, J. Am. Chem. Soc., 1977, 99, 1104 CrossRef CAS.
- L. M. Clarkson, W. Clegg, D. C. R. Hockless and N. C. Norman, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 236 CrossRef.
- For appropriate studies on [HFe(CO)4]− and [HFe(CO)3(PR3)]− see: (a) M. Y. Darensbourg, H. J. Darensbourg and H. L. C. Barros, Inorg. Chem., 1978, 17, 297 CrossRef CAS; (b) C. E. Ash, T. Delord, D. Simmons and M. Y. Darensbourg, Organometallics, 1986, 5, 17 CrossRef CAS.
- B. Zhou and J. M. Goicoechea, Chem. – Eur. J., 2010, 16, 11145 CrossRef CAS PubMed.
- (a) M. Zhou and L. Andrews, J. Chem. Phys., 1999, 110, 10370 CrossRef CAS PubMed; (b) G. Wang, C. Chi, J. Cui, X. Xing and M. Zhou, J. Phys. Chem. A, 2012, 116, 2484 CrossRef CAS PubMed.
- Raman data: P. S. Braterman, J. Mol. Spectrosc., 1977, 65, 334 CrossRef CAS.
- J. E. Ellis and Y.-S. Chen, Organometallics, 1989, 8, 1350 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS PubMed.
- J. E. M. N. Klein, B. Miehlich, M. S. Holzwarth, M. Bauer, M. Milek, M. M. Khusniyarov, G. Knizia, H.-J. Werner and B. Plietker, Angew. Chem., Int. Ed., 2014, 53, 1790 CrossRef CAS PubMed.
- J.-J. Brunet, R. Chauvin and D. Neibecker, Synth. Commun., 1997, 27, 1433 CrossRef CAS.
- (a) R. Mathieu, A.-M. Caminade, J.-P. Majoral, S. Attali and M. Banchez, Organometallics, 1986, 5, 1914 CrossRef CAS; (b) R. Mathieu, A.-M. Caminade, J.-P. Majoral and J.-C. Daran, J. Am. Chem. Soc., 1986, 108, 8007 CrossRef CAS; (c) A.-M. Caminade, J.-P. Majoral, M. Sanchez, R. Mathieu, S. Attali and A. Grand, Organometallics, 1987, 6, 1459 CrossRef CAS; (d) H. Westermann, M. Nieger, E. Niecke, J.-P. Majoral, A.-M. Caminade and R. Mathieu, Organometallics, 1989, 8, 244 CrossRef CAS.
- For a first such complex see: J. P. Bezombes, P. B. Hitchcock, M. F. Lappert and J. E. Nycz, Dalton Trans., 2004, 499 RSC.
- A. Poletti, A. Santucci and A. Foffani, J. Mol. Struct., 1969, 3, 311 CrossRef CAS.
- H. Nakazawa, Y. Yamaguchi, T. Mizuta, S. Ichimura and K. Miyoshi, Organometallics, 1995, 14, 4635 CrossRef CAS.
- W. A. Herrmann, K. Öfele and C. E. Zybill, in Synthetic Methods in Organometallic and Inorganic Chemistry, ed. W. A. Herrmann, Thieme, Stuttgart, 1997, vol. 7, 24ff Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision E.01, Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- D. Feller, J. Comput. Chem., 1996, 17, 1571 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 CrossRef CAS PubMed (http://bse.pnl.gov/bse/portal).
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1 Search PubMed.
- G. Schaftenaar and J. H. Noordik, J. Comput. Aided Mol. Des., 2000, 14, 123 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Representation of NMR data and computational results. CCDC 1032390–1032394. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00008d |
| This journal is © The Royal Society of Chemistry 2015 |