
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tp*Cu(I)–CN–SiL2–NC–Cu(I)Tp* – a hexacoordinate Si-complex as connector for redox active metals via π-conjugated ligands†‡
Alexander
Kämpfe
a,
Erica
Brendler
b,
Edwin
Kroke
a and
Jörg
Wagler
*a
aInstitut für Anorganische Chemie, TU Bergakademie Freiberg, D-09596 Freiberg, Germany. E-mail: joerg.wagler@chemie.tu-freiberg.de; Fax: (+49) 3731 39 4058
bInstitut für Analytische Chemie, TU Bergakademie Freiberg, D-09596 Freiberg, Germany
First published on 30th January 2015
Abstract
Hexacoordinate silicon complexes L2SiX2 (L = a 2-benzoylpyrrol-1-yl derivative, X = CN, NCS) were synthesized from L2SiCl2 by ligand exchange with trimethylsilyl reagents Me3SiX. In the presence of [Tp*CuNCMe] and Me3SiCN the silicon complex L2Si(NC(CuTp*))2 was obtained, which contains a linear Cu–CN–Si–NC–Cu unit (Tp* = hydrotris(3,5-dimethylpyrazolyl)borato).
There is considerable interest in the creation and exploration of metal complexes with long-range intramolecular electronic communication.1,2 Compounds such as Prussian Blue3 or the Creutz–Taube salt,4 compounds with C/N ligand backbones (cyanide and pyrazine, respectively) served as early motivators. Further explorations of oligonuclear complexes with intramolecular communication between the metal atoms included carbon based bridging ligands such as (hetero)arenediyl groups and polyynes (e.g., in I1a and II,1bScheme 1). Hypercoordinate silicon chemistry, i.e., the chemistry of silicon compounds with Si coordination numbers greater than four, is another field of coordination chemistry which attracts the interest of various chemists.5,6 In this context, we reported electronic communication via trans-disposed ligand sites in hexacoordinate silicon complexes (e.g., III, Scheme 1), that is, inter-ligand charge-transfer between an electron rich amido ligand and an imine moiety as electron acceptor through the near linear N–Si–N moiety.7 Hence, hexacoordinate silicon complexes with suitable ambidentate ligands (e.g., C,N based ligands with conjugated π-systems) should represent another class of potential linkers for oligonuclear metal complexes which show intramolecular electronic communication. Thus, for further studies of electronic communication via trans-ligands in silicon complexes we strived for creating redox-active silicon-bridged dimetallic complexes. Only few hexacoordinate Si complexes with ligand bridges to transition metals (TM) have been reported so far (e.g., IV8 and V9). Whereas IV combines more than just two transition metals in this complex (and thus has metal–ligand–Si–ligand–metal connections in both trans and cis fashion), the compounds with exclusive trans connections published so far (like V and others with Si–O–[spacer]–TM connection10–12) seem less likely of connecting the metal atoms via π-conjugation. In the particular example of V the authors reported cyclovoltammetric features characteristic of ferrocenecarboxylate (1 peak) and the macrocyclic ligand (3 peaks) but none of a novel “Fe2” redox system in this complex.
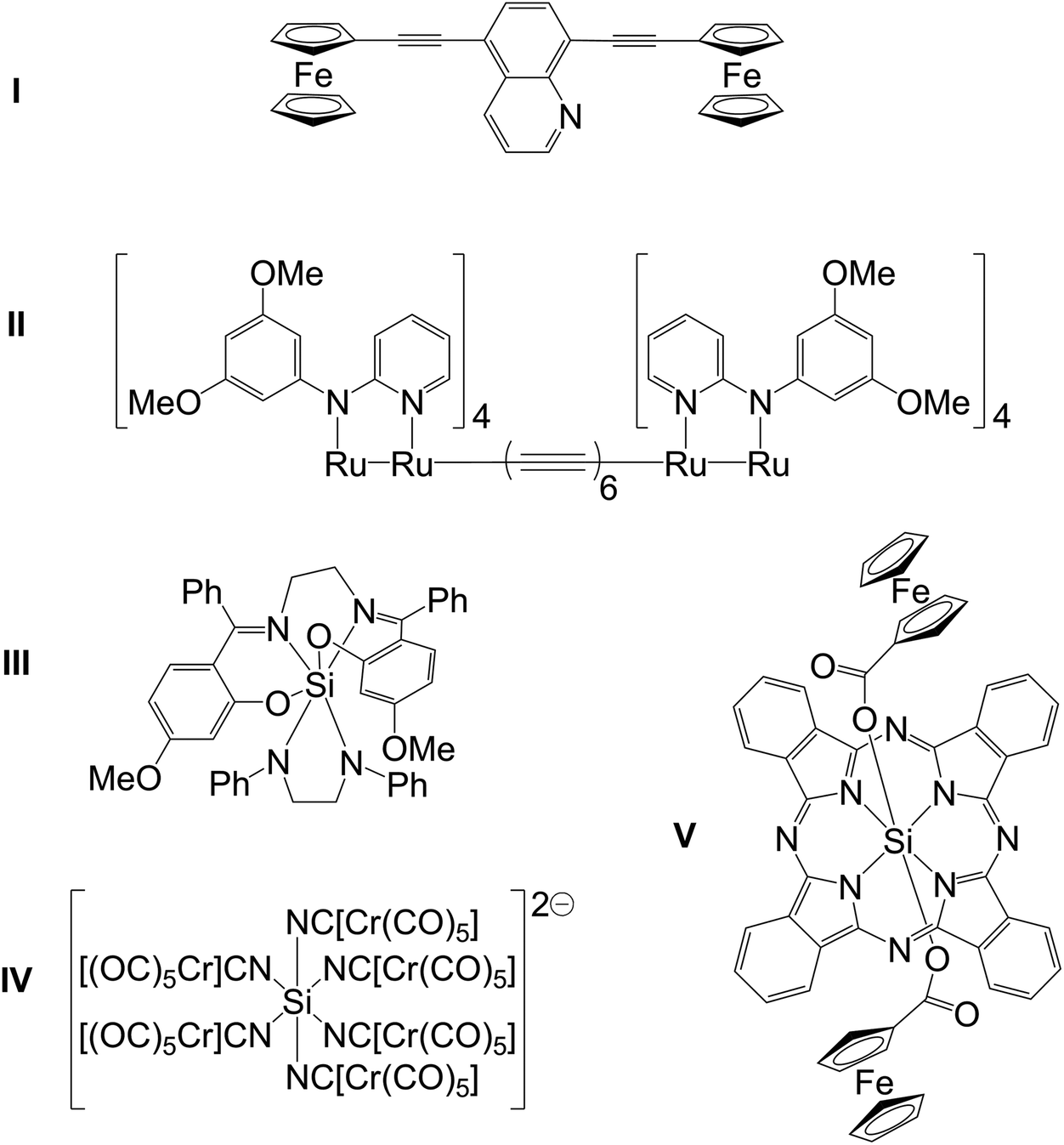 | ||
| Scheme 1 Selected hexacoordinate Si complexes. | ||
Our recently reported Si-complexes with 2-benzoylpyrrolyl scaffold offered ideal prerequisites with the tendency of forming all-trans complexes of the type L2SiX′2 (L = 2-benzoylpyrrol-1-yl, X′2 = Cl2, (OTf)2, ClPh).13 Thus, we started introducing metal complexes into the Si coordination sphere of this type of complexes via Si–N coordination.
Out of this series, the chloro complex 1 (Scheme 2, 29Si CP/MAS NMR δ = −171.0 ppm)13 was chosen as starting material for ligand exchange with Me3SiX (X = CN, NCS). These reagents, which already have proven to be suitable cyano and thiocyanato synthons in hypercoordinate silicon chemistry,14,15 afforded the desired complexes 2 and 3 (Scheme 2, 29Si CP/MAS NMR δ = −198.4 and −194.0 ppm, respectively). Their molecular structures, confirmed by single-crystal X-ray diffraction, reveal the target configuration all-trans (Fig. 1). In accord with findings of other groups, the cyanide in 2 is C-bound and the thiocyanate in 3 exhibits Si–N coordination.14,15
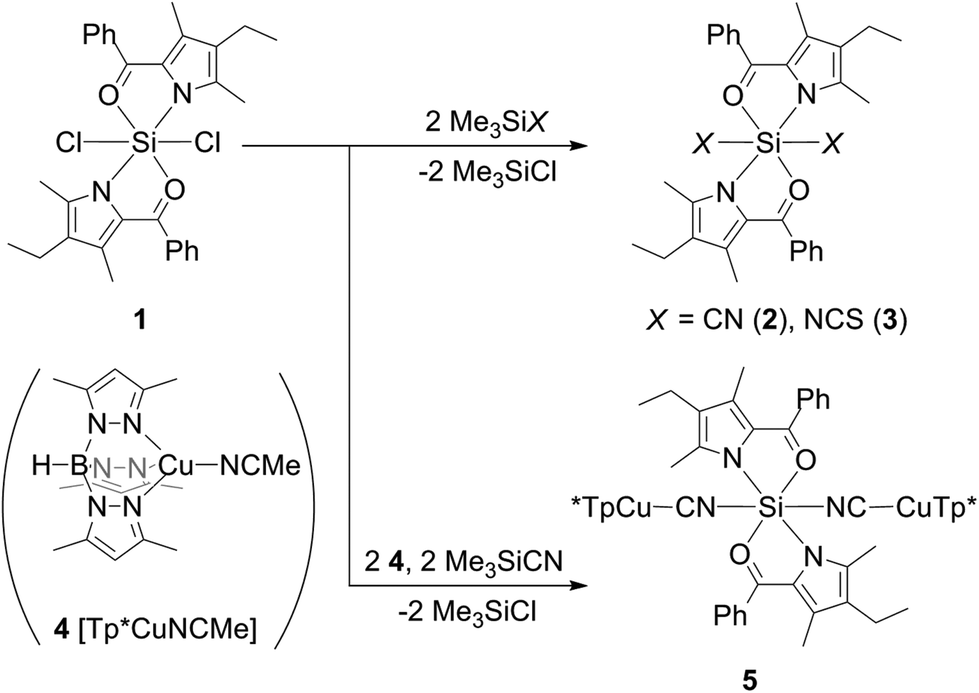 | ||
| Scheme 2 Syntheses of compounds 2, 3 and 5. | ||
 | ||
| Fig. 1 Molecular structures of (from left) 2 (in 2·(CHCl3)2) and 3 in the crystal (selected atoms are labelled, displacement ellipsoids show 50% probability, H atoms are omitted for clarity). In both cases the asymmetric unit consists of half a formula moiety, Si1 is located on a centre of inversion. Selected bond lengths [Å] and angles [°] for 2: Si1–O1 1.779(1), Si1–N1 1.877(2), Si1–C16 1.963(2), N2–C16 1.138(3); Si1–C16–N2 176.8(2); for 3: Si1–O1 1.766(1), Si1–N1 1.861(1), Si1–N2 1.859(1), N2–C16 1.163(2); S1–C16–N2 179.2(2), Si1–N2–C16 167.0(1). | ||
Cyanide and thiocyanate are known to behave as ambidentate ligands16,17 and might thus give rise to possible coordination isomerism. Whereas the Si-bound thiocyanate already exhibits the soft sulphur atom as a terminal group, suitable for binding to a soft transition metal atom, the cyanide in 2 binds to silicon via the rather soft C atom, a coordination mode which might switch to the alternative Si–N coordination as soon as a soft competitor Lewis acid is available. Thus, we analysed the alternative Si–C and Si–N coordination modes of the different octahedral isomers of 2 by quantum chemical calculations (Table 1).
| 2CN | 2NC | |
|---|---|---|
| all-trans | 0.0 (0.1) | 7.0 (11.6) |
| X,X-trans | 10.5 (8.6) | 17.9 (20.6) |
| N,N-trans | 0.1 (0.3) | 7.0 (12.0) |
| O,O-trans | 2.8 (0.4) | 9.4 (11.9) |
| all-cis | 1.9 (0.0) | 8.7 (11.7) |
They confirmed the experimentally encountered all-trans coordination with Si–C bound cyanide as an energetically favourable isomer (although only slightly lower in energy than the N,N-trans, O,O-trans and all-cis isomers), but the alternative silyl isonitrile (2NC) should be considerably less stable than the corresponding silyl cyanide complex (2CN) in all cases. For the all-trans isomers of 3 chosen as a representative example (i.e., 3NCS and 3SCN) total energy differences of 35.0/25.0 kcal mol−1 (DFT/MP2, same methods and basis sets used as for the isomers of 2) were calculated, thus highlighting the pronounced stability of 3NCS over 3SCN.
For further functionalisation of compounds 2 and 3 we considered tris(3,5-dimethylpyrazolyl)borato-Cu(I) (Tp*Cu) a suitable moiety which comprises an in general redox active soft metal centre within a stable coordinative scaffold with one vacant site, which can be occupied either by a neutral ligand (solvent) or alternatively by the dangling donor atoms of the ambidentate ligands in 2 and 3. Thus, complex [Tp*Cu-NCMe] (4, Scheme 2) with the labile acetonitrile ligand18 should serve as a convenient precursor (for its crystallographic characterisation see the ESI‡). Comparative cyclovoltammetric measurements on [Tp*Cu-NCMe] and on the corresponding Cu(II) complex [Tp*2Cu]19 have shown that [Tp*Cu-NCMe] undergoes irreversible oxidation and reduction processes in the range between −1.0 and +1.0 V. Thus, the CV features of [Tp*Cu-NCMe] are different from the cyclovoltammetric behaviour of [Tp*2Cu], which does not reveal any striking redox chemistry in the range between −2.0 and +2.0 V (for CV traces see the ESI‡). We attribute the irreversible nature of the CV characteristics of [Tp*Cu-NCMe] to the labile MeCN ligand, which might turn into reversible redox features upon introducing a less labile ligand.
For complex 3 reaction with 4·MeCN failed. Both the treatment of 3 with this copper(I) complex and the one-pot reaction of 1 and Me3SiNCS in the presence of 4·MeCN only afforded compound 3.
In contrast, the analogous one-pot reaction of 1, 4·MeCN and Me3SiCN led to the formation of complex 5 (Scheme 2). This compound reveals much better solubility in organic solvents and its formation was first indicated by a 29Si NMR signal at −197.6 ppm in toluene. Crystallisation from toluene eventually afforded single-crystals suitable for X-ray diffraction analysis (Fig. 2). Although two solvates were obtained, i.e., 5·(toluene)4 and 5·(toluene) (and a ferrocene solvate had also been obtained, vide infra), in all of them the molecular structure of 5 exhibits an octahedrally all-trans coordinated Si atom in an almost linear Cu–CN–Si–NC–Cu sequence. Crystal packing effects lead to some variations in the Si–N–C (168.3(2)–174.7(3)°) and Cu–C–N angles (171.9(2)–177.1(2)°), whereas Si–N (1.841(4)–1.866(2) Å) and Cu–C bond lengths (1.829(5)–1.851(2) Å) are very similar and C–N bond lengths (1.157(3)–1.163(5) Å) are basically identical within this set of crystallographically characterised related molecular structures.
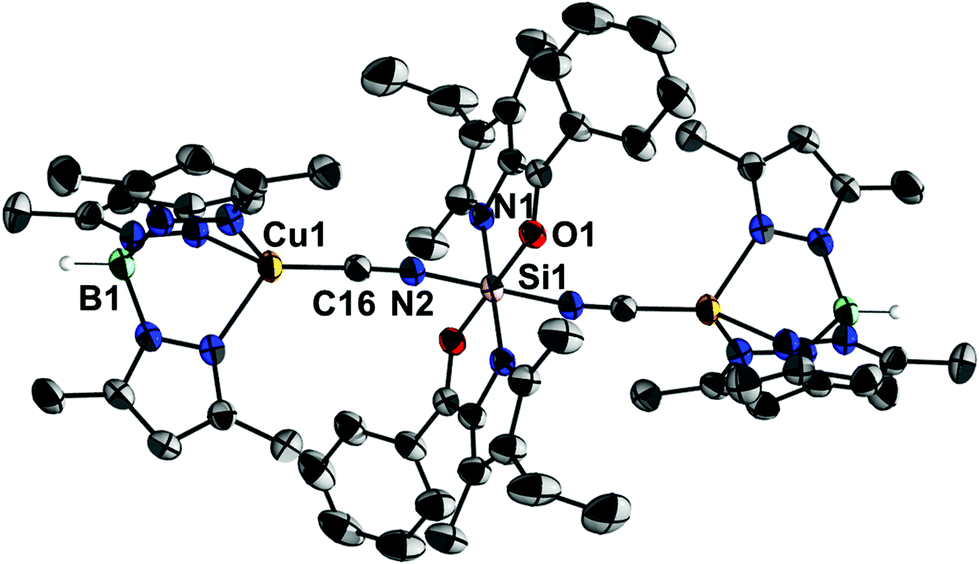 | ||
| Fig. 2 Molecular structure of 5 in the crystal of 5·(toluene)4 (selected atoms are labelled, displacement ellipsoids show 30% probability, H atoms are omitted for clarity). The asymmetric unit consists of a half formula moiety, Si1 is located on a centre of inversion. Selected bond lengths [Å] and angles [°]: Cu1–C16 1.835(2), Si1–O1 1.782(1), Si1–N1 1.866(2), Si1–N2 1.848(2), N2–C16 1.157(3); Cu1–C16–N2 177.1(2), Si1–N2–C16 168.3(2). | ||
To the best of our knowledge compound 5 is the first hexacoordinate silicon complex which holds two electron rich and redox active metal atoms connected via bridging ligands L′ capable of π-conjugation. Only two further examples of crystallographically characterised complexes of the type trans-M–CN–MM6–NC–M (where M is any metal and MM6 is a hexacoordinate group 14 metal or metalloid atom)20 were found in the literature, one of them being complex IV8 and the other one the trinuclear complex {[(PhO)3P](dppm)(CO)2Mn-CN}2SnCl4 (VI), where dppm = bis(diphenylphosphino)methane.21 Related structures for MM6 being a hexacoordinate group 13 or group 15 element have not been found.20 The C–N bond lengths in IV and VI are the same as in 5 (in both compounds 1.16 Å within the standard deviation). Furthermore, in IV the Si–N bond lengths (1.80–1.83 Å) are only slightly shorter than those in 5 and Si–N–C angles (169–177°) are variable within the same range. With respect to other hexacoordinate silicon complexes which contain the Si6–N2–C2–X moiety (i.e., containing two-coordinate N and C atom), derivatives with X = O (13 hits), S (19 hits), Se (1 hit) were found.20 The Si–N bond lengths cover the ranges of 1.7822–1.8523 Å (for X = O), 1.8115a–1.9724 Å (for X = S) and 1.89 Å (for X = Se25). The very long Si–NCS bond of 1.97 Å24 is caused by the influence of the electron releasing trans-substituent, i.e., a methyl group. This set of structures of Si6–N2–C2–X compounds reveals even greater variability of the Si–N–C angles, which cover the ranges of 180–146°26 (X = O), 180–151°24 (X = S) and 176° (X = Se25).
In order to elucidate the electronic communication between the two Cu-centres in complex 5 that are formally connected by a five atom C–N–Si–N–C “wire” we performed quantum chemical calculations on a simplified model complex 5′ (with all alkyl substituents replaced by hydrogens). The highest occupied molecular orbitals (in particular HOMO, −1, −2, −3, −4, −5, −8, −9, −10, −11) are located at both Cu atoms and their ligand atoms with equal orbital contributions, as shown for HOMO and HOMO−4 as representative examples (Fig. 3), i.e., they are indeed delocalised about the Cu–CN–Si–NC–Cu axis. To confirm this communication between the copper atoms in 5 experimentally, we aimed at generating a compound 5+ with mixed oxidation states at the copper atoms, i.e. Cu(I)/Cu(II). On a synthetic route we thus applied ferrocenium hexafluorophosphate (FcPF6, Fc+ as oxidant)27 in an equimolar ratio.
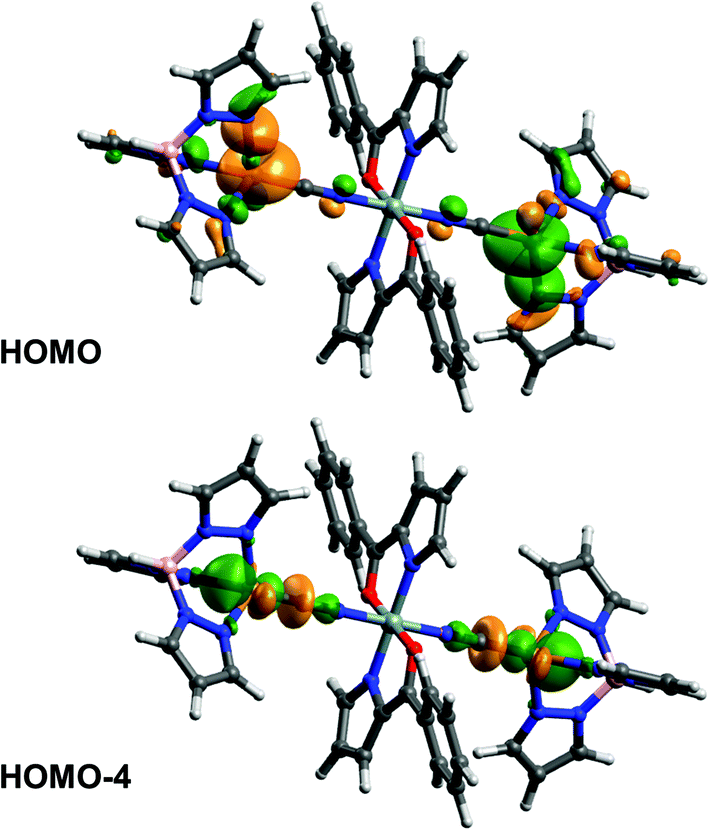 | ||
| Fig. 3 HOMO and HOMO−4 calculated for 5′. | ||
Unfortunately, we only were able to isolate some crystals of a co-crystallisate of 5 with two equivalents of ferrocene instead of the desired compound and without any hints at the fate of the potential reducing agent. Apparently, one fraction of the starting material 5 must have reacted with Fc+ in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio, and the other fraction remained intact and thus available for co-crystallisation with the ferrocene formed. For in situ monitoring of the redox process we therefore applied cyclic voltammetry (CV) to an acetonitrile solution of 10−4 M 5 (Bu4NPF6 0.1 M). The CV features of [Tp*CuNCMe] (in the range between −0.5 and +0.5 V) are absent, instead we found an irreversible process at +1.1 V. This shift of the first oxidative process to more positive potential strongly hints at the stabilisation of Cu(I) in complex 5 and, together with the irreversibility of this redox step, at the instability of a hypothetical mono-cationic complex 5+. Even though tetrahedral coordination is frequently encountered with both Cu(I) and Cu(II), the degenerate orbital situation associated with complex 5 and the degenerate SOMO resulting therefrom in case of formation of a Cu(I)/Cu(II) complex 5+ apparently gives rise to the more positive oxidation potential and supports decomposition upon oxidation.
:1 molar ratio, and the other fraction remained intact and thus available for co-crystallisation with the ferrocene formed. For in situ monitoring of the redox process we therefore applied cyclic voltammetry (CV) to an acetonitrile solution of 10−4 M 5 (Bu4NPF6 0.1 M). The CV features of [Tp*CuNCMe] (in the range between −0.5 and +0.5 V) are absent, instead we found an irreversible process at +1.1 V. This shift of the first oxidative process to more positive potential strongly hints at the stabilisation of Cu(I) in complex 5 and, together with the irreversibility of this redox step, at the instability of a hypothetical mono-cationic complex 5+. Even though tetrahedral coordination is frequently encountered with both Cu(I) and Cu(II), the degenerate orbital situation associated with complex 5 and the degenerate SOMO resulting therefrom in case of formation of a Cu(I)/Cu(II) complex 5+ apparently gives rise to the more positive oxidation potential and supports decomposition upon oxidation.
Conclusions
Ligand exchange between trimethylsilyl cyanide and complex 1 (L2SiCl2, L = O,N bidentate ligand) in the presence of the Cu(I) complex 4 (Tp*CuNCMe) furnished the hexacoordinate silicon complex 5 with an almost linear Cu–C–N–Si–N–C–Cu sequence. Although electronic communication is evident from the delocalisation of MOs about the two Cu atoms, attempts at one-electron-oxidising this “Cu2 entity” failed with decomposition of the oxidation product. In principle, our synthetic approach is promising for the generation of further oligonuclear complexes with linear M–C–N–Si–N–C–M sequence for further investigations of redox, electronic, magnetic and related properties.Experimental – general considerations
All manipulations were carried out using standard Schlenk and Glovebox techniques under an atmosphere of dry argon. Compound 1 (L2SiCl2) was prepared as reported earlier.13 Acetonitrile was purchased from Roth in an Ultra LC-MS grade quality (99.98%) and stored over activated molecular sieves 3 Å. Toluene was distilled from sodium and stored over sodium wire. CDCl3 was purchased from Armar Chemicals and stored over activated molecular sieves 3 Å. Potassium hydrotris(3,5-dimethylpyrazolyl)borate (KTp*) and CuCl were prepared according to established methods.28,29 Trimethylsilylcyanide was purchased from Aldrich and used without further purification. Trimethylsilylthiocyanate was prepared from dry NaNCS and chlorotrimethylsilane in a modified procedure related to an earlier report.30 Ferrocenium hexafluorophosphate (FcPF6) was prepared according to a literature procedure.27Quantum Chemical calculations were carried out using the Gaussian09 package.31 Molecules were optimized at the DFT MPW1PW91/6-311G(d,p) level of theory. Additional single point energy calculations for the optimized structures were performed at the MP2/6-31G(d) level. For compound 5′ population analyses were carried out after optimization using DFT MPW1PW91/6-311G(d,p) for the s and p block elements and a Stuttgart–Dresden effective core potential (SDD) for the copper atoms.
Elemental analyses were carried out with a Vario Micro Cube analyser (Elementar, Hanau, Germany). 1H, 11B, 13C and 29Si NMR spectra were recorded on a Bruker DPX 400 spectrometer or on a Bruker AVANCE 500 spectrometer. Chemical shifts are given in ppm relative to SiMe4 (as internal standard for 1H, 13C and 29Si) or BF3·Et2O (externally referenced for 11B). 29Si CP/MAS NMR spectra were recorded on a Bruker Avance 400 WB spectrometer (using a 4 mm ZrO2 rotor or a 7 mm ZrO2 rotor with KelF insert) and referenced against external Q8M8 (−109 ppm for the most upfield peak).
X-ray crystallography
Single-crystal X-ray diffraction data were collected on a Stoe IPDS-2/2 T diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Crystals were selected under an inert oil and mounted on a glass capillary which was coated with a thin layer of silicone grease. The structures were solved by direct methods (Shelxs-97)32,33 and refined with full-matrix least-squares methods (refinement of F2 against all reflections with Shelxl-97).33,34 All non-hydrogen atoms were refined anisotropically. C bound H-atoms were refined in idealised positions, the coordinates of the BH-hydrogen atoms were refined isotropically without restraints. For the cyanosilicon compounds 2·(CHCl3)2 and 5·(toluene) we have probed the Si–C (in 2) and Si–N coordination mode (in 5) by a test refinement of the cyano moiety (C16–N2) together with an alternative moiety of inverted atomic sites (N2a–C16a), same coordinates and anisotropic displacement parameters for C16 and N2a, N2 and C16a, and a free variable for the site occupancy with the alternative orientations of the CN moiety. We found, within the 3-fold standard deviation, full site occupancies for the herein reported coordination modes (0.94(3) of Si–CN in 2 and 0.99(5) Si–NC in 5).Syntheses and characterisation
1H NMR (500 MHz, 40 °C, CDCl3): δ 1.10 (t, 3J(H,H) = 7.5 Hz, 6H, CH2CH3), 2.13 (s, 6H, CH3), 2.45 (q, 3J(H,H) = 7.5 Hz, 4H, CH2CH3), 2.68 (s, 6H, CH3), 7.53–7.56 (m, 4H, m-Ph), 7.60–7.63 (m, 2H, p-Ph), 7.84–7.86 (m, 4H, o-Ph). 13C NMR (125 MHz, 40 °C, CDCl3): δ 12.4, 14.0, 14.2 (CH3), 17.9 (CH2), 128.5, 129.7 (o/m-Ph), 132.4 (p-Ph), 132.3, 133.7, 136.2, 138.6, 157.1 (i-Ph, pyr), 135.2 (CN), 169.4 (CO); 29Si NMR (99.4 MHz, 40 °C, CDCl3): δ −199.5.
29Si CP/MAS NMR (79.5 MHz, νspin = 4 kHz): δiso −198.4.
C/H/N microanalysis calcd (%) for C32H32N4O2Si: C 72.15, H 6.05, N 10.52; found: C 71.98, H 6.13, N 10.22.
A sample (ca. 20 mg) of 2 was dissolved in a mixture of chloroform (4 mL)–toluene (1 mL) upon heating. Storage at −24 °C afforded crystals of 2·(CHCl3)2 suitable for X-ray diffraction analysis.
1H NMR (500 MHz, 40 °C, CDCl3): δ 1.11 (t, 3J(H,H) = 7.7 Hz, 6H, CH2CH3), 2.11 (s, 6H, CH3), 2.44 (q, 3J(H,H) = 7.7 Hz, 4H, CH2CH3), 2.63 (s, 6H, CH3), 7.53–7.56 (m, 4H, m-Ph), 7.60–7.63 (m, 2H, p-Ph), 7.82–7.83 (m, 4H, o-Ph). 13C NMR (125 MHz, 40 °C, CDCl3): δ 12.4, 14.0, 14.2 (CH3), 17.9 (CH2), 128.6, 129.8 (o/m-Ph), 132.4 (p-Ph), 129.4, 132.1, 135.8, 138.1, 157.2 (i-Ph, pyr), 169.2 (CO), resonance signal for SCN not observed; δ29Si signal not observed due to poor solubility.
29Si CP/MAS NMR (79.5 MHz, νspin = 4 kHz): δiso −192.1.
C/H/N microanalysis calcd (%) for C32H32N4O2SiS2: C 64.40, H 5.40, N 9.39; found: C 64.14, H 5.39, N 9.43.
1H NMR (400 MHz, 23 °C, C6D6): δ 0.62 (s, 6H, CH3CN), 1.84 (s, 9H, pzCH3), 1.29 (s, 9H, pzCH3), 5.76 (s, 3H, pzH), ∼4.9 (very broad, 1H, HBpz3); 13C NMR (100.1 MHz, 23 °C, C6D6): δ 0.11 (CH3CN), 13.6 (pzCH3), 13.8 (pzCH3), 105.4 (4-pz), 116.0 (CH3CN), 144.9, 148.7 (3,5-pz).
C/H/N microanalysis calcd (%) for C19H28BN8Cu: C 48.46, H 5.99, N 23.79; found: C 48.73, H 6.31, N 23.60.
1H NMR (500 MHz, 23 °C, CDCl3): δ 1.10 (t, 3J(H,H) = 7.4 Hz, 6H, CH2CH3), 1.97 (s, 18H, pz-CH3), 2.13 (s, 6H, pyr-CH3), 2.24 (s, 18H, pz-CH3), 2.46 (q, 3J(H,H) = 7.4 Hz, 4H, CH2CH3), 2.69 (s, 6H, pyr-CH3), ∼4.5 (s, very broad, 2H, B-H), 5.51 (s, 6H, pz-H) 7.52–7.55 (m, 4H, m-Ph), 7.60–7.63 (m, 2H, p-Ph), 7.82–7.83 (m, 4H, o-Ph). 13C NMR (125 MHz, 23 °C, CDCl3): δ 12.3 (pyr-alkyl), 12.4 (pz-CH3), 13.5 (pz-CH3), 13.8, 14.3, 17.8 (pyr-alkyl), 103.7 (pz-CH), 128.5 o/m-Ph, 129.4 (o/m-Ph), 132.2, 132.2, 132.3 (o-,i-Ph, pyr), 133.0, 135.7 (pyr), 142.4, 146.7 (pz), 157.0 (pyr), 169.3 (CO) resonance signal for CN not observed; 11B NMR (160.4 MHz, 23 °C, CDCl3): δ −9.8 (br); 29Si NMR (99.4 MHz, 23 °C, CDCl3): δ −197.9.
29Si CP/MAS NMR (79.5 MHz, νspin = 5 kHz): δiso −197.5.
C/H/N microanalysis calcd (%) for C62H76B2Cu2N16O2Si·C7H8: C 61.56, H 6.29, N 16.65; found: C 61.27, H 6.58, N 16.98.
Storage of the supernatant at −24 °C afforded further crystals, this time of the composition 5·(toluene)4, which dissolved upon removal from the freezer.
Acknowledgements
This work was performed within the Cluster of Excellence “Structure Design of Novel High-Performance Materials via Atomic Design and Defect Engineering (ADDE)” that is financially supported by the European Union (European regional development fund) and by the Ministry of Science and Art of Saxony (SMWK).Notes and references
- (a) H. H. Shah, R. A. Al-Balushi, M. K. Al-Suti, M. S. Khan, C. H. Woodall, K. C. Molloy, P. R. Raithby, T. P. Robinson, S. E. C. Dale and F. Marken, Inorg. Chem., 2013, 52, 4898 CrossRef CAS PubMed; (b) Z. Cao, B. Xi, D. S. Jodoin, L. Zhang, S. P. Cummings, Y. Gao, S. F. Tyler, P. E. Fanwick, R. J. Crutchley and T. Ren, J. Am. Chem. Soc., 2014, 136, 12174 CrossRef CAS PubMed.
- E. Goto, R. A. Begum, S. Zhan, T. Tanase, K. Tanigaki and K. Sakai, Angew. Chem., Int. Ed., 2004, 43, 5029 CrossRef CAS PubMed.
- H. J. Buser, D. Schwarzenbach, W. Petter and A. Ludi, Inorg. Chem., 1977, 16, 2704 CrossRef CAS.
- (a) C. Creutz and H. Taube, J. Am. Chem. Soc., 1969, 91, 3988 CrossRef CAS; (b) U. Fürholz, S. Joss, H. B. Bürgi and A. Ludi, Inorg. Chem., 1985, 24, 943 CrossRef.
- For reviews on hypercoordinate silicon compounds see, for example: (a) J. Wagler, U. Böhme and E. Kroke, in Structure and Bonding, ed. D. Scheschkewitz, Springer, 2014, vol. 155, p. 29 Search PubMed; (b) W. Levason, G. Reid and W. Zhang, Coord. Chem. Rev., 2011, 255, 1319 CrossRef CAS; (c) D. Kost and I. Kalikhman, Acc. Chem. Res., 2009, 42, 303 CrossRef CAS PubMed; (d) R. Tacke and O. Seiler, in Silicon Chemistry, ed. P. Jutzi and U. Schubert, Wiley-VCH, Weinheim, 2003, p. 324 Search PubMed; (e) C. Chuit, R. J. P. Corriu and C. Reye, in Chemistry of Hypervalent Compounds, ed. K. Akiba, Wiley-VCH, Weinheim, 1999, p. 81 Search PubMed; (f) D. Kost and I. Kalikhman, in The Chemistry of Organo Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, John Wiley & Sons Ltd, Chichester, 1998, vol. 2, p. 1339 Search PubMed.
- For recent contributions on hypercoordinate silicon chemistry see, for example: (a) J. Weiß, K. Sinner, J. A. Baus, C. Burschka and R. Tacke, Eur. J. Inorg. Chem., 2014, 475 CrossRef; (b) J. Weiß, B. Theis, J. A. Baus, C. Burschka, R. Bertermann and R. Tacke, Z. Anorg. Allg. Chem., 2014, 640, 300 CrossRef; (c) S. Jähnigen, E. Brendler, U. Böhme, G. Heide and E. Kroke, New J. Chem., 2014, 38, 744 RSC; (d) B. M. Kraft and W. W. Brennessel, Organometallics, 2014, 33, 158 CrossRef CAS; (e) M. Yamamura, M. Albrecht, M. Albrecht, Y. Nishimura, T. Arai and T. Nabeshima, Inorg. Chem., 2014, 53, 1355 CrossRef CAS PubMed; (f) E. Wächtler, A. Kämpfe, K. Krupinski, D. Gerlach, E. Kroke, E. Brendler and J. Wagler, Z. Naturforsch., B: Chem. Sci., 2014, 69, 1402 CrossRef; (g) S. Wahlicht, E. Brendler, T. Heine, L. Zhechkov and J. Wagler, Organometallics, 2014, 33, 2479 CrossRef CAS; (h) K. Junold, J. A. Baus, C. Burschka, M. Finze and R. Tacke, Eur. J. Inorg. Chem., 2014, 5099 CrossRef CAS; (i) K. Junold, M. Nutz, J. A. Baus, C. Burschka, C. Fonseca Guerra, F. M. Bickelhaupt and R. Tacke, Chem. – Eur. J., 2014, 20, 9319 CrossRef CAS PubMed; (j) C. Peureux and V. Jouikov, Chem. – Eur. J., 2014, 20, 9290 CrossRef CAS PubMed; (k) T. Breiding, J. Henker, C. Fu, Y. Xiang, S. Glöckner, P. Hofmann, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2014, 2924 CrossRef CAS; (l) H. Kameo, T. Kawamoto, S. Sakaki, D. Bourissou and H. Nakazawa, Organometallics, 2014, 33, 6557 CrossRef CAS; (m) N. Kano, K. Sasaki, H. Miyake and T. Kawashima, Organometallics, 2014, 33, 2358 CrossRef CAS.
- (a) J. Wagler, D. Gerlach, U. Böhme and G. Roewer, Organometallics, 2006, 25, 2929 CrossRef CAS; (b) J. Wagler and G. Roewer, Inorg. Chim. Acta, 2007, 360, 1717 CrossRef CAS.
- M. Fritz, D. Rieger, E. Bär, G. Beck, J. Fuchs, G. Holzmann and W. P. Fehlhammer, Inorg. Chim. Acta, 1992, 198–200, 513 CrossRef CAS.
- J. Silver, J. L. Sosa-Sanchez and C. S. Frampton, Inorg. Chem., 1998, 37, 411 CrossRef CAS PubMed.
- M. T. M. Choi, C.-F. Choi and D. K. P. Ng, Tetrahedron, 2004, 60, 6889 CrossRef CAS.
- W. Kläui, N. Mocigemba, A. Weber-Schuster, R. Bell, W. Frank, D. Mootz, W. Poll and H. Wunderlich, Chem. – Eur. J., 2002, 8, 2335 CrossRef.
- U. Kölle, T. Rüther, N. Le Narvor, U. Englert and W. Kläui, Angew. Chem., Int. Ed. Engl., 1994, 33, 991 CrossRef.
- A. Kämpfe, E. Brendler, E. Kroke and J. Wagler, Chem. – Eur. J., 2014, 20, 9409 CrossRef PubMed.
- I. Kalikhman, B. Gostevskii, E. Kertsnus, M. Botoshansky, C. A. Tessier, W. J. Youngs, S. Deuerlein, D. Stalke and D. Kost, Organometallics, 2007, 26, 2652 CrossRef CAS.
- (a) O. Seiler, R. Bertermann, N. Buggisch, C. Burscka, M. Penka, D. Tebbe and R. Tacke, Z. Anorg. Allg. Chem., 2003, 629, 1403 CrossRef CAS; (b) J. A. Baus, C. Burschka, R. Bertermann, C. Fonseca Guerra, F. M. Bickelhaupt and R. Tacke, Inorg. Chem., 2013, 52, 10664 CrossRef CAS PubMed; (c) S. Metz, B. Theis, C. Burschka and R. Tacke, Chem. – Eur. J., 2010, 16, 6844 CrossRef CAS PubMed.
- J. Wang, Z.-R. Sun, L. Deng, Z.-H. Wei, W.-H. Zhang, Y. Zhang and J.-P. Lang, Inorg. Chem., 2007, 46, 11381 CrossRef CAS PubMed.
- D. L. Smith and V. I. Saunders, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1981, 37, 1807 CrossRef.
- H.-C. Liang, E. Kim, C. D. Incarvito, A. L. Rheingold and K. D. Karlin, Inorg. Chem., 2002, 41, 2209 CrossRef CAS PubMed.
- S. Trofimenko, J. Am. Chem. Soc., 1967, 89, 3170 CrossRef CAS.
- ConQuest 1.16 (released December 2013) Search of the Cambridge crystal structure database (CSD).
- K. M. Anderson, N. G. Connelly, N. J. Goodwin, G. R. Lewis, M. T. Moreno, A. G. Orpen and A. J. Wood, Dalton Trans., 2001, 1421 RSC.
- P. Portius and M. Davis, Dalton Trans., 2010, 39, 527 RSC.
- S. Metz, C. Burschka and R. Tacke, Chem. – Asian J., 2009, 4, 581 CrossRef CAS PubMed.
- J. Wagler, Private Communication to the Cambridge Structural Database, 2009, CCDC 733464 Search PubMed.
- W. Heininger, K. Polborn and G. Nagorsen, Z. Naturforsch., B: Chem. Sci., 1988, 43, 857 CrossRef CAS.
- S. Cota, M. Beyer, R. Bertermann, C. Burschka, K. Götz, M. Kaupp and R. Tacke, Chem. – Eur. J., 2010, 16, 6582 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- M. Bochmann, in Inorganic Experiments, ed. J. D. Woollins, VCH Weinheim, Germany, 1994, p. 28 Search PubMed.
- G. Pizarro and C. Fighetti, Rev. Ing. Quim., 1947, 7, 51 Search PubMed.
- British Thomson-Houston Ltd. GB19480015570 19480609, Patent 643941 (A), 1950.
- M. J. Frisch, et al., GAUSSIAN 09, Revision B.01, Gaussian, Inc., Wallingford, CT, 2010 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for the Solution of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELXL-97, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
Footnotes |
| † Dedicated to Prof. Gerhard Roewer on the occasion of his 75th birthday. |
| ‡ Electronic supplementary information (ESI) available: Cartesian coordinates and energies of the optimized molecular structures of the isomers of 2 listed in Table 1 and of all-trans3NCS and 3SCN, graphical representations and energies of the higest occupied MOs calculated for 5′, CV traces of [Tp*Cu(NCMe)], [Tp*2Cu] and 5, tables with parameters of data collection and structure refinement of the crystal structures reported in this paper and CIF. CCDC 999708 (2·(CHCl3)2), 999703 (3), 999704 (4·MeCN), 999705 (5·(ferrocene)2), 999706 (5·toluene) and 999707 (5·(toluene)4). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03903c |
| This journal is © The Royal Society of Chemistry 2015 |