
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntermolecular hydroaminoalkylation of alkenes and dienes using a titanium mono(formamidinate) catalyst†
Jaika
Dörfler
,
Till
Preuß
,
Christian
Brahms
,
Dennis
Scheuer
and
Sven
Doye
*
Institut für Chemie, Universität Oldenburg, Carl-von-Ossietzky-Strasse 9-11, D-26111 Oldenburg, Germany. E-mail: doye@uni-oldenburg.de
First published on 13th February 2015
Abstract
An easily accessible formamidinate ligand-bearing titanium complex initially synthesized by Eisen et al. is used as catalyst for intermolecular hydroaminoalkylation reactions of unactivated, sterically demanding 1,1- and 1,2-disubstituted alkenes and styrenes with secondary amines. The corresponding reactions, which have never been achieved with titanium catalysts before, take place with excellent regioselectivity (up to 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and in addition, corresponding reactions of 1,3-butadienes with N-methylbenzylamine are also described for the first time.
:1) and in addition, corresponding reactions of 1,3-butadienes with N-methylbenzylamine are also described for the first time.
Introduction
The direct addition of α-C(sp3)–H bonds of primary or secondary amines across C–C double bonds, the so-called hydroaminoalkylation of alkenes, represents a very promising and atom-efficient approach towards the synthesis of various nitrogen-containing molecules.1 Due to the fact that the formed α-alkylated amines are of great importance in the agrochemical, fine chemical and pharmaceutical industries2 a plethora of suitable hydroaminoalkylation catalysts has already been identified in recent years.3–7 Although hydroaminoalkylation reactions can be achieved in the presence of ruthenium,3 iridium,4 group 5 metal,5 zirconium,6 or titanium catalysts7 a number of limitations in scope still exists. For example, ruthenium and iridium catalysts can only be used for hydroaminoalkylation reactions of amine substrates bearing a directing 2-pyridinyl substituent bound to the nitrogen atom of the amine.3,4 Furthermore, zirconium complexes only catalyze intramolecular hydroaminoalkylation reactions of primary aminoalkenes.6 On the other hand, group 5 metal5 and titanium catalysts7 have successfully been applied for the intermolecular hydroaminoalkylation of alkenes, styrenes and 1,3-butadienes. For example, the conversion of styrene (1) into the corresponding hydroaminoalkylation products 3a and 3b (Scheme 1) could be achieved in the presence of tantalum or titanium catalysts.5m,7d,g,i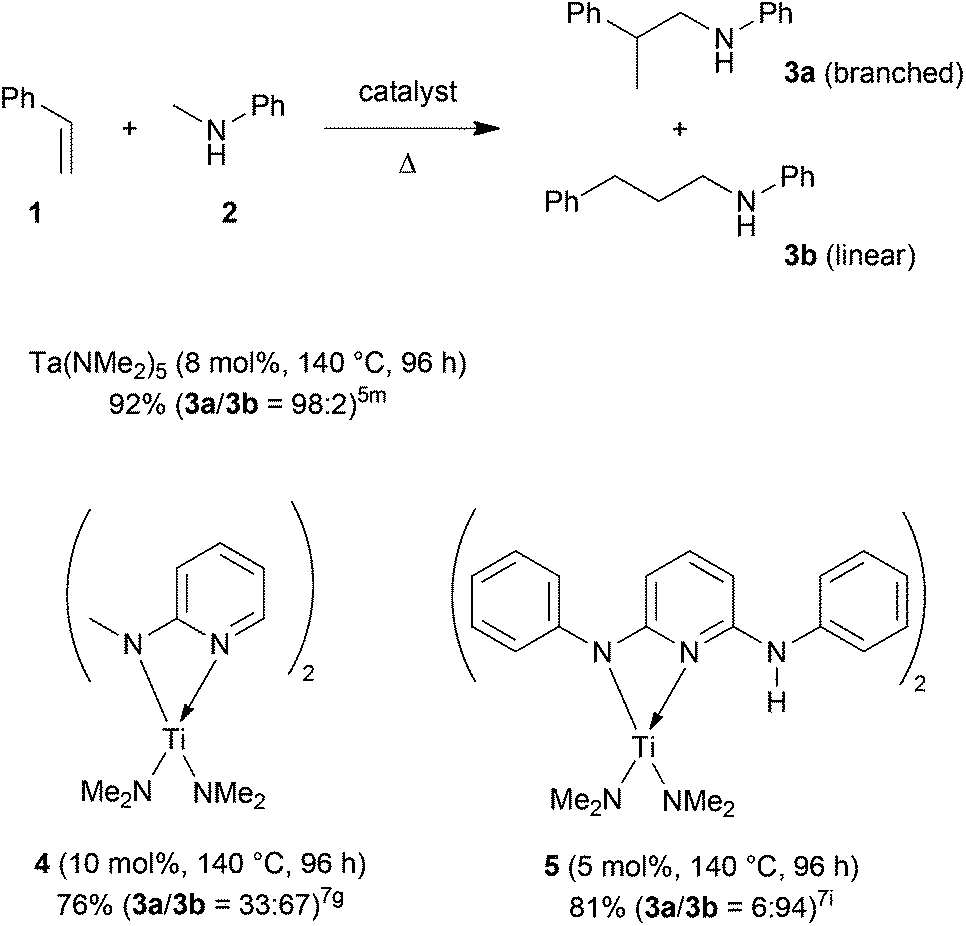 | ||
| Scheme 1 Intermolecular hydroaminoalkylation of styrene (1) with N-methylaniline (2) catalyzed by group 4 and group 5 metal catalysts. | ||
In this context, it is worth mentioning that the regioselectivity of hydroaminoalkylation reactions of styrenes can simply be controlled by the nature of the catalyst. Whereas in the presence of Ta(NMe2)5, the branched product 3a is formed almost exclusively,5m the aminopyridinato titanium catalysts 4 and 5 favor the formation of the corresponding linear hydroaminoalkylation product 3b.7g,i However, so far, the use of titanium catalysts is limited to hydroaminoalkylation reactions of sterically less demanding mono-substituted alkenes, styrenes, or 1,3-butadienes with N-alkylanilines or dialkylamines. Among the class of sterically more demanding disubstituted alkenes, only norbornene7b–d,g and methylenecyclohexane7c,g were found to undergo hydroaminoalkylation reactions in the presence of selected titanium catalysts. On the other hand, hydroaminoalkylation reactions of sterically demanding 1,1- and 1,2-disubstituted alkenes are well known for group 5 metal catalysts.5c,e–g,i,k,l,n Most impressively, very recently, Schafer et al. reported a 2-pyridonate tantalum catalyst that is able to catalyze high-yielding hydroaminoalkylation reactions of sterically demanding E- and Z-internal alkenes like cyclohexene, (E)- and (Z)-3-hexene, (Z)-β-methylstyrene or (E)-1-(para-methoxyphenyl)propene.5n In the latter two cases, hydroaminoalkylation at the β-carbon of the β-substituted styrene derivatives is favored but only modest regioselectivities (up to 4.4:1) could be achieved.
The mechanism of group 4 and 5 metal-catalyzed hydroaminoalkylation reactions has already been studied1,5c,d,i,6,7e and it is generally accepted that the amine substrate undergoes N–H and C–H bond activation in the presence of the catalyst precursor to give a strained metallaaziridine as the catalytically active species. Subsequent alkene insertion into the M–C bond of the metallaaziridine which determines the regioselectivity of the over-all transformation then results in the formation of a 2-metallapyrrolidine. Finally, aminolysis of the M–C bond of the five membered intermediate and product-forming C–H bond activation regenerate the catalytically active metallaaziridine.
Herein, we present the first example of a titanium-based catalyst for intermolecular hydroaminoalkylation reactions of unactivated, sterically demanding 1,1- and 1,2-disubstituted alkenes and styrenes with secondary amines. The corresponding reactions take place with excellent regioselectivity (up to 99:1) and the formamidinate ligand-bearing catalyst is also capable of catalyzing reactions of 1,3-butadienes with N-methylbenzylamine.
Results and discussion
In order to develop a more efficient catalyst system that is able to expand the substrate scope of the titanium-catalyzed intermolecular hydroaminoalkylation of alkenes, we recently focused on ligand motifs which are structurally similar to the aminopyridinato ligands of the second-generation hydroaminoalkylation catalysts 4 and 5 (Scheme 1). In this context, it must be mentioned that the chelating heteroallylic N–C–N unit of the aminopyridinato ligands that binds to the titanium center can also be found in the well-known class of amidinate ligands [R1-C(NR2)2]−. A variety of corresponding group 4 amidinate complexes has already been used as catalysts in the polymerization of olefins by Eisen et al.8 and a particular advantage of this class of catalysts is that the ligands are easily prepared and modified.8a On the other hand, corresponding catalysts have never been used for hydroaminoalkylation reactions of alkenes.During an investigation of various titanium formamidinate catalysts we found that complex 6 (Scheme 2) which bears 2,6-diisopropylphenyl groups as the amidinate N-substituents8a is a highly active catalyst for the hydroaminoalkylation of styrene (1) with N-methylaniline (2). Under usually applied conditions (10 mol% 6, n-hexane, 140 °C, 96 h, sealed Schlenk tube),7g,i the reaction took place smoothly to give a mixture of the two regioisomeric hydroaminoalkylation products 3a and 3b in an excellent total yield of 96%. Unfortunately, the regioselectivity of the reaction which in this case, favors the formation of the branched product 3a was only modest (3a/3b = 73:27). However, an interesting finding is the fact that in comparison to the structurally related aminopyridinato catalysts 4 and 5, the regioselectivity is reversed with the formamidinate catalyst 6.
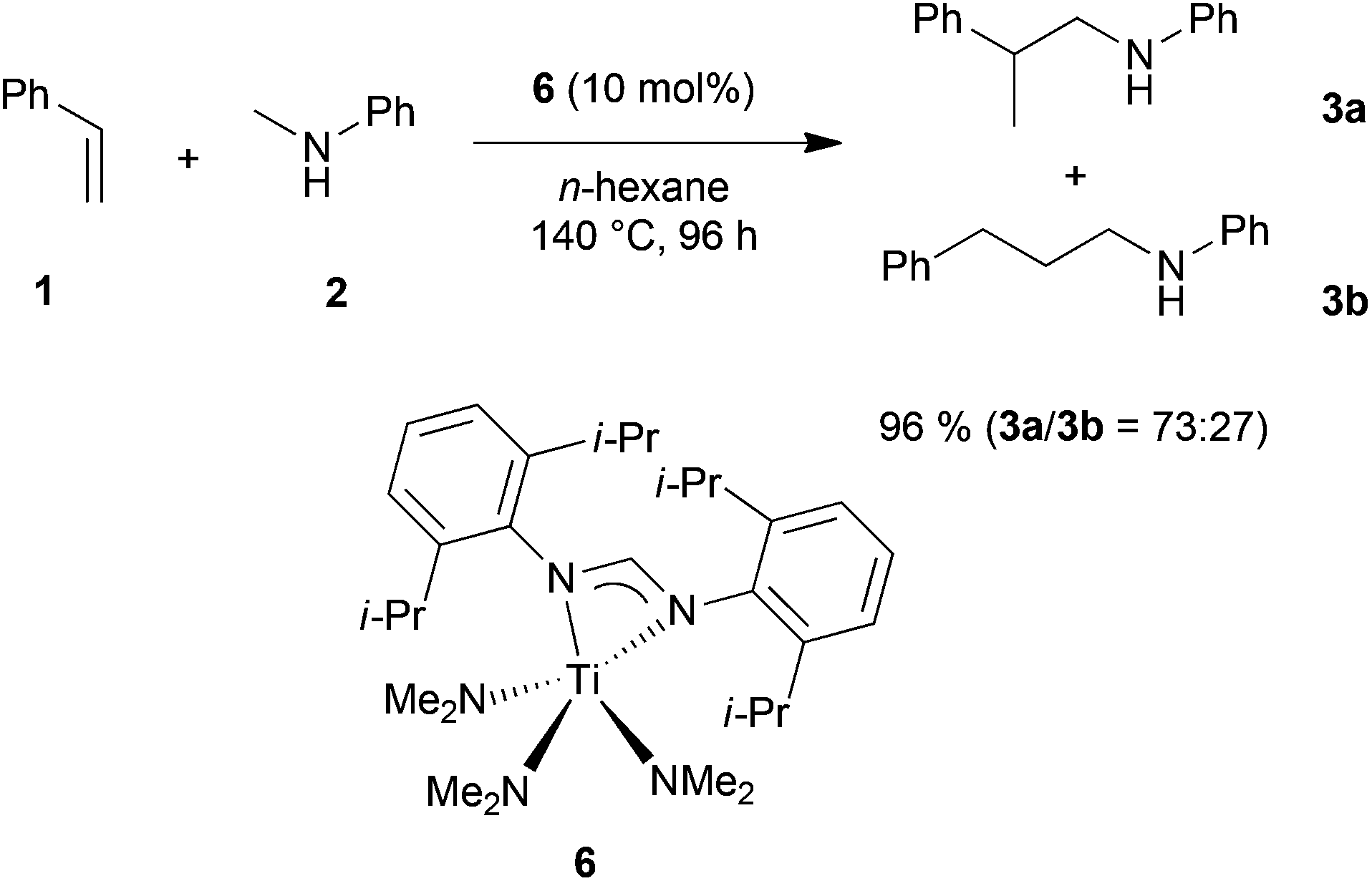 | ||
| Scheme 2 Intermolecular hydroaminoalkylation of styrene (1) with N-methylaniline (2) catalyzed by titanium mono(formamidinate) complex 6. | ||
Impressed by the high catalytic activity of complex 6 and the clean formation of the desired hydroaminoalkylation products 3a and 3b, we further investigated whether complex 6 is also able to catalyze intermolecular hydroaminoalkylation reactions of sterically more demanding disubstituted alkenes with N-methylaniline (2, Table 1).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Alkene | T (°C) | Isolated product(s) | Yielda (%) | Selectivity a/bb |
| a Reaction conditions: N-methylaniline (2, 2.0 mmol, 214 mg), alkene (3.0 mmol), 6 (109 mg, 0.2 mmol, 10 mol%), n-hexane (1 mL), 140 °C or 180 °C, 96 h, sealed Schlenk tube. Yields refer to the yield of the isolated product (a or b). b GC analysis prior to chromatography. c A yield of 89% was obtained after a reaction time of 24 h. d Yields of 76% and 67% were obtained after reaction times of 48 h and 24 h, respectively. e The yield refers to the total yield of isolated products (a + b). f A comparable yield of 69% was obtained after a reaction time of 192 h. | |||||
| 1 |
|
140 |
|
90 | 99:1 |
| 2 | 180 | 90c | 99:1 |
||
| 3 |
|
140 | — | — | — |
| 4 | 180 | — | — | — | |
| 5 |
|
140 |
|
43 | 99:1 |
| 6 | 180 | 86d | 99:1 |
||
| 7 |
|
140 |
|
38 | 99:1 |
| 8 | 180 | 91 | 99:1 |
||
| 9 |
|
140 |
|
17 | 1:99 |
| 10 | 180 | 64 | 1:99 |
||
| 11 |
|
180 |
|
97e | 34:66 |
| 12 |
|
180 | — | — | — |
| 13 |
|
180 | — | — | — |
| 14 |
|
180 | — | — | — |
| 15 |
|
180 | — | — | — |
| 16 |
|
180 |
|
16e | 35:65 |
| 17 |
|
180 |
|
75 | — |
| 18 |
|
180 |
|
72f | — |
| 19 |
|
180 |
|
93 | — |
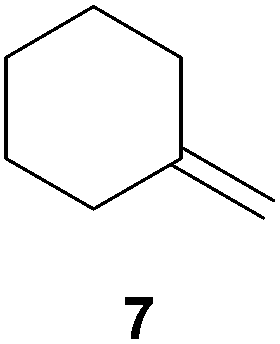
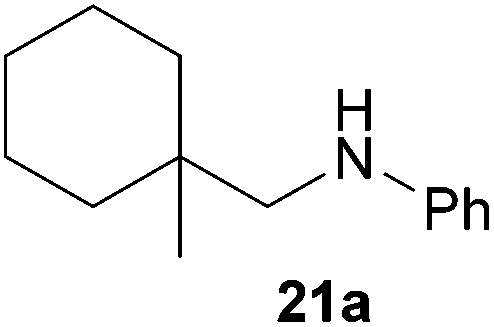
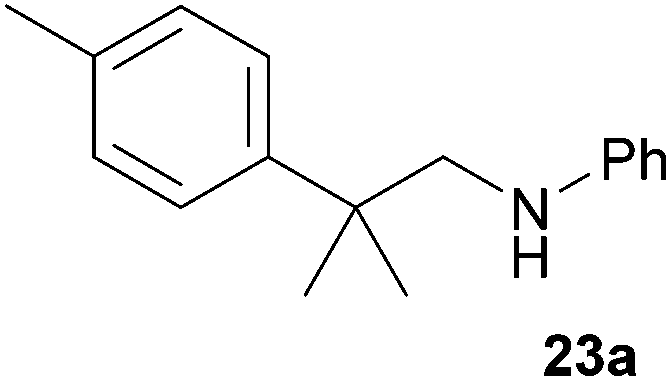
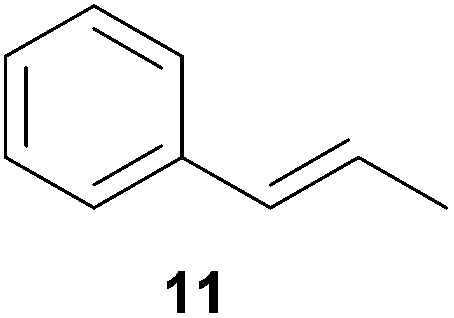
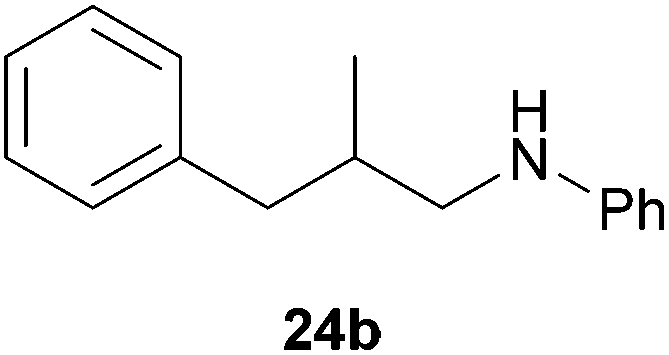
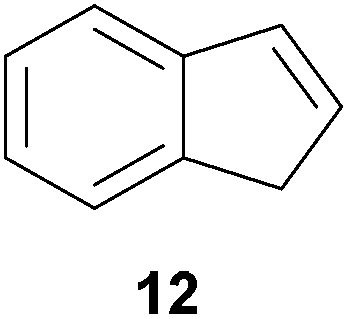
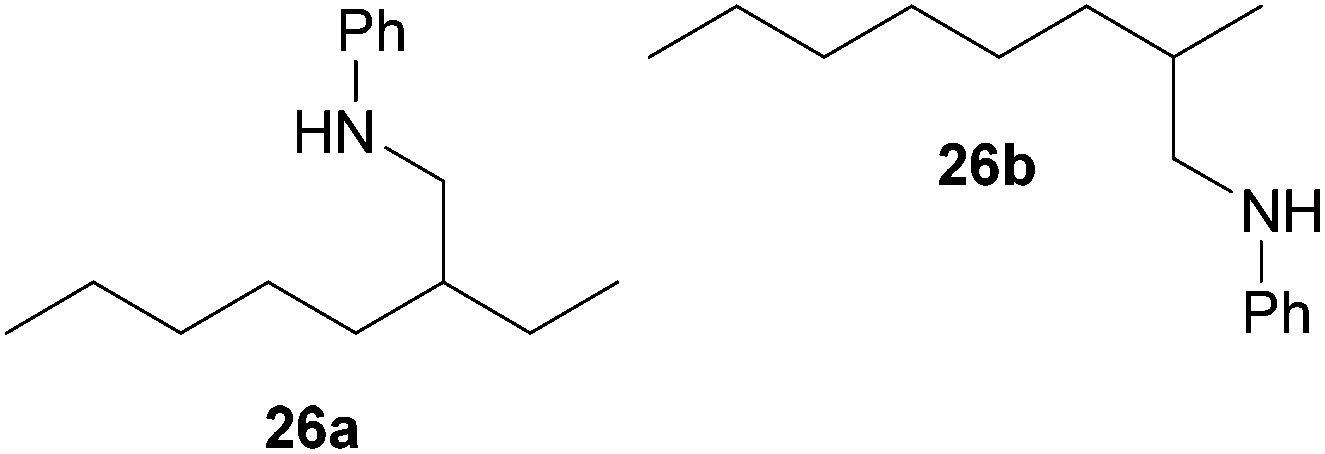
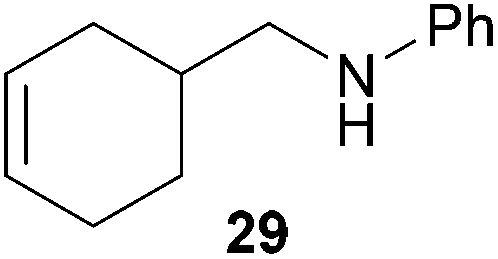
As can be seen from Table 1, a number of successful hydroaminoalkylation reactions of 1,1- and 1,2-disubstituted alkenes and styrenes with N-methylaniline (2) can be achieved at elevated temperatures (140–180 °C) in the presence of 10 mol% of catalyst 6. For example, methylenecyclohexane (7, Table 1, entries 1 and 2) regioselectively reacted to the corresponding branched amine 21a which could be isolated in excellent yield of 90%. Even though bulky 1,1-diphenylethylene (8) did not react at all under the applied conditions (Table 1, entries 3 and 4), α-methylstyrene (9) and para-α-dimethylstyrene (10) could be converted regioselectively to the corresponding branched hydroaminoalkylation products 22a and 23a, respectively (Table 1, entries 5–8). In these cases, the yields of the reactions were significantly improved by increasing the reaction temperature from 140 °C to 180 °C. Finally, it was possible to isolate the branched products 22a and 23a in 86% and 91% yield, respectively. Although a corresponding dependence on the reaction temperature had not been observed during the initially performed reactions of methylenecyclohexane (7, Table 1, entries 1 and 2) we performed most additional reactions presented in Table 1 at 180 °C. A similar observation was made when reactions of 7 or 9 were conducted with reduced reaction times of 24 h or 48 h. While the reduced reaction times did not affect the conversion of methylenecyclohexane (7, 89% yield of product 21a after 24 h), corresponding reactions of α-methylstyrene (9) gave product 22a in significantly reduced yields (76% after 48 h and 67% after 24 h). As a consequence, we performed all additional experiments with a reaction time of 96 h. To the best of our knowledge, the results obtained with styrenes 9 and 10 represent the first successful examples of group 4 metal-catalyzed hydroaminoalkylation reactions of α-substituted styrene derivatives. In addition, it is worth to mention that only two examples of a corresponding reaction catalyzed by a group 5 metal catalyst have been described in the literature5k,m and in this case, the yield was only modest (61%). An additional finding that underlines the high catalytic activity of 6 is the fact that with this catalyst, it was even possible to react the 1,2-disubstituted substrate (E)-β-methylstyrene (11) with N-methylaniline (2, Table 1, entries 9 and 10). Interestingly and in contrast to the reaction of α-methylstyrene, the hydroaminoalkylation took place regioselectively at the sterically less shielded β-carbon of the double bond and correspondingly, the regioisomer 24b could be isolated as the sole product of the reaction in 64% yield. Most impressively, the regioselectivity of the reaction was found to be 24a/24b = 1:99. Previously, only two successful hydroaminoalkylations of β-substituted styrene derivatives had been achieved with a tantalum catalyst5n and in these cases, the regioselectivities were only modest (1:2.0 and 1:4.4). The well-established fact that hydroaminoalkylation product 24b can be obtained from allylbenzene under much milder conditions5e is strongly underlined by a hydroaminoalkylation reaction between allylbenzene and N-methylaniline (2) performed at 140 °C in the presence of 10 mol% of catalyst 6. While under these conditions, (E)-β-methylstyrene (11) gave 24b only in 17% yield (Table 1, entry 9), 24b was obtained in 87% yield from allylbenzene. Interestingly, isomerization of (E)-β-methylstyrene (11) to allylbenzene during the reactions of 11 could not be observed by GC analysis. Additional experiments with indene (12, Table 1, entry 11) then revealed that even this challenging substrate undergoes successful hydroaminoalkylation with 2. Although the reaction delivered the two regioisomers 25a and 25b in 97% combined yield, the regioselectivity was found to be surprisingly low (25a/25b = 34:66). However, as can be seen from Table 1, entries 12–15, sterically more demanding internal alkenes like 13–16 failed to react in the presence of catalyst 6. On the other hand, it was possible to convert (E)-2-octene (17) to a mixture of the corresponding hydroaminoalkylation products 26a and 26b (Table 1, entry 16) but in this case, the yield and the regioselectivity of the reaction turned out to be poor. In contrast to the disappointing behavior of the 1,2-dialkyl-substituted (E)-alkenes 15–17, the (Z)-double bond-containing substrates cyclopentene (18) and cyclohexene (19) gave access to the corresponding hydroaminoalkylation products 27 and 28 in reasonable yields of 75% and 72%, respectively (Table 1, entries 17 and 18). In this context, it can be noted that additional alkyl-substituted cyclohexenes like 4-methyl- or 3-methylcyclohexene also underwent high-yielding hydroaminoalkylation reactions with 2 but unfortunately, formation of four inseparable regio- and diastereomers was observed in both reactions. As the result, all attempts on a precise structure assignment of the isomers failed so far. With regard to catalyst stability it can be stated that longer reaction times do not necessarily lead to improved yields. For example, reactions of cylohexene (19) with N-methylaniline (2) gave comparable yields of product 28 after 96 h (72%, Table 1, entry 18) and 192 h (69%). Interestingly, the additional double bond present in 1,4-cyclohexadiene (20) seems to facilitate the hydroaminoalkylation reaction and as a consequence, an excellent yield of 93% of product 29 was obtained in this case (Table 1, entry 19). The remarkable fact that only the monoalkylated product 29 could be isolated from the reaction mixture is in good agreement with results from Schafer et al.5n and the remaining double bond offers a promising chance for further functionalization. Finally, 3-bromocyclohexene turned out to be unreactive under the reaction conditions while 3-methoxycyclohexene (30) underwent a nucleophilic substitution reaction that delivered the tertiary amine 31 in 61% yield (Scheme 3). The assumption that the latter process does not require the presence of a titanium catalyst is strongly supported by the fact that product 31 could also be isolated in comparable yield (58%) from a control experiment performed in the absence of 6.
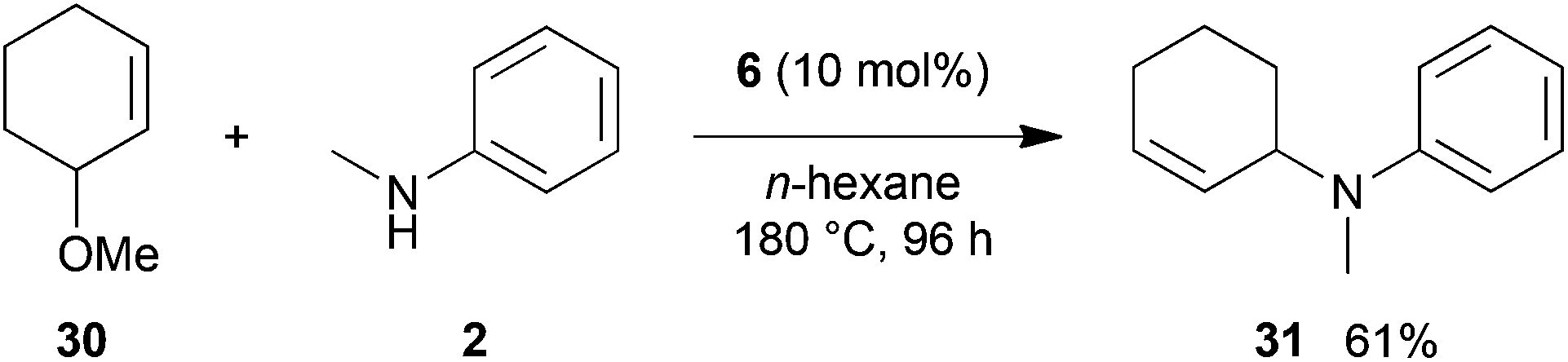 | ||
| Scheme 3 Intermolecular reaction of 3-methoxycyclohexene (30) with N-methylaniline (2) catalyzed by titanium mono(formamidinate) complex 6. | ||
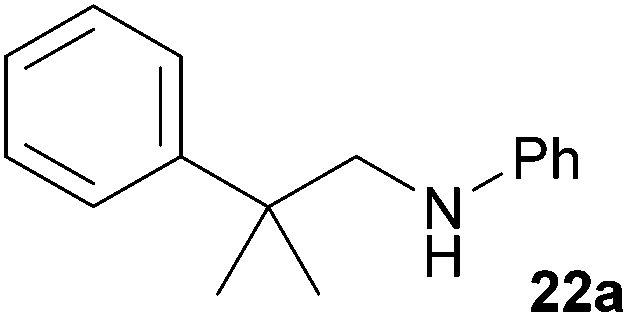
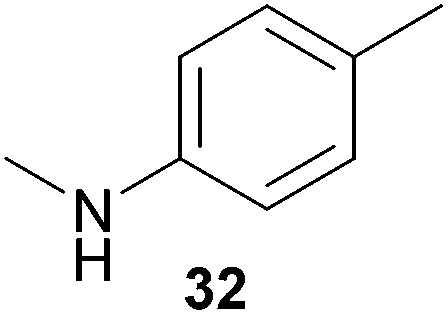
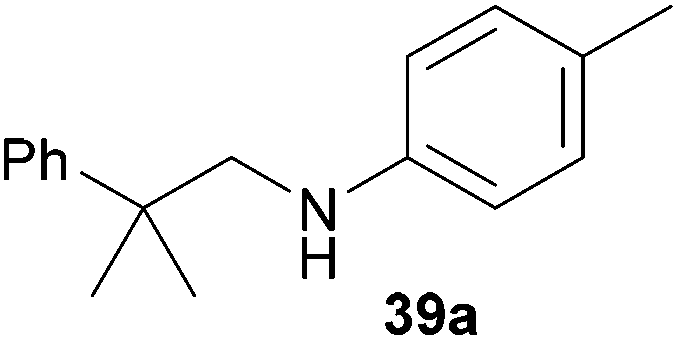
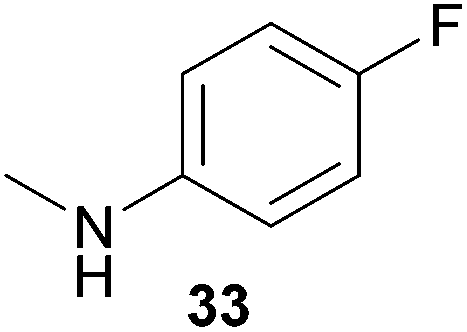
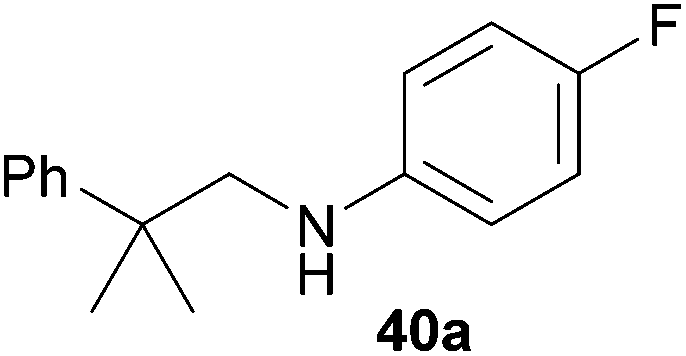
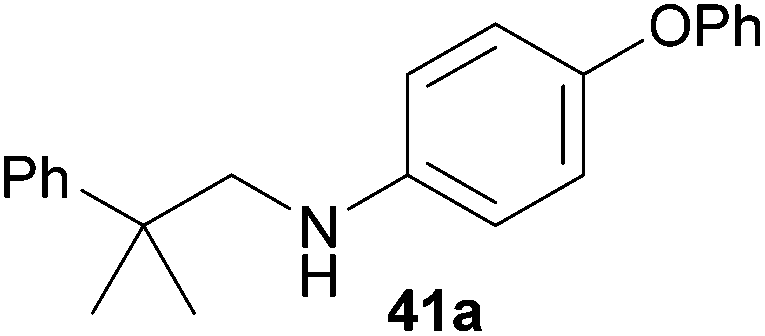
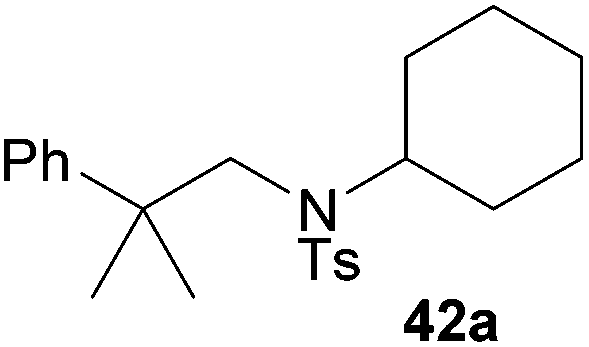
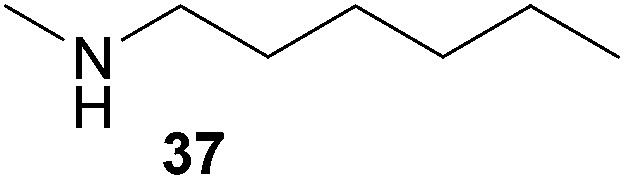
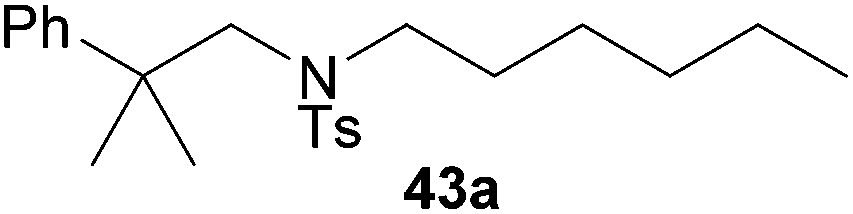
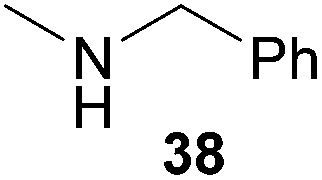
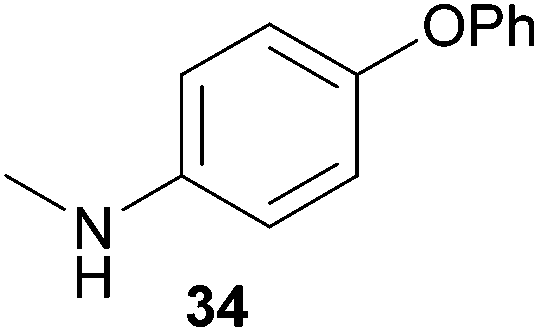
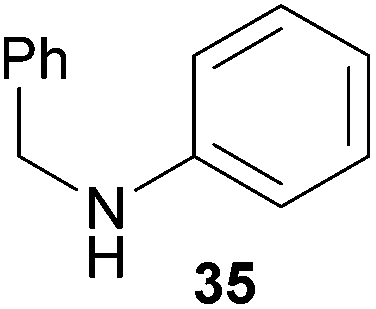
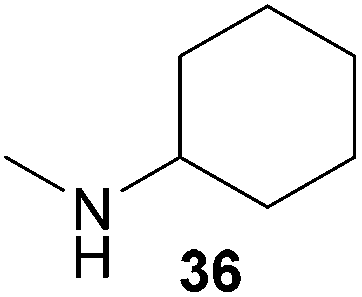
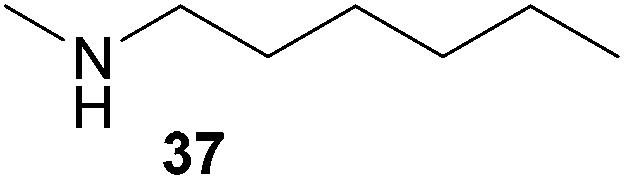
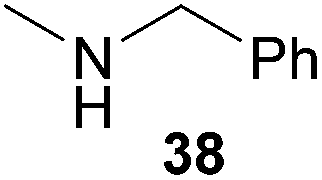
Encouraged by the promising results obtained with N-methylaniline (2) we next turned our attention towards hydroaminoalkylation reactions of α- and (E)-β-methylstyrene with various other secondary amines (Table 2). The unsymmetrically substituted styrenes were chosen as substrates to get further information about the regioselectivity of the hydroaminoalkylation. Initial reactions of α-methylstyrene (9) with the donor- and acceptor-substituted N-methylanilines 32–34 (Table 2, entries 1–3) performed at 180 °C in the presence of 10 mol% 6 showed that both classes of substituents are generally tolerated and the para-substituents do not affect the regioselectivity of the hydroaminoalkylation. In all cases, the regioselectivity was determined to be 99:1 in favor of the branched product a which is formed by hydroaminoalkylation at the sterically more hindered α-carbon of 9. On the other hand, the nature of the substituent on the benzene ring of the N-methylaniline seems to influence the efficiency of the reaction significantly. For example, N-methylaniline 33 bearing an electron-withdrawing para-fluoro substituent on the benzene ring gave the hydroaminoalkylation product 40a in 98% yield (Table 2, entry 2) while the corresponding donor-substituted substrate para-phenoxy-N-methylaniline (34) delivered the corresponding product 41a only in significantly reduced yield of 67% (Table 2, entry 3). This trend in reactivity is further supported by the observation that para-methyl-N-methylaniline (32) with its weak electron-donating methyl substituent gave the hydroaminoalkylation product 39a in 88% yield (Table 2, entry 1) which is comparable to the result obtained with N-methylaniline (2, 86%, Table 1, entry 6). The well-established fact that the efficiency of titanium-catalyzed hydroaminoalkylation reactions of alkenes is strongly influenced by the steric bulk of the amine7b,d,i is in good agreement with our observation that the sterically more demanding amines N-benzylaniline (35, Table 2, entry 4), N-ethylaniline, N-propylaniline, 1,2,3,4-tetrahydroquinoline, and ortho-methyl-N-methylaniline failed to react with α-methylstyrene (9) in the presence of catalyst 6. On the other hand, the dialkylamines N-methylcylohexylamine (36) and N-methylhexylamine (37) (Table 2, entries 5 and 6) were found to undergo highly regioselective hydroaminoalkylation with 9 under identical reaction conditions. Unfortunately, the branched products which were always formed with excellent regioselectivity (a/b = 99:1) could only be obtained in modest to poor yields (14–49%) after derivatization as the corresponding para-toluenesulfonamides 42a and 43a. The observation that both reactions took place at the methyl group of the amines exclusively is in good agreement with results obtained before with N-methylcylohexylamine (36) and N-methylhexylamine (37).5e,j,k,n,7f,g,i Finally, it must be noted that no successful alkylation of N-methylbenzylamine (38, Table 2, entry 7) could be achieved with α-methylstyrene (9). Additional hydroaminoalkylation reactions performed with (E)-β-methylstyrene (11, Table 2, entries 8–14) then revealed that 11 is significantly less reactive than α-methylstyrene (9) and as a result, the corresponding hydroaminoalkylation products could only be obtained in modest to poor yields (14–65%). However, the trend in reactivity observed with the N-methylanilines 32–34 (Table 2, entries 8–10) is in good agreement with the results obtained with α-methylstyrene (9, Table 2, entries 1–3). Again, the para-fluoro-substituted aniline 33 gave the best result (65% yield) while the electron-donating para-phenoxy substituent present in 34 caused a severe drop in yield (14%). Among the dialkylamines 36–38 (Table 2, entries 12–14), again only N-methylcylohexylamine (36) and N-methylhexylamine (37) reacted successfully with 11 and in these cases, the products could be isolated after derivatization as the corresponding para-toluenesulfonamides 47b and 48b in 38% and 41% yield, respectively. On the other hand, no reaction could be achieved with N-methylbenzylamine (38) and the sterically more demanding substrate N-benzylaniline (35, Table 2, entries 11 and 14). However, as described above (Table 1, entry 9), all successful hydroaminoalkylation reactions of (E)-β-methylstyrene (11) took place with excellent regioselectivities (≥98:2) at the sterically less hindered β-carbon of 11 giving access to 3-phenylpropylamine derivatives.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Styrene | Amine | Isolated product | Yielda (%) | Selectivity a/bb |
| a Reaction conditions: amine (2.0 mmol), alkene (3.0 mmol), 6 (109 mg, 0.2 mmol, 10 mol%), n-hexane (1 mL), 180 °C, 96 h, sealed Schlenk tube. Yields refer to the yield of the isolated product (a or b). b GC analysis prior to chromatography. c The product was isolated after derivatization as the corresponding para-toluenesulfonamide. d The reaction took place at the methyl group of the amine exclusively. | |||||
| 1 |
|
|
|
88 | 99:1 |
| 2 |
|
|
98 | 99:1 |
|
| 3 |
|
|
67 | 99:1 |
|
| 4 |
|
— | — | — | |
| 5 |
|
|
14c,d | 99:1 |
|
| 6 |
|
|
49c,d | 99:1 |
|
| 7 |
|
— | — | — | |
| 8 |
|
|
|
31 | 1:99 |
| 9 |
|
|
65 | 2:98 |
|
| 10 |
|

|
14 | 1:99 |
|
| 11 |
|
— | — | — | |
| 12 |
|
|
38c,d | 1:99 |
|
| 13 |
|
|
41c,d | 1:99 |
|
| 14 |
|
— | — | — | |
Owing to the electronic similarities between styrenes and conjugated dienes, we next turned our attention towards hydroaminoalkylation reactions of substituted 1,3-butadienes. Corresponding reactions are extremely rare in the literature4,7h,i and so far, Ind2TiMe2 (Ind = η5-indenyl)7h and aminopyridinato titanium complex 57i represent the only suitable early transition metal catalysts for the intermolecular hydroaminoalkylation of 1-aryl- and 1-alkyl-substituted 1,3-butadienes. Unfortunately, the titanium-catalyzed reaction is limited to selected N-methylanilines and the efficiency of the reaction strongly depends on the substituents bound to the 1,3-diene or the benzene ring of the N-methylaniline. For example, reactions performed with the chloro-substituted substrates para-chloro-N-methylaniline or (E)-1-(para-chlorophenyl)-1,3-butadiene gave the corresponding hydroaminoalkylation products only in poor yields (≤37%) and in addition, dialkylamines or N-alkylanilines bearing alkyl groups larger than a methyl group did not react at all in the presence of Ind2TiMe2. Initial hydroaminoalkylation reactions of (E)-1-phenyl-1,3-butadiene (49) with various para-substituted N-methylanilines were performed in the presence of 10 mol% 6 at 120 °C. The relatively low reaction temperature was chosen because it has already been observed that dimerization of the 1,3-butadiene by Diels–Alder cycloaddition becomes a yield-reducing side reaction at temperatures above 130 °C.7h As can be seen from Table 3, entries 1–7, successful hydroaminoalkylation could be achieved with electron donor- (34, 55–58) and electron acceptor-substituted N-methylanilines (59) and in all cases, only the terminal double bond of the diene reacted. Owing to their better separation from other nitrogen-containing molecules in the reaction mixture, the hydroaminoalkylation products were isolated after conversion into their corresponding acetamides (AcCl, NEt3, CH2Cl2, rt). Although the products 60–66a/b could be obtained with good to modest yields (45–82%), the regioselectivity of the hydroaminoalkylation was always close to 1:1 which is worse compared to typical regioselectivities obtained with the catalysts 5 or Ind2TiMe2. With the latter catalyst, formation of the branched regioisomer was usually favored and selectivities between 2:1 and 3:17h could be achieved while 5 produced linear regioisomers almost exclusively.7i In this context, it should be noted that separation of the regioisomers is possible by chromatography. On the other hand, it is worth mentioning that with several substrates significantly improved yields were obtained with the new catalyst 6. For example, para-chloro-N-methylaniline (59) gave a mixture of the corresponding products 66a and 66b in 65% combined yield (Table 3, entry 7) while the corresponding reaction performed with Ind2TiMe2 gave the same products only in 6% yield.7h In addition, a significantly improved yield (46% versus 23%7h) was obtained with para-methoxy-N-methylaniline (55) and it was also possible to perform the hydroaminoalkylation with para-trifluoromethoxy-N-methylaniline (56, Table 3, entry 4). Amine 56 represents a highly promising substrate because the para-trifluoromethoxy group is often present in synthetic agrochemicals and pharmaceuticals.9 Impressively, even para-thiomethyl-N-methylaniline (57) which does not undergo hydroaminoalkylation with 49 in the presence of Ind2TiMe2 did react in the presence of catalyst 6 (Table 3, entry 5). Additional reactions between N-methylaniline (2) and 1,3-butadienes 50–54 (Table 3, entries 8–12) delivered mixtures of the branched and the linear hydroaminoalkylation products 67–71a/b in good to modest yields but again, only with poor regioselectivities. In this context, it must be mentioned that a third product was formed during reactions of the 1-alkyl-substituted dienes (E)-1,3-decadiene (52) and (E)-6-phenyl-1,3-hexadiene (53, Table 3, entries 10 and 11). These additionally formed products were identified to be branched hydroaminoalkylation products with an isomerized double bond. The formation of two branched isomeric olefins from 2 and 52 was confirmed by hydrogenation of a mixture of 69a and its isomer with Pd/C which gave the corresponding saturated compound N-phenyl-N-(2-methyldecyl)acetamide (72) as the sole hydrogenation product in 85% yield. On the other hand, formation of additional linear hydroaminoalkylation products with isomerized double bond was not observed in the presence of catalyst 6. This is in sharp contrast to the finding that linear products with isomerized double bond are formed during hydroaminoalkylation reactions of 1,3-butadienes catalyzed by aminopyridinato titanium catalyst 5.7i Finally, it was found that even the 1,2-disubstituted 1,3-diene 54 undergoes successful hydroaminoalkylation with 2. The corresponding products 71a and 71b were obtained in 73% combined yield and the regioselectivity was determined to be 51:49 (Table 3, entry 12). In this context, it is worth mentioning that 54 and 2 are exclusively converted into the branched hydroaminoalkylation product with the catalyst Ind2TiMe2.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Diene | R1 | R2 | Amine | R3 | Products | Yielda (%) | Selectivity a/bb |
| a Reaction conditions: (1) amine (2.0 mmol), diene (3.0 mmol), 6 (109 mg, 0.2 mmol, 10 mol%), n-hexane (1 mL), 120 °C, 48 h, sealed Schlenk tube; (2) acetyl chloride (1 M in CH2Cl2, 4.0 mmol), NEt3 (4.0 mmol), CH2Cl2 (30 mL), 25 °C, 16 h. Yields refer to the total yield of isolated products (a + b). b GC analysis prior to acetylation and chromatography. c The reaction was carried out on a 4 mmol scale. d Formation of an additional branched product with an isomerized double bond was observed. The selectivity is given as branched (a)/branched isomerized/linear (b). For details, see the experimental section. e Directly isolated as the corresponding amine (without acetylation). | ||||||||
| 1 | 49 | Ph | H | 2 | H | 60a/b | 73 | 55:45 |
| 2 | 49 | Ph | H | 34 | p-OPh | 61a/b | 45 | 54:46 |
| 3 | 49 | Ph | H | 55 | p-OMe | 62a/b | 46 | 54:46 |
| 4 | 49 | Ph | H | 56 | p-OCF3 | 63a/b | 82 | 53:47 |
| 5 | 49 | Ph | H | 57 | p-SMe | 64a/b | 53 | 54:46 |
| 6 | 49 | Ph | H | 58 | p-iso-Pr | 65a/b | 64 | 53:47 |
| 7 | 49 | Ph | H | 59 | p-Cl | 66a/b | 65 | 50:50 |
| 8 | 50 | p-Cl-C6H4 | H | 2 | H | 67a/b | 52 | 50:50 |
| 9 | 51 | p-F-C6H4 | H | 2 | H | 68a/b | 53 | 50:50 |
| 10 | 52 | n-C6H13 | H | 2 | H | 69a/b | 68c | 50:27:23d |
| 11 | 53 | C6H5-(CH2)2 | H | 2 | H | 70a/be | 87e | 47:28:25d |
| 12 | 54 | Ph | CH3 | 2 | H | 71a/b | 73 | 51:49 |
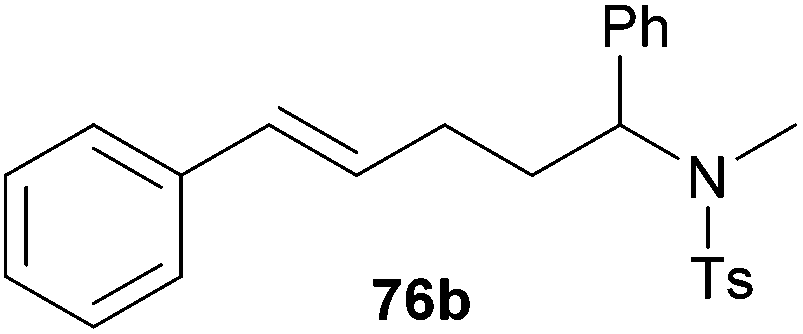
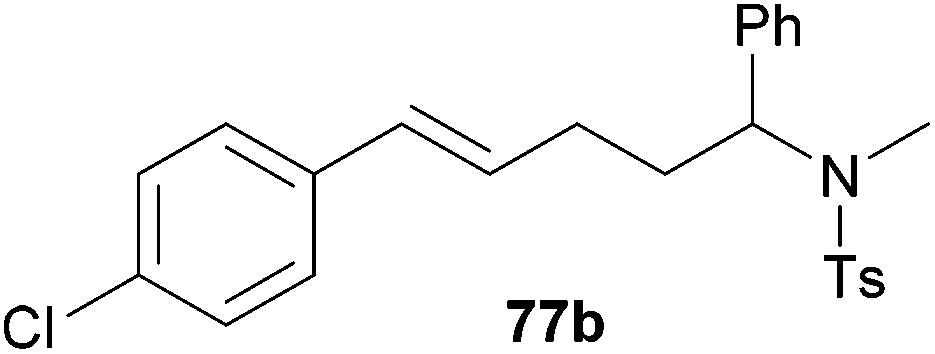
Because hydroaminoalkylation reactions of styrenes with N-alkylanilines catalyzed by aminopyridinato complex 4 have already revealed that the regioselectivity in favor of the linear product increases with increasing size of the N-alkyl substituent,7g we additionally tried to react (E)-1-phenyl-1,3-butadiene (49) with the sterically more demanding amines N-benzylaniline (35), N-methylcylohexylamine (36), N-methylhexylamine (37), and N-methylbenzylamine (38). All reactions were performed with 10 mol% 6 in n-hexane at 120 °C in sealed Schlenk tubes and after 48 h, the reaction mixtures were analyzed by GC. During this study, it was found that among the four amines, only N-methylbenzylamine (38) underwent successful hydroaminoalkylation with 49 in the presence of catalyst 6 (Table 4, entry 1). Interestingly and in good agreement with the behavior of aminopyridinato catalyst 4, a high regioselectivity of 93:7 in favor of the linear hydroaminoalkylation product was observed and after conversion into the corresponding para-toluenesulfonamide and purification by chromatography, the linear product 76b was isolated in 58% yield. Due to the high regioselectivity of the reaction, the branched product could not be isolated but the NMR spectra of 76b showed very weak signal sets, which could be assigned to trace impurities of the branched product. In this context, it must also be mentioned that the isolated material additionally contained minor amounts of tosylated N-methylbenzylamine formed from unreacted amine 38. Interestingly and in contrast to the reactions of N-methylcylohexylamine (36) and N-methylhexylamine (37) with α- and (E)-β-methylstyrene (9 and 11, Table 2, entries 5, 6, 12 and 13), alkylation of 38 occurred at the benzyl position of the amine exclusively. However, this observation is in good agreement with the fact that group 47g,i and 55e metal-catalyzed hydroaminoalkylation reactions of sterically less demanding mono-substituted alkenes and styrenes performed with 38 usually take place selectively at the benzyl position of 38. Selective alkylation at the methyl group of N-methylbenzylamine (38) has only been achieved with vinylcyclohexane in the presence of tantalum and niobium binaphtholate catalysts developed by the Hultzsch group.5h,i With these results in hand, we finally used the para-substituted 1-phenyl-1,3-butadienes 50, 51, and 73 as well as the ortho-substituted dienes 74 and 75 for hydroaminoalkylation reactions with amine 38 (Table 4, entries 2–6). The results obtained with these substrates clearly prove that electron-withdrawing chloro or fluoro substituents (Table 4, entries 2 and 3) and electron-donating methyl or methoxy groups (Table 4, entries 4–6) as well as ortho-substitution of the phenyl ring (Table 4, entries 5 and 6) are tolerated with no significant impact on the yield (60–71%) and the regioselectivity (13:87–8:92) of the reaction. The latter finding is in good agreement with the remote position of the phenyl substituent relative to the reacting double bond of the diene.7h However, in all cases, again only the linear hydroaminoalkylation products 77–81b could be isolated from the reaction mixtures and the branched products were only detected by GC or as trace impurities in the NMR spectra of the isolated linear products. In this context, it is worth mentioning that corresponding δ-alkenylamine derivatives are promising substrates for the synthesis of cis-N-methyl-2,5-disubstituted pyrrolidines.10 Although Table 4 summarizes the first successful examples of hydroaminoalkylation reactions of conjugated dienes with N-methylbenzylamine (38), complex 6 unfortunately failed to catalyze a number of closely related reactions. For example, 1-phenyl-substituted 1,3-butadienes bearing additional methyl or chloro substituents in the 2-position of the diene system did not undergo the reaction and the 1-alkyl-substituted 1,3-butadiene 52 delivered only trace amounts of the expected hydroaminoalkylation products which could only be detected and characterized by GC/MS analysis. In addition, even a small variation of the N-methylbenzylamine motif resulted in a lack of reactivity. Neither N-ethyl-, N-iso-propyl-, N-cyclohexyl-, N-benzyl-, nor N-(2-phenylethyl)-substituted benzylamines underwent successful hydroaminoalkylation with (E)-1-phenyl-1,3-butadiene (49) in the presence of catalyst 6.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Diene | R | Isolated linear producta | Yieldb (%) | Selectivity a/bc |
| a Only the isolated, linear product is presented. b Reaction conditions: (1) N-methylbenzylamine (38, 242 mg, 2.0 mmol), diene (3.0 mmol), 6 (109 mg, 0.2 mmol, 10 mol%), n-hexane (1 mL), 120 °C, 48 h, sealed Schlenk tube; (2) para-toluenesulfonyl chloride (763 mg, 4.0 mmol), pyridine (10 mL), 25 °C, 16 h. Yields refer to the yield of the isolated linear product (b). c GC analysis prior to tosylation and chromatography. | |||||
| 1 | 49 | H |
|
58 | 7:93 |
| 2 | 50 | p-Cl |
|
67 | 8:92 |
| 3 | 51 | p-F |
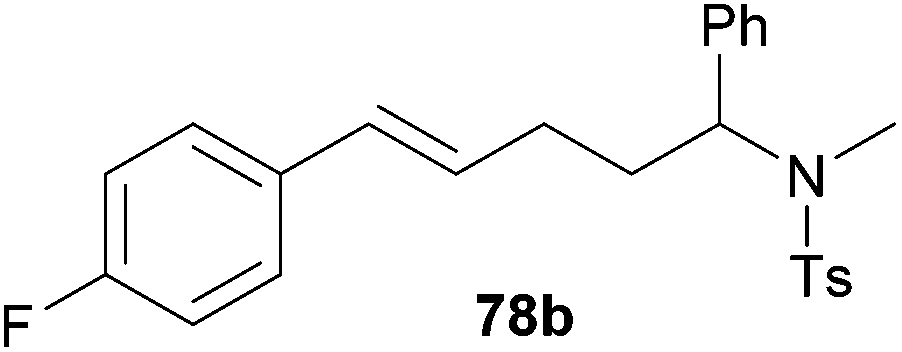
|
71 | 9:91 |
| 4 | 73 | p-MeO |
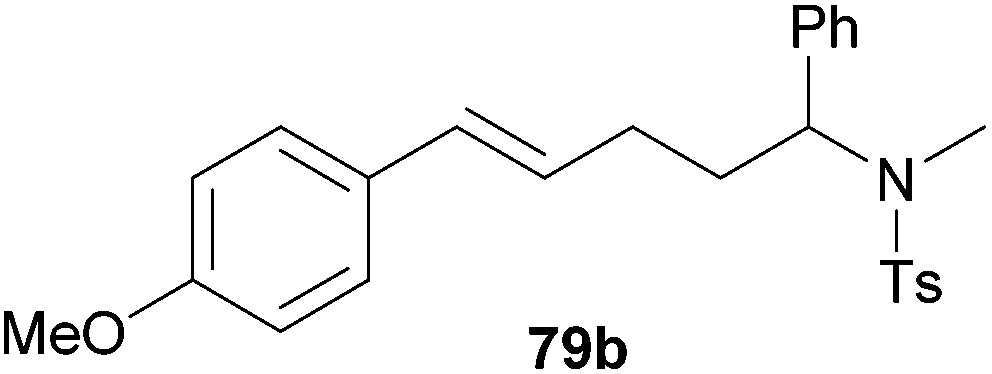
|
60 | 10:90 |
| 5 | 74 | o-Me |
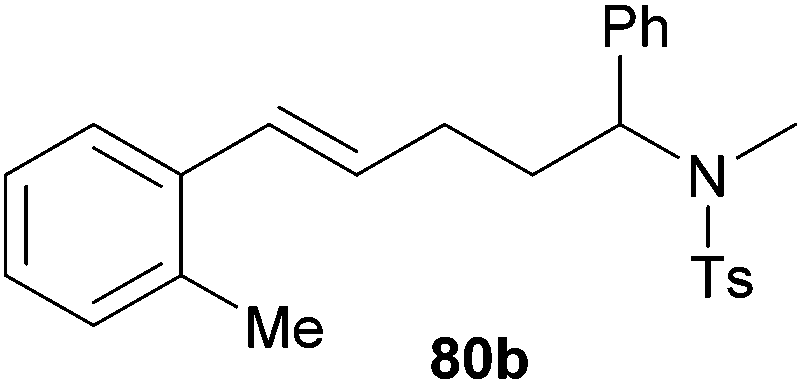
|
69 | 13:87 |
| 6 | 75 | o-MeO |
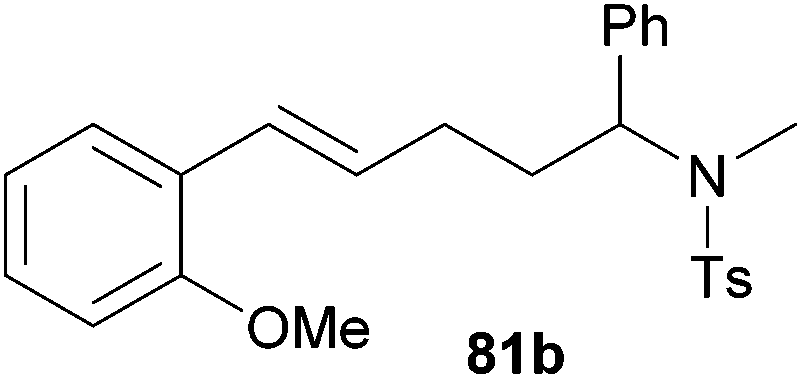
|
66 | 11:89 |
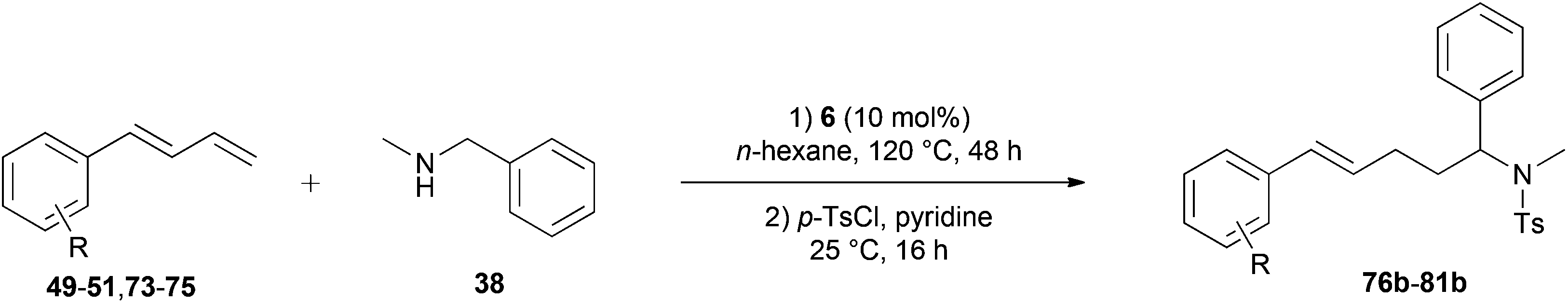
Conclusions
In summary, we have shown that the easily accessible titanium mono(formamidinate) complex 68a which bears 2,6-diisopropylphenyl groups as the amidinate N-substituents is a highly active catalyst for the regioselective intermolecular hydroaminoalkylation of unactivated, sterically demanding 1,1- and 1,2-disubstituted alkenes and styrenes with secondary amines. While corresponding reactions of 1,1-disubstituted alkenes and styrenes give access to the branched hydroaminoalkylation products exclusively, (E)-β-methylstyrene selectively reacts at the sterically less hindered β-carbon. In addition, with catalyst 6, it is also possible for the first time to convert 1-phenyl-substituted (E)-1,3-butadienes and N-methylbenzylamine into δ-alkenylamine derivatives.Experimental
General information
All reactions were performed under an inert atmosphere of nitrogen in oven-dried Schlenk tubes (Duran glassware, 100 mL, ø = 30 mm) equipped with Teflon stopcocks and magnetic stirring bars (15 × 4.5 mm). Prior to use, all alkenes, styrenes, and amines were distilled and degassed. n-Hexane was purified by distillation from sodium wire and degassed. The alkenes, styrenes, amines, and n-hexane were stored in a nitrogen-filled glove box. Catalyst 68a and the dienes7h,i were synthesized according to literature procedures and stored in a nitrogen-filled glove box. All other chemicals were purchased from commercial sources and were used without further purification. Unless otherwise noted, the ratio of regioisomers was determined by gas chromatography prior to flash column chromatography. For thin layer chromatography, silica on TLC aluminium foils with fluorescent indicator 254 nm was used. The substances were detected with UV light and/or iodine. For flash column chromatography, silica gel (particle size 0.037–0.063 mm) was used. Light petroleum ether (b.p. 40–60 °C, PE), tert-butyl methyl ether (MTBE) and ethyl acetate (EtOAc) used for chromatography were distilled via Raschig columns at ambient pressure. Other solvents used for chromatography were distilled prior to use. All products that have already been reported in the literature were identified by comparison of the obtained 1H NMR and 13C NMR spectra with those reported in the literature. New compounds were additionally characterized by infrared spectroscopy, mass spectrometry, and high resolution mass spectrometry (HRMS). 1H, 13C and 19F NMR spectra were recorded at 500, 470, 300, 125 and 75 MHz. All 1H NMR spectra are reported in δ units (ppm) relative to the signal of TMS at 0.00 ppm or relative to the residual solvent signal of CDCl3 at 7.26 ppm. All 13C NMR spectra are reported in δ units (ppm) relative to the central line of the triplet for CDCl3 at 77.0 ppm. All 19F NMR spectra are reported in δ units (ppm) relative to the signal of CFCl3 at 0.00 ppm. Infrared spectra were recorded either via KBr pellets or on an ATR spectrometer. Mass spectra and high resolution mass spectra (HRMS) were recorded either in EI (with an ionization potential of 70 eV) or in ESI mode (ESI, TOF).General procedure A for the hydroaminoalkylation of disubstituted alkenes and styrenes with N-substituted arylamines (Tables 1 and 2)
An oven dried Schlenk tube equipped with a Teflon stopcock and a magnetic stirring bar was transferred into a nitrogen-filled glovebox and charged with complex 6 (109 mg, 0.2 mmol, 10 mol%) and n-hexane (0.5 mL). Afterwards the N-substituted arylamine (2.0 mmol), the alkene or styrene (3.0 mmol), and n-hexane (0.5 mL) were added. After heating the mixture to 180 °C for 96 h, the mixture was cooled to room temperature and dichloromethane (40 mL) was added. Then the regioselectivity of the reaction was determined by GC (if applicable) and finally, the crude product was purified by flash column chromatography (SiO2).General procedure B for the hydroaminoalkylation of disubstituted styrenes with dialkylamines (Table 2)
An oven dried Schlenk tube equipped with a Teflon stopcock and a magnetic stirring bar was transferred into a nitrogen-filled glovebox and charged with complex 6 (109 mg, 0.2 mmol, 10 mol%) and n-hexane (0.5 mL). Afterwards the dialkylamine (2.0 mmol), the styrene (3.0 mmol), and n-hexane (0.5 mL) were added. After heating the mixture to 180 °C for 96 h, the crude product was transferred to a 100 mL round-bottomed flask and the Schlenk tube was rinsed with dichloromethane (4 × 10 mL). The regioselectivity of the reaction was determined by GC and subsequently, the combined solutions were concentrated under vacuum to a total volume of 10 mL. The resulting mixture was then treated with 2 N aqueous NaOH (5 mL, 10.0 mmol) and para-toluenesulfonyl chloride (572 mg, 3.0 mmol). The resulting biphasic mixture was stirred vigorously at room temperature for 20 h before the solution was partitioned between dichloromethane (50 mL) and water (20 mL). The formed layers were separated, the aqueous one was extracted with dichloromethane (3 × 10 mL) and the combined organic layers were dried with MgSO4. After concentration under vacuum, the product was isolated by flash column chromatography (SiO2).General procedure C for the hydroaminoalkylation of 1,3-dienes with N-methylanilines (Table 3)
An oven dried Schlenk tube equipped with a Teflon stopcock and a magnetic stirring bar was transferred into a nitrogen-filled glovebox and charged with 6 (109 mg, 0.20 mmol, 10 mol%), the diene (3.0 mmol), the amine (2.0 mmol), and n-hexane (1 mL). Then the tube was sealed and the resulting mixture was heated to 120 °C for 48 h. After the mixture had been cooled to room temperature, tert-butyl methyl ether (30 mL) was added and the regioselectivity of the reaction was determined by GC. To achieve a better separation of the products from remaining N-methylanilines, acetyl chloride (1 M in CH2Cl2, 4 mL, 4 mmol), and triethylamine (405 mg, 4 mmol) were added and the reaction mixture was stirred at room temperature for 16 h. Finally, the solvent was removed in the presence of Celite® and the residue was purified by flash column chromatography (SiO2).General procedure D for the hydroaminoalkylation of 1,3-dienes with N-methylbenzylamine (38, Table 4)
An oven dried Schlenk tube equipped with a Teflon stopcock and a magnetic stirring bar was transferred into a nitrogen-filled glovebox and charged with 6 (109 mg, 0.20 mmol, 10 mol%), the diene (3.0 mmol), N-methylbenzylamine (38, 242 mg, 2.0 mmol), and n-hexane (1 mL). Then the tube was sealed and the resulting mixture was heated to 120 °C for 48 h. After the mixture had been cooled to room temperature, tert-butyl methyl ether (30 mL) was added and the regioselectivity of the reaction was determined by GC. Then the solvent was removed under reduced pressure and pyridine (10 mL) and para-toluenesulfonyl chloride (763 mg, 4.0 mmol) were added. After the resulting mixture had been stirred overnight at room temperature, dichloromethane (50 mL) was added and the organic mixture was extracted with aqueous HCl (4 M, 50 mL). Then the aqueous layer was extracted with dichloromethane (3 × 15 mL) and the combined organic layers were dried with MgSO4 and concentrated under reduced pressure in the presence of silica gel. (Note: The use of Celite® instead of silica gel leads to significantly reduced yields.) Finally, the product was purified by flash column chromatography (SiO2). Due to the viscosity of the products, removal of remaining solvents required storage of the products under high vacuum for several hours.:1), a mixture of 3a and 3b (406 mg, 1.92 mmol, 96%) was isolated as a yellow oil. Prior to chromatography, the ratio 3a/3b was determined to be 73:27. For characterization, it was possible to separate the regioisomers by chromatography (PE–EtOAc, 40:1). 3a: 1H NMR (300 MHz, CDCl3, TMS) δ = 7.25 (t, J = 7.3 Hz, 2H), 7.19–7.13 (m, 3H), 7.09 (t, J = 7.7 Hz, 2H), 6.63 (t, J = 7.3 Hz, 1H), 6.51 (d, J = 7.8 Hz, 2H), 3.26 (dd, J = 12.4, 6.2 Hz, 1H), 3.16 (dd, J = 12.3, 8.3 Hz, 1H), 2.99 (sext, J = 7.0 Hz, 1H), 1.26 (d, J = 7.0 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 147.8 (C), 144.4 (C), 129.2 (CH), 128.6 (CH), 127.2 (CH), 126.6 (CH), 117.5 (CH), 113.1 (CH), 51.0 (CH2), 39.1 (CH), 19.7 (CH3) ppm. 3b: 1H NMR (300 MHz, TMS) δ = 7.34–7.24 (m, 2H), 7.24–7.09 (m, 5H), 6.69 (t, J = 7.3 Hz, 1H), 6.57 (d, J = 8.1 Hz, 2H), 3.59 (br. s, 1H), 3.14 (t, J = 7.0 Hz, 2H), 2.73 (t, J = 7.6 Hz, 2H), 1.94 (quin, J = 7.3 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.1 (C), 141.6 (C), 129.2 (CH), 128.4 (CH), 128.4 (CH), 125.9 (CH), 117.4 (CH), 112.9 (CH), 43.5 (CH2), 33.4 (CH2), 31.0 (CH2) ppm.
:1), 21a (364 mg, 1.79 mmol, 90%) was isolated as a yellow oil. Prior to chromatography, the ratio 21a/21b was determined to be 99:1. 21a: 1H NMR (300 MHz, CDCl3, TMS) δ = 7.15 (t, J = 7.4 Hz, 2H), 6.70–6.56 (m, 3H), 3.59 (br. s, 1H), 2.92 (s, 2H), 1.59–1.18 (m, 11H), 0.97 (s, 3H) ppm. 13C NMR (75 MHz, CDCl3) δ = 149.1 (C), 129.1 (CH), 116.7 (CH), 112.6 (CH), 54.6 (CH2), 35.8 (CH2), 34.2 (C), 26.4 (CH2), 23.5 (CH3), 22.0 (CH2) ppm.
:1), 22a (387 mg, 1.72 mmol, 86%) was isolated as a yellow oil. Prior to chromatography, the ratio 22a/22b was determined to be 99:1. 22a: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.42–7.38 (m, 2H), 7.35 (t, J = 7.0 Hz, 2H), 7.25–7.21 (m, 1H), 7.13 (br. t, J = 7.3 Hz, 2H), 6.66 (br. t, J = 7.3 Hz, 1H), 6.53 (br. d, J = 8.5 Hz, 2H), 3.28 (s, 2H), 1.41 (s, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.7 (C), 146.7 (C), 129.1 (CH), 128.5 (CH), 126.2 (CH), 126.1 (CH), 117.1 (CH), 112.7 (CH), 55.7 (CH2), 38.9 (C), 27.4 (CH3) ppm.
:1), 23a (435 mg, 1.82 mmol, 91%) was isolated as a yellow oil. Prior to chromatography, the ratio 23a/23b was determined to be 99:1. 23a: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.28 (br. d, J = 8.2 Hz, 2H), 7.22–7.09 (m, 4H), 6.65 (t, J = 7.3 Hz, 1H), 6.53 (d, J = 7.7 Hz, 2H), 3.32 (br. s, 1H), 3.25 (s, 2H), 2.33 (s, 3H), 1.39 (s, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.7 (C), 143.6 (C), 135.7 (C), 129.2 (CH), 129.1 (CH), 126.0 (CH), 117.0 (CH), 112.7 (CH), 55.6 (CH2), 38.5 (C), 27.5 (CH3), 20.9 (CH3) ppm.
:1), 24b (288 mg, 1.28 mmol, 64%) was isolated as a yellow oil. Prior to chromatography, the ratio 24a/24b was determined to be 1:99. 24b: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.28 (t, J = 7.4 Hz, 2H), 7.22–7.11 (m, 5H), 6.67 (br. t, J = 7.3 Hz, 1H), 6.53 (d, J = 7.7 Hz, 2H), 3.66 (br. s, 1H), 3.08 (dd, J = 12.5, 6.1 Hz, 1H), 2.93 (dd, J = 12.5, 7.0 Hz, 1H), 2.74 (dd, J = 13.5, 6.4 Hz, 1H), 2.49 (dd, J = 13.5, 7.8 Hz, 1H), 2.06 (octet, J = 6.8 Hz, 1H), 0.96 (d, J = 6.7 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.3 (C), 140.5 (C), 129.2 (CH), 129.1 (CH), 128.3 (CH), 125.9 (CH), 117.0 (CH), 112.7 (CH), 49.8 (CH2), 41.3 (CH2), 35.0 (CH), 18.1 (CH3) ppm.
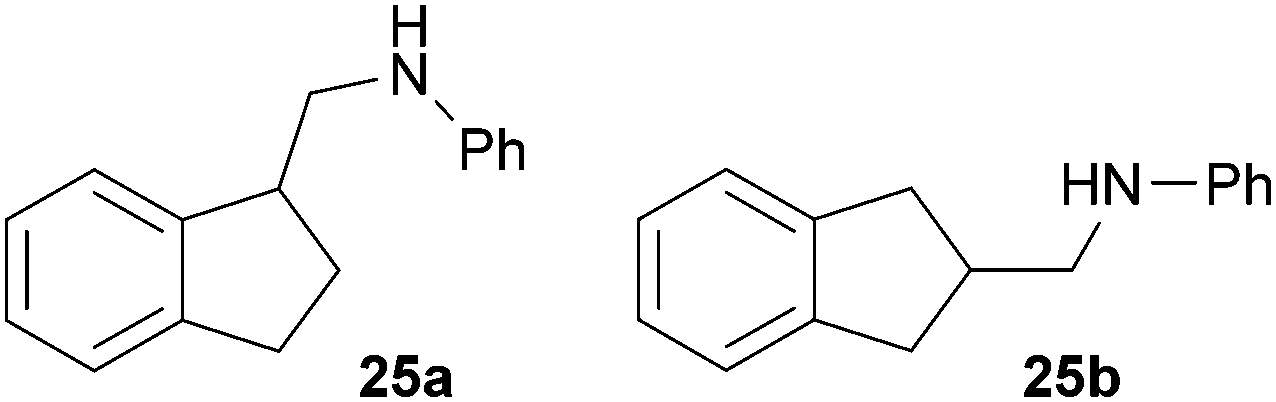
:1) gave three fractions as oils. Fraction 1 contained pure compound 25a (30 mg, 0.13 mmol, 7%), fraction 2 contained a mixture of 25a and 25b (203 mg, 0.91 mmol, 45%) and fraction 3 contained pure compound 25b (200 mg, 0.90 mmol, 45%) as yellow oils. Prior to chromatography, the ratio 25a/25b was determined to be 34:66. 25a: Rf = 0.13 (PE–MTBE, 80:1). 1H NMR (500 MHz, CDCl3) δ = 7.33–7.27 (m, 2H), 7.25–7.18 (m, 4H), 6.74 (t, J = 7.3 Hz, 1H), 6.67 (d, J = 7.7 Hz, 2H), 3.75 (br. s, 1H), 3.54–3.44 (m, 2H), 3.29 (dd, J = 11.8, 7.3 Hz, 1H), 3.06–2.98 (m, 1H), 2.98–2.89 (m, 1H), 2.39–2.30 (m, 1H), 1.99–1.91 (m, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.4 (C), 144.7 (C), 144.4 (C), 129.3 (CH), 126.9 (CH), 126.3 (CH), 124.7 (CH), 124.0 (CH), 117.4 (CH), 112.8 (CH), 48.0 (CH2), 44.6 (CH), 31.3 (CH2), 30.0 (CH2) ppm. IR (ATR) vmax = 3417 (w), 3047 (w), 3019 (w), 2939 (w), 2847 (w), 1601 (s), 1504 (s), 1319 (m), 1260 (m), 744 (vs), 691 (s) cm−1. GC/MS m/z = 223 ([M]+), 106 ([C7H8N]+), 77 ([C6H5]+). HRMS (ESI+) m/z calcd for C16H18N 224.1439 ([M + H]+), found 224.1438. 25b: Rf = 0.10 (PE–MTBE, 80:1). 1H NMR (500 MHz, CDCl3) δ = 7.25–7.15 (m, 6H), 6.72 (t, J = 7.3 Hz, 1H), 6.64 (d, J = 7.7 Hz, 2H), 3.74 (br. s, 1H), 3.22 (d, J = 6.7 Hz, 2H), 3.19–3.11 (m, 2H), 2.87–2.75 (m, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.4 (C), 142.6 (C), 129.2 (CH), 126.3 (CH), 124.6 (CH), 117.3 (CH), 112.7 (CH), 48.8 (CH2), 39.1 (CH), 37.2 (CH2) ppm. IR (ATR) vmax = 3414 (w), 3048 (w), 3020 (w), 2929 (w), 2900 (w), 2841 (w), 1601 (s), 1505 (s), 1316 (m), 1256 (m), 740 (vs), 690 (s) cm−1. GC/MS m/z = 223 ([M]+), 106 ([C7H8N]+), 93 ([C6H7N]+), 77 ([C6H5]+). HRMS (ESI+) m/z calcd for C16H18N 224.1439 ([M + H]+), found 224.1436.
:1), a mixture of 26a and 26b (69 mg, 0.32 mmol, 16%) was isolated as a yellow oil. Prior to chromatography, the ratio 26a/26b was determined to be 35:65. 1H NMR (500 MHz, CDCl3, TMS, mixture of two regioisomers) signals of the major regioisomer 26b: δ = 7.09 (t, J = 8.4 Hz, 2H), 6.60 (t, J = 7.3 Hz, 1H), 6.53 (d, J = 7.6 Hz, 2H), 3.59 (br. s, 1H), 3.01–2.92 (m, 1H), 2.80 (dd, J = 12.2, 7.3 Hz, 1H), 1.66 (octet, J = 6.6 Hz, 1H), 1.41–1.05 (m, 13H), 0.89 (d, J = 6.7 Hz, 3H), 0.81 (t, J = 6.8 Hz, 3H) ppm; important signals of the minor regioisomer 26a: δ = 3.01–2.92 (m, 2H), 1.49 (sept, J = 6.1 Hz, 1H), 0.84 (t, J = 7.4 Hz, 3H), 0.82 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3, mixture of two regioisomers) signals of the major regioisomer 26b: δ = 148.7 (C), 129.2 (CH), 116.9 (CH), 112.6 (CH), 50.3 (CH2), 34.8 (CH2), 32.9 (CH), 31.9 (CH2), 29.6 (CH2), 26.9 (CH2), 22.7 (CH2), 18.1 (CH3), 14.1 (CH3) ppm; signals of the minor regioisomer 26a: δ = 148.6 (C), 129.2 (CH), 116.9 (CH), 112.6 (CH), 47.0 (CH2), 39.1 (CH), 32.3 (CH2), 31.6 (CH2), 26.4 (CH2), 24.5 (CH2), 22.7 (CH2), 14.1 (CH3), 10.9 (CH3) ppm. IR (neat, mixture of two regioisomers) vmax = 3421 (s), 3052 (w), 3021 (w), 2957 (s), 2926 (s), 2856 (s), 1603 (s), 1506 (s), 1465 (m), 1320 (m), 1260 (m), 747 (s), 691 (s) cm−1. GC/MS m/z = 219 ([M]+), 106 ([C7H8N]+), 77 ([C6H5]+).
:1), 27 (262 mg, 1.49 mmol, 75%) was isolated as a yellow oil. 27: 1H NMR (300 MHz, CDCl3, TMS) δ = 7.17 (br. t, J = 7.9 Hz, 2H), 6.68 (br. t, J = 7.3 Hz, 1H), 6.61 (br. d, J = 8.1 Hz, 2H), 3.64 (br. s, 1H), 3.02 (d, J = 7.2 Hz, 2H), 2.15 (sept, J = 7.5 Hz, 1H), 1.91–1.72 (m, 2H), 1.67–1.47 (m, 4H), 1.37–1.17 (m, 2H) ppm. 13C NMR (75 MHz, CDCl3) δ = 148.6 (C), 129.2 (CH), 117.0 (CH), 112.6 (CH), 49.5 (CH2), 39.4 (CH), 30.7 (CH2), 25.3 (CH2) ppm.
:1), 28 (272 mg, 1.44 mmol, 72%) was isolated as a yellow oil. 28: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.18–7.13 (m, 2H), 6.67 (tt, J = 7.3, 1.0 Hz, 1H), 6.61–6.56 (m, 2H), 3.71 (br. s, 1H), 2.95 (d, J = 6.7 Hz, 2H), 1.85–1.78 (m, 2H), 1.78–1.71 (m, 2H), 1.70–1.65 (m, 1H), 1.64–1.52 (m, 1H), 1.31–1.13 (m, 3H), 0.98 (qd, J = 12.1, 3.1 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.6 (C), 129.2 (CH), 116.9 (CH), 112.7 (CH), 50.6 (CH2), 37.6 (CH), 31.3 (CH2), 26.6 (CH2), 26.0 (CH2) ppm.
:1), 29 (348 mg, 1.86 mmol, 93%) was isolated as a yellow oil. 29: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.17 (br. t, J = 6.9 Hz, 2H), 6.68 (br. t, J = 7.3 Hz, 1H), 6.61 (br. d, J = 8.5 Hz, 2H), 5.73–5.63 (m, 2H), 3.73 (br. s, 1H), 3.10–3.01 (m, 2H), 2.24–2.15 (m, 1H), 2.13–2.03 (m, 2H), 1.96–1.83 (m, 2H), 1.83–1.74 (m, 1H), 1.39–1.28 (m, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.5 (C), 129.2 (CH), 127.2 (CH), 125.8 (CH), 117.0 (CH), 112.6 (CH), 49.7 (CH2), 33.5 (CH), 29.7 (CH2), 26.8 (CH2), 24.7 (CH2) ppm. IR (neat) vmax = 3418 (m), 3051 (w), 3021 (m), 2916 (s), 2837 (m), 1603 (s), 1507 (s), 1433 (m), 1323 (s), 1254 (s), 1179 (w), 748 (s), 692 (s) cm−1. GC/MS m/z = 187 ([M]+), 106 ([C7H8N]+), 77 ([C6H5]+). HRMS (ESI+) m/z calcd for C13H18N 188.1439 ([M + H]+), found 188.1435.
:1 → PE–Et2O, 40:1), 31 (228 mg, 1.22 mmol, 61%) was isolated as a yellow oil. 31: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.27–7.18 (m, 2H), 6.79 (d, J = 8.3 Hz, 2H), 6.69 (t, J = 7.2 Hz, 1H), 5.94–5.88 (m, 1H), 5.63 (br. d, J = 10.2 Hz, 1H), 4.48–4.41 (m, 1H), 2.78 (s, 3H), 2.12–1.95 (m, 2H), 1.91–1.77 (m, 2H), 1.71–1.52 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 149.7 (C), 130.6 (CH), 129.9 (CH), 129.1 (CH), 116.2 (CH), 112.8 (CH), 54.9 (CH), 32.4 (CH3), 25.3 (CH2), 24.9 (CH2), 21.5 (CH2) ppm.
:1), 39a (422 mg, 1.76 mmol, 88%) was isolated as a yellow oil. Prior to chromatography, the ratio 39a/39b was determined to be 99:1. 39a: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.39 (br. d, J = 7.8 Hz, 2H), 7.34 (t, J = 7.8 Hz, 2H), 7.22 (br. t, J = 7.3 Hz, 1H), 6.94 (d, J = 8.0 Hz, 2H), 6.45 (d, J = 8.4 Hz, 2H), 3.25 (s, 2H), 3.17 (br. s, 1H), 2.21 (s, 3H), 1.40 (s, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ 146.8 (C), 146.5 (C), 129.6 (CH), 128.5 (CH), 126.2 (C), 126.1 (CH), 126.1 (CH), 112.8 (CH), 56.1 (CH2), 38.9 (C), 27.4 (CH3), 20.3 (CH3) ppm. IR (neat) vmax = 3413 (m), 3021 (m), 2964 (m), 2920 (m), 1619 (s), 1521 (s), 1480 (m), 1317 (m), 1294 (m), 1251 (m), 806 (s), 764 (m), 700 (s) cm−1. GC/MS m/z = 239 ([M]+), 120 ([C8H10N]+), 91 ([C7H7]+). HRMS (ESI+) m/z calcd for C17H21NNa 262.1572 ([M + Na]+), found 262.1567.
:1), 40a (475 mg, 1.95 mmol, 98%) was isolated as a yellow oil. Prior to chromatography, the ratio 40a/40b was determined to be 99:1. 40a: 1H NMR (300 MHz, CDCl3, TMS) δ = 7.43–7.30 (m, 4H), 7.23 (tt, J = 6.8, 1.7 Hz, 1H), 6.87–6.77 (m, 2H), 6.49–6.39 (m, 2H), 3.22 (s, 2H), 3.21 (br. s, 1H), 1.41 (s, 6H) ppm. 13C NMR (75 MHz, CDCl3) δ = 155.6 (d, 1JC,F = 234.5 Hz, C), 146.6 (C), 145.1 (C), 128.5 (CH), 126.2 (CH), 126.0 (CH), 115.5 (d, 2JC,F = 22.2 Hz, CH), 113.5 (d, 3JC,F = 7.4 Hz, CH), 56.5 (CH2), 38.8 (C), 27.4 (CH3) ppm. IR (neat) vmax = 3412 (w), 3059 (w), 2965 (m), 1612 (w), 1511 (s), 1481 (m), 1221 (s), 819 (s), 765 (s), 701 (s) cm−1. GC/MS m/z = 243 ([M]+), 124 ([C7H7NF]+), 91 ([C7H7]+). HRMS (ESI+) m/z calcd for C16H19NF 244.1502 ([M + H]+), found 244.1503.
:1), 41a (424 mg, 1.34 mmol, 67%) was isolated as a yellow solid. Prior to chromatography, the ratio 41a/41b was determined to be 99:1. 41a: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.40 (br. d, J = 8.3 Hz, 2H), 7.37–7.32 (m, 2H), 7.27–7.21 (m, 3H), 6.98 (t, J = 7.4 Hz, 1H), 6.90 (d, J = 8.4 Hz, 2H), 6.85 (br. d, J = 8.8 Hz, 2H), 6.51 (br. d, J = 8.8 Hz, 2H), 3.26 (br. s, 3H), 1.42 (s, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ = 159.1 (C), 147.3 (C), 146.6 (C), 145.4 (C), 129.4 (CH), 128.5 (CH), 126.2 (CH), 126.0 (CH), 121.8 (CH), 121.2 (CH), 116.9 (CH), 113.7 (CH), 56.4 (CH2), 38.8 (C), 27.4 (CH3) ppm. IR (neat) vmax = 3412 (s), 3053 (w), 3034 (w), 2965 (m), 2924 (w), 2854 (w), 1613 (s), 1517 (s), 1489 (m), 1233 (s), 1163 (m), 832 (m), 752 (m), 704 (m), 692 (m) cm−1. GC/MS m/z = 317 ([M]+), 198 ([C13H12NO]+), 91 ([C7H7]+). HRMS (ESI+) m/z calcd for C22H23NONa 340.1677 ([M + Na]+), found 340.1681.
:1 → PE–EtOAc, 40:1 → PE–EtOAc, 20:1), 42a (107 mg, 0.28 mmol, 14%) was isolated as a colorless solid. Prior to tosylation and chromatography, the ratio of the branched and the linear product was determined to be 99:1. 42a: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.68 (d, J = 8.3 Hz, 2H), 7.40–7.35 (m, 2H), 7.32 (br. t, J = 7.8 Hz, 2H), 7.26–7.24 (m, 2H), 7.21 (br. t, J = 7.3 Hz, 1H), 3.44 (s, 2H), 2.53–2.45 (m, 1H), 2.41 (s, 3H), 1.39 (s, 6H), 1.35–1.12 (m, 7H), 0.74–0.59 (m, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ 147.9 (C), 142.7 (C), 139.2 (C), 129.4 (CH), 128.2 (CH), 127.3 (CH), 126.3 (CH), 126.1 (CH), 60.9 (CH), 59.4 (CH2), 39.0 (C), 31.4 (CH2), 27.3 (CH3), 26.6 (CH2), 25.1 (CH2), 21.5 (CH3) ppm. IR (neat) vmax = 3029 (w), 2940 (m), 2859 (m), 1601 (s), 1494 (m), 1371 (s), 1161 (s), 1045 (w), 816 (m), 761 (s), 705 (s) cm−1. MS (ESI+) m/z = 408 ([M + Na]+). HRMS (ESI+) m/z calcd for C23H31NO2SNa 408.1973 ([M + Na]+), found 408.1980.
:1 → PE–EtOAc, 40:1 → PE–EtOAc, 30:1), a mixture of 43a and tosylated N-methylhexylamine (421 mg) was isolated as a colorless oil. According to 1H NMR spectroscopy, the mixture contained 380 mg of 43a (0.98 mmol, 49%) and 41 mg of tosylated N-methylhexylamine (0.15 mmol). Prior to tosylation and chromatography, the ratio of the branched and the linear product was determined to be 99:1. 43a: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.65 (d, J = 8.3 Hz, 2H), 7.37 (br. d, J = 8.1 Hz, 2H), 7.33–7.24 (m, 4 H), 7.19 (br. t, J = 7.3 Hz, 1H), 3.39 (s, 2H), 2.54–2.47 (m, 2H), 2.41 (s, 3H), 1.41 (s, 6H), 1.12–0.98 (m, 3H), 0.95–0.85 (m, 3H), 0.78 (t, J = 7.3 Hz, 3H), 0.62 (pent, J = 7.6 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 147.4 (C), 142.9 (C), 137.3 (C), 129.5 (CH), 128.2 (CH), 127.2 (CH), 126.1 (CH), 126.0 (CH), 59.7 (CH2), 49.3 (CH2), 39.0 (C), 31.1 (CH2), 27.1 (CH3), 27.1 (CH2), 26.3 (CH2), 22.4 (CH2), 21.5 (CH3), 13.9 (CH3) ppm. IR (neat) vmax = 2958 (s), 2929 (s), 2858 (m), 1599 (m), 1495 (m), 1465 (m), 1340 (s), 1160 (s), 1091 (m), 815 (m), 757 (m), 702 (s), 655 (s) cm−1. MS (ESI+) m/z = 410 ([M + Na]+). HRMS (ESI+) m/z calcd for C23H33NO2SNa 410.2130 ([M + Na]+), found 410.2123.
:1), 44b (150 mg, 0.63 mmol, 31%) was isolated as a yellow oil. Prior to chromatography, the ratio 44a/44b was determined to be 1:99. 44b: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.28 (t, J = 7.4 Hz, 2H), 7.22–7.13 (m, 3H), 6.96 (d, J = 8.1 Hz, 2 H), 6.47 (br. d, J = 8.4 Hz, 2 H), 3.53 (br. s, 1H), 3.06 (dd, J = 12.4, 6.0 Hz, 1H), 2.92 (dd, J = 12.4, 6.9 Hz, 1H), 2.74 (dd, J = 13.4, 6.4 Hz, 1H), 2.49 (dd, J = 13.5, 7.9 Hz, 1H), 2.22 (s, 3H), 2.06 (oct, J = 6.7 Hz, 1H), 0.96 (d, J = 6.7 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 146.1 (C), 140.6 (C), 129.7 (CH), 129.1 (CH), 128.2 (CH), 126.3 (C), 125.9 (CH), 112.9 (CH), 50.2 (CH2), 41.4 (CH2), 35.0 (CH), 20.3 (CH3), 18.1 (CH3) ppm.
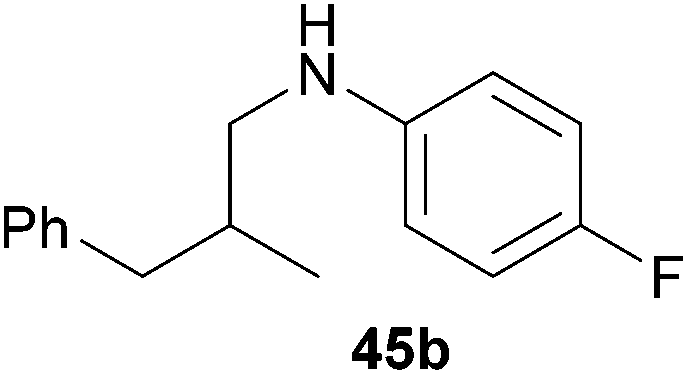
:1), 45b (316 mg, 1.30 mmol, 65%) was isolated as a red oil. Prior to chromatography, the ratio 45a/45b was determined to be 2:98. 45b: 1H NMR (300 MHz, CDCl3, TMS) δ = 7.37–7.11 (m, 5H), 6.91–6.79 (m, 2H), 6.50–6.38 (m, 2H), 3.52 (br. s, 1H), 3.03 (dd, J = 12.3, 6.1 Hz, 1H), 2.90 (dd, J = 12.3, 6.9 Hz, 1H), 2.73 (dd, J = 13.5, 6.5 Hz, 1H), 2.51 (dd, J = 13.5, 7.7 Hz, 1H), 2.05 (oct, J = 6.7 Hz, 1H), 0.97 (d, J = 6.7 Hz, 3H) ppm. 13C NMR (75 MHz, CDCl3) δ = 155.6 (d, 1JC,F = 234.4 Hz, C), 144.7 (C), 140.4 (C), 129.1 (CH), 128.3 (CH), 126.0 (CH), 115.6 (d, 2JC,F = 22.2 Hz, CH), 113.4 (d, 3JC,F = 7.4 Hz, CH), 50.5 (CH2), 41.4 (CH2), 35.0 (CH), 18.1 (CH3) ppm. IR (neat) vmax = 3422 (m), 3061 (w), 3027 (m), 2958 (m), 2925 (m), 1612 (m), 1511 (s), 1454 (m), 1319 (w), 1256 (m), 1221 (s), 819 (s), 741 (s), 701 (s) cm−1. GC/MS m/z = 243 ([M]+), 124 ([C7H7NF]+), 91 ([C7H7]+). HRMS (ESI+) m/z calcd for C16H19FN 244.1502 ([M + H]+), found 244.1496.
:1 → PE–EtOAc, 20:1), 46b (88 mg, 0.28 mmol, 14%) was isolated as a dark yellow solid. Prior to chromatography, the ratio 46a/46b was determined to be 1:99. 46b: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.31–7.24 (m, 4H), 7.23–7.14 (m, 3H), 6.99 (br. t, J = 7.4 Hz, 1H), 6.92 (br. d, J = 7.7 Hz, 2H), 6.87 (br. d, J = 8.8 Hz, 2H), 6.54 (br. d, J = 8.9 Hz, 2H), 3.58 (br. s, 1H), 3.08 (dd, J = 12.3, 6.0 Hz, 1H), 2.94 (dd, J = 12.3, 7.0 Hz, 1H), 2.75 (dd, J = 13.5, 6.5 Hz, 1H), 2.52 (dd, J = 13.5, 7.7 Hz, 1H), 2.08 (oct, J = 6.7 Hz, 1H), 0.99 (d, J = 6.7 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 159.2 (C), 147.4 (C), 145.1 (C), 140.5 (C), 129.5 (CH), 129.1 (CH), 128.3 (CH), 126.0 (CH), 121.9 (CH), 121.2 (CH), 117.1 (CH), 113.7 (CH), 50.5 (CH2), 41.4 (CH2), 35.1 (CH), 18.1 (CH3) ppm. IR (neat) vmax = 3026 (m), 2957 (m), 2925 (m), 2870 (m), 1613 (m), 1589 (m), 1512 (s), 1488 (s), 1232 (s), 869 (s), 832 (s), 745 (s), 700 (s), 692 (s) cm−1. GC/MS m/z = 317 ([M]+), 198 ([C13H12NO]+), 91 ([C7H7]+). HRMS (ESI+) m/z calcd for C22H24NO 318.1858 ([M + H]+), found 318.1854.
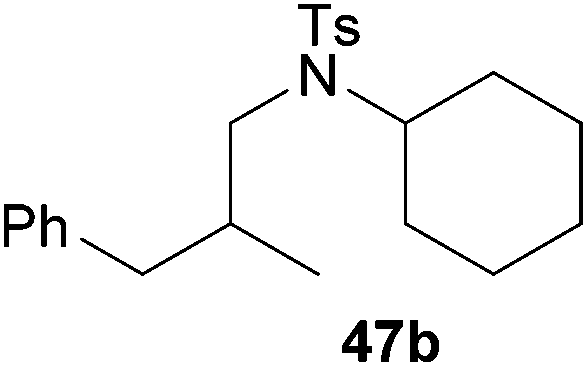
:1 → PE–EtOAc, 40:1 → PE–EtOAc, 20:1), 47b (294 mg, 0.76 mmol, 38%) was isolated as a colorless solid. Prior to tosylation and chromatography, the ratio of the α- and the β-alkylated product was determined to be 1:99. 47b: 1H NMR (300 MHz, CDCl3, TMS) δ = 7.64 (d, J = 8.3 Hz, 2H), 7.35–7.10 (m, 7H), 3.66–3.53 (m, 1H), 3.06 (dd, J = 14.4, 6.9, 1H), 2.97 (dd, J = 14.4, 7.7, 1H), 2.82 (dd, J = 13.0, 5.0 Hz, 1H), 2.40 (s, 3H), 2.36–2.11 (m, 2H), 1.77–1.49 (m, 5H), 1.38–1.12 (m, 5H), 0.90 (d, J = 6.4 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 142.7 (C), 140.8 (C), 138.0 (C), 129.5 (CH), 129.0 (CH), 128.2 (CH), 126.9 (CH), 125.8 (CH), 58.5 (CH), 50.2 (CH2), 41.0 (CH2), 35.8 (CH), 31.9 (CH2), 31.8 (CH2), 26.2 (CH2), 26.2 (CH2), 25.4 (CH2), 21.4 (CH3), 17.5 (CH3) ppm. IR (neat) vmax = 3027 (m), 2935 (s), 2856 (s), 1599 (s), 1494 (s), 1453 (s), 1334 (s), 1155 (s), 1090 (m), 1005 (m), 988 (m), 849 (m), 814 (m), 742 (m), 702 (s), 661 (s) cm−1. MS (ESI+) m/z = 408 ([M + Na]+). HRMS (ESI+) m/z calcd for C23H31NO2NaS 408.1973 ([M + Na]+), found 408.1967.
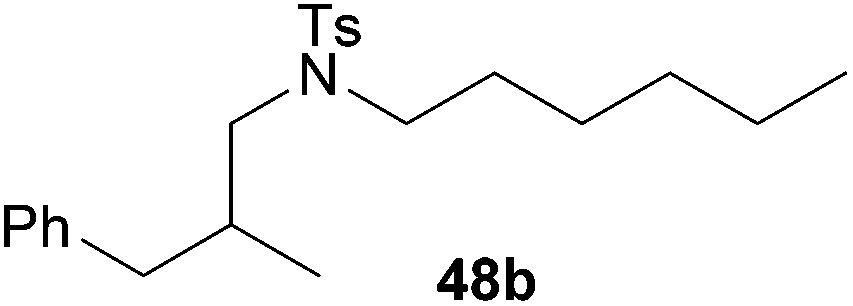
:1 → PE–EtOAc, 40:1 → PE–EtOAc, 20:1), 48b (322 mg, 0.83 mmol, 41%) was isolated as a colorless oil. Prior to tosylation and chromatography, the ratio of the α- and the β-alkylated product was determined to be 1:99. 48b: 1H NMR (500 MHz, CDCl3, TMS) δ = 7.64 (d, J = 8.3 Hz, 2H), 7.30–7.25 (m, 4H), 7.19 (t, J = 7.4 Hz, 1H), 7.13 (d, J = 7.1 Hz, 2H), 3.12–2.96 (m, 3H), 2.92 (dd, J = 13.7, 8.0 Hz, 1H), 2.77 (dd, J = 13.6, 5.7 Hz, 1H), 2.41 (s, 3H), 2.34 (dd, J = 13.6, 8.8 Hz, 1H), 2.12–2.01 (m, 1H), 1.51–1.40 (m, 2H), 1.30–1.15 (m, 6H), 0.89 (d, J = 6.6 Hz, 3H), 0.87 (t, J = 7.0 Hz, 3 H) ppm. 13C NMR (125 MHz, CDCl3) δ = 142.9 (C), 140.4 (C), 136.7 (C), 129.5 (CH), 129.0 (CH), 128.2 (CH), 127.2 (CH), 125.9 (CH), 54.6 (CH2), 49.2 (CH2), 40.9 (CH2), 34.3 (CH), 31.3 (CH2), 28.4 (CH2), 26.5 (CH2), 22.5 (CH2), 21.5 (CH3), 17.5 (CH3), 14.0 (CH3) ppm. IR (neat) vmax = 3027 (m), 2928 (s), 2870 (s), 1599 (m), 1495 (m), 1455 (s), 1340 (s), 1161 (s), 1191 (m), 981 (m), 815 (m), 741 (s), 701 (m), 655 (s) cm−1. MS (ESI+) m/z = 410 ([M + Na]+). HRMS (ESI+) m/z calcd for C23H33NO2SNa 410.2130 ([M + Na]+), found 410.2126.
:45. 60a: Rf = 0.16 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.39 (t, J = 7.5 Hz, 2H), 7.35–7.27 (m, 5H), 7.22–7.20 (m, 1H), 7.17 (d, J = 7.4 Hz, 2H), 6.37 (d, J = 15.8 Hz, 1H), 6.06 (dd, J = 15.8, 8.5 Hz, 1H), 3.80 (dd, J = 13.5, 8.8 Hz, 1H), 3.73 (dd, J = 13.5, 6.5 Hz, 1H), 2.62 (hept, J = 6.9 Hz, 1H), 1.81 (s, 3H), 1.08 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.6 (C), 143.4 (C), 137.5 (C), 133.9 (CH), 129.9 (CH), 129.6 (CH), 128.5 (CH), 128.3 (CH), 127.7 (CH), 127.0 (CH), 126.1 (CH), 54.5 (CH2), 36.6 (CH), 22.8 (CH3), 18.4 (CH3) ppm. IR (ATR) vmax = 3057 (w), 3025 (w), 2967 (w), 2927 (w), 2871 (w), 1656 (vs), 1594 (m), 1494 (s), 1394 (s), 1292 (m), 1277 (m), 966 (m), 749 (s), 695 (vs) cm−1. MS (ESI+) m/z = 302 ([M + Na]+). HRMS (ESI+) m/z calcd for C19H21NONa 302.1521 ([M + Na]+), found 302.1514. 60b: Rf = 0.11 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.43 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 7.31–7.25 (m, 4H), 7.19–7.15 (m, 3H), 6.33 (d, J = 15.8 Hz, 1H), 6.16 (dt, J = 15.8, 6.8 Hz, 1H), 3.78–3.73 (m, 2H), 2.25–2.18 (m, 2H), 1.84 (s, 3H), 1.70 (pent, J = 7.6 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.3 (C), 143.1 (C), 137.6 (C), 130.3 (CH), 129.7 (CH), 129.6 (CH), 128.4 (CH), 128.1 (CH), 127.8 (CH), 126.9 (CH), 125.9 (CH), 48.7 (CH2), 30.3 (CH2), 27.4 (CH2), 22.8 (CH3). IR (ATR) vmax = 3060 (w), 3027 (w), 2931 (w), 2860 (w), 1655 (vs), 1596 (m), 1496 (s), 1397 (s), 1296 (m), 1280 (m), 968 (m), 744 (m), 698 (vs) cm−1. MS (ESI+) m/z = 302 ([M + Na]+). HRMS (ESI+) m/z calcd for C19H21NONa 302.1521 ([M + Na]+), found 302.1511.
:1) gave three fractions as oils. Fraction 1 contained pure compound 61a (62 mg, 0.17 mmol, 8%) and fraction 2 contained a mixture of 61a and 61b (198 mg, 0.53 mmol, 27%). Fraction 3 contained pure compound 61b (74 mg, 0.20 mmol, 10%). Prior to acetylation and chromatography, the ratio of the branched and the linear product was determined to be 54:46. 61a: Rf = 0.22 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.40–7.35 (m, 2H), 7.33–7.27 (m, 4H), 7.22–7.14 (m, 2H), 7.11 (br. d, J = 8.8 Hz, 2H), 7.05 (br. d, J = 7.7 Hz, 2H), 6.99 (br. d, J = 8.8 Hz, 2H), 6.38 (d, J = 15.9 Hz, 1H), 6.07 (dd, J = 15.9, 8.5 Hz, 1H), 3.78 (dd, J = 13.4, 8.9 Hz, 1H), 3.72 (dd, J = 13.4, 6.4 Hz, 1H), 2.70–2.60 (m, 1H), 1.84 (s, 3H), 1.10 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.7 (C), 156.8 (C), 156.3 (C), 138.1 (C), 137.4 (C), 133.9 (CH), 129.9 (CH), 129.9 (CH), 129.5 (CH), 128.5 (CH), 127.0 (CH), 126.1 (CH), 124.0 (CH), 119.4 (CH), 119.0 (CH), 54.5 (CH2), 36.6 (CH), 22.8 (CH3), 18.4 (CH3) ppm. IR (ATR) vmax = 3059 (w), 3025 (w), 2964 (w), 2926 (w), 2869 (w), 1656 (vs), 1588 (m), 1504 (s), 1488 (vs), 1393 (m), 1234 (vs), 967 (m), 870 (m), 844 (m), 749 (s), 692 (s) cm−1. MS (ESI+) m/z = 372 ([M + H]+). HRMS (ESI+) m/z calcd for C25H25NO2Na 394.1783 ([M + Na]+), found 394.1787. 61b: Rf = 0.14 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.41–7.36 (m, 2H), 7.34–7.26 (m, 4H), 7.21–7.15 (m, 2H), 7.12 (br. d, J = 8.8 Hz, 2H), 7.07 (br. d, J = 7.7 Hz, 2H), 7.02 (br. d, J = 8.8 Hz, 2H), 6.36 (d, J = 15.9 Hz, 1H), 6.19 (dt, J = 15.9, 6.9 Hz, 1H), 3.76–3.71 (m, 2H), 2.24 (q, J = 7.4 Hz, 2H), 1.87 (s, 3H), 1.72 (pent, J = 7.6 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.3 (C), 157.0 (C), 156.2 (C), 137.8 (C), 137.5 (C), 130.3 (CH), 129.9 (CH), 129.6 (CH), 129.3 (CH), 128.4 (CH), 126.8 (CH), 125.9 (CH), 124.0 (CH), 119.4 (CH), 119.1 (CH), 48.7 (CH2), 30.2 (CH2), 27.4 (CH2), 22.8 (CH3) ppm. IR (ATR) vmax = 3062 (w), 3028 (w), 2926 (w), 2851 (w), 1658 (s), 1506 (s), 1489 (s), 1397 (m), 1294 (m), 1233 (vs), 968 (m), 750 (m), 693 (s) cm−1. MS (ESI+) m/z = 372 ([M + H]+). HRMS (ESI+) m/z calcd for C25H25NO2Na 394.1783 ([M + Na]+), found 394.1778.
:1) gave three fractions as oils. Fraction 1 contained pure compound 62a (82 mg, 0.27 mmol, 13%) and fraction 2 contained a mixture of 62a and 62b (142 mg, 0.46 mmol, 23%). Fraction 3 contained pure compound 62b (59 mg, 0.19 mmol, 10%). Prior to acetylation and chromatography, the ratio of the branched and the linear product was determined to be 54:46. 62a: Rf = 0.24 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.30–7.24 (m, 4H), 7.19–7.15 (m, 1H), 7.05 (br. d, J = 8.8 Hz, 2H), 6.86 (br. d, J = 8.8 Hz, 2H), 6.34 (d, J = 15.9 Hz, 1H), 6.04 (dd, J = 15.8, 8.4 Hz, 1H), 3.79 (s, 3H), 3.73 (dd, J = 13.4, 8.4 Hz, 1H), 3.66 (dd, J = 13.4, 6.5 Hz, 1H), 2.62–2.54 (m, 1H), 1.77 (s, 3H), 1.05 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.9 (C), 158.7 (C), 137.5 (C), 136.1 (C), 133.9 (CH), 129.8 (CH), 129.2 (CH), 128.4 (CH), 127.0 (CH), 126.1 (CH), 114.6 (CH), 55.4 (CH3), 54.5 (CH2), 36.5 (CH), 22.7 (CH3), 18.3 (CH3) ppm. 62b: Rf = 0.21 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.32–7.24 (m, 4H), 7.19–7.15 (m, 1H), 7.08 (br. d, J = 8.9 Hz, 2H), 6.92 (br. d, J = 8.9 Hz, 2H), 6.33 (d, J = 15.8 Hz, 1H), 6.16 (dt, J = 15.8, 6.9 Hz, 1H), 3.83 (s, 3H), 3.73–3.69 (m, 2H), 2.21 (q, J = 7.6 Hz, 2H), 1.82 (s, 3H), 1.68 (pent, J = 7.6 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.6 (C), 158.9 (C), 137.6 (C), 135.8 (C), 130.2 (CH), 129.7 (CH), 129.1 (CH), 128.4 (CH), 126.9 (CH), 125.9 (CH), 114.7 (CH), 55.4 (CH3), 48.7 (CH2), 30.3 (CH2), 27.4 (CH2), 22.8 (CH3) ppm.
:1) gave three fractions as oils. Fraction 1 contained pure compound 63a (76 mg, 0.21 mmol, 11%) and fraction 2 contained a mixture of 63a and 63b (412 mg, 1.14 mmol, 57%). Fraction 3 contained pure compound 63b (107 mg, 0.30 mmol, 14%). Prior to acetylation and chromatography, the ratio of the branched and the linear product was determined to be 53:47. 63a: Rf = 0.28 (PE–MTBE, 1:1). 1H NMR (500 MHz, CDCl3) δ = 7.31–7.27 (m, 4H), 7.24–7.17 (m, 5H), 6.36 (d, J = 15.9 Hz, 1H), 6.01 (dd, J = 15.8, 8.6 Hz, 1H), 3.79–3.70 (m, 2H), 2.68–2.58 (m, 1H), 1.82 (s, 3H), 1.08 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.4 (C), 148.2 (C), 142.0 (C), 137.3 (C), 133.7 (CH), 130.2 (CH), 129.8 (CH), 128.5 (CH), 127.2 (CH), 126.1 (CH), 122.0 (CH), 120.4 (q, 1JC,F = 258.0 Hz, CF3), 154.7 (CH2), 36.7 (CH), 22.8 (CH3), 18.4 (CH3) ppm. 19F NMR (500 MHz, CDCl3) δ = −58.0 ppm. IR (ATR) vmax = 3061 (w), 3026 (w), 2965 (w), 2929 (w), 2872 (w), 1662 (s), 1506 (s), 1392 (m) 1252 (vs), 1198 (vs), 1160 (vs), 967 (m), 858 (m), 749 (s), 694 (s) cm−1. MS (EI) m/z = 363 ([M]+). HRMS (EI) m/z calcd for C20H20O2NF3 363.1441 ([M]+), found 363.1449. 63b: Rf = 0.20 (PE–MTBE, 1:1). 1H NMR (500 MHz, CDCl3) δ = 7.30–7.26 (m, 5H), 7.23–7.17 (m, 4H), 6.34 (d, J = 15.9 Hz, 1H), 6.15 (dt, J = 15.7, 6.8 Hz, 1H), 3.78–3.71 (m, 2H), 2.25–2.18 (m, 2H), 1.84 (s, 3H), 1.70 (pent, J = 7.5 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.0 (C), 148.4 (C), 141.7 (C), 137.5 (C), 130.5 (CH), 129.6 (CH), 129.4 (CH), 128.5 (CH), 127.0 (CH), 126.0 (CH), 122.1 (CH), 120.4 (q, 1JC,F = 258.0 Hz, CF3), 48.8 (CH2), 30.3 (CH2), 27.5 (CH2), 22.8 (CH3) ppm. 19F NMR (500 MHz, CDCl3) δ = −58.0 ppm. IR (ATR) vmax = 3063 (w), 3027 (w), 2931 (w), 2866 (w), 1660 (s), 1506 (s), 1448 (m), 1396 (m), 1252 (vs), 1203 (vs), 1161 (vs), 967 (m), 857 (m), 747 (m), 695 (m) cm−1. MS (EI) m/z = 363 ([M]+). HRMS (EI) m/z calcd for C20H20O2NF3 363.1441 ([M]+), found 363.1444.
:1) gave three fractions as oils. Fraction 1 contained pure compound 64a (123 mg, 0.38 mmol, 19%) and fraction 2 contained a mixture of 64a and 64b (149 mg, 0.46 mmol, 23%). Fraction 3 contained pure compound 64b (73 mg, 0.23 mmol, 11%). Prior to acetylation and chromatography, the ratio of the branched and the linear product was determined to be 54:46. 64a: 1H NMR (500 MHz, CDCl3) δ = 7.31–7.28 (m, 4H), 7.23 (br. d, J = 8.5 Hz, 2H), 7.21–7.18 (m, 1H), 7.07 (br. d, J = 8.5 Hz, 2H), 6.35 (d, J = 15.8 Hz, 1H), 6.04 (dd, J = 15.9, 8.5 Hz, 1H), 3.76 (dd, J = 13.4, 8.8 Hz, 1H), 3.70 (dd, J = 13.4, 6.5 Hz, 1H), 2.64–2.57 (m, 1H), 2.49 (s, 3H), 1.81 (s, 3H), 1.07 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.6 (C), 140.2 (C), 138.4 (C), 137.4 (C), 133.8 (CH), 129.9 (CH), 128.6 (CH), 128.5 (CH), 127.1 (CH), 126.1 (CH), 54.4 (CH2), 36.6 (CH), 22.8 (CH3), 18.4 (CH3), 15.6 (CH3) ppm. IR (ATR) vmax = 3025 (w), 2963 (w), 2921 (m), 2851 (w), 1656 (vs), 1492 (vs), 1391 (s), 1286 (m), 1098 (m), 966 (s), 829 (m), 747 (s), 694 (s) cm−1. MS (ESI+) m/z = 348 ([M + Na]+). HRMS (ESI+) m/z calcd for C20H23NOSNa 348.1398 ([M + Na]+), found 348.1401. 64b: 1H NMR (500 MHz, CDCl3) δ = 7.24–7.18 (m, 6H), 7.13–7.09 (m, 1H), 7.01 (br. d, J = 8.4 Hz, 2H), 6.25 (d, J = 15.7 Hz, 1H), 6.08 (dt, J = 15.8, 6.9 Hz, 1H), 3.68–3.62 (m, 2H), 2.43 (s, 3H), 2.17–2.11 (m, 2H), 1.77 (s, 3H), 1.61 (pent, J = 7.6 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.3 (C), 139.9 (C), 138.6 (C), 137.5 (C), 130.3 (CH), 129.6 (CH), 128.5 (CH), 128.4 (CH), 127.2 (CH), 126.9 (CH), 125.9 (CH), 48.6 (CH2), 30.3 (CH2), 27.4 (CH2), 22.8 (CH3), 15.6 (CH3) ppm. IR (ATR) vmax = 3024 (w), 2920 (m), 2851 (w), 1654 (vs), 1492 (s), 1438 (m), 1392 (s), 1291 (m), 1098 (m), 966 (s), 828 (m), 747 (m), 693 (s) cm−1. MS (ESI+) m/z = 348 ([M + Na]+). HRMS (ESI+) m/z calcd for C20H23NOSNa 348.1398 ([M + Na]+), found 348.1397.
:1) gave three fractions as oils. Fraction 1 contained pure compound 65a (74 mg, 0.23 mmol, 12%) and fraction 2 contained a mixture of 65a and 65b (278 mg, 0.87 mmol, 43%). Fraction 3 contained pure compound 65b (61 mg, 0.19 mmol, 9%). Prior to acetylation and chromatography, the ratio of the branched and the linear product was determined to be 53:47. 65a: 1H NMR (500 MHz, CDCl3) δ = 7.31–7.27 (m, 4H), 7.22 (d, J = 8.3 Hz, 2H), 7.21–7.18 (m, 1H), 7.07 (d, J = 8.3 Hz, 2H), 6.36 (d, J = 15.9 Hz, 1H), 6.06 (dd, J = 15.8, 8.4 Hz, 1H), 3.77 (dd, J = 13.4, 8.7 Hz, 1H), 3.71 (dd, J = 13.4, 6.5 Hz, 1H), 2.93 (hept, J = 6.9 Hz, 1H), 2.67–2.58 (m, 1H), 1.81 (s, 3H), 1.26 (d, J = 6.9 Hz, 6H), 1.08 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.8 (C), 148.4 (C), 140.9 (C), 137.6 (C), 134.0 (CH), 129.7 (CH), 128.4 (CH), 128.0 (CH), 127.4 (CH), 127.0 (CH), 126.1 (CH), 54.4 (CH2), 36.5 (CH), 33.7 (CH), 23.9 (CH3), 22.9 (CH3), 18.3 (CH3) ppm. IR (ATR) vmax = 3027 (w), 2963 (m), 2929 (w), 2872 (w), 1658 (vs), 1511 (s), 1395 (s), 1289 (m), 1215 (w), 968 (m), 845 (m), 749 (s), 696 (s) cm−1. MS (ESI+) m/z = 344 ([M + Na]+). HRMS (ESI+) m/z calcd for C22H27NONa 344.1990 ([M + Na]+), found 344.1982. 65b: 1H NMR (500 MHz, CDCl3) δ = 7.31–7.24 (m, 7H), 7.07 (d, J = 8.3 Hz, 2H), 6.33 (d, J = 15.9 Hz, 1H), 6.16 (dt, J = 15.8, 6.8 Hz, 1H), 3.75–3.71 (m, 2H), 2.94 (hept, J = 7.0 Hz, 1H), 2.24–2.18 (m, 2H), 1.83 (s, 3H), 1.70 (pent, J = 7.6 Hz, 2H), 1.27 (d, J = 6.9 Hz, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.5 (C), 148.6 (C), 140.7 (C), 137.6 (C), 130.2 (CH), 129.8 (CH), 128.4 (CH), 127.8 (CH), 127.6 (CH), 126.9 (CH), 125.9 (CH), 48.7 (CH2), 33.7 (CH), 30.3 (CH2), 27.4 (CH2), 23.9 (CH3), 22.8 (CH3) ppm. IR (ATR) vmax = 3028 (w), 2962 (m), 2930 (w), 2872 (w), 1658 (vs), 1511 (s), 1397 (s), 1294 (m), 968 (m), 845 (m), 749 (m), 696 (s) cm−1. MS (ESI+) m/z = 344 ([M + Na]+). HRMS (ESI+) m/z calcd for C22H28NO 322.2165 ([M + H]+), found 322.2168.
:1) gave three fractions as oils. Fraction 1 contained pure compound 66a (111 mg, 0.36 mmol, 18%) and fraction 2 contained a mixture of 66a and 66b (206 mg, 0.66 mmol, 33%). Fraction 3 contained compound 66b (88 mg, 0.28 mmol, 14%) with trace impurities of 66a. Prior to acetylation and chromatography, the ratio of the branched and the linear product was determined to be 50:50. 66a: Rf = 0.23 (PE–MTBE, 1:1). 1H NMR (500 MHz, CDCl3) δ = 7.36 (d, J = 8.6 Hz, 2H), 7.32–7.28 (m, 4H), 7.24–7.19 (m, 1H), 7.10 (d, J = 8.5 Hz, 2H), 6.35 (d, J = 15.9 Hz, 1H), 6.02 (dd, J = 15.8, 8.6 Hz, 1H), 3.76 (dd, J = 13.5, 9.0 Hz, 1H), 3.70 (dd, J = 13.5, 6.4 Hz, 1H), 2.65–2.56 (m, 1H), 1.81 (s, 3H), 1.07 (d, J = 6.7 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.3 (C), 141.9 (C), 137.3 (C), 133.6 (CH), 133.5 (C), 130.1 (CH), 129.8 (CH), 129.6 (CH), 128.5 (CH), 127.2 (CH), 126.1 (CH), 54.5 (CH2), 36.6 (CH), 22.8 (CH3), 18.4 (CH3) ppm. 66b: Rf = 0.19 (PE–MTBE, 1:1). 1H NMR (500 MHz, CDCl3) δ = 7.36 (d, J = 8.6 Hz, 2H), 7.28–7.21 (m, 4H), 7.16–7.12 (m, 1H), 7.07 (d, J = 8.6 Hz, 2H), 6.29 (d, J = 15.8 Hz, 1H), 6.10 (dt, J = 15.7, 6.9 Hz, 1H), 3.71–3.66 (m, 2H), 2.20–2.14 (m, 2H), 1.80 (s, 3H), 1.63 (pent, J = 7.6 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.0 (C), 141.6 (C), 137.5 (C), 133.7 (C), 130.4 (CH), 129.9 (CH), 129.4 (CH), 128.4 (CH), 126.9 (CH), 125.9 (CH), 48.7 (CH2), 30.2 (CH2), 27.4 (CH2), 22.8 (CH3) ppm. IR (ATR) vmax = 3059 (w), 3025 (w), 2929 (m), 2864 (w), 1657 (vs), 1489 (vs), 1446 (m), 1392 (s), 1289 (m), 1091 (s), 1014 (m), 966 (s), 838 (m), 746 (s) 693 (s) cm−1. MS (ESI+) m/z = 336 ([M + Na]+). HRMS (ESI+) m/z calcd for C19H20NOClNa 336.1131 ([M + Na]+), found 336.1130.
:1) gave three fractions as oils. Fraction 1 contained pure compound 67a (65 mg, 0.21 mmol, 10%) and fraction 2 contained a mixture of 67a and 67b (150 mg, 0.48 mmol, 25%). Fraction 3 contained pure compound 67b (109 mg, 0.35 mmol, 17%). Prior to acetylation and chromatography, the ratio of the branched and the linear product was determined to be 50:50. 67a: Rf = 0.19 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.38 (t, J = 7.6 Hz, 2H), 7.32 (t, J = 7.4 Hz, 1H), 7.25–7.19 (m, 4H), 7.14 (d, J = 7.7 Hz, 2H), 6.31 (d, J = 15.9 Hz, 1H), 6.03 (dd, J = 15.8, 8.4 Hz, 1H), 3.79 (dd, J = 13.5, 8.8 Hz, 1H), 3.70 (dd, J = 13.4, 6.4 Hz, 1H), 2.66–2.56 (m, 1H), 1.80 (s, 3H), 1.07 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.6 (C), 143.3 (C), 135.9 (C), 134.6 (CH), 132.6 (C), 129.6 (CH), 128.6 (CH), 128.5 (CH), 128.2 (CH), 127.7 (CH), 127.3 (CH), 54.4 (CH2), 36.6 (CH), 22.8 (CH3), 18.3 (CH3) ppm. IR (ATR) vmax = 3028 (w), 2965 (w), 2926 (m), 2871 (w), 1655 (vs), 1594 (s), 1491 (s), 1394 (s), 1291 (s), 1276 (s), 1089 (s), 967 (m), 806 (m), 776 (m), 700 (s) cm−1. MS (ESI+) m/z = 314 ([M + H]+). HRMS (ESI+) m/z calcd for C19H20NOClNa 336.1131 ([M + Na]+), found 336.1122. 67b: Rf = 0.10 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.42 (t, J = 7.6 Hz, 2H), 7.35 (t, J = 7.4 Hz, 1H), 7.23–7.19 (m, 4H), 7.16 (d, J = 7.5 Hz, 2H), 6.27 (d, J = 15.9 Hz, 1H), 6.13 (dt, J = 15.8, 6.8 Hz, 1H), 3.77–3.72 (m, 2H), 2.20 (q, J = 7.0 Hz, 2H), 1.83 (s, 3H), 1.68 (pent, J = 7.5 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.3 (C), 143.0 (C), 136.0 (C), 132.3 (C), 130.4 (CH), 129.7 (CH), 129.1 (CH), 128.5 (CH), 128.0 (CH), 127.8 (CH), 127.1 (CH), 48.6 (CH2), 30.2 (CH2), 27.3 (CH2), 22.7 (CH3) ppm. IR (ATR) vmax = 3026 (w), 2924 (m), 2850 (w), 1652 (vs), 1595 (m), 1491 (s), 1396 (s), 1294 (m), 1089 (m), 967 (m), 775 (m), 700 (vs) cm−1. MS (ESI+) m/z = 336 ([M + Na]+). HRMS (ESI+) m/z calcd for C19H20NOClNa 336.1131 ([M + Na]+), found 336.1124.
:1) gave three fractions as oils. Fraction 1 contained pure compound 68a (100 mg, 0.34 mmol, 18%) and fraction 2 contained a mixture of 68a and 68b (151 mg, 0.51 mmol, 25%). Fraction 3 contained pure compound 68b (62 mg, 0.21 mmol, 10%). Prior to acetylation and chromatography, the ratio of the branched and the linear product was determined to be 50:50. 68a: Rf = 0.19 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.31 (t, J = 7.5 Hz, 2H), 7.25 (t, J = 7.4 Hz, 1H), 7.19–7.15 (m, 2H), 7.08 (d, J = 7.4 Hz, 2H), 6.89 (t, J = 8.7 Hz, 2H), 6.25 (d, J = 15.9 Hz, 1H), 5.89 (dd, J = 15.9, 8.5 Hz, 1H), 3.72 (dd, J = 13.4, 8.9 Hz, 1H), 3.63 (dd, J = 13.4, 6.4 Hz, 1H), 2.57–2.49 (m, 1H), 1.73 (s, 3H), 0.99 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.6 (C), 161.9 (d, 1JC,F = 246.0 Hz, C), 143.3 (C), 133.6 (CH), 129.5 (CH), 128.6 (CH), 128.2 (CH), 127.7 (CH), 127.5 (d, 3JC,F = 7.9 Hz, CH), 115.3 (d, 2JC,F = 21.4 Hz, CH), 54.4 (CH2), 36.5 (CH), 22.8 (CH3), 18.3 (CH3) ppm. 19F NMR (500 MHz, CDCl3) δ = −115.4 ppm. IR (ATR) vmax = 3038 (w), 2967 (w), 2927 (w), 2871 (w), 1655 (vs), 1594 (m), 1507 (m), 1495 (m), 1395 (m), 1293 (m), 1277 (m), 1224 (s), 1158 (m), 967 (m), 849 (m), 813 (m), 772 (m), 700 (s) cm−1. MS (ESI+) m/z = 298 ([M + H]+). HRMS (ESI+) m/z calcd for C19H21FNO 298.1607 ([M + H]+), found 298.1606. 68b: Rf = 0.12 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.34 (t, J = 7.6 Hz, 2H), 7.27 (t, J = 7.4 Hz, 1H), 7.19–7.14 (m, 2H), 7.09 (d, J = 7.9 Hz, 2H), 6.87 (t, J = 8.7 Hz, 2H), 6.21 (d, J = 15.9 Hz, 1H), 5.99 (dt, J = 15.9, 6.9 Hz, 1H), 3.69–3.64 (m, 2H), 2.12 (q, J = 7.4 Hz, 2H), 1.76 (s, 3H), 1.61 (pent, J = 7.5 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.2 (C), 161.8 (d, 1JC,F = 245.7 Hz, C), 143.1 (C), 133.7 (d, 4JC,F = 3.3 Hz, C), 129.7 (CH), 129.4 (CH), 129.1 (CH), 128.1 (CH), 127.8 (CH), 127.3 (d, 3JC,F = 7.9 Hz, CH), 115.2 (d, 2JC,F = 21.4 Hz, CH), 48.6 (CH2), 30.2 (CH2), 27.4 (CH2), 22.8 (CH3) ppm. 19F NMR (500 MHz, CDCl3) δ = −115.7 ppm. IR (ATR) vmax = 3038 (w), 2926 (m), 2853 (w), 1652 (vs), 1595 (s), 1506 (s), 1495 (s), 1395 (s), 1224 (s), 967 (m), 838 (m), 770 (m), 700 (vs) cm−1. MS (ESI+) m/z = 298 ([M + H]+). HRMS (ESI+) m/z calcd for C19H21NOF 298.1607 ([M + H]+), found 298.1602.
:1) gave three fractions as oils. Fraction 1 contained compound 69a and a derivative with isomerized double bond (526 mg, 1.83 mmol, 46%). Fraction 2 contained compound 69a, its isomerized derivative as well as compound 69b (143 mg, 0.50 mmol, 12%). Fraction 3 contained compound 69b (112 mg, 0.39 mmol, 10%) with trace impurities of 69a. Prior to acetylation and chromatography, the ratio of the branched, the branched isomerized, and the linear product was determined to be 50:27:23. 69a: 1H NMR (500 MHz, CDCl3) δ = 7.44–7.37 (m, 2H), 7.35–7.29 (m, 1H), 7.16 (d, J = 7.5 Hz, 2H), 5.43–5.33 (m, 1H), 5.26 (dd, J = 15.4, 8.0 Hz, 1H), 3.66 (dd, J = 13.5, 8.4 Hz, 1H), 3.59 (dd, J = 13.3, 6.9 Hz, 1H), 2.33 (hept, J = 7.3 Hz, 1H), 2.95 (q, J = 7.0 Hz, 2H), 1.81 (s, 3H), 1.35–2.26 (m, 8H), 0.95 (d, J = 6.8 Hz, 3H), 0.87 (t, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.5 (C), 143.4 (C), 133.2 (CH), 130.7 (CH), 129.5 (CH), 128.2 (CH), 127.6 (CH), 54.4 (CH2), 35.7 (CH), 32.5 (CH2), 31.7 (CH2), 29.5 (CH2), 28.8 (CH2), 22.9 (CH3), 22.6 (CH2), 14.1 (CH3) ppm. IR (ATR) vmax = 2957 (m), 2925 (m), 2855 (m), 1661 (vs), 1595 (m), 1495 (m), 1393 (s), 1276 (m), 1217 (w), 1073 (w), 968 (m), 776 (w), 700 (vs). MS (ESI+) m/z = 310 ([M + Na]+). HRMS (ESI+) m/z calcd for C19H29NONa 310.2147 ([M + Na]+), found 310.2145. 69b: 1H NMR (500 MHz, CDCl3) δ = 7.39 (t, J = 7.7 Hz, 2H), 7.32 (t, J = 7.3 Hz, 1H), 7.14 (d, J = 8.0 Hz, 2H), 5.36–5.26 (m, 2H), 3.69–3.64 (m, 2H), 1.95 (q, J = 7.0 Hz, 2H), 1.91 (q, J = 6.2 Hz, 2H), 1.80 (s, 3H), 1.55 (pent, J = 7.5 Hz, 2H), 1.30–1.19 (m, 8H), 0.85 (t, J = 6.7 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.1 (C), 143.2 (C), 131.1 (CH), 129.6 (CH), 128.9 (CH), 128.1 (CH), 127.7 (CH), 48.7 (CH2), 32.5 (CH2), 31.7 (CH2), 29.8 (CH2), 29.4 (CH2), 28.7 (CH2), 27.6 (CH2), 22.8 (CH3), 22.6 (CH2), 14.0 (CH3) ppm. IR (ATR) vmax = 2955 (w), 2924 (m), 2854 (m), 1659 (vs), 1596 (m), 1405 (m), 1394 (s), 1294 (m), 967 (m), 774 (w), 700 (vs). MS (ESI+) m/z = 310 ([M + Na]+). HRMS (ESI+) m/z calcd for C19H29NONa 310.2147 ([M + Na]+), found 310.2143.
:1) gave three fractions as oils. Fraction 1 contained compound 70a and its double bond isomer (E)-N-(2-methyl-6-phenylhex-4-en-1-yl)aniline (301 mg, 1.13 mmol, 57%) in a ratio of 2:1 and fraction 2 contained a mixture of 70a and 70b (88 mg, 0.33 mmol, 17%). Fraction 3 contained pure compound 70b (72 mg, 0.27 mmol, 13%). Prior to chromatography, the ratio of the branched (70a), the branched isomerized, and the linear product (70b) was determined to be 47:28:25. 70a: Rf = 0.24 (PE–MTBE, 70:1). 1H NMR (500 MHz, CDCl3) δ = 7.31–7.26 (m, 2H), 7.23–7.15 (m, 5H), 6.70 (t, J = 7.3 Hz, 1H), 6.62–6.55 (m, 2H), 5.57–5.50 (m, 1H), 5.30 (dd, J = 15.4, 8.0 Hz, 1H), 3.66 (br. s, 1H), 3.06 (dd, J = 11.9, 5.4 Hz, 1H), 2.84 (dd, J = 11.8, 8.5 Hz, 1H), 2.71 (t, J = 7.5 Hz, 2H), 2.37 (m, 2H), 1.05 (d, J = 6.8 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.3 (C), 141.8 (C), 134.3 (CH), 130.3 (CH), 129.2 (CH), 128.5 (CH), 128.3 (CH), 125.8 (CH), 117.1 (CH), 112.9 (CH), 49.6 (CH2), 36.7 (CH), 35.9 (CH2), 34.3 (CH2), 18.1 (CH3) ppm. 70b: Rf = 0.23 (PE–MTBE, 70:1). 1H NMR (500 MHz, CDCl3) δ = 7.29 (t, J = 7.5 Hz, 2H), 7.23–7.16 (m, 5H), 6.71 (t, J = 7.3 Hz, 1H), 6.61 (d, J = 7.7 Hz, 2H), 5.56–5.42 (m, 2H), 3.63 (br. s, 1H), 3.10 (t, J = 7.1 Hz, 2H), 2.73–2.66 (m, 2H), 2.34 (q, J = 7.4 Hz, 2H), 2.11 (q, J = 7.0 Hz, 2H), 1.68 (pent, J = 7.2 Hz, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 148.4 (C), 142.0 (C), 130.2 (CH), 130.0 (CH), 129.2 (CH), 128.4 (CH), 128.2 (CH), 125.7 (CH), 117.1 (CH), 112.7 (CH), 43.3 (CH2), 36.0 (CH2), 34.4 (CH2), 30.0 (CH2), 29.2 (CH2) ppm. IR (ATR) vmax = 3412 (w), 3052 (w), 3024 (w), 2925 (m), 2852 (w), 1602 (vs), 1505 (vs), 1453 (m), 1319 (m), 1257 (m), 1179 (w), 968 (m), 867 (w), 745 (s), 691 (s) cm−1. MS (ESI+) m/z = 266 ([M + H]+). HRMS (ESI+) m/z calcd for C19H24N 266.1909 ([M + H]+), found 266.1901.
:1) gave three fractions as oils. Fraction 1 contained pure compound 71a (159 mg, 0.54 mmol, 27%) and fraction 2 contained a mixture of 71a and 71b (236 mg, 0.81 mmol, 41%). Fraction 3 contained pure compound 71b (30 mg, 0.10 mmol, 5%). Prior to acetylation and chromatography, the ratio of the branched and the linear product was determined to be 51:49. 71a: Rf = 0.26 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.40–7.36 (m, 2H), 7.33–7.28 (m, 3H), 7.23–7.16 (m, 5H), 6.30 (s, 1H), 3.87 (dd, J = 13.6, 9.1 Hz, 1H), 3.79 (dd, J = 13.6, 6.1 Hz, 1H), 2.66–2.57 (m, 1H), 1.83 (s, 3H), 1.68 (s, 3H), 1.08 (d, J = 7.0 Hz, 3H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.3 (C), 143.1 (C), 140.4 (C), 138.0 (C), 129.3 (CH), 128.7 (CH), 128.1 (CH), 127.9 (CH), 127.5 (CH), 126.0 (CH), 125.9 (CH), 52.7 (CH2), 42.1 (CH), 22.7 (CH3), 17.1 (CH3), 13.7 (CH3) ppm. IR (ATR) vmax = 3057 (w), 3025 (w), 2967 (w), 2933 (w), 2874 (w), 1658 (vs), 1596 (m), 1496 (s), 1395 (s), 1295 (m), 1277 (m), 779 (m), 745 (m), 700 (vs) cm−1. MS (ESI+) m/z = 316 ([M + Na]+). HRMS (ESI+) m/z calcd for C20H23NONa 316.1677 ([M + Na]+), found 316.1682. 71b: Rf = 0.17 (PE–MTBE, 2:1). 1H NMR (500 MHz, CDCl3) δ = 7.43 (t, J = 7.6 Hz, 2H), 7.36 (t, J = 7.4 Hz, 1H), 7.29 (t, J = 7.6 Hz, 2H), 7.21–7.15 (m, 5H), 6.21 (s, 1H), 3.77–3.71 (m, 2H), 2.20–2.14 (m, 2H), 1.85 (s, 3H), 1.80 (d, J = 1.3 Hz, 3H), 1.79–1.73 (m, 2H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.2 (C), 143.2 (C), 138.4 (C), 138.1 (C), 129.7 (CH), 128.8 (CH), 128.1 (CH), 128.0 (CH), 127.8 (CH), 125.8 (CH), 125.2 (CH), 48.9 (CH2), 37.8 (CH2), 26.1 (CH2), 22.8 (CH3), 17.6 (CH3) ppm. IR (ATR) vmax = 3057 (w), 3025 (w), 2932 (w), 2859 (w), 1656 (s), 1597 (s), 1496 (s), 1397 (s), 1298 (m), 1076 (w), 743 (m), 700 (vs) cm−1. MS (ESI+) m/z = 316 ([M + Na]+). HRMS (ESI+) m/z calcd for C20H23NONa 316.1677 ([M + Na]+), found 316.1682.
:1), compound 72 (301 mg, 1.04 mmol, 85%) was isolated as a colorless oil. 72: 1H NMR (500 MHz, CDCl3) δ = 7.41 (t, J = 7.7 Hz, 2H), 7.33 (t, J = 7.3 Hz, 1H), 7.16 (d, J = 7.5 Hz, 2H), 3.65 (dd, J = 13.2, 8.2 Hz, 1H), 3.54 (dd, J = 13.4, 6.7 Hz, 1H), 1.83 (s, 3H), 1.60–1.51 (m, 1H), 1.32–1.11 (m, 14 H), 0.89–0.84 (m, 6H) ppm. 13C NMR (125 MHz, CDCl3) δ = 170.6 (C), 143.4 (C), 129.6 (CH), 128.0 (CH), 127.6 (CH), 54.7 (CH2), 34.2 (CH2), 31.9 (CH2), 31.5 (CH), 29.8 (CH2), 29.5 (CH2), 29.3 (CH2), 26.8 (CH2), 22.9 (CH3), 22.6 (CH2), 17.3 (CH3), 14.1 (CH3) ppm. IR (ATR) vmax = 2956 (m), 2924 (s), 2854 (m), 1662 (vs), 1595 (m), 1495 (m), 1455 (m), 1395 (s), 1290 (m), 1276 (m), 776 (w), 700 (s) cm−1. MS (ESI+) m/z = 312 ([M + Na]+). HRMS (ESI+) m/z calcd for C19H31NONa 312.2303 ([M + Na]+), found 312.2294.
:1) gave one fraction (504 mg) as an oil of high viscosity. According to 1H NMR spectroscopy, the fraction contained a mixture of 76b (466 mg, 1.15 mmol, 58%) and tosylated N-methylbenzylamine (35 mg, 0.13 mmol). Prior to tosylation and chromatography, the ratio of the branched and the linear product was determined to be 7:93. 76b: Rf = 0.25 (PE–MTBE, 12:1). 1H NMR (300 MHz, CDCl3) δ = 7.62 (d, J = 8.3 Hz, 2H), 7.31–7.16 (m, 12H), 6.27 (d, J = 15.8 Hz, 1H), 6.13 (dt, J = 15.8, 6.4 Hz, 1H), 5.11 (t, J = 7.3 Hz, 1H), 2.62 (s, 3H), 2.37 (s, 3H), 2.18–1.99 (m, 3H), 1.88–1.75 (m, 1H) ppm. 13C NMR (75 MHz, CDCl3) δ = 143.0 (C), 138.0 (C), 137.5 (C), 137.2 (C), 130.7 (CH), 129.5 (CH), 129.1 (CH), 128.5 (CH), 128.4 (CH), 128.0 (CH), 127.8 (CH), 127.2 (CH), 127.0 (CH), 125.9 (CH), 59.5 (CH), 30.3 (CH2), 29.9 (CH2), 28.8 (CH3), 21.5 (CH3) ppm. IR (ATR) vmax = 3062 (w), 3028 (w), 2941 (w), 2872 (w), 1598 (m), 1494 (m), 1452 (m), 1333 (s), 1131 (vs), 1088 (s), 933 (s), 813 (m), 736 (s), 712 (vs) cm−1. MS (ESI+) m/z = 428 ([M + Na]+). HRMS (ESI+) m/z calcd for C25H27NO2SNa 428.1660 ([M + Na]+), found 428.1666.
:1) gave one fraction (615 mg) as an oil of high viscosity. According to 1H NMR spectroscopy, the fraction contained a mixture of 77b (590 mg, 1.34 mmol, 67%) and tosylated N-methylbenzylamine (25 mg, 0.09 mmol). Prior to tosylation and chromatography, the ratio of the branched and the linear product was determined to be 8:92. 77b: Rf = 0.22 (PE–MTBE, 12:1). 1H NMR (500 MHz, CDCl3) δ = 7.62 (d, J = 8.4 Hz, 2H), 7.30–7.17 (m, 11H), 6.28 (d, J = 15.8 Hz, 1H), 6.14 (dt, J = 15.8, 6.7 Hz, 1H), 5.13 (t, J = 7.6 Hz, 1H), 2.63 (s, 3H), 2.38 (s, 3H), 2.22–2.03 (m, 3H), 1.90–1.82 (m, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 143.0 (C), 138.0 (C), 137.2 (C), 136.0 (C), 132.5 (C), 129.9 (CH), 129.5 (CH), 129.4 (CH), 128.6 (CH), 128.4 (CH), 128.0 (CH), 127.8 (CH), 127.1 (CH), 59.4 (CH), 30.2 (CH2), 29.8 (CH2), 28.7 (CH3), 21.4 (CH3) ppm. IR (ATR) vmax = 3064 (w), 3032 (w), 2939 (w), 1599 (m), 1493 (m), 1455 (m), 1335 (s), 1157 (vs), 1090 (s), 935 (s), 812 (s), 701 (s), 659 (vs) cm−1. MS (ESI+) m/z = 462 ([M + Na]+). HRMS (ESI+) m/z calcd for C25H26NO2SClNa 462.1270 ([M + Na]+), found 462.1267.
:1) gave one fraction (661 mg) as an oil of high viscosity. According to 1H NMR spectroscopy, the fraction contained a mixture of 78b (597 mg, 1.41 mmol, 71%) and tosylated N-methylbenzylamine (64 mg, 0.23 mmol). Prior to tosylation and chromatography, the ratio of the branched and the linear product was determined to be 9:91. 78b: Rf = 0.24 (PE–MTBE, 12:1). 1H NMR (500 MHz, CDCl3) δ = 7.64 (d, J = 8.3 Hz, 2H), 7.33–7.25 (m, 6H), 7.24–7.19 (m, 3H), 6.99 (br. t, J = 8.7 Hz, 2H), 6.30 (d, J = 15.9 Hz, 1H), 6.08 (dt, J = 15.9, 6.8 Hz, 1H), 5.15 (t, J = 7.6 Hz, 1H), 2.65 (s, 3H), 2.40 (s, 3H), 2.23–2.14 (m, 2H), 2.12–2.04 (m, 1H), 1.90–1.83 (m, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 161.9 (d, 1JC,F = 245.9 Hz, CF), 143.0 (C), 138.0 (C), 137.2 (C), 133.6 (d, 4JC,F = 3.2 Hz, C), 129.4 (CH), 128.4 (CH), 128.0 (CH), 127.3 (d, 3JC,F = 7.8 Hz, CH), 127.1 (CH), 115.3 (d, 2JC,F = 21.5 Hz, CH), 59.4 (CH), 30.3 (CH2), 29.8 (CH2), 28.7 (CH3), 21.4 (CH3) ppm. 19F NMR (500 MHz, CDCl3) δ = −115.5 ppm. IR (ATR) vmax = 3059 (w), 3031 (w), 2948 (w), 2920 (w), 1598 (m), 1508 (m), 1452 (m), 1336 (s), 1228 (m), 1154 (s), 1088 (m), 972 (m), 927 (s), 820 (m), 699 (s), 656 (s) cm−1. MS (ESI+) m/z = 446 ([M + Na]+). HRMS (ESI+) m/z calcd for C25H26NO2SFNa 446.1566 ([M + Na]+), found 446.1560.
:1) gave one fraction as an oil of high viscosity. According to 1H NMR spectroscopy, the fraction contained 79b (524 mg, 1.20 mmol, 60%) and trace amounts of the corresponding branched product. Prior to tosylation and chromatography, the ratio of the branched and the linear product was determined to be 10:90. 79b: Rf = 0.30 (PE–MTBE, 6:1). 1H NMR (500 MHz, CDCl3) δ = 7.65 (d, J = 8.3 Hz, 2H), 7.29–7.24 (m, 5H), 7.24–7.21 (m, 4H), 6.85 (d, J = 8.7 Hz, 2H), 6.27 (d, J = 15.9 Hz, 1H), 6.02 (dt, J = 15.8, 6.6 Hz, 1H), 5.14 (t, J = 7.4 Hz, 1H), 3.81 (s, 3H), 2.65 (s, 3H), 2.40 (s, 3H), 2.22–2.04 (m, 3H), 1.87–1.79 (m, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 158.8 (C), 143.0 (C), 138.1 (C), 137.3 (C), 130.3 (C), 130.1 (CH), 129.4 (CH), 128.4 (CH), 128.0 (CH), 127.7 (CH), 127.2 (CH), 127.0 (CH), 126.9 (CH), 59.5 (CH), 55.3 (CH3), 30.4 (CH2), 29.9 (CH2), 28.8 (CH3), 21.5 (CH3) ppm. IR (ATR) vmax = 3062 (w), 3032 (w), 2956 (w), 2923 (m), 2853 (m), 1608 (m), 1512 (s), 1457 (m), 1335 (m), 1248 (s), 1156 (vs), 1089 (m), 1033 (m), 934 (m), 809 (s), 701 (s), 660 (vs) cm−1. MS (ESI+) m/z = 458 ([M + Na]+). HRMS (ESI+) m/z calcd for C26H29NO3SNa 458.1766 ([M + Na]+), found 458.1758.
:1) gave one fraction (605 mg) as an oil of high viscosity. According to 1H NMR spectroscopy, the fraction contained a mixture of 80b (575 mg, 1.37 mmol, 69%), tosylated N-methylbenzylamine (30 mg, 0.11 mmol), and trace amounts of the corresponding branched product. Prior to tosylation and chromatography, the ratio of the branched and the linear product was determined to be 13:87. 80b: Rf = 0.18 (PE–MTBE, 9:1). 1H NMR (500 MHz, CDCl3) δ = 7.65 (d, J = 8.3 Hz, 2H), 7.39 (d, J = 6.6 Hz, 1H), 7.30–7.26 (m 3H), 7.25–7.20 (m, 4H), 7.19–7.12 (m, 3H), 6.53 (d, J = 15.9 Hz, 1H), 6.01 (dt, J = 15.8, 6.8 Hz, 1H), 5.16 (t, J = 7.5 Hz, 1H), 2.67 (s, 3H), 2.40 (s, 3H), 2.32 (s, 2H), 2.29–2.08 (m, 3H), 1.90–1.80 (m, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 143.0 (C), 138.0 (C), 137.3 (C), 136.6 (C), 135.0 (C), 130.5 (CH), 130.1 (CH), 129.4 (CH), 128.7 (CH), 128.4 (CH), 128.1 (CH), 127.8 (CH), 127.2 (CH), 127.0 (CH), 126.0 (CH), 125.4 (CH), 59.4 (CH), 30.3 (CH2), 30.2 (CH2), 28.8 (CH3), 21.5 (CH3), 19.8 (CH3) ppm. IR (ATR) vmax = 3064 (w), 3031 (w), 2928 (w), 2868 (w), 1600 (w), 1496 (w), 1456 (m), 1336 (s), 1157 (vs), 1090 (m), 936 (s), 815 (m), 736 (s), 701 (vs), 660 (vs) cm−1. MS (ESI+) m/z = 442 ([M + Na]+). HRMS (ESI+) m/z calcd for C26H29NO2SNa 442.1817 ([M + Na]+), found 442.1814.
:1) gave one fraction as an oil of high viscosity. According to 1H NMR spectroscopy, the fraction contained 81b (574 mg, 1.32 mmol, 66%) and trace amounts of the corresponding branched product. Prior to tosylation and chromatography, the ratio of the branched and the linear product was determined to be 11:89. 81b: Rf = 0.44 (PE–MTBE, 9:1) 1H NMR (500 MHz, CDCl3) δ = 7.63 (d, J = 8.3 Hz, 2H), 7.36 (dd, J = 7.6, 1.4 Hz, 1H), 7.28–7.15 (m, 8H), 6.89 (t, J = 7.3 Hz, 1H), 6.84 (d, J = 8.2, 1H), 6.62 (d, J = 16.0 Hz, 1H), 6.13 (dt, J = 15.9, 6.7 Hz, 1H), 5.13 (t, J = 7.4 Hz, 1H), 3.82 (s, 3H), 2.63 (s, 3H), 2.37 (s, 3H), 2.20–2.03 (m, 3H), 1.85–1.76 (m, 1H) ppm. 13C NMR (125 MHz, CDCl3) δ = 156.3 (C), 142.9 (C), 138.2 (C), 137.3 (C), 129.9 (CH), 129.5 (CH), 128.4 (CH), 128.1 (CH), 127.7 (CH), 127.2 (CH), 126.5 (C), 126.5 (CH), 125.4 (CH), 120.6 (CH), 110.8 (CH), 59.6 (CH), 55.4 (CH3), 30.4 (CH2), 28.8 (CH3), 21.5 (CH3) ppm. IR (ATR) vmax = 3063 (w), 3031 (w), 2924 (w), 2851 (w), 1597 (m), 1489 (m), 1455 (m), 1333 (s), 1242 (s), 1155 (vs), 1088 (m), 1027 (m), 933 (s), 813 (m), 751 (s), 700 (vs), 658 (vs) cm−1. MS (ESI+) m/z = 458 ([M + Na]+). HRMS (ESI+) m/z calcd for C26H29NO3SNa 458.1766 ([M + Na]+), found 458.1778.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft for financial support of our research as well as Jessica Reimer and Leoni Fritsche for experimental assistance.Notes and references
- For reviews on the hydroaminoalkylation of alkenes, see: (a) P. W. Roesky, Angew. Chem., Int. Ed., 2009, 48, 4892 CrossRef CAS PubMed; (b) T.-Q. He, X.-J. Zheng, H. Cai and Z.-L. Xue, Chin. J. Inorg. Chem., 2014, 30, 53 CAS; (c) E. Chong, P. Garcia and L. L. Schafer, Synthesis, 2014, 46, 2884 CrossRef PubMed.
- R. N. Salvatore, C. H. Yon and K. W. Jung, Tetrahedron, 2001, 57, 7785 CrossRef CAS.
- Ruthenium catalysts: (a) C.-H. Jun, D.-C. Hwang and S.-J. Na, Chem. Commun., 1998, 1405 RSC; (b) N. Chatani, T. Asaumi, S. Yorimitsu, T. Ikeda, F. Kakiuci and S. Murai, J. Am. Chem. Soc., 2001, 123, 10935 CrossRef CAS PubMed.
- Iridium catalysts: S. Pan, K. Endo and T. Shibata, Org. Lett., 2011, 13, 4692 CrossRef CAS PubMed.
- Group 5 metal catalysts: (a) M. G. Clerici and F. Maspero, Synthesis, 1980, 305 CrossRef CAS; (b) W. A. Nugent, D. W. Ovenall and S. J. Holmes, Organometallics, 1983, 2, 161 CrossRef CAS; (c) S. B. Herzon and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 6690 CrossRef CAS PubMed; (d) S. B. Herzon and J. F. Hartwig, J. Am. Chem. Soc., 2008, 130, 14940 CrossRef CAS PubMed; (e) P. Eisenberger, R. O. Ayinla, J. M. P. Lauzon and L. L. Schafer, Angew. Chem., Int. Ed., 2009, 48, 8361 CrossRef CAS PubMed; (f) P. Eisenberger and L. L. Schafer, Pure Appl. Chem., 2010, 82, 1503 CrossRef CAS; (g) G. Zi, F. Zhang and H. Song, Chem. Commun., 2010, 46, 6296 RSC; (h) A. L. Reznichenko, T. J. Emge, S. Audörsch, E. G. Klauber, K. C. Hultzsch and B. Schmidt, Organometallics, 2011, 30, 921 CrossRef CAS; (i) A. L. Reznichenko and K. C. Hultzsch, J. Am. Chem. Soc., 2012, 134, 3300 CrossRef CAS PubMed; (j) P. Garcia, Y. Y. Lau, M. R. Perry and L. L. Schafer, Angew. Chem., Int. Ed., 2013, 52, 9144 CrossRef CAS PubMed; (k) Z. Zhang, J.-D. Hamel and L. L. Schafer, Chem. – Eur. J., 2013, 19, 8751 CrossRef CAS PubMed; (l) P. Garcia, P. R. Payne, E. Chong, R. L. Webster, B. J. Baron, A. C. Behrle, J. A. R. Schmidt and L. L. Schafer, Tetrahedron, 2013, 69, 5737 CrossRef CAS PubMed; (m) J. Dörfler and S. Doye, Eur. J. Org. Chem., 2014, 2790 CrossRef; (n) E. Chong, J. W. Brandt and L. L. Schafer, J. Am. Chem. Soc., 2014, 136, 10898 CrossRef CAS PubMed.
- Zirconium catalysts: J. A. Bexrud, P. Eisenberger, D. C. Leitch, P. R. Payne and L. L. Schafer, J. Am. Chem. Soc., 2009, 131, 2116 CrossRef CAS PubMed.
- Titanium catalysts: (a) C. Müller, W. Saak and S. Doye, Eur. J. Org. Chem., 2008, 2731 CrossRef; (b) R. Kubiak, I. Prochnow and S. Doye, Angew. Chem., Int. Ed., 2009, 48, 1153 CrossRef CAS PubMed; (c) I. Prochnow, R. Kubiak, O. N. Frey, R. Beckhaus and S. Doye, ChemCatChem, 2009, 1, 162 CrossRef CAS; (d) R. Kubiak, I. Prochnow and S. Doye, Angew. Chem., Int. Ed., 2010, 49, 2626 CrossRef CAS PubMed; (e) I. Prochnow, P. Zark, T. Müller and S. Doye, Angew. Chem., Int. Ed., 2011, 50, 6401 CrossRef CAS PubMed; (f) D. Jaspers, W. Saak and S. Doye, Synlett, 2012, 23, 2098 CrossRef CAS PubMed; (g) J. Dörfler and S. Doye, Angew. Chem., Int. Ed., 2013, 52, 1806 CrossRef PubMed; (h) T. Preuß, W. Saak and S. Doye, Chem. – Eur. J., 2013, 19, 3833 CrossRef PubMed; (i) J. Dörfler, T. Preuß, A. Schischko, M. Schmidtmann and S. Doye, Angew. Chem., Int. Ed., 2014, 53, 7918 CrossRef PubMed; (j) E. Chong and L. L. Schafer, Org. Lett., 2013, 15, 6002 CrossRef CAS PubMed.
- (a) T. Elkin, N. V. Kulkarni, B. Tumanskii, M. Botoshansky, L. J. W. Shimon and M. S. Eisen, Organometallics, 2013, 32, 6337 CrossRef CAS; (b) T. Elkin, M. Botoshansky, R. M. Waymouth and M. S. Eisen, Organometallics, 2013, 33, 840 CrossRef; (c) L. R. Groom, A. F. Russell, A. D. Schwarz and P. Mountford, Organometallics, 2014, 33, 1002 CrossRef CAS; (d) N. V. Kulkarni, T. Elkin, B. Tumanskii, M. Botoshansky, L. J. W. Shimon and M. S. Eisen, Organometallics, 2014, 33, 3119 CrossRef CAS and references cited therein.
- F. R. Leroux, B. Manteau, J.-P. Vors and S. Pazenok, Beilstein J. Org. Chem., 2008, 4, 13 Search PubMed.
- H. Fujita, M. Tokuda, M. Nitta and H. Suginome, Tetrahedron Lett., 1992, 33, 6359 CrossRef CAS.
- D. Weickmann, W. Frey and B. Plietker, Chem. – Eur. J., 2013, 19, 2741 CrossRef CAS PubMed.
- R. T. Gephart III, D. L. Huang, M. J. B. Aguila, G. Schmidt, A. Shahu and T. H. Warren, Angew. Chem., Int. Ed., 2012, 51, 6488 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of the 1H and 13C NMR spectra of all products. See DOI: 10.1039/c4dt03916e |
| This journal is © The Royal Society of Chemistry 2015 |