
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCyclometallated gold(III) aryl-pyridine complexes as efficient catalysts for three-component synthesis of substituted oxazoles†
Henrik
von Wachenfeldt
,
Alexey V.
Polukeev
,
Nagarajan
Loganathan
,
Filip
Paulsen
,
Philipp
Röse
,
Marion
Garreau
,
Ola F.
Wendt
and
Daniel
Strand
*
Centre for Analysis and Synthesis, Department of Chemistry, Lund University, Box 124, 221 00 Lund, Sweden. E-mail: daniel.strand@chem.lu.se
First published on 9th February 2015
Abstract
Cyclometallated aryl-pyridine gold(III) complexes are shown to be efficient catalysts for the multicomponent reaction between N-benzyl imines, alkynes, and acyl chlorides to form trisubstituted oxazoles. The reaction typically proceeds in good yields (up to over 80%) and short reaction times (∼15 minutes). The high stability of the investigated cyclometallated catalysts enables a retained efficiency for this reaction in terms of rate and yield using as little as 0.5 mol% catalyst, a reduction by an order of magnitude compared to previously used Au(III)–salen complexes. An attractive feature of the present catalytic system is that active catalysts can be formed from simple pre-catalysts under the reaction conditions. Both cyclometallated and non-cyclometallated complexes were characterized in the solid state by single crystal X-ray diffraction.
Introduction
Gold catalysis has been the subject of a growing interest over the past two decades. For applications in homogenous catalysis, these efforts have been dominated by reports relying on Au(I).1 Studies on Au(III) catalysis are more sparse and have primarily focused on the application of simple salts such as AuCl3, and AuBr3.2 In more recent contributions, also Au(III) complexes with organic ligands such as salen,3 N-heterocyclic carbenes,4 phosphines,5 picolinate,6 and tethered aryl-pyridines,7,8 have been investigated. The role of the ligands in such systems however remains underexplored and it is often unclear whether the ligated system is the actual catalyst or if it primarily serves a precursor for more active species formed under the reaction conditions.9 To this end, the straightforward access to structural variants of aryl-pyridine ligands constitutes an attractive entry to tuning the catalytic properties of cyclometallated Au(III) salts for use in carbon–carbon bond forming reactions such as A3 couplings between alkynes, aldehydes and amines,10 and multicomponent processes to form aromatic heterocycles (Fig. 1).11,12 | ||
| Fig. 1 Three-component reactions to form propargyl amines or oxazoles under AuIII catalysis. | ||
In particular, the previously demonstrated efficiency of cyclometallated 2-benzylpyridine-(bnpy)AuCl23 in the A3 coupling10d drew our attention as variations on this theme would be of value also in the context of our recently reported gold catalyzed synthesis of tri-substituted oxazoles from imines, alkynes and acyl chlorides.3 Hence, herein we present an investigation of a series of cyclometallated Au(III) complexes and their non-cyclometallated precursors as catalysts for this transformation. Compared to the previously used N,N′-ethylenebis(salicylimine)-(salen) AuPF6 or AuCl3, the more stable cyclometallated complexes (e.g. (bnpy)AuCl23) enabled a reduction of the catalyst loading by a factor of ten with retained high yields and short reaction times for a number of substrate combinations. An additional attractive feature of this system is that a simple pre-catalyst, the non-cyclometallated precursor of (bnpy)AuCl23, (bnpyH)AuCl32, readily formed in minutes from commercially available KAuCl4 and 2-benzylpyridine (1), can be used without significant loss of efficiency, presumably as cyclometallation occurs, at least in part, under the reaction conditions. A series of aryl-pyridine complexes with varying tether lengths and electronic properties of the aryl moiety were also investigated and gave similar results in terms of yield; qualitatively a methylene bridge between the aryl and pyridyl moieties of the ligand was beneficial for catalysis, and electron-withdrawing substituents on the aryl moiety gave a marginally reduced efficiency compared to using 2-phenylpyridine (4a) as ligand.
Results and discussion
Synthesis and characterisation of cyclometallated gold catalysts
A series of Au(III) complexes were selected for investigation as catalysts and synthesized as shown in Scheme 1. The previously known LAuCl3 complexes (bnpyH)AuCl32 and 2-phenylpyridine-(ppyH) AuCl35a as well as a new complex, 2-(2,4-difluorophenyl)pyridine-(dfppyH) AuCl35b were all prepared from KAuCl4 and the respective pyridine ligands in mixtures of MeCN and H2O (Scheme 1) using a modification of the literature procedure for the synthesis of 2.13 Complexes 2 and 5a were subsequently cyclometallated in water using microwave heating as reported by Shaw et al.14 to give the known complexes (bnpy)AuCl23 and (ppy)AuCl26a respectively. Pleasingly, this method could also be extended to cyclometallation of 5b to give the known (dfppy)AuCl26b. Compared to the previously described transmetallation procedure for the synthesis of this complex,15 the present procedure provided a slightly higher yield conveniently avoiding toxic mercury salts.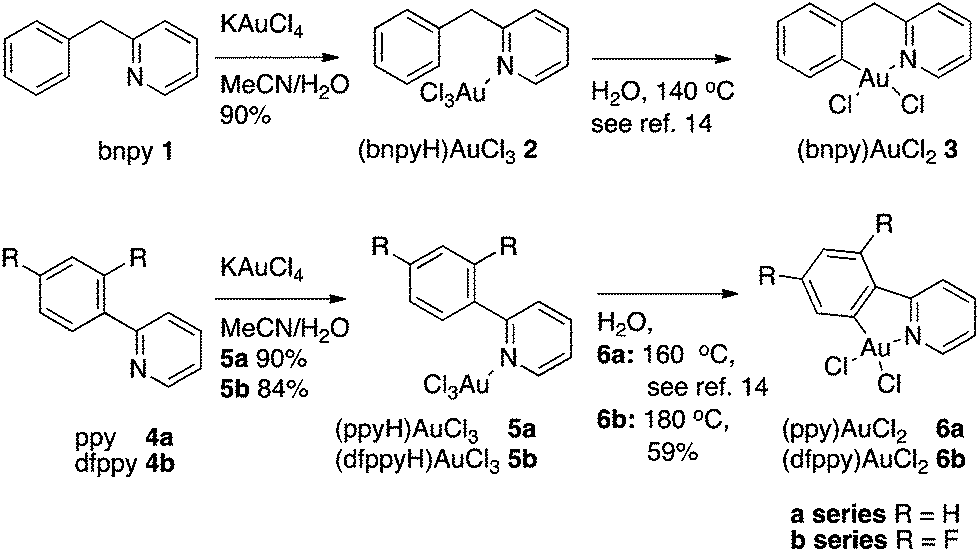 | ||
| Scheme 1 Synthesis of Au-pyridine complexes 2, 3, 5a/b, and 6a/b. | ||
Mechanistically, we interpret the cyclometallation processes for this class of compounds as initiated by a nucleophilic attack of the aryl moiety on the electron deficient gold center, similar to that seen in electrophilic aromatic substitution reactions, followed by an re-aromatization of the ring with an overall loss of HCl. This is supported by a recent contribution from Wendt and co-workers wherein naphthylpyridine-Au(III) salts were shown to exclusively cyclometalate in the more nucleophilic naphthyl 8-position rather than in the 2-position;16 a complete reversal in selectivity compared with for instance Pd(II) mediated C–H activation of naphthylpyridine.17
The 1H NMR spectrum of (dfppyH)AuCl35b reveals the expected seven resonances with well-resolved long-range couplings; the resonance from H5 of the pyridine ring is shifted downfield (9.39 ppm); consistent with coordination of nitrogen to AuCl3. In the 13C{1H} NMR spectrum, all carbons of the phenyl ring are split by the two non-equivalent fluorine atoms.
Single crystal XRD structures of (bnpy)AuCl23, (bnpyH)AuCl32, (dfppy)AuCl26b and (dfppyH)AuCl35b have not been described previously.18 Pleasingly, single crystals of these compounds suitable for XRD analysis could be grown by slow evaporation of acetonitrile (3, 2, and 6b) or acetone (5b) solutions. The respective molecular structures are given together with crystallographic data in Table 1.
|
|
||||
|---|---|---|---|---|
| Compound | (bnpy)AuCl23 | (bnpyH)AuCl32 | (dfppy)AuCl26b | (dfppyH)AuCl35b |
| Crystal data | ||||
| Chemical formula | C12H10AuCl2N | C12H11AuCl3N | C11H6AuCl2F2N | C11H7AuCl3F2N |
| M r | 436.08 | 472.53 | 458.03 | 494.49 |
| Crystal system, space group | Monoclinic, P21/n | Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Monoclinic, P21/c | Monoclinic, P21/c |
| Temperature (K) | 293 | 293 | 293 | 293 |
| a, b, c (Å) | 8.0841 (5), 8.5351 (4), 17.4924 (11) | 7.816 (5), 8.605 (5), 11.558 (5) | 8.058 (6), 16.703 (2), 9.025 (5) | 13.139 (4), 7.796 (6), 13.139 (3) |
| α, β, γ (°) | 90, 91.388 (6), 90 | 85.425 (5), 72.501 (5), 71.714 (5) | 90, 106.149 (3), 90 | 90, 100.440 (11), 90 |
| V (Å3) | 1206.60 (12) | 703.8 (7) | 1166.8 (11) | 1323.6 (11) |
| Z | 4 | 2 | 4 | 4 |
| Radiation type | Mo Kα | Mo Kα | Mo Kα | Mo Kα |
| μ (mm−1) | 12.6 | 11 | 13.07 | 11.72 |
| Crystal size (mm) | 0.14 × 0.14 × 0.12 | 0.23 × 0.18 × 0.13 | 0.18 × 0.17 × 0.12 | 0.33 × 0.28 × 0.19 |
| Data collection | ||||
| Absorption correction | Multi-scan | Multi-scan | Multi-scan | Multi-scan |
| T min, Tmax | 0.271, 0.313 | 0.187, 0.329 | 0.202, 0.303 | 0.113, 0.214 |
| No. of measured, independent and observed [I > 2σ(I)] reflections | 23878, 2610, 2179 | 5281, 2969, 2722 | 8421, 2540, 1870 | 26607, 2863, 2514 |
| R int | 0.126 | 0.014 | 0.069 | 0.101 |
| Refinement | ||||
| R[F2 > 2σ(F2)], wR(F2), S | 0.078, 0.194, 1.15 | 0.017, 0.042, 1.02 | 0.053, 0.128, 1.02 | 0.032, 0.079, 1.02 |
| No. of reflections | 2610 | 2969 | 2540 | 2863 |
| No. of parameters | 145 | 154 | 154 | 163 |
| No. of restraints | 0 | 0 | 0 | 0 |
| Δρmax, Δρmin (e Å−3) | 4.42, −1.92 | 1.03, −0.62 | 3.83, −1.21 | 1.59, −2.31 |
| CCDC | 1022615 | 1022616 | 1022617 | 1022618 |
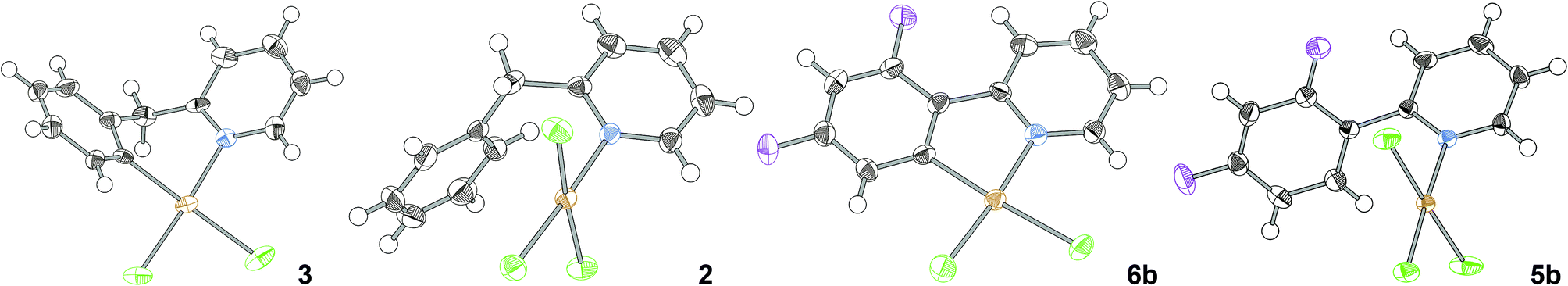
All structures display distorted square-planar geometries with the expected cyclometallation seen in (bnpy)AuCl23 and (dfppy)AuCl26b. In the LAuCl3 crystals, the coordinated pyridine is almost perpendicular to the coordination plane. In the cyclometallated complex (bnpy)AuCl23, the six-membered metallacycle forces the ligand out of the coordination plane with a highly puckered conformation as a result. On the other hand, the constraints imposed by the five member metallacycle in (dfppy)AuCl26b result in the ligand adapting a co-planar orientation with the coordination plane giving an almost perfectly planar molecule similarly to what was found in the structure of the parent phenylpyridine complex 6a.18b Compounds 6a and 6b are thus set-up for a strong π-interaction between ligand and metal, which is reflected in the substantially shorter Au–N and Au–C distances in these structures compared to the corresponding distances in (bnpy)AuCl23 (cf.ref 18b). The planarity of (dfppy)AuCl26b also forces the C–Au–N angle to below 90° with an increase of the corresponding Cl–Au–Cl angle as a result, whereas in (bnpy)AuCl23 the bond angles around gold are closer to the ideal.
Catalytic performance of various cyclometallated Au complexes and their pre-catalysts in oxazole synthesis
We recently reported that a cationic Au(III)–salen complex catalyzes the addition of terminal alkynes to in situ generated acyl iminium ions in an event that triggers a cycloisomerization domino reaction that ultimately results in oxazoles as products.19 The merger of the otherwise incompatible A3 coupling and cycloisomerization manifolds into a single domino process is enabled by the loss of a sacrificial benzyl group on the imine nitrogen.20 Synthetically, the method is attractive, as it generates building blocks for ligands and bioactive structures in a single step from simple, often commercially available, components.21 The cyclometallated Au-complexes 3, 6a/b as well as their non-cyclometallated homologs 2, 5a/b were investigated as catalysts for this transformation (Table 2). The latter class of pre-catalysts are particularly interesting in this context as in situ cyclometallation under the reaction conditions would release a small amount of HCl, which should facilitate oxazole formation by promoting both the debenzylation and cycloisomerization steps in the reaction mechanism. The catalytic performance was benchmarked using the reaction between N-Bn imine 8a, benzoyl chloride (9a), and phenyl acetylene (10a) to form oxazole 11a. For comparative purposes, commercially available pyridine (py)AuCl3722 was also included as catalyst in the study.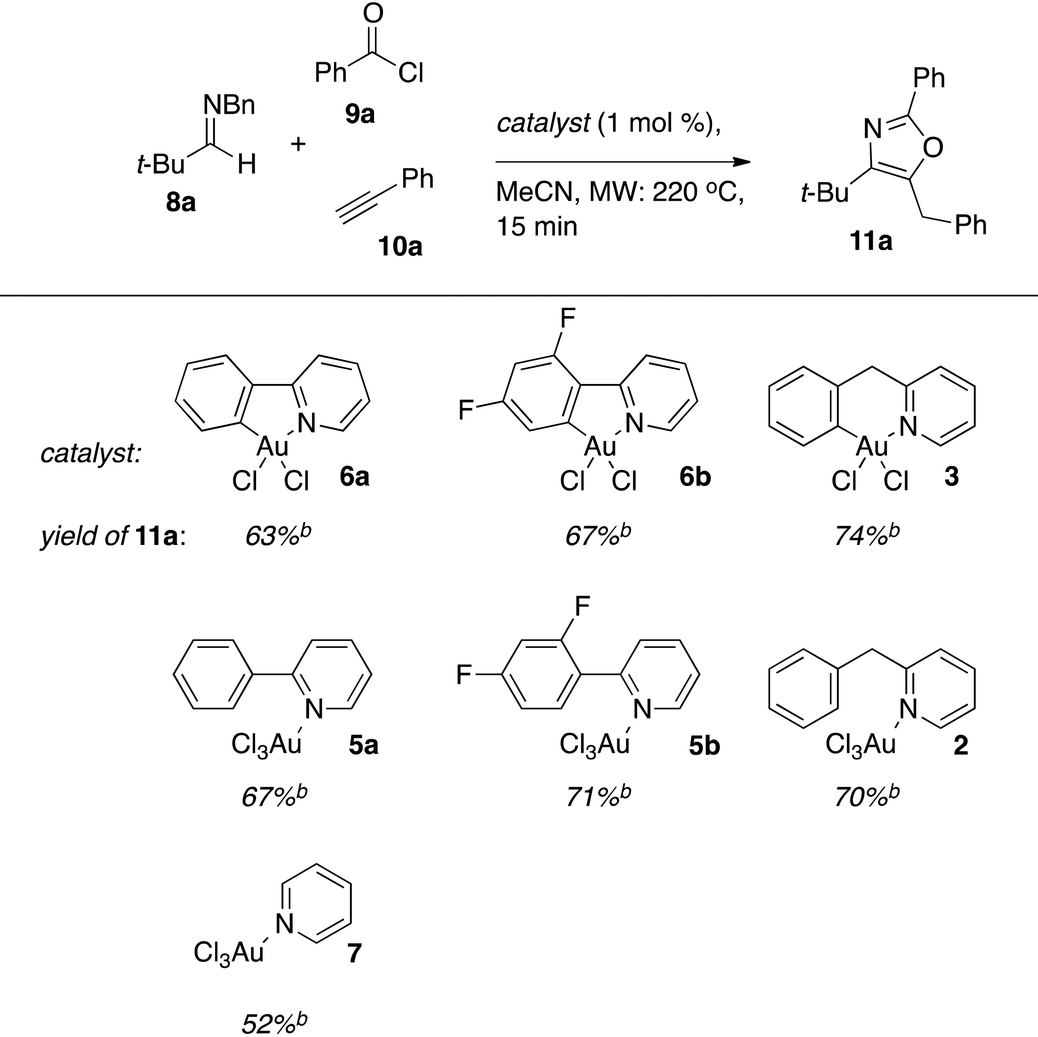
The cyclometallated aryl-pyridine complexes 3 and 6a/b gave comparable yields although a slight advantage was found for the (bnpy)AuCl23. This is in line with the earlier observation, that six-membered Au-metallacycles are more efficient than five-membered in the A3-coupling.10d It is noteworthy that each of the non-cyclometallated catalysts 2 and 5a/b gave similar results to that of the corresponding cyclometallated structures, which suggested that the active species in catalysis were the same in each pair of catalysts.
When employing (bnpyH)AuCl32 as a pre-catalyst, we were not able to detect the corresponding cyclometallated structure (bnpy)AuCl23 in the crude reaction mixture by ESI-MS or 1H NMR. However, this is not surprising, as (bnpy)AuCl23 could be shown to decompose during the course of the reaction (15 min, 240 °C) (vide infra). We also observed that although we were unable to detect 3 in the reaction mixture, addition of additional starting material after a complete catalysis reaction, followed by re-heating of the resulting mixture to 240 °C for 15 minutes gave an additional 65% yield of oxazole 11a (based on the second addition), demonstrating that the mixture was still catalytically active. An interpretation of this result is that either small amounts of highly active 3 remains in the mixture and/or that decomposition occurs at high temperatures to produce new active species such as gold nanoparticles that can contribute to catalysis. The latter notion is supported by the observed deposition of metallic gold in catalysis experiments using high catalyst loadings of 3 (>10 mol%).
In catalysis experiments using the non-cyclometalated complex 2 in acetonitrile-d3, the 1H NMR spectra indicated ligand exchange upon addition of the acyl chloride/imine components. To investigate whether cyclometallation is possible under the reaction conditions, (bnpyH)AuCl32 was heated to 240 °C in acetonitrile-d3. After 1 minute of heating, formation of ∼10 mol% of the cyclometallated complex 3 was indeed observed in the 1H NMR spectrum of the mixture, along with uncyclized precursor 2. After an additional three minutes of heating however, only trace amounts of 3 remained, and after 15 minutes, no signals attributed to complex 3 could be detected.
We note however that a marked difference in efficiency remains between the pre-catalysts that can cyclometallate ((ppy)AuCl35a, (dfppy)AuCl35b, and (bnppy)AuCl32) and (py)AuCl37, which gives oxazole 11a in 52% yield in the benchmark reaction. Cyclometallation under the reaction conditions thus appears viable, but the precise nature of the actual active catalytic species under the reaction conditions used remains ambiguous.
As the (bnpy)AuCl23 gave the best results in catalysis, oxazole formation was optimized for this complex (Table 3). An increase in the temperature to 240 °C was found to be beneficial giving a yield of 78% of oxazole 11a. It is noteworthy that at this temperature, as little as 0.25 mol% catalyst could be used and still allow for formation of 11a in 72% yield. The reaction also proceeded neat and in process-friendly 2-Me-THF with this catalyst, but with a lower yield as a result.
|
|
|||||
|---|---|---|---|---|---|
| Entry | T/°C | Catalyst-loading/mol% | t/min | Solvent | Yieldb/% |
| a General conditions: reactions run on a 1.0 mmol scale (imine 8a) with 1.0 equiv. benzoyl chloride (9a), 2.0 equiv. phenyl acetylene (10a), and 0.5 mol% of (bnpy)AuCl23 using microwave heating (240 °C, 15 min). b Measured by 1H NMR spectroscopy using mesitylene as an internal standard. | |||||
| 1 | 240 | 1 | 15 | MeCN | 78 |
| 2 | 250 | 1 | 15 | MeCN | 78 |
| 3 | 240 | 0.5 | 15 | MeCN | 77 |
| 4 | 240 | 0.25 | 15 | MeCN | 70 |
| 5 | 240 | 0.25 | 30 | MeCN | 72 |
| 6 | 240 | 0.5 | 20 | MeCN | 77 |
| 7 | 240 | 0.5 | 15 | Neat | 54 |
| 8 | 225 | 0.5 | 15 | 2-Me-THF | 53 |

Catalytic performance of (dfppy)AuCl26b and (dfppyH)AuCl35b in the A3 coupling
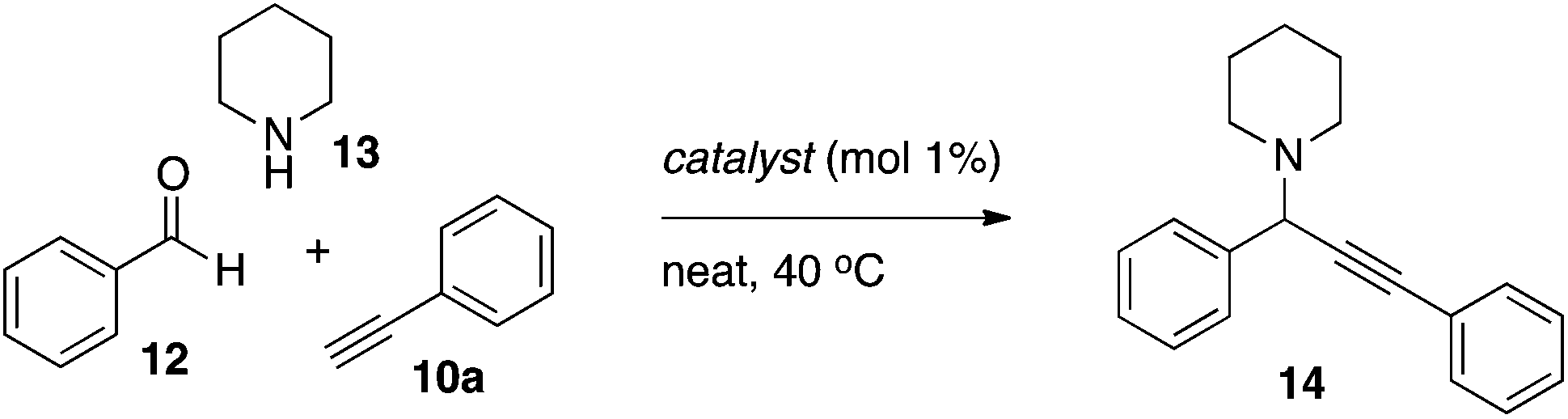
Since the new fluorinated catalysts (dfppy)AuCl26b and (dfppyH)AuCl35b gave oxazole product in the three-component reaction, these complexes were also evaluated in the parent A3 coupling under solvent-free conditions (Table 4). Both the cyclometallated complex and its non-cyclometallated counterpart gave clean formation of propargyl amine 14 using 1 mol% catalyst at 40 °C. The yield using the non-cyclometallated catalyst 5b was essentially quantitative which is in line with what is found for (py)AuCl3 or AuCl3 under the same conditions. An immediate color change was seen upon addition of the amine component in these experiments suggesting a ligand exchange, and that (dfppyH)AuCl35b acts primarily as a pre-catalyst. The (dfppy)AuCl26b was also active in catalysis, however the isolated yield was lower than that reported for (bnpy)AuCl23 (83%)10d in the same transformation.|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb/% |
| a General conditions: (a) reactions run at 40 °C using 1.0 mol% of the catalyst indicated on a 1.0 mmol scale (aldehyde), with 1.5 equiv. alkyne and 1.1 equiv. amine. The reactions were run for up to 24 hours or to full consumption of aldehyde (1H NMR control). b Measured by 1H NMR spectroscopy of the reaction crude using mesitylene as an internal standard. | |||
| 1. | (dfppyH)AuCl35b | — | 100 |
| 2. | (dfppy)AuCl26b | — | 71 |
Substrate scope for oxazole formation catalyzed by (bnpy)AuCl23
Outgoing from the optimized conditions for oxazole formation with (bnpy)AuCl23, the substrate scope was investigated by varying the imine, alkyne and acyl chloride components.Under these conditions, a series of acyl chlorides including heterocyclic-, aliphatic- and aryl substituted examples all competently participated in oxazole formation with moderate to good yields as a result (Table 5). A catalyst loading of 0.5 mol% was sufficient to attain full conversion of the starting materials in each case as evident by 1H NMR spectroscopy of the crude reaction mixtures. The good turn over numbers with (bnpy)AuCl23 as the catalyst extended also to reactions with varying alkyne and imine components (Table 6). Similarly to what was previously seen with the Au–salen system, quaternary substitution at the α-position of the imine was however necessary for efficient reactions. Aromatic imines give at most traces of isolatable oxazole products with the majority of the mass after the reaction comprising unspecific decomposition. A difference compared to the Au–salen system is that lower yields are consistently obtained with linear aliphatic alkynes (Table 6, entries 8 and 10). The reason for this difference remains unclear.
| a General conditions: reactions run on a 1.0 mmol scale (imine 8a) with 1.0 equiv. acyl chlorides 9b–f, 2.0 equiv. phenyl acetylene (10a), and 0.5 mol% of (bnpy)AuCl23 using microwave heating (240 °C, 15 min). |
|---|
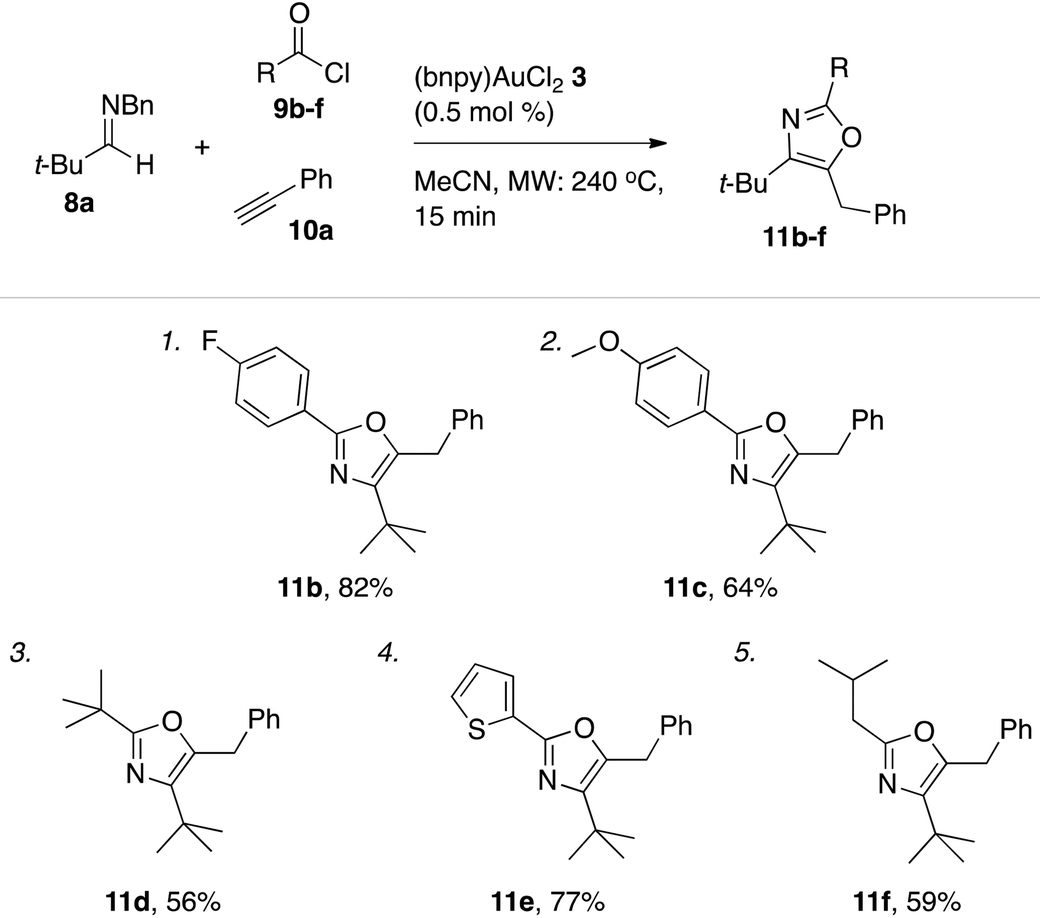
|
| a General conditions: reactions run on a 1.0 mmol scale (imines 8a–d) with 1.0 equiv. benzyl chloride 9a, 2.0 equiv. alkynes 10a–h, and 0.5 mol% of catalyst (bnpy)AuCl23 using microwave heating (240 °C, 15 min). b The yields of oxazole 11m and 11p was measured by 1H NMR spectroscopy using mesitylene as an internal standard. |
|---|
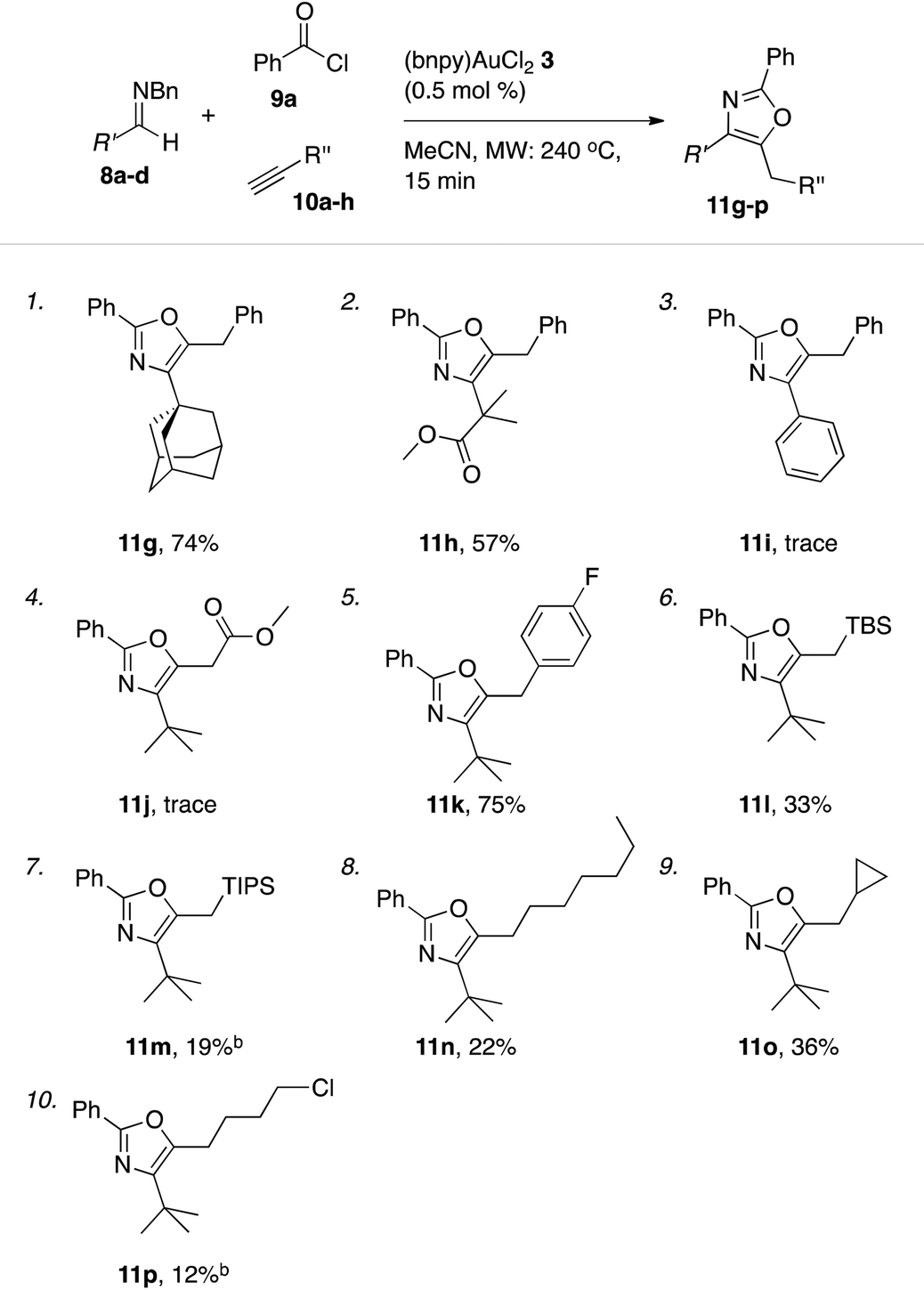
|
Conclusions
In conclusion, a series of cyclometallated aryl-pyridine Au(III) complexes have been shown to efficiently catalyze oxazole formation in the three-component reaction between N-Bn imines, alkynes, and acyl chlorides. In particular, (bnpy)AuCl23 enables the use of down to 0.25 mol% of catalyst with good yields of the oxazole products in 15 minutes. The conditions are amenable to the synthesis of oxazoles varying in substitution at all positions (15 examples demonstrated) using down to one-tenth of the catalyst loading needed for the previously employed Au–salen complex. Importantly, as the reaction occurs under elevated temperatures, readily available non-cyclometallated pre-catalysts can also be exploited in this reaction without a significant loss of efficiency. Further studies on applications of this methodology are under way and will be reported in due course.Methods
General experimental methods
All reactions were carried out in oven-dried glassware under an atmosphere of nitrogen gas unless otherwise stated. Microwave reactions were performed using a Biotage Initiator. Imines 8a–d were synthesized according to literature procedures.3a Catalysts (bnpy)AuCl23 and (ppy)AuCl26a were synthesized following literature procedures.14 Acetonitrile was distilled from CaH2. All other solvents and reagents were bought from commercial suppliers and used as received. Yields are reported for isolated products after chromatographic purification unless otherwise stated. Spectroscopic data for known compounds ((bnpyH)AuCl32,23 (ppyH)AuCl35a,13 (dfppy)AuCl26b,15 oxazoles 11b–h, 11k–l and 11n–o,3a and propargyl amine 8a24) were in agreement with literature data. 1H, 19F, and 13C NMR spectra were recorded on a Bruker Avance 400 NMR spectrometer, with the residual solvent peak used as an internal reference. IR spectra were recorded on a Bruker Alpha spectrometer. Elemental analyses were performed by H. Kolbe Microanalytisches Laboratorium, Mülheim an der Ruhr, Germany.General procedure for synthesis of LAuCl3 complexes
The respective pyridine ligand (1.1 equiv.), dissolved in MeCN (10 mL), was added in one portion to KAuCl4 in H2O (10 mL). The reaction mixture was then stirred at room temperature for 2 h after which the formed yellow precipitate was collected by filtration. The precipitate was washed with water and MeCN and dried under reduced pressure.General procedure for A3 coupling reactions
The respective gold complex (0.01 mmol) was charged in a vial under an air atmosphere and phenylacetylene (165 μl, 1.5 mmol) was added. Piperidine (109 μl, 1.1 mmol) and benzaldehyde (101 μl, 1.0 mmol) was then added sequentially and the resulting homogenous mixture was heated to 40 °C for 24 h. The reaction mixture was then cooled to ambient temperature and mesitylene (70 μl, 0.5 mmol) was added. The product to mesitylene ratio was determined through integration of the propargyl (product) and aryl (mesitylene) signals in the 1H NMR spectra.General procedure for optimization of Au-catalyzed oxazole formation
To a microwave vial, charged with the respective gold catalyst and flushed with nitrogen, was added the solvent indicated (0.5 mL) followed by addition of N-Bn-imine 8a (0.21 mL, 1.0 mmol), phenylacetylene (10a) (0.23 mL, 2.0 mmol), and benzoyl chloride (9a) (0.12 mL, 1.0 mmol) in short sequence. The resulting mixture was immediately heated by microwave irradiation to the temperature indicated (sample absorption set to “high”). After the time indicated, the reaction was cooled to room temperature and mesitylene (0.5 equiv.) was added as an internal standard. A sample of the reaction mixture was diluted with CDCl3 and the 1H NMR yield was quantified by measuring the product-to-mesitylene ratio through integration of the tert-butyl signal of the oxazole 11a (1H NMR δ = 1.37 ppm) and methyl signal in mesitylene (1H NMR δ = 2.28 ppm) in the 1H NMR spectra.General procedure for (bnpy)AuCl23 catalyzed oxazole synthesis
To a microwave vial, charged with (bnpy)AuCl23 (2.2 mg, 0.005 mmol) and flushed with nitrogen gas, was added anhydrous MeCN (0.5 mL). Imine (1.0 mmol), alkyne (2.0 mmol) and acyl chloride (1.0 mmol) were then added in short sequence. The resulting mixture was immediately heated for 15 min by microwave irradiation using a ceiling temperature of 240 °C and a sample absorption set to “high”. The reaction mixture was then cooled to room temperature. The mixture was diluted with CH2Cl2 and a small amount of silica gel was added. The resulting slurry was concentrated under reduced pressure to yield a dry powder. Purification by flash chromatography (elution with EtOAc–petroleum ether) gave the corresponding oxazole products.Crystallography
Intensity data were collected with an Oxford Diffraction Excalibur 3 system, using ω-scans and Mo Kα (λ = 0.71073 Å) radiation.25 The data were extracted and integrated using Crysalis RED.26 The structures were solved by direct methods and refined by full-matrix least-squares calculations on F2 using SHELXTL.27 Molecular graphics were generated using CrystalMaker 9.0.3. CCDC deposition numbers 1022615–1022618.Acknowledgements
We thank the Swedish Research Council, the Carl Trygger Foundation, Knut and Alice Wallenberg foundation, The Royal Physiographical Society, and Lund University for generous support.References
- For recent reviews see:
(a) M. Rudolpha and A. S. K. Hashmi, Chem. Soc. Rev., 2012, 41, 2448 RSC
; (b) D. J. Gorin and F. D. Toste, Nature, 2007, 446, 395 CrossRef CAS PubMed
-
(a) H. Schmidbaur and A. Schier, Arabian J. Sci. Eng., 2012, 37, 1187 CrossRef CAS PubMed
-
(a) H. v. Wachenfeldt, P. Röse, F. Paulsen, N. Loganathan and D. Strand, Chem. – Eur. J., 2013, 19, 7982 CrossRef PubMed
- M. Pažický, A. Loos, M. J. Ferreira, D. Serra, N. Vinokurov, F. Rominger, C. Jäkel, A. S. K. Hashmi and M. Limbach, Organometallics, 2010, 29, 4448 CrossRef
- T. E. Müller, M. Grosche, E. Herdtweck, A.-K. Pleyer, E. Walter and Y.-K. Yan, Organometallics, 2000, 19, 170 CrossRef
-
(a) A. S. K. Hashmi, J. P. Weyrauch, M. Rudolph and E. Kurpejović, Angew. Chem., Int. Ed., 2004, 43, 6545 CrossRef CAS PubMed
-
(a) E. Langseth, C. H. Görbitz, R. H. Heyn and M. Tilset, Organometallics, 2012, 31, 6567 CrossRef CAS
- For recent reviews see:
(a) Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239 CrossRef CAS PubMed
- For recent review see: D. J. Gorin, B. D. Sherry and F. D. Toste, Chem. Rev., 2008, 108, 3351 CrossRef CAS PubMed
- For recent reviews see:
(a) V. A. Peshkov, O. P. Pereshivkoa and E. V. Van der Eycken, Chem. Soc. Rev., 2012, 41, 3790 RSC
- For recent review, see:
(a) S. Bresciani and N. C. O. Tomkinson, Heterocycles, 2014, 89, 2479 CrossRef CAS PubMed
- For lead examples of multicomponent oxazole synthesis see:
(a) E. Merkul, O. Grotkopp and T. J. J. Müller, Synthesis, 2009, 502 CAS
- C. Constable and T. A. Leese, J. Organomet. Chem., 1989, 363, 419 CrossRef
- A. P. Shaw, M. Tilset, R. H. Heyn and S. Jakobsen, J. Coord. Chem., 2011, 64, 38 CrossRef CAS
- J. A. Garg, O. Blacque and K. Venkatesan, Inorg. Chem., 2011, 50, 5430 CrossRef CAS PubMed
-
(a) M. Kondrashov, S. Raman and O. F. Wendt, Chem. Commun., 2015, 51, 911 RSC
- D. Kalyani, A. R. Dick, W. Q. Anani and M. S. Sanford, Tetrahedron, 2006, 62, 11483 CrossRef CAS PubMed
- For SC-XRD structure of 5a see:
(a) X.-P. Zhang, G. Yang, L. Wang and S. W. Ng, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m1582 CAS
- A. D. Black and B. A. Arndtsen, Org. Lett., 2004, 6, 1107 CrossRef PubMed
- For a detailed mechanistic discussion on this process, see ref. 3a and 3b.
- X. Sun, P. Janvier, G. Zhao, H. Bienayme and J. Zhu, Org. Lett., 2001, 3, 877 CrossRef CAS PubMed
- L. Cattalini and M. L. Tobe, Inorg. Chem., 1966, 5, 1145 CrossRef CAS
- M. A. Cinellu, A. Zucca, S. Stoccoro, G. Minghetti, M. Manassero and M. Sansoni, J. Chem. Soc., Dalton Trans., 1995, 2865 RSC
- P.-H. Li and L. Wang, Chin. J. Chem., 2005, 23, 1076 CrossRef CAS
-
Crysalis CCD, Oxford Diffraction Ltd, Abingdon, Oxfordshire, UK, 2005 Search PubMed
-
Crysalis RED, Oxford Diffraction Ltd, Abingdon, Oxfordshire, UK, 2005 Search PubMed
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H NMR, 13C NMR, and 19F NMR spectra for (dfppyH)AuCl35b. SC-XRD data for compounds 2, 3, 5b, and 6b in cif format. CCDC 1022615–1022618. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03806a |
| This journal is © The Royal Society of Chemistry 2015 |