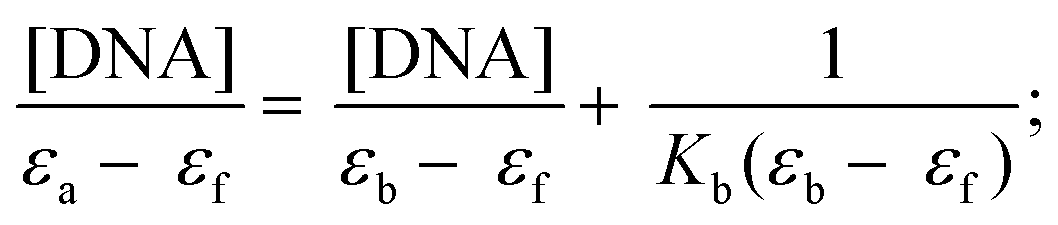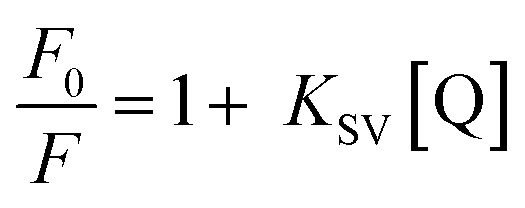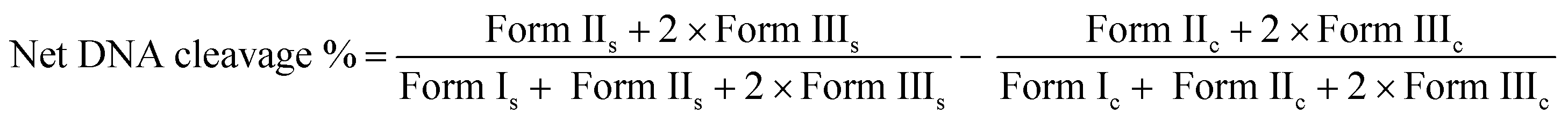DOI:
10.1039/C4DT03764B
(Paper)
Dalton Trans., 2015,
44, 6140-6157
Synthesis, X-ray structure and in vitro cytotoxicity studies of Cu(I/II) complexes of thiosemicarbazone: special emphasis on their interactions with DNA†
Received
9th December 2014
, Accepted 14th February 2015
First published on 18th February 2015
Abstract
4-(p-X-phenyl)thiosemicarbazone of napthaldehyde {where X = Cl (HL1) and X = Br (HL2)}, thiosemicarbazone of quinoline-2-carbaldehyde (HL3) and 4-(p-fluorophenyl)thiosemicarbazone of salicylaldehyde (H2L4) and their copper(I) {[Cu(HL1)(PPh3)2Br]·CH3CN (1) and [Cu(HL2)(PPh3)2Cl]·DMSO (2)} and copper(II) {[(Cu2L32Cl)2(μ-Cl)2]·2H2O (3) and [Cu(L4)(Py)] (4)} complexes are reported herein. The synthesized ligands and their copper complexes were successfully characterized by elemental analysis, cyclic voltammetry, NMR, ESI-MS, IR and UV-Vis spectroscopy. Molecular structures of all the Cu(I) and Cu(II) complexes have been determined by X-ray crystallography. All the complexes (1–4) were tested for their ability to exhibit DNA-binding and -cleavage activity. The complexes effectively interact with CT-DNA possibly by groove binding mode, with binding constants ranging from 104 to 105 M−1. Among the complexes, 3 shows the highest chemical (60%) as well as photo-induced (80%) DNA cleavage activity against pUC19 DNA. Finally, the in vitro antiproliferative activity of all the complexes was assayed against the HeLa cell line. Some of the complexes have proved to be as active as the clinical referred drugs, and the greater potency of 3 may be correlated with its aqueous solubility and the presence of the quinonoidal group in the thiosemicarbazone ligand coordinated to the metal.
Introduction
Cisplatin (cis-diamminedichloroplatinum(II)) is a well-known metal based drug for cancer; despite its wide application as a chemotherapeutic agent, cisplatin exhibits severe side effects, such as nausea, kidney and liver failure, typical of heavy metal toxicity.1–5 Therefore attempts are constantly made to replace it with suitable alternatives; hence various transition metal complexes have been synthesized and tried for their anticancer properties.
Metal complexes which efficiently bind and cleave DNA under physiological conditions are considered to have the potential to be used as therapeutic agents for medicinal applications and for genomic research.6–9 Depending on the exact nature of the metal and the ligand, the complexes can bind with nucleic acid covalently or non-covalently.10,11 Non-covalent interactions between transition-metal complexes and DNA can occur by intercalation, groove binding, or external electrostatic binding. Therefore, the study of the interaction of transition metal complexes with DNA is of great significance for the design of new drugs and their applications.
Among the transition metals, the coordination chemistry of copper has attracted increasing interest because of the use of many copper complexes as models for biological functions, for example, amine oxidases,12 catechol oxidase,13 nitrite reductase,14 superoxide dismutase,15 and tyrosinase.16 Copper complexes have been extensively utilized in metal ion mediated DNA cleavage through hydrogen ion abstraction by activated oxygen species.17 In recent years, a large number of biocompatible Cu(II) complexes have been investigated for their anticancer properties.18
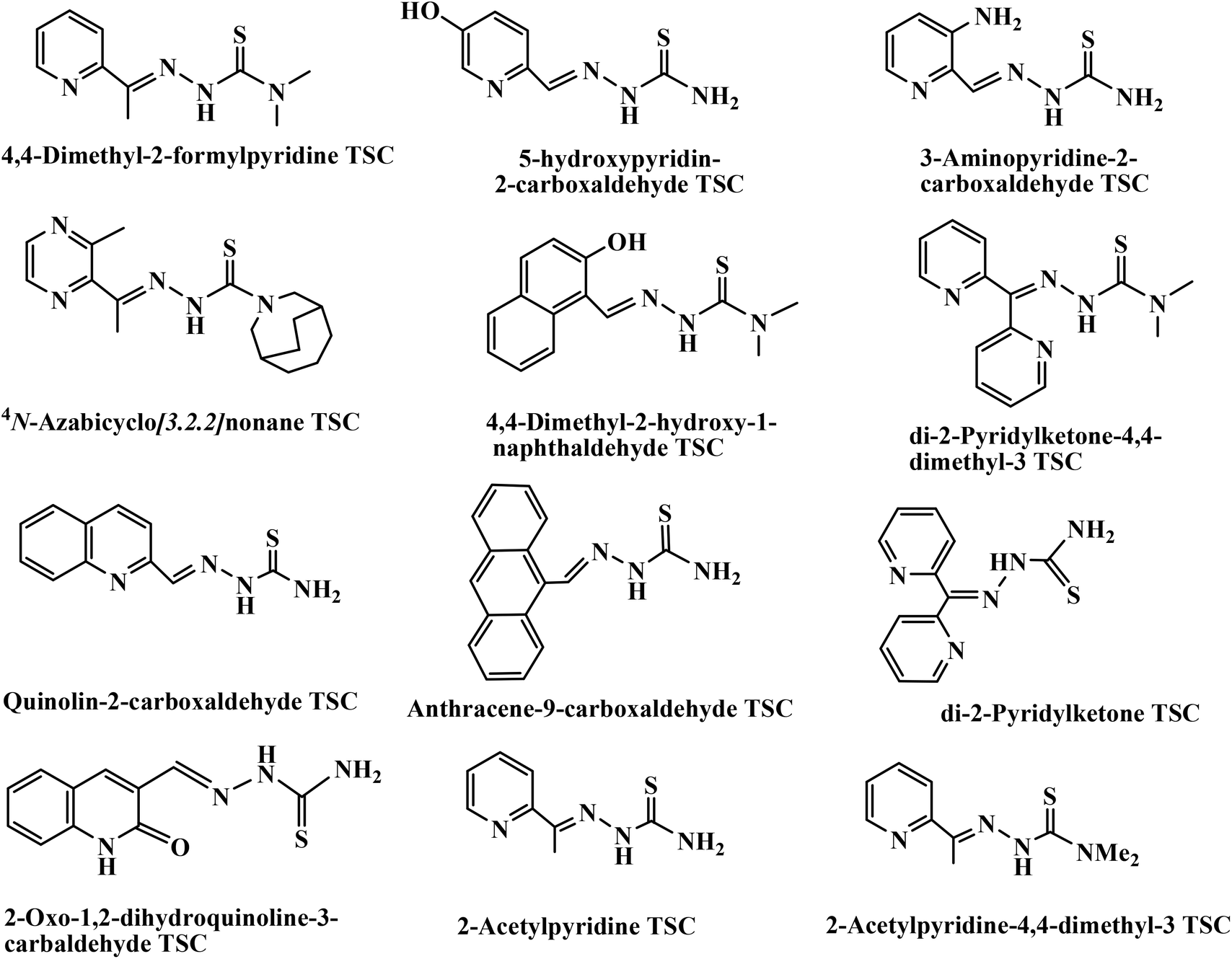
Additionally, thiosemicarbazones (TSCs) are a class of Schiff bases which are considered to be one of the most important scaffolds and are embedded in many biologically active compounds.19 Brockman et al. first reported that 2-formylpyridine TSC possesses antileukemic activity in mice.20 Following this report, various aliphatic, aromatic, and heteroaromatic carbaldehyde TSCs were synthesized and evaluated for their antitumor activity against a wide range of transplanted murine neoplasms.21–25 The list of TSC derivatives that have been found to exhibit intense anticancer activities is shown in Chart 1.18b,26 Again, transition metal complexes with TSCs as ligands have kindled interest amongst many researchers, and continue to be the subject of many studies, especially as anticancer chemotherapeutic agents27–29 and as DNA-binding and -cleaving agents.18b,30 TSC complexes have also demonstrated significant activity as antitumor, antiviral, antimicrobial, anti-amoebic and anti-inflammatory agents.31–33 Many Cu complexes of TSCs have demonstrated efficient antitumor potential.18b,c,26a,b,34–38 Although the chemistry of Cu(II) TSC complexes is well developed,30g,39–41 relatively less information is available for Cu(I) complexes,42–46 particularly about their pharmacological properties.
 |
| | Chart 1 List of TSC derivatives exhibiting intense anticancer activities. | |
Again, while many TSC complexes exhibit good biological activities, their water solubility is still unsatisfactory, which may restrict their application. Hence, it seemed of interest to synthesize some new water-soluble transition metal complexes of TSCs which may have significant pharmacological effects.
Considering these facts and as a continuation of our ongoing research on the study of pharmacological properties47 of transition metal complexes, in this report, two new Cu(I) complexes {[Cu(HL1)(PPh3)2Br]·CH3CN (1) and [Cu(HL2)(PPh3)2Cl]·DMSO (2)}, a novel tetranuclear copper(II) complex [(Cu2L32Cl)2(μ-Cl)2]·2H2O (3) and a new Cu(II) monomeric complex [Cu(L4)(Py)] (4) were synthesized and fully characterized. The interactions of these complexes with calf-thymus DNA (CT-DNA) utilizing UV-Vis absorption titration, competitive DNA binding fluorescence experiments, circular dichroism and thermal denaturation studies were performed. Their chemical as well as photo-induced cleavage activities with pUC19 supercoiled plasmid DNA were investigated. Furthermore, the cytotoxicity of the complexes against the HeLa cell line was surveyed by the MTT assay.
Experimental
Materials and methods
All chemicals were purchased from commercial sources and were used without further purification. Reagent grade solvents were dried and distilled prior to use. The thiosemicarbazides were prepared from distilled substituted aniline by a known method reported earlier.48 The ligands 4-(p-X-phenyl)thiosemicarbazone of napthaldehyde {where X = Cl (HL1) and X = Br (HL2)}, thiosemicarbazone of quinoline-2-carbaldehyde (HL3) and 4-(p-fluorophenyl) thiosemicarbazone of salicylaldehyde (H2L4) were prepared by reported methods.47c,49 MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium) and DAPI (4′,6-diamidino-2-phenylindole dihydrochloride) were purchased from Sigma Aldrich (USA). Minimal essential medium (MEM) was purchased from Gibco, India. The supercoiled (SC) pUC19 DNA was purified from E. coli cells with the aid of a GeneJET Plasmid Isolation kit (Thermo Scientific, USA). Calf thymus (CT) DNA was purchased from SRL (India) (biochemistry grade). Elemental analyses were performed on a Vario ELcube CHNS elemental analyzer. IR spectra were recorded on a Perkin-Elmer Spectrum RXI spectrometer. 1H, 13C and 31P NMR spectra were recorded with a Bruker Ultrashield 400 MHz spectrometer using SiMe4 as an internal standard. Electronic spectra were recorded on a Perkin-Elmer Lambda25 spectrophotometer. Mass spectra were recorded on a SQ-300 MS instrument operating in ESI mode. Electrochemical data were collected using a PAR electrochemical analyzer and a PC-controlled potentiostat/galvanostat (PAR 273A) at 298 K under a dry nitrogen atmosphere. Cyclic voltammetry experiments were carried out with Pt working and auxiliary electrodes and Ag/AgCl as the reference electrode and TEAP as the supporting electrolyte. Commercially available TEAP (tetra ethyl ammonium perchlorate) was properly dried and used as a supporting electrolyte for recording cyclic voltammograms of the complexes.
Synthesis of complexes {[Cu(HL1)(PPh3)2Br]·CH3CN (1) and [Cu(HL2)(PPh3)2Cl]·DMSO (2)}
Cu(I)X (X = Br/Cl) (1.0 mmol) was added to a solution of the ligand HL1–2 (1.0 mmol) in 20 mL of CH3CN, and the contents were refluxed for 1 h, followed by the addition of PPh3 (1.0 mmol) and continued refluxing for another 1 h. The resulting yellow solution was filtered, and slow evaporation of the filtrate over 4–5 days produced a yellow crystalline product. Crystals suitable for X-ray analysis were isolated for complex 1. X-ray quality crystals of complex 2 were obtained by recrystallizing in DMSO.
[Cu(HL1)(PPh3)2Br]·CH3CN (1).
Yield: 67%. Anal. calc. for C56H47BrClCuN4P2S: C, 64.12; H, 4.52; N, 5.34. Found: C, 64.13; H, 4.54; N, 5.38. Main IR peaks (KBr, cm−1): 3285 m ν(N(1)–H), 3049 m ν(N(2)–H), 2901 m ν(C(8)–H), 1632 s ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1547 s ν(–C(8)N(3)), 1096 s ν(P–CPh), 770 s ν(C(7)S). 1H NMR (DMSO-d6, 400 MHz) δ: 12.03 (s, 1H, –C(7)–N(1)H), 10.25 (s, 1H, –C(7)–N(2)H), 9.094 (s, 1H, –N(3)C(8)H), 8.42–7.27 (m, 26H, Ph + PPh3). 13C NMR (DMSO-d6, 100 MHz) δ: 175.39 (C(7), C–S), 138.6 (C(8), NCH), 136.72, 136.32, 135.87, 135.23, 134.85, 134.43, 133.81, 133.12, 132.83, 132.25, 131.91, 131.33, 130.78, 130.26, 130.02, 129.83 (16C, C6H6), 128.96, 128.47, 127.80 (PPh3). 31P NMR (DMSO-d6, 162 MHz) δ: 46.26 and 44.79 (2 s, 2PPh3). ESI MS (CH3OH): m/z 1047.74 (100%, [M − H]+); m/z 1071.95 (30%, [M + Na]+); m/z 1087.69 (65%, [M + K]+).
C), 1547 s ν(–C(8)N(3)), 1096 s ν(P–CPh), 770 s ν(C(7)S). 1H NMR (DMSO-d6, 400 MHz) δ: 12.03 (s, 1H, –C(7)–N(1)H), 10.25 (s, 1H, –C(7)–N(2)H), 9.094 (s, 1H, –N(3)C(8)H), 8.42–7.27 (m, 26H, Ph + PPh3). 13C NMR (DMSO-d6, 100 MHz) δ: 175.39 (C(7), C–S), 138.6 (C(8), NCH), 136.72, 136.32, 135.87, 135.23, 134.85, 134.43, 133.81, 133.12, 132.83, 132.25, 131.91, 131.33, 130.78, 130.26, 130.02, 129.83 (16C, C6H6), 128.96, 128.47, 127.80 (PPh3). 31P NMR (DMSO-d6, 162 MHz) δ: 46.26 and 44.79 (2 s, 2PPh3). ESI MS (CH3OH): m/z 1047.74 (100%, [M − H]+); m/z 1071.95 (30%, [M + Na]+); m/z 1087.69 (65%, [M + K]+).
[Cu(HL2)(PPh3)2Cl]·DMSO (2).
Yield: 67%. Anal. calc. for C56H50BrClCuN3OP2S2: C, 61.93; H, 4.64; N, 3.87. Found: C, 61.90; H, 4.67; N, 3.88. Main IR peaks (KBr, cm−1): 3284 m ν(N(1)–H), 3047 m ν(N(2)–H), 2908 m ν(C(8)–H), 1627 s ν(CC), 1551 s ν(–C(8)N(3)), 1090 s ν(P–CPh), 768 s ν(C(7)S). 1H NMR (DMSO-d6, 400 MHz) δ: 12.47 (s, 1H, –C(7)–N(1)H), 10.29 (s, 1H, –C(7)–N(2)H), 9.09 (s, 1H, –N(3)C(8)H), 8.07–7.25 (m, 26H, Ph + PPh3), 2.53 (s, 6H, DMSO). 13C NMR (DMSO-d6, 100 MHz) δ:178.18 (C(7), C–S), 140.51 (C(8), NCH), 137.82, 137.12, 136.87, 135.73, 134.95, 134.41, 133.85, 133.19, 132.79, 132.41, 131.86, 131.23, 130.96, 130.26, 130.12, 129.92 (16C, C6H6), 129.12, 128.87, 128.17 (PPh3). 31P NMR (DMSO-d6, 162 MHz) δ: 46.85 and 44.72 (2 s, 2PPh3). ESI MS (CH3OH): m/z 1086.70 (12%, [M + H]+); m/z 1051.92 (20%, [M − Cl]).
Synthesis of the complex [(Cu2L32Cl)2(μ-Cl)2]·2H2O (3)
CuCl2·2H2O (1.0 mmol) was added to a solution of the ligand HL3 (1.0 mmol) in 20 mL of hot methanol and the mixture was refluxed for 2 h. The resulting dark green solution was filtered and slow evaporation of the filtrate over 4–5 days produced deep green crystals suitable for X-ray analysis.
[(Cu2L32Cl)2(μ-Cl)2]·2H2O.
Yield: 58%. Anal. calc. for C44H40Cl4Cu4N16S4O2: C, 39.17; H, 2.99; N, 16.61. Found: C, 39.19; H, 2.97; N, 16.63. Main IR peaks (KBr, cm−1): 3228 m ν(–N(1)–H2), 3047 m ν(C(2)–H), 1635 s ν(CC), 1557 s, ν(–C(2)N(3)), 752 s ν(C(1)–S). ESI MS (CH3OH): m/z 1318.80 (68%, [(M − 2H2O) + 5H]+); m/z 1352.55 (100% [(M + 3H]+).
Synthesis of the complex [Cu(L4)(Py)] (4)
CuCl2·2H2O (1.0 mmol) was added to a solution of H2L4 (1.0 mmol) in 20 ml of hot methanol followed by the addition of pyridine (1.0 mmol). The mixture was refluxed for 3 h and a clear bluish green solution was obtained, which was filtered, and slow evaporation of the filtrate over 3–4 days produced bluish green crystals suitable for X-ray analysis.
[Cu(L4)(Py)].
Yield: 67%. Anal. calc. for C19H15CuFN4OS: C, 53.08; H, 3.52; N, 13.03. Found: C, 53.11; H, 3.56; N, 13.07. Main IR peaks (KBr, cm−1): 3224 s ν(N(1)–H), 2356 m ν(C(8)–H), 1602 s ν(CC), 1531 s ν(–C(8)N(3)), 748 s ν(C(7)–S(1)). ESI MS (CH3OH): m/z 430.07 (100%, [M]+); m/z 431.72 (50%, [M + H]+); m/z 351.14 (46%, [M − Py]+).
Crystallography
Single crystals of complexes were mounted on a Stoe IPDS 2 diffractometer equipped with an Oxford Cryosystem open flow cryostat (1 and 2) and on a Bruker Smart Apex CCD diffractometer (3 and 4) equipped with a graphite monochromator and a Mo Kα radiator (λ) 0.71073 Å. Crystallographic data and details of refinement of 1–4 are given in Table 1. The unit cell dimensions and intensity data were measured at 200(2) K for 1 and 2, 273(2) K for 3 and 296(2) for 4. Absorption correction was partially integrated into the data reduction procedure for crystals of 1 and 2.50 The intensity data were corrected for Lorentz, polarization and absorption effects. Absorption corrections were applied using SADABS51 and the structures were solved by direct methods using the program SHELXS-9752 and refined using least squares with the SHELXL-9752 software program. Hydrogens were either found or placed in calculated positions and isotropically refined using a riding model. The non-hydrogen atoms were refined anisotropically.
Table 1 Crystal and refinement data of complexes 1–4
| Compound |
1
|
2
|
3
|
4
|
| Formula |
C56H47BrClCuN4P2S |
C56H50BrClCuN3OP2S2 |
C44H40Cl4Cu4N16S4O2 |
C19H15CuFN4OS |
|
M
|
1048.88 |
1085.95 |
1349.12 |
429.95 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P21/c |
|
a (Å) |
9.4984(4) |
9.6604(5) |
8.9472(15) |
13.3041(5) |
|
b (Å) |
13.1221(5) |
13.1537(6) |
9.8129(16) |
5.8394(2) |
|
c (Å) |
20.6583(9) |
20.3412(10) |
14.928(2) |
23.1869(9) |
|
α (°) |
100.185(3) |
99.305(4) |
85.086(3) |
90 |
|
β (°) |
95.359(3) |
94.696(4) |
72.973(3) |
102.115(2) |
|
γ (°) |
96.502(3) |
93.570(4) |
80.782(3) |
90 |
|
V (Å3) |
2500.96(18) |
2534.7(2) |
1236.0(4) |
1761.22(11) |
|
Z
|
2 |
2 |
1 |
4 |
|
D
calc (Mg cm−3) |
1.393 |
1.423 |
1.813 |
1.621 |
|
F(000) |
1076 |
1116 |
680 |
876 |
|
μ(Mo-Kα) (mm−1) |
1.436 |
1.460 |
2.142 |
1.386 |
| max./min. trans. |
0.9460 and 0.8576 |
0.8921 and 0.7026 |
0.9873 and 0.8343 |
0.9728 and 0.7787 |
| 2θ(max.) (°) |
25.00 |
25.00 |
21.99 |
30.5 |
| Reflections collected/unique |
48![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 158/8790 [R(int) = 0.0383] 158/8790 [R(int) = 0.0383] |
48925/8938 [R(int) = 0.0576] |
8969/4328 [R(int) = 0.0406] |
33365/5407 [R(int) = 0.0410] |
|
R
1[I > 2σ(I)] |
R
1 = 0.0301, wR2 = 0.0788 |
R
1 = 0.0477, wR2 = 0.1269 |
R
1 = 0.0743, wR2 = 0.1738 |
R
1 = 0.0345, wR2 = 0.0796 |
| wR2[all data] |
R
1 = 0.0353, wR2 = 0.0814 |
R
1 = 0.0754, wR2 = 0.1400 |
R
1 = 0.1261, wR2 = 0.1963 |
R
1 = 0.0581, wR2 = 0.0885 |
|
S[goodness of fit] |
1.040 |
1.034 |
1.018 |
1.015 |
| min./max. res. (e Å−3) |
0.788 and −0.755 |
0.824 and −0.867 |
2.059 and −0.897 |
0.315 and −0.295 |
DNA binding experiments
Absorption spectral studies.
The DNA binding experiments were performed using a Perkin-Elmer Lambda35 spectrophotometer as described previously.47e Briefly, the absorption titration experiments were performed by varying the concentration of CT-DNA from 0 to 70 μM and keeping the metal complex concentration constant at 25 μM in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF. The binding constant Kb was computed from the data obtained using the following equation:47e| | 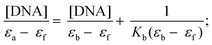 | (1) |
where [DNA] is the concentration of DNA base pairs, and εa, εf and εb correspond to apparent extinction coefficients for the complex i.e. Abs/[complex] in the presence of DNA, in the absence of DNA and to fully bound DNA respectively. A plot of [DNA]/(εa − εf) vs. [DNA] gave a slope and the intercept equal to 1/(εb − εf) and 1/Kb(εb − εf), respectively. The binding constant Kb was calculated from the ratio of the slope to the intercept. Ligand interactions with CT-DNA were also studied by titrating a fixed concentration of the ligand (25 μM) with variable CT-DNA concentration ranging from 0 to 350 μM in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF.
Competitive DNA binding fluorescence measurements.
The apparent binding constant (Kapp) values for the complexes were determined by fluorescence measurements using an ethidium bromide (2 μM) (EB) bound CT-DNA (50 μM) solution in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF with the aid of a Fluoromax 4P spectrofluorimeter (Horiba Jobin Mayer, USA). The fluorescence intensities of EB at 597 nm (excitation 510 nm) with an increase of the complex concentration (0−60 μM) were measured. In the presence of DNA, EB showed enhanced emission intensity due to intercalative binding with DNA. A competitive binding of metal complexes with CT-DNA leads to the decrease in the emission intensity due to emission quenching or the displacement of bound EB to CT-DNA by the complexes. The quenching constant was calculated using the following Stern–Volmer equation:53| | 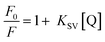 | (2) |
where F0 and F are the emission intensities of EB bound CT-DNA in the absence and presence of the quencher (complexes) concentration [Q], respectively, which gave the Stern–Volmer quenching constant (KSV). The apparent binding constant (Kapp) was calculated from the following equation:| | | KEB × [EB] = Kapp × [complex]50 | (3) |
where Kapp is the apparent binding constant of the complex, [complex]50 is the concentration of the complex at 50% quenching of the emission intensity of EB bound CT-DNA, KEB is the binding constant of EB (KEB = 1.0 × 107 M−1) and [EB] is the concentration of ethidium bromide (2 μM).53
Thermal melting studies.
Thermal melting studies of CT-DNA (100 μM) in the absence and presence of complexes (50 μM) were carried out by monitoring the absorbance at 260 nm in the temperature range of 30–90 °C with a ramp rate of 0.5 °C min−1 in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF. The experiments were carried out using a Chirascan CD spectropolarimeter (Applied Photophysics, UK) in absorbance mode equipped with a temperature controller. The melting temperature (Tm) was determined from the derivative plot (dA260/dT vs. T) of the melting profile.47e
Circular dichroism studies.
The circular dichroism (CD) spectroscopic studies were performed using a Chirascan CD spectropolarimeter (Applied Photophysics, UK) at 25 °C. CD spectra of CT-DNA (50 μM) in the absence and presence of complexes (10 μM) were obtained in the wavelength range of 240–400 nm in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF, using a quartz cell with 10 mm path length.47e
DNA cleavage experiments
DNA cleavage was carried out as previously reported.47e The chemical-induced and photo-induced DNA cleavage experiments were done with 300 ng supercoiled (SC) pUC19 DNA in 50 mM Tris–HCl buffer (pH 8.0) containing 1% DMF.
Chemical-induced DNA cleavage.
In order to study the chemical nuclease activity of the complexes, reactions were performed in the dark using hydrogen peroxide (0.5 mM) as the oxidising agent in the absence and presence of complexes (1–300 μM). The solutions were incubated at 37 °C for 1 h and analysed for DNA cleaved products by agarose gel electrophoresis.
Photo-induced DNA cleavage.
The photo-induced DNA cleavage activity was performed as described previously.47e Briefly, the photo-induced DNA cleavage experiments were carried out using a UVA source at 350 nm (Luzchem Photoreactor Model LZC-1, Ontario, Canada) fitted with 14 UVA tubes (84 W) for 1 h, on supercoiled (SC) pUC19 DNA (300 ng) with complexes (1–300 μM) in 50 mM Tris–HCl buffer (pH 8.0) containing 1% DMF. DNA cleavage was indicated by the decrease in the supercoiled pUC19 DNA (Form I) and subsequent formation of nicked circular DNA (Form II) and linear DNA (Form III). The percentage of net DNA cleavage was calculated using the following equation:| | 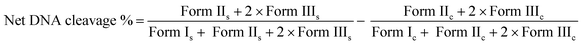 | (4) |
The subscripts “s” and “c” refers to the sample and the control respectively.54 Appropriate DNA controls were taken to calculate the net DNA cleavage percent. The observed error in measuring the band intensities ranged between 3%–6%.
For mechanistic investigations of both hydrolytic and photolytic DNA cleavage, experiments were carried out with singlet oxygen quenchers such as sodium azide (NaN3) and L-histidine, while for hydroxyl radical scavengers, potassium iodide (KI) and D-mannitol were used. Each of the additives was used at a concentration of 0.5 mM.
Anticancer activity
Cell culture.
Human cervical cells HeLa were obtained from the National Centre of Cell Science (NCCS), Pune, India and were maintained in minimal essential medium supplemented with 10% fetal bovine serum, penicillin–streptomycin solution and incubated at 37 °C in a 5% CO2 and 95% humidified incubator. The complexes were dissolved in DMSO at a concentration of 100 mM as the stock solution, and diluted in culture medium at concentrations of 12.5, 25.0, 50.0 and 100.0 μM as the working solution. To avoid DMSO toxicity, the concentration of DMSO was less than 0.1% (v/v) in all experiments.
Cytotoxic assay.
HeLa cells were harvested from maintenance cultures in the logarithmic phase, after counting in a hemocytometer using a trypan blue solution. The cell concentration was adjusted to 5 × 104 cells ml−1 and the cells were plated in 96 well flat bottom culture plates and incubated for 72 h with various concentrations of the test compounds. The effect of the drugs on cancer cell viability was studied using an MTT dye reduction assay by measuring the optical density at 595 nm using a micro-plate reader spectrophotometer (Perkin-Elmer 2030).55
Nuclear staining.
Nuclear staining using the DAPI stain was performed according to the method previously described.56 Briefly, HeLa cells either treated or untreated with test compounds were smeared on a clean glass slide, cells were fixed with 3.7% formaldehyde for 15 minutes, permeabilized with 0.1% Triton X-100 and stained with 1 μg ml−1 DAPI for 5 min at 37 °C. The cells were then washed with PBS and examined by fluorescence microscopy (Olympus IX 71) to ascertain any condensation or fragmentation of the nuclei indicating cells undergoing apoptosis.
Results and discussion
Synthesis
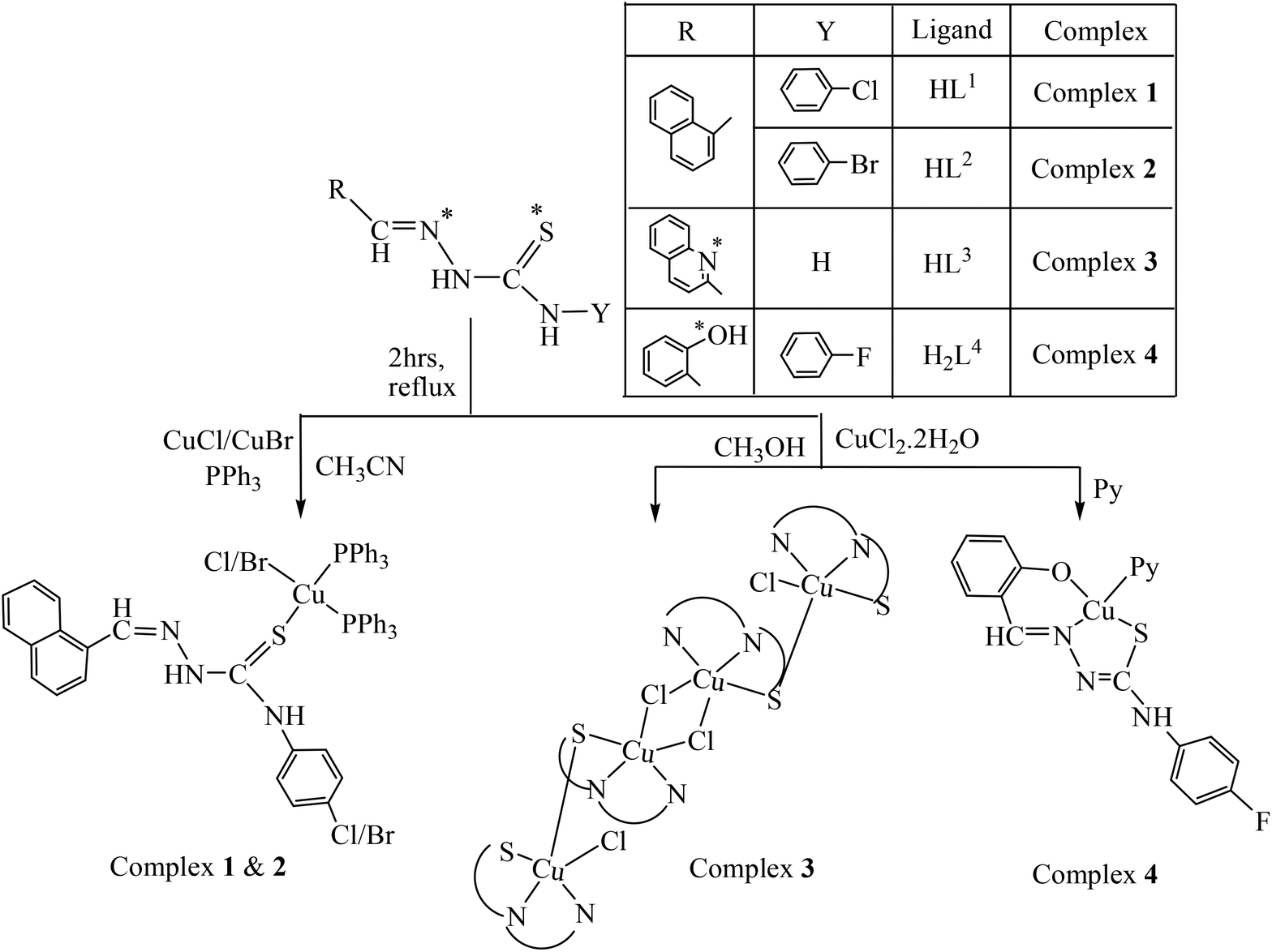
Reaction of Cu(I)X (X = Cl, Br) with 4-(p-X-phenyl) thiosemicarbazone of napthaldehyde {X = Cl (HL1); X = Br (HL2)} in the molar ratio of 1:1 in CH3CN formed an insoluble product of stoichiometry [CuX(HL1–2)], which after addition of two moles of PPh3 yielded light yellow colored monomeric complexes [CuX(HL1–2)(PPh3)2]·Solvent (X = Br, 1; Cl, 2). Reaction of copper(II) chloride with quinoline-2-carbaldehyde thiosemicarbazone (HL3) in a molar ratio of 1:1 in CH3OH yielded a dark green colored tetrameric complex [(Cu2L32Cl)2(μ-Cl)2]·2H2O (3), whereas that with 4-(p-F-phenyl) thiosemicarbazone of salicylaldehyde (H2L4) in the presence of pyridine as a coligand yielded a dark green monomeric complex [Cu(L4)(Py)] (4). The electrospray mass spectra (ESI MS) and NMR spectra were consistent with the X-ray structures. The purity of these compounds was further confirmed by elemental analyses. The synthetic methods of all the complexes are illustrated in Scheme 1. All complexes were soluble in MeOH, MeCN, DMF and DMSO. Complex 3 was completely and the other three complexes (1, 38%; 2, 35%; and 4, 45%, H2O–DMSO solution) were partially soluble in H2O. All the complexes were stable in both solid and solution phases. The solution phase stability of the complexes was confirmed by electronic absorption, NMR and ESI-MS spectral studies. The representative spectra are given in ESI† Fig. S1, Fig. S2 and Fig. S3.
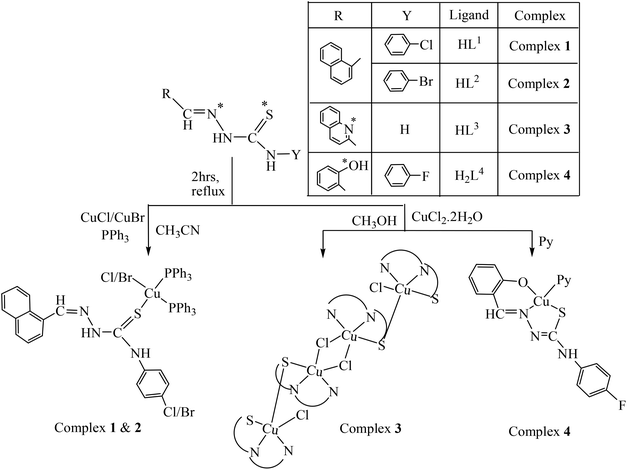 |
| | Scheme 1 Schematic representation of ligands and syntheses of copper complexes. | |
Structure
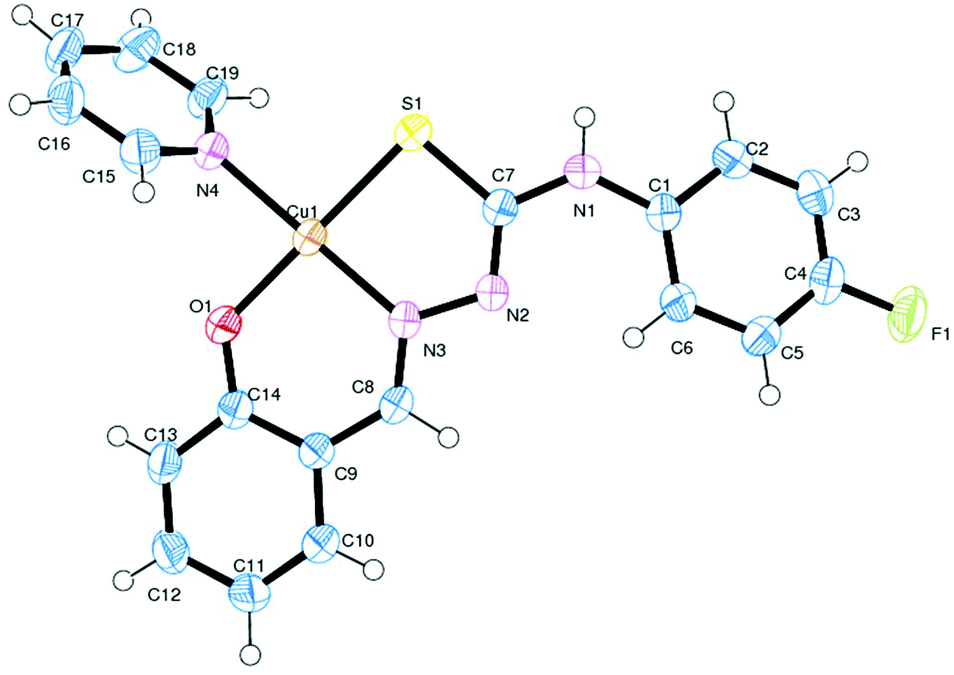
The observed elemental (C, H, N) analytical data of all the complexes (1–4) are in consistent with their composition. It appears from the formulation of 1 and 2 that the TSC is serving as a monodentate ligand, whereas in 3 and 4 it is serving as a tridentate ligand. In order to authenticate the coordination mode of the TSC in the complexes, the structures have been determined by X-ray crystallography.
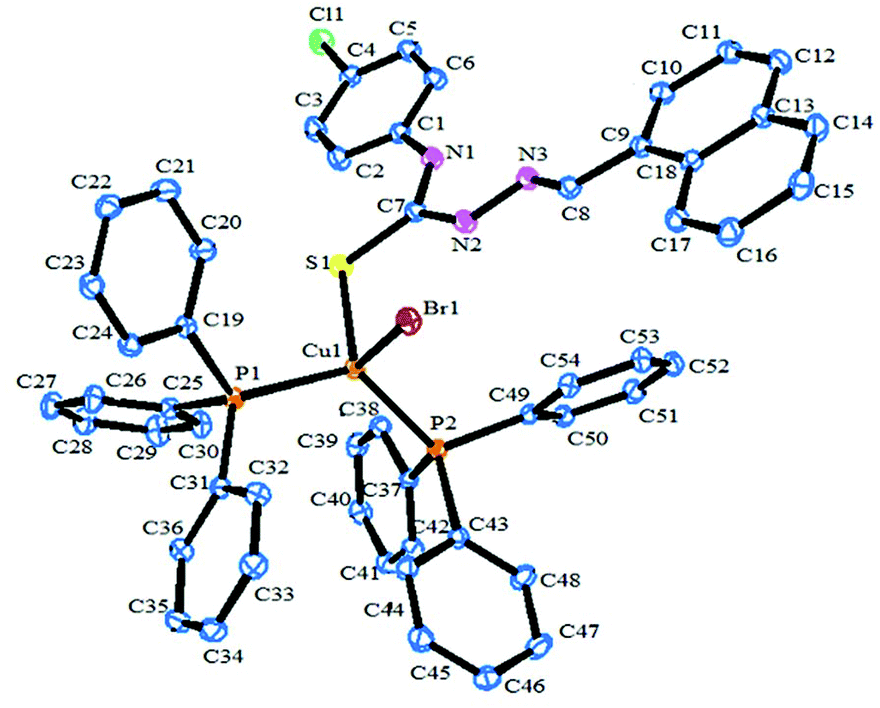
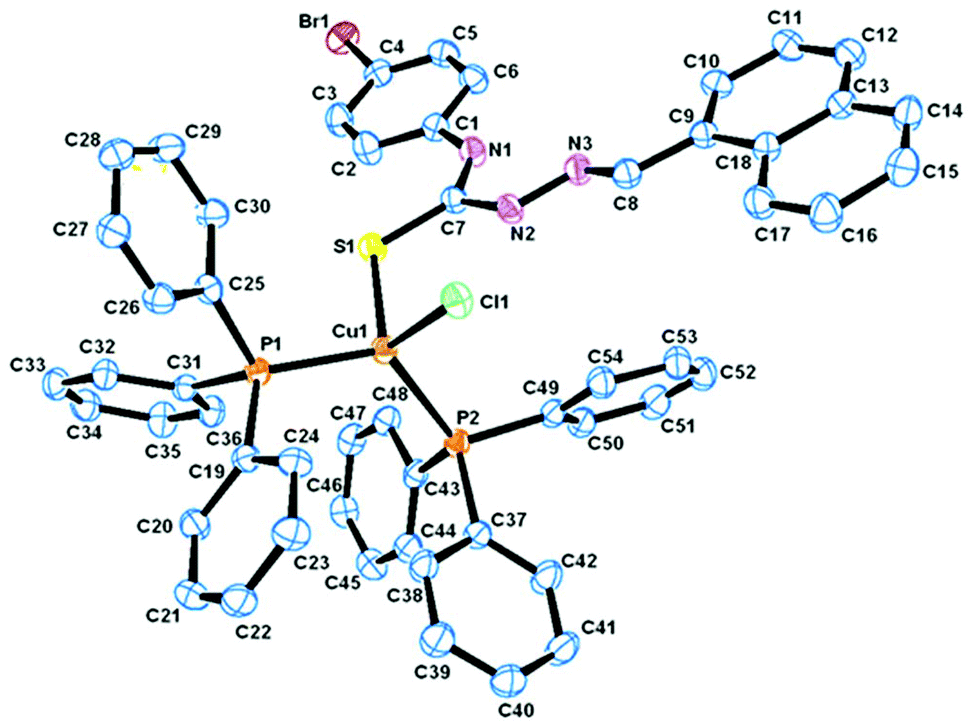
Description of X-ray structures of [Cu(HL1)(PPh3)2Br]·CH3CN (1) and [Cu(HL2)(PPh3)2Cl]·DMSO (2)
The molecular structure and the atom numbering scheme for the complexes [Cu(HL1)(PPh3)2Br]·CH3CN (1) and [Cu(HL2)(PPh3)2Cl]·DMSO (2) are shown in Fig. 1 and 2 respectively; the relevant bond distances and angles are collected in Table 2. Compounds 1 and 2 contain CH3CN and DMSO as a solvent of crystallization respectively. The coordination geometry around the Cu(I) atom in 1 and 2 reveals a distorted tetrahedral environment with an SXP2 [X = Br (1) and Cl (2)] coordination sphere as the bond angles around the copper atom vary from ca. 100 to 124° in 1 and 2, with P–Cu–P being the largest angle.49,57 The ligand HL1–2 acts as a monodentate ligand coordinating through the S atom. The other positions of the tetrahedron are occupied by one halogen atom and two triphenylphosphine ligands. In the compound, the Cu–S bond lengths are 2.401(7) Å for 1 and 2.387(1) Å for 2, while the Cu–halogen bond distances lie in the range 2.374(1)–2.517(4) Å as is usually found for tetrahedrally coordinated copper(I) and S atom donors.44,49 The Cu–P distances [2.276(6), 2.290(7) Å for 1, 2.274(1), 2.295(1) Å for 2] are comparable to those found in similar complexes.44,49
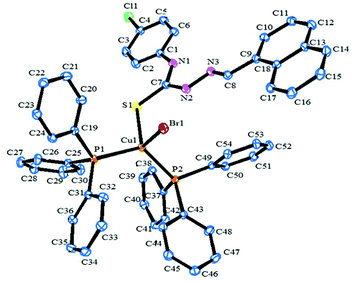 |
| | Fig. 1 ORTEP diagram of [Cu(HL1)(PPh3)2Br]·CH3CN (1) with the atom labeling scheme. | |
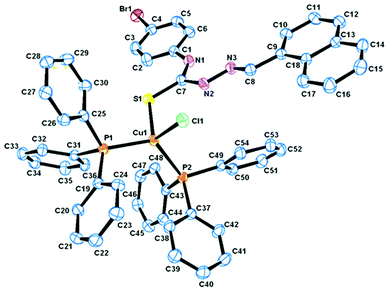 |
| | Fig. 2 ORTEP diagram of [Cu(HL2)(PPh3)2Cl]·DMSO (2) with the atom labeling scheme. | |
Table 2 Selected bond distances (Å) and bond angles (°) for [Cu(HL1)(PPh3)2Br]·CH3CN (1) and [Cu(HL2)(PPh3)2Cl]·DMSO (2)
| |
Complex (1) |
Complex (2) |
| Bond distance |
| Cu(1)–S(1) |
2.401(7) |
2.387(1) |
| Cu(1)–P(1) |
2.277(6) |
2.274(1) |
| Cu(1)–P(2) |
2.290(7) |
2.295(1) |
| Br(1)–Cu(1) |
2.517(4) |
— |
| Cl(1)–Cu(1) |
— |
2.374(1) |
| |
| Bond angles |
| P(2)–Cu(1)–P(1) |
124.51(2) |
122.72(4) |
| P(2)–Cu(1)–S(1) |
109.26(2) |
107.07(4) |
| P(1)–Cu(1)–S(1) |
104.48(2) |
105.19(4) |
| P(2)–Cu(1)–Br(1) |
108.41 (2) |
— |
| P(2)–Cu(1)–Cl(1) |
— |
108.07(4) |
| P(1)–Cu(1)–Br(1) |
100.29(2) |
— |
| P(1)–Cu(1)–Cl(1) |
— |
104.81(4) |
| S(1)–Cu(1)–Br(1) |
108.97(2) |
— |
| Cl(1)–Cu(1)–S(1) |
— |
108.35(4) |
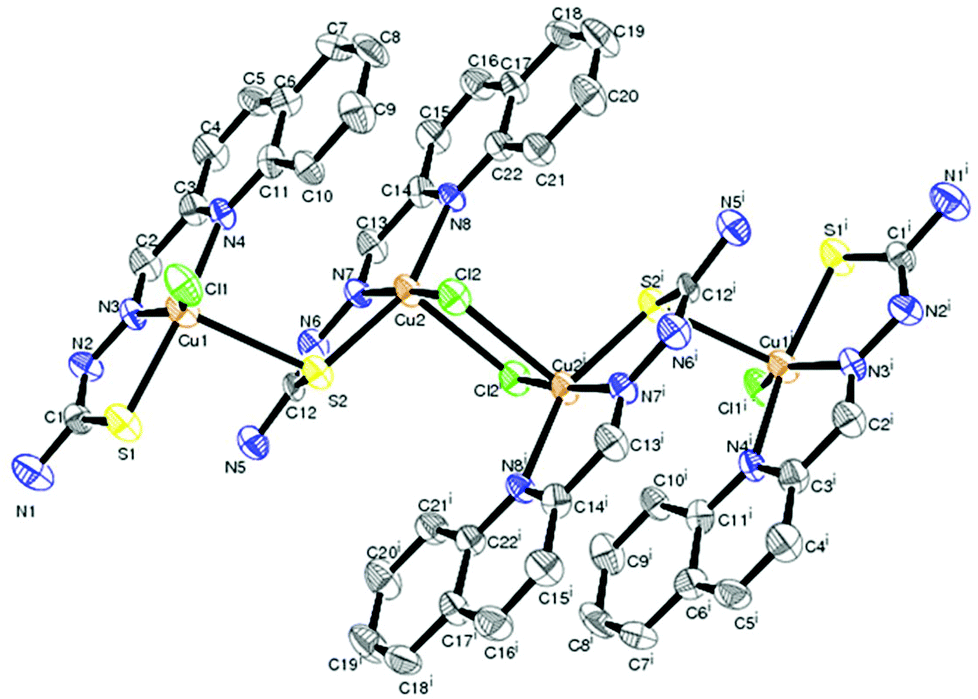
Description of X-ray structure of [(Cu2L32Cl)2(μ-Cl)2]·2H2O (3)
The structure of the tetranuclear Cu(II) complex [(Cu2L32 Cl)2(μ-Cl)2]·2H2O is illustrated in Fig. 3 and selected bond parameters are collected in Table 3. Compound 3 contains two H2O molecules as a solvent of crystallization. The structure contains four units composed of two identical Cu(1)LCl outer units and two identical Cu(2)LCl inner units. In other words, the tetranuclear Cu(II) species is formed by the dimerisation of two binuclear Cu2L2Cl2 units bridged by two reciprocal coordinated chlorine atoms of individual Cu2L2Cl2 units. Each copper atom in the outer unit is coordinated by a quinoline nitrogen, azomethine nitrogen and thiolate sulfur of the thiosemicarbazone moiety and a chlorine group. The Cu–Nquinoline bonds are ∼0.131 Å farther away than Cu–Nimine bonds, indicating the strength of the azomethine nitrogen coordination. The length of the other metal coordinated bonds (Cu–S and Cu–Cl) is usual like similar systems reported earlier.58,59 The bond angles are also in conformity with a distorted square pyramidal structure around the copper centers. Each copper atom in the inner subunit is pentacoordinate with the bonds Cu(2)–S(2), Cu(2)–Cl(2), Cu(2)–N(8), Cu(2)–N(7) and Cu(2)–Cl(2)# adapting a distorted square pyramidal geometry with bridging Cl(2)# of the other inner moiety at the apical site. The quinoline nitrogen N(8), the imino nitrogen N(7), and the thiolate sulfur S(2) atom, together with Cl(2), constitute the basal plane. The bond lengths in the basal plane agree with those found in copper(II) complexes containing thiosemicarbazones which act as uninegative tridentate ligands.58,59 The bond lengths and bond angles reveal a distorted square pyramidal geometry around Cu(2).
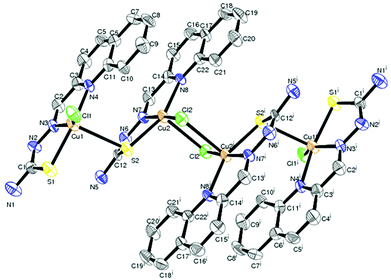 |
| | Fig. 3 ORTEP diagram of [(Cu2L32Cl)2(μ-Cl)2]·2H2O (3) with the atom labeling scheme. | |
Table 3 Selected bond distances (Å) and bond angles (°) for [(Cu2L32Cl)2(μ-Cl)2]·2H2O (3) and [Cu(L4)(Py)] (4)
| |
Complex (3) |
Complex (4) |
| Bond distance |
| Cu(1)–Cl(1) |
2.253(3) |
— |
| Cu(1)–O(1) |
— |
1.906 (2) |
| Cu(1)–S(1) |
2.304(3) |
2.247(6) |
| Cu(1)–S(2) |
2.715(2) |
— |
| Cu(1)–N(3) |
1.975(7) |
1.932(1) |
| Cu(1)–N(4) |
2.107(7) |
2.013(1) |
| Cu(2)–Cl(2) |
2.290(2) |
— |
| Cu(2)–S(2) |
2.303(2) |
— |
| Cu(2)–N(7) |
1.973(6) |
— |
| Cu(2)–N(8) |
2.146(6) |
— |
| Cu(2)–Cl(2)1 |
2.691(2) |
— |
| |
| Bond angles |
| Cl(1)–Cu(1)–S(1) |
92.60(1) |
— |
| O(1)–Cu(1)–N(3) |
— |
94.04(7) |
| Cl(1)–Cu(1)–S(2) |
101.30(1) |
— |
| O(1)–Cu(1)–N(4) |
— |
86.40(7) |
| Cl(1)–Cu(1)–N(3) |
150.60(2) |
— |
| O(1)–Cu(1)–S(1) |
— |
177.41(5) |
| Cl(1)–Cu(1)–N(4) |
104.10(2) |
— |
| S(1)–Cu(1)–S(2) |
94.15(8) |
— |
| S(1)–Cu(1)–N(3) |
82.30(2) |
85.92(5) |
| S(1)–Cu(1)–N(4) |
161.90(2) |
93.72(5) |
| S(2)–Cu(1)–N(3) |
107.90(2) |
— |
| S(2)–Cu(1)–N(4) |
89.40(2) |
— |
| N(3)–Cu(1)–N(4) |
79.70(3) |
178.10(7) |
| Cl(2)–Cu(2)–S(2) |
89.88(8) |
— |
| Cl(2)–Cu(2)–N(7) |
171.20(2) |
— |
| Cl(2)–Cu(2)–N(8) |
108.60(2) |
— |
| Cl(2)–Cu(2)–Cl(2)1 |
87.25(7) |
— |
| S(2)–Cu(2)–N(7) |
81.60(2) |
— |
| S(2)–Cu(2)–N(8) |
155.20(2) |
— |
| Cl(2)1–Cu(2)–S(2) |
102.01(9) |
— |
| N(7)–Cu(2)–N(8) |
79.20(3) |
— |
| Cl(2)1–Cu(2)–N(7) |
96.30(2) |
— |
| Cl(2)1–Cu(2)–N(8) |
95.50(2) |
— |
| Cu(2)–Cl(2)–Cu(2)1 |
92.80(9) |
— |
Description of X-ray structure of [Cu(L4)(Py)] (4)
The atom numbering scheme for complex 4 is given in Fig. 4 with the relevant bond distances and angles collected in Table 3. The structure shows that the thiosemicarbazone ligand (L2−) is coordinated to copper in the expected tridentate fashion (Scheme 1), forming a six- and a five-membered chelate ring with O(1)–Cu(1)–N(3) and S(1)–Cu(1)–N(3) bite angles of 94.04(7)° and 85.92(5)° respectively. The coligand pyridine is coordinated to the metal center and is trans to the nitrogen atom N(3). The rather large Cu(1)–N(4) distance of 2.013(1) Å revealed that the pyridine moiety is weakly coordinated to the Cu-center.60 Copper is thus nested in a NOSN core, which is slightly distorted from an ideal square-planar geometry, as reflected in the bond parameters around the metal center. The Cu–N(3), Cu–O(1), Cu–N(4) (coligand) and Cu–S(1) distances are normal, as observed in other structurally characterized complexes of Cu containing these bonds.60,61
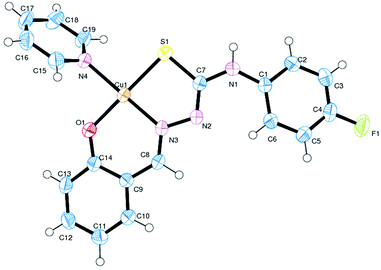 |
| | Fig. 4 ORTEP diagram of [Cu(L4)(Py)] (4) with the atom labeling scheme. | |
Spectral characteristics
The IR spectra of 1 and 2 showed the presence of ν(N–H) bands in the ranges 3284–3285 cm−1 for –N(1)–H and 3047–3049 cm−1 for –N(2)–H stretching, which suggests that the thiosemicarbazone ligand is coordinated to the Cu(I) centre in the neutral form. In all the complexes (1–4), ν(CN) and ν(CC) vibrational modes appeared in the range 1635–1531 cm−1, while the thioamide bands ν(C–S (1 and 2) and CS (3 and 4)) appeared in the range 770–748 cm−1 (compared to free ligands, 854–785 cm−1).47c The characteristic ν(P–CPh) bands at 1096–1090 cm−1 indicate the presence of Ph3P in 1 and 2.
The electronic spectra of all the complexes (Table 4) were recorded in methanol solutions. In the spectra of 1–4 three strong absorptions are observed in the wavelength range 448–220 nm. The lower energy absorptions at around 448–349 nm are ascribable to metal to ligand (1 and 2) and ligand to metal (3 and 4) charge transfer transitions whereas the higher energy absorptions are likely to be due to ligand centered transitions.46,47c Weak absorptions in the range 676–668 nm are also observed for 3 and 4 (Cu(II) complexes), which are assigned to d–d transitions.62
Table 4 Electronic spectra for complexes 1–4 in CH3OH
| Complex |
λ
max/nm (ε/dm3 mol−1 cm−1) |
| [Cu(HL1)(PPh3)2Br]·CH3CN (1) |
239 (51814), 349 (26982) |
| [Cu(HL2)(PPh3)2Cl]·DMSO (2) |
227 (45121), 385 (27637) |
| [(Cu2L32Cl)2(μ-Cl)2]·2H2O (3) |
230 (29790), 306 (23258), 364 (12917), 448 (11768), 676 (17788) |
| [Cu(L4)(Py)] (4) |
220 (5132), 256 (8526), 332 (3310), 395 (2574), 668 (4353) |
The NMR spectra (1H, 13C and 31P) of 1 and 2 were recorded using DMSO-d6. The 1H NMR spectrum exhibits three singlets in the range 12.47–9.09 ppm due to NH (–C(7)–N(1)H), NH (–C(7)–N(2)H) and CH (–N(3)C(8)H) groups respectively. Signals for aromatic protons found as multiplets in the 8.42–7.25 ppm range.47c The 13C NMR spectra of complexes (1 and 2) showed a sharp singlet appearing at 178.18–175.39 ppm due to C–S carbon. The peak for the azomethine (–CHN) carbon exhibited a peak in the region 140.51–138.6 ppm. The peaks observed in the 137.82–129.83 ppm region have been assigned to aromatic carbons. The PPh3 peaks are assigned in the range 129.12–127.80 ppm.6331P NMR spectra were recorded in order to confirm the presence of a triphenyl phosphine group. The two signals appeared at 46.85–44.72 ppm and indicated that the two triphenyl phosphine ligands were cis to each other in these complexes.63 The detailed NMR data have been included in the Experimental section.
ESI mass spectra of 1–4 have been recorded in a methanol solution. Mass spectral analysis for 1 and 2 shows peaks at m/z 1047.34 [(M + H)+] and 1086.70 [(M + H)+] respectively, whereas 3 shows the molecular ion peak [(M + 3H)+] at m/z 1352.55. The ESI-MS peak for 4 shows the characteristic molecular ion peak (M+) at m/z 430.07. ESI† Fig. S4 shows a representative ESI mass spectrum of 4.
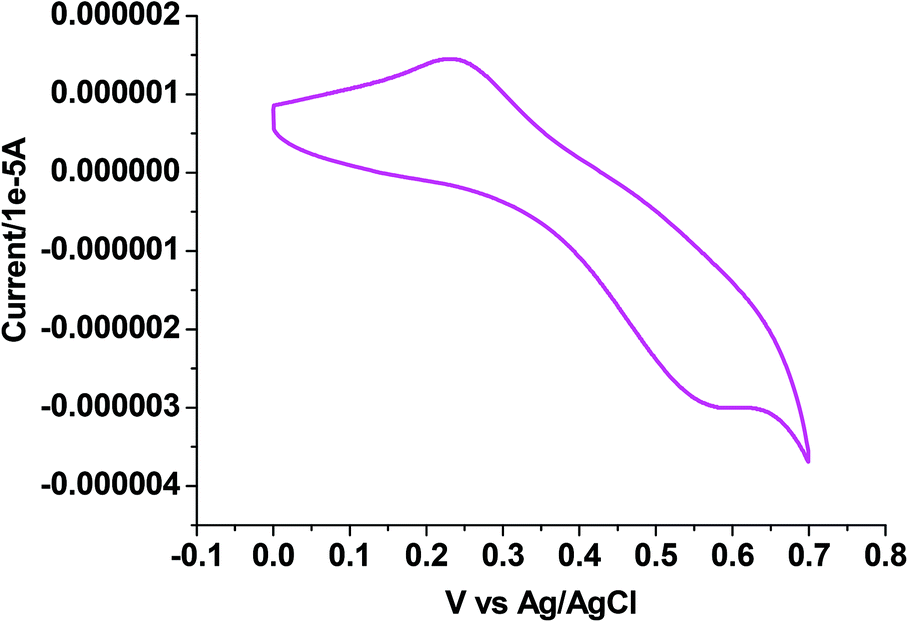
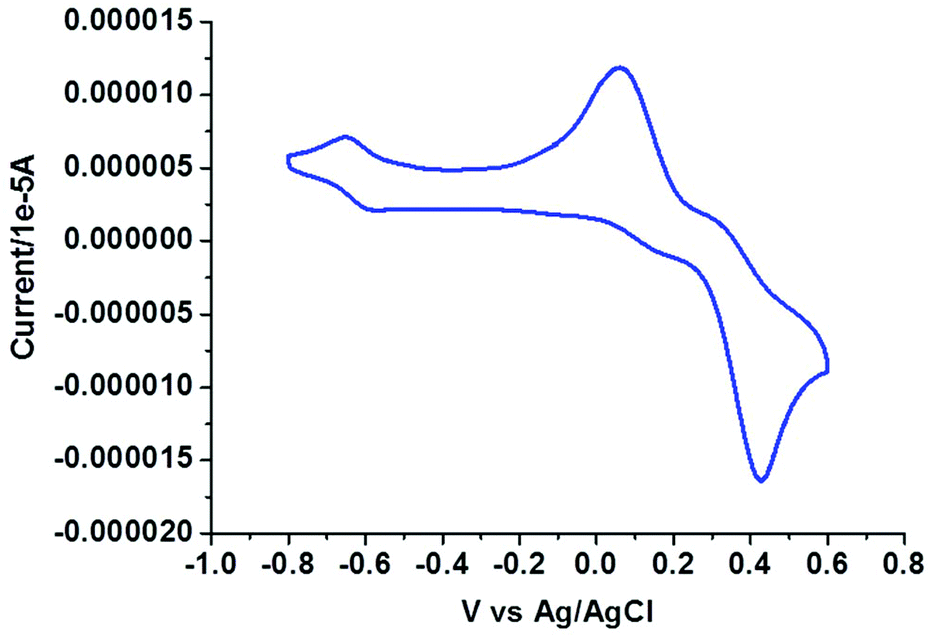
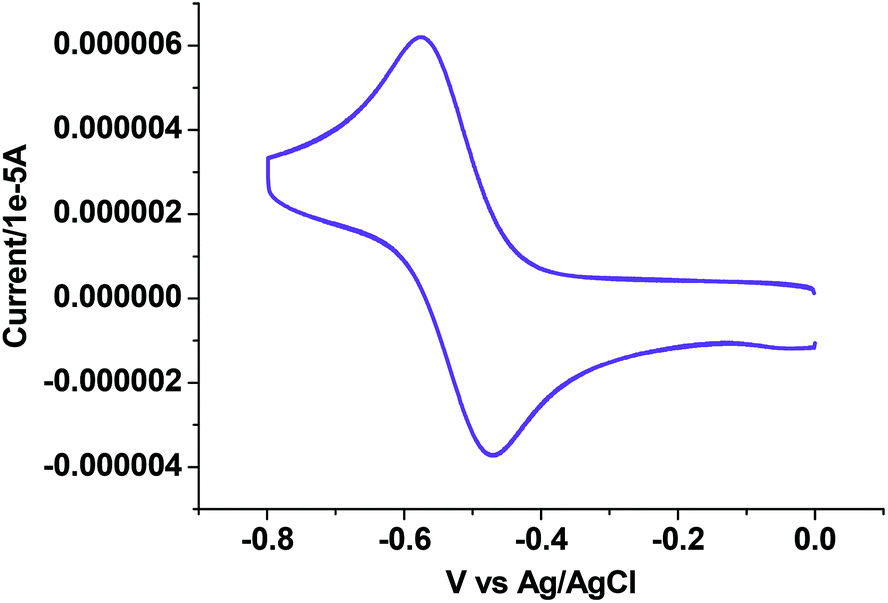
Electrochemical properties
The electrochemical properties of 1–4 were examined in CH3CN solution (0.1 M TEAP) by cyclic voltammetry using a platinum working electrode, a platinum auxiliary electrode and an Ag/AgCl reference electrode. The potential data are listed in Table 5. Fig. 5–7 show the representative voltammograms of 1 {Cu(I)}, 3 {Cu(II)} and 4 {Cu(II)} respectively. The voltammograms of all four complexes include both oxidation and reduction processes. The voltammogram pattern is similar for 1 and 2, which includes a quasireversible (Fig. 5) process at E1/2 values in the range 0.37–0.40 V, corresponding to the Cu(I)/Cu(II) redox couple,46 whereas in the cathodic region (ESI† Fig. S5), Cu(I) is reduced to Cu(0) showing an irreversible single electron wave at Epc values within the potential window −0.70 to −0.72 V.64
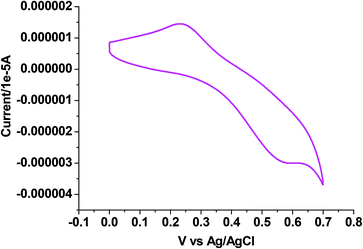 |
| | Fig. 5 Cyclic voltammogram of complex 1. | |
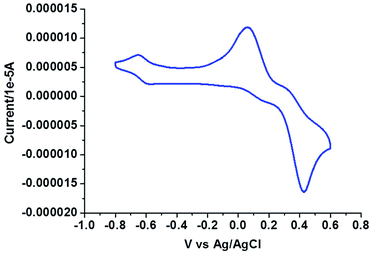 |
| | Fig. 6 Cyclic voltammogram of complex 3. | |
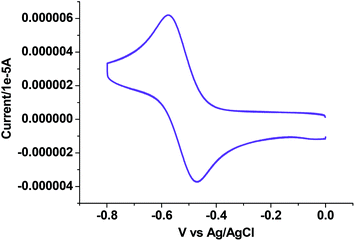 |
| | Fig. 7 Cyclic voltammogram of complex 4. | |
Table 5 Cyclic voltammetrica results for complexes 1–4 at 298 K
| Complex |
Cu(I)/Cu(II) Ea1/2(ΔEaP) |
Cu(I)/Cu(0) Epc |
Potentials (V) versus Ag/AgCl |
| Ligand-centered oxidation Ea1/2(ΔEaP) |
Ligand-centered reduction Epc |
Cu(II)/Cu(I) Ec/a1/2(ΔEc/aP) |
|
In CH3CN at a scan rate of 100 mV s−1. E1/2 = (Epa + Epc)/2, where Epa and Epc are anodic and cathodic peak potentials vs. Ag/AgCl, respectively. ΔEP = Epa − Epc.
|
| [Cu(HL1)(PPh3)2Br]·CH3CN (1) |
0.40 (320) |
−0.72 |
0.89 (240) |
−1.41, −1.63 |
— |
| [Cu(HL2)(PPh3)2Cl]·DMSO (2) |
0.37 (326) |
−0.70 |
0.87 (156) |
−1.45, −1.65 |
— |
| [(Cu2L32Cl)2(μ-Cl)2]·2H2O (3) |
— |
— |
0.88 (190) |
−1.39, −1.63 |
−0.62 (50), 0.10 (77), 0.36 (113), |
| [Cu(L4)(Py)] (4) |
— |
— |
0.91 (264) |
−1.37, −1.61 |
−0.52 (100) |
In the voltammogram of tetrameric Cu(II) complex (3), there are three quasireversible/reversible (Fig. 6) processes at Ec/a1/2 values of −0.62, 0.10 and 0.36 V corresponding to Cu(II)/Cu(I) redox couples65 of four different Cu(II) centers, whereas for the monomeric Cu(II) complex (4) a quasireversible (Fig. 7) process for the above couple appears at an Ec1/2 value of −0.52 V.
For all four complexes (1–4) an oxidation peak (ESI† Fig. S6) in the range 0.87–0.91 V66 and two reduction peaks (ESI† Fig. S5) in the range −1.37 to −1.45 and −1.61 to −1.65 V31,47c belong to ligand centered processes respectively. A representative oxidative voltammogram of a free ligand HL3 is given in ESI† Fig. S7.
DNA binding studies
Absorption spectroscopic studies.
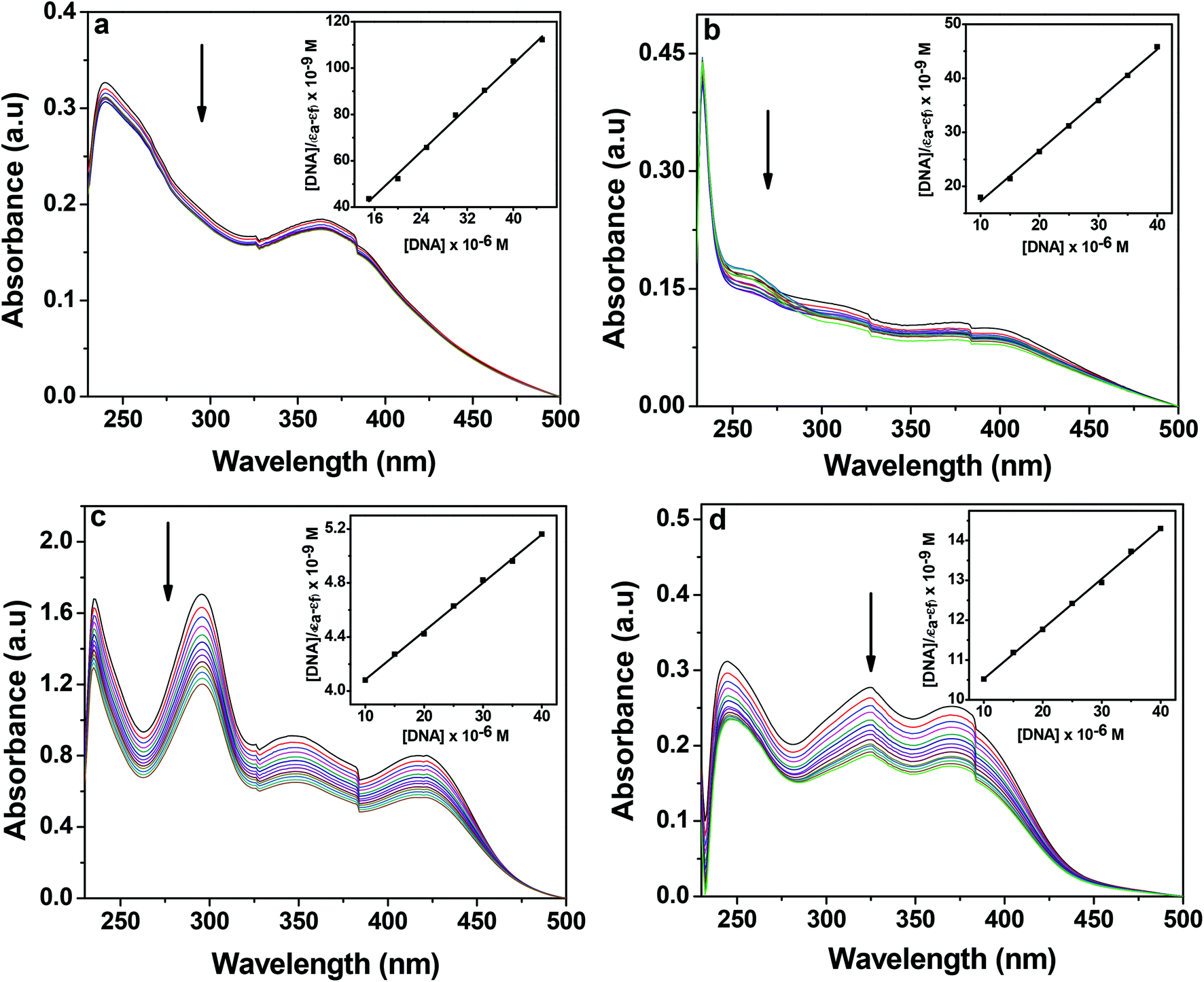
DNA is often a vital target to mediate apoptosis or necrosis to a cell. Therefore the binding affinity of the complexes to CT-DNA was studied using different spectral methods. UV-Vis titration experiments were carried out to determine the binding constant (Kb) of the complexes to CT-DNA (Fig. 8 and Table 6). Complexes 1–4 show absorption bands in the region 448–349 nm which are attributed to metal to ligand (1 and 2) and ligand to metal (3 and 4) charge transfer transitions whereas the absorption bands at higher energy are due to intra-ligand transitions. The binding of complexes to DNA either leads to hypochromism or hyperchromism, which provides a measure of the strength of the intercalative or groove binding respectively.67
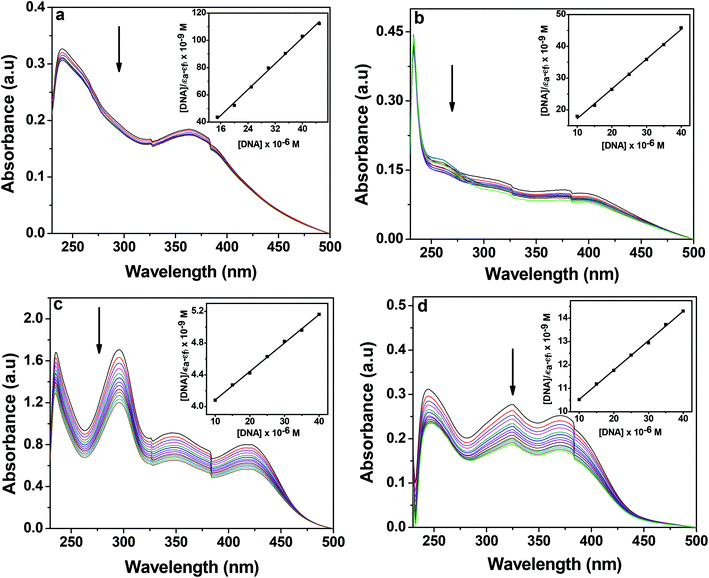 |
| | Fig. 8 Electronic absorption spectra of 1 (a), 2 (b), 3 (c) and 4 (d) (25 μM each) upon the titration of CT-DNA (0–70 μM) in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF. The arrow shows the changes in absorbance with respect to an increase in the CT-DNA concentration. The inset shows the linear fit of [DNA]/(εa − εf) vs. [DNA] and the binding constant (Kb) was calculated using eqn (1). | |
Table 6 DNA binding parameters for complexes 1–4
| Complex |
Binding constant (Kb)a (M−1) |
ΔTmb (°C) |
Stern–Volmer quenching constant (KSV)c (M−1) |
K
appd (M−1) |
|
DNA binding constant by the UV-vis spectral method.
Change in the melting temperature of CT-DNA.
Stern–Volmer quenching constant for the CT-DNA–EB complex.
The apparent DNA binding constant.
|
|
1
|
3.40 × 105 |
+1.05 |
3.06 × 103 |
6.87 × 105 |
|
2
|
1.20 × 105 |
+1.65 |
2.22 × 103 |
6.70 × 105 |
|
3
|
9.60 × 105 |
+1.83 |
5.36 × 104 |
7.34 × 105 |
|
4
|
1.30 × 104 |
+1.11 |
1.32 × 103 |
5.79 × 105 |
In order to quantify the binding affinity of the interaction between CT-DNA and each of 1–4, the binding constant (Kb) was calculated using eqn (1) (Experimental section). The binding constant (Kb) of the complexes was in the range of 1.30 × 104 to 9.60 × 105 M−1. Copper(I) complexes 1 and 2 exhibited higher binding affinity than copper(II) complexes 3 and 4. The binding propensities of ligands to CT-DNA were also estimated. All the ligands showed lower DNA binding affinity than their respective complexes, yielding Kb values of the order of 103 M−1 (ESI† Fig. S8 and Table S1).
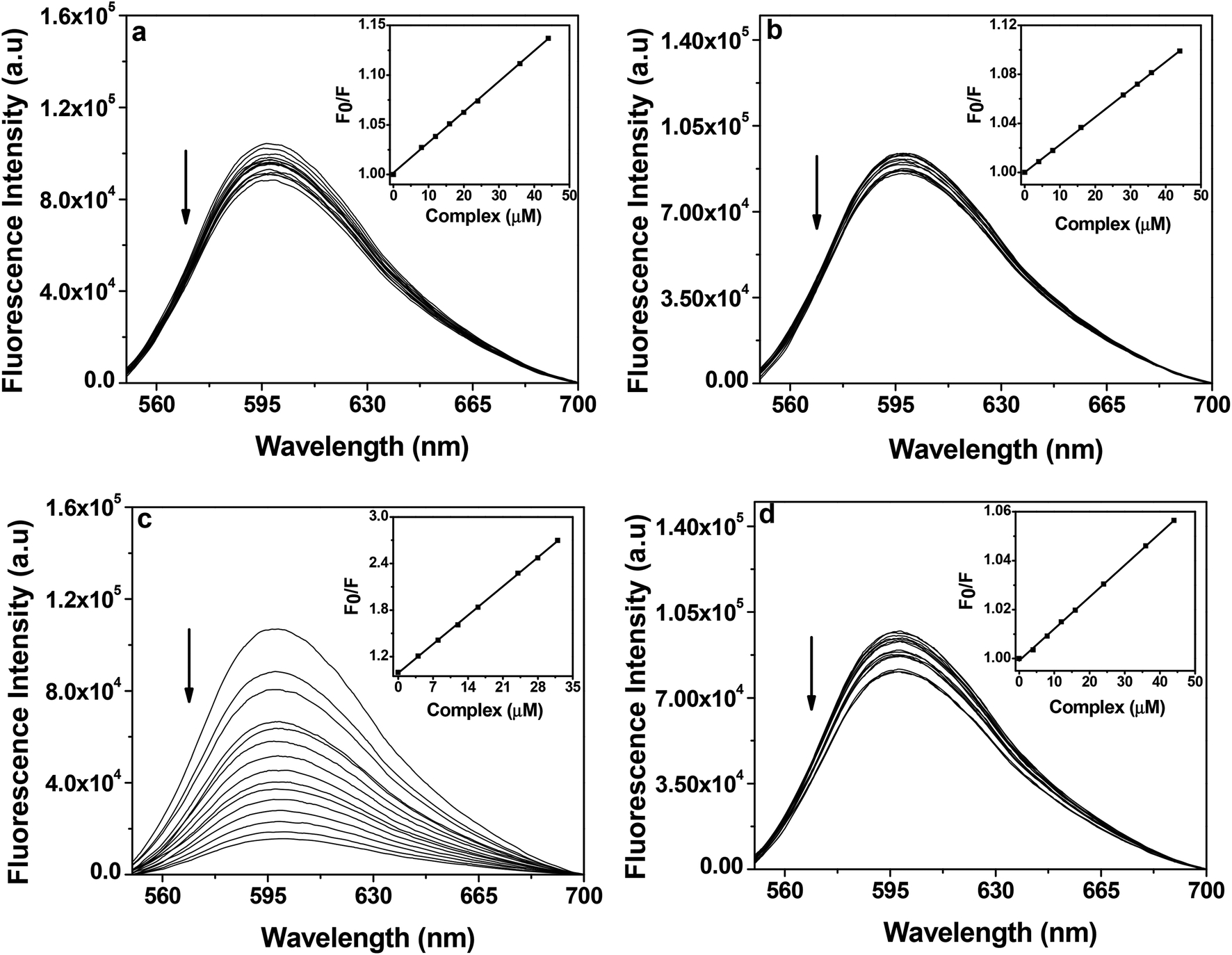
Competitive DNA binding fluorescence studies.
Ethidium dibromide (EB) is a standard intercalating agent and exhibits fluorescence upon binding to DNA. The relative binding of complexes 1–4 to CT-DNA was also investigated by monitoring the quenching of the fluorescence emission from EB bound CT-DNA on successive addition of the complexes. EB is nonemissive in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF due to fluorescence quenching of free EB by solvent molecules, while in the presence of DNA, EB shows enhanced emission intensity due to intercalative binding.68 On addition of the copper complexes to EB bound CT-DNA, the emission intensity at 597 nm was quenched by ∼14% and ∼13% for copper(I) complexes 1 and 2 respectively, whereas copper(II) complexes 3 and 4 exhibited a decrease of ∼82% and ∼10% respectively (Fig. 9). The quenching of emission intensity of ethidium bromide upon addition of 1–4 showed that the complexes probably compete with EB for the binding with DNA. Copper(I) complexes 1 and 2 exhibited a KSV value of 3.06 × 103 and 2.22 × 103 M−1 as calculated from eqn (2) and a Kapp value of 6.87 × 105 and 6.70 × 105 M−1 as calculated from eqn (3) respectively (Table 6). Similarly, for copper(II) complexes, 3 exhibited the highest decrease in emission intensity of EB which is well reflected in its KSV and Kapp values of 5.36 × 104 and 7.34 × 105 M−1 respectively. Complex 4 showed the least decrease in emission intensity of EB, which is consistent with its lower KSV and Kapp values of 1.32 × 103 and 5.79 × 105 M−1 respectively (Table 6). The higher KSV and Kapp values of 3 than the other complexes may be attributed to its higher solubility in aqueous medium and the presence of the quinonoidal group in the thiosemicarbazone ligand coordinated to the metal.
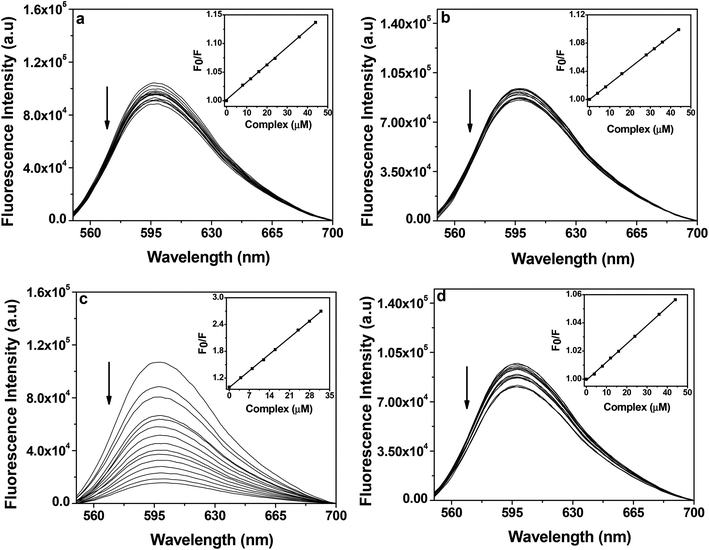 |
| | Fig. 9 Fluorescence emission spectra of ethidium bromide (2 μM) bound to CT-DNA (50 μM) in the presence of complex 1 (a), 2 (b), 3 (c) and 4 (d) (0–60 μM) in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF. The arrow indicates the effect of increasing the concentration of the complex on the fluorescence emission of ethidium bromide bound CT-DNA. The inset shows the linear fit of F0/F vs. [complex] and the Stern–Volmer quenching constant (KSV) was calculated using eqn (2). | |
The Kapp of the complexes were of the order of ∼102 lower than the classical intercalator EB (i.e. 1.00 × 107 M−1), which suggests that the interactions between the complexes and CT-DNA were possibly groove binding in nature. The trend in Kapp values are in line with the trend in the Kb values which were obtained from the UV-Vis absorption spectral studies (Table 6). Control competitive DNA binding experiments with ligands showed that ligands have lower KSV and Kapp values than their corresponding complexes (ESI† Fig. S9 and Table S1).
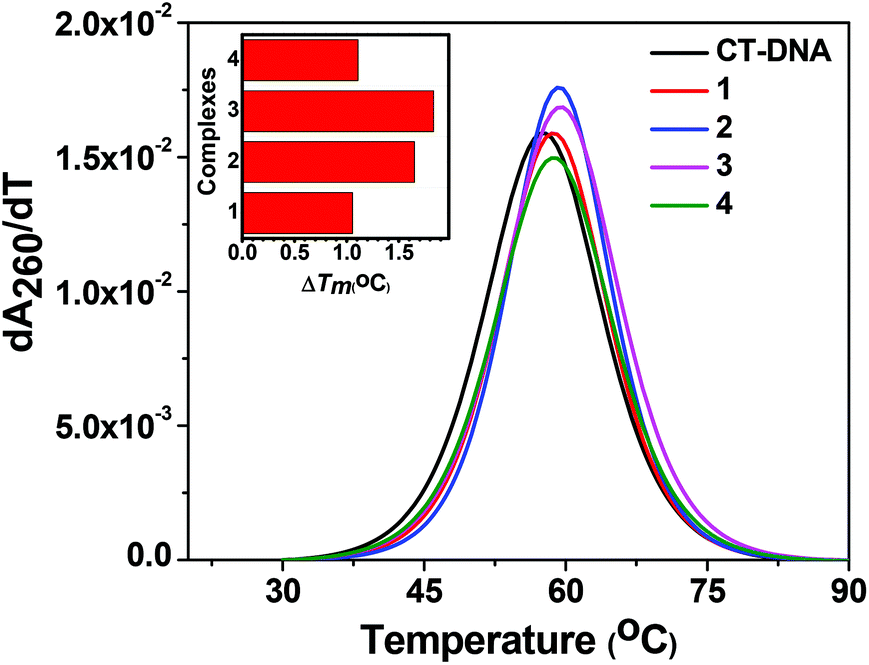
Thermal melting studies.
In order to gain an insight into the nature of interactions and the conformational changes brought about by the complexes on interaction with CT-DNA, thermal denaturation experiments were performed.69 The melting temperature of CT-DNA (Tm) in the absence of any complexes was ∼65.7 °C (Fig. 10). In the presence of the copper complexes the DNA melting temperature (Tm) showed a slight increase from ∼1.05 °C to 1.83 °C (Table 6). Among all the complexes, 3 showed the highest shift of the DNA melting temperature (ΔTm) of +1.83 °C which may be attributed to its better interaction with CT-DNA as evidenced from UV-Vis absorption and competitive DNA binding studies. The lower ΔTm values suggest that the complexes interact with CT-DNA primarily through a groove binding mode rather than an intercalative mode of binding to DNA which generally results in a higher positive shift in the Tm of CT-DNA.69,70
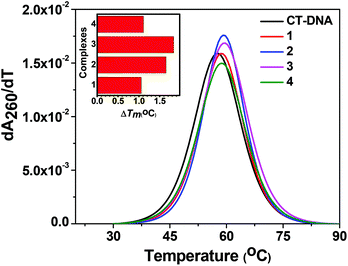 |
| | Fig. 10 Derivative plot of thermal denaturation of CT-DNA (100 μM) in the absence and presence of 1–4 (50 μM). The experiment was done in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF. The inset shows the ΔTm (°C) of the complexes as compared to CT-DNA. | |
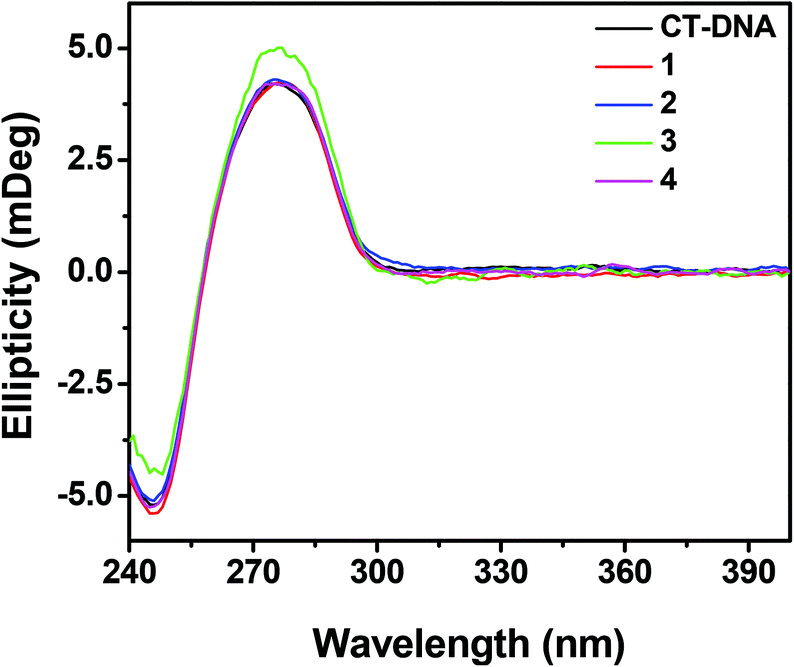
Circular dichroism studies.
Circular dichroism was used to investigate the conformational changes in CT-DNA due to the interaction with the complexes. CT-DNA shows two conserved bands in the UV region, a positive band at 275 nm due to base stacking interaction and a negative band at 245 nm due right handed helicity.71 The interaction of 1, 2 and 4 showed marginal changes in the CD spectra of CT-DNA, whereas the interaction of 3 with CT-DNA induced a decrease in the intensity for the negative ellipticity at 245 nm and an increase in the positive ellipticity band at 275 nm (Fig. 11). These results suggest that interaction of 1, 2 and 4 did not bring about any conformational changes in CT-DNA while 3 perturbed the stacking interaction as well as the right handed helicity of CT-DNA.
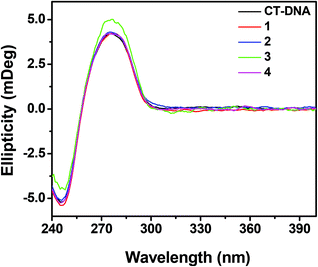 |
| | Fig. 11 Circular dichroism spectra of CT-DNA (50 μM) in the presence and absence of 1–4 (20 μM) in 10 mM Tris–HCl buffer (pH 8.0) containing 1% DMF. The path length of the cuvette was 10 mm. | |
DNA cleavage studies
Chemical-induced DNA cleavage.
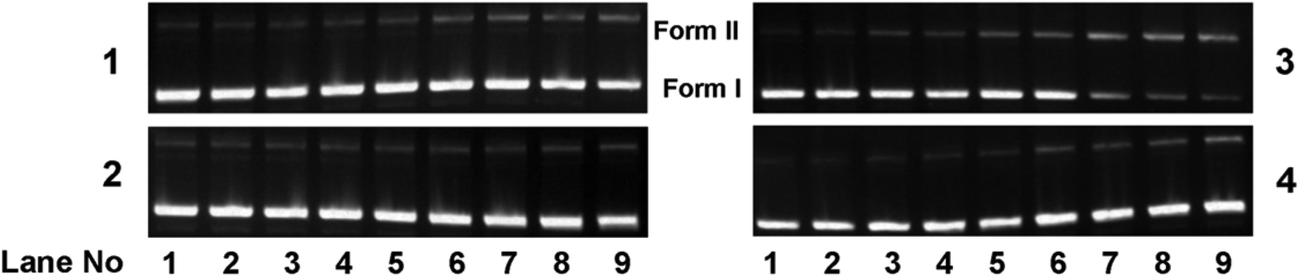
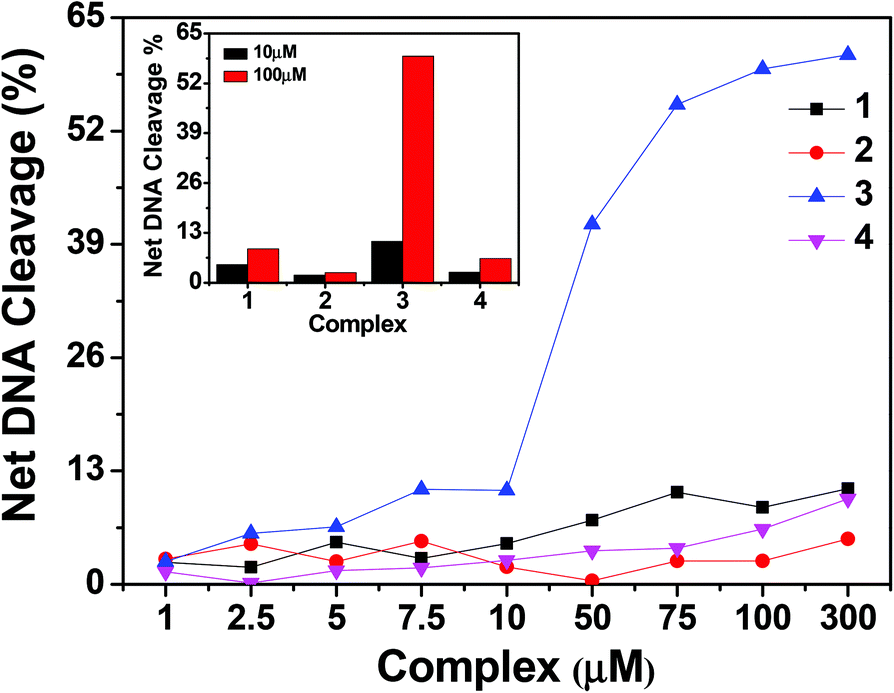
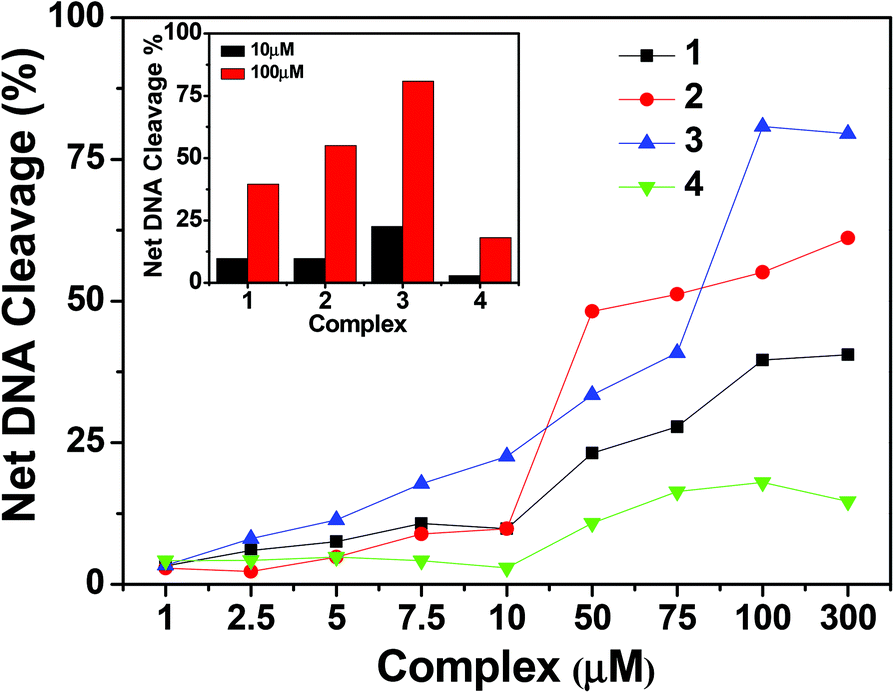
To assess whether the DNA binding properties of the complexes are associated with the chemical nuclease activity, 300 ng of pUC19 DNA was incubated in the presence of hydrogen peroxide as an oxidising agent, with different concentrations of the complexes (1–300 μM) in 50 mM Tris–HCl buffer (pH 8.0) containing 1% DMF in the dark for 1 h. Upon gel electrophoresis, complexes 1, 2 and 4 showed slight DNA cleavage activity ranging from ∼2 to 10%, whereas complex 3 exhibited a maximum chemical nuclease activity of ∼60% at a complex concentration of 100 μM (Fig. 12 and 13). This enhanced chemical nuclease activity of 3 can be possibly rationalized on the basis of its higher binding affinity towards CT-DNA as observed from the DNA binding studies. Control experiments using the oxidizing agent hydrogen peroxide and the ligands showed that neither hydrogen peroxide nor the ligands were cleavage active under similar experimental conditions (ESI† Fig. S10). All the complexes, in the absence of the oxidising agent, were cleavage inactive under dark conditions.
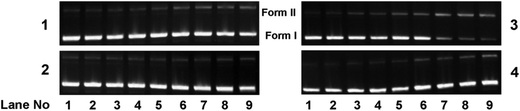 |
| | Fig. 12 Gel diagram showing the concentration dependent chemical nuclease activity by 1–4; 300 ng of SC pUC19 DNA at different concentrations of the complexes [1–300 μM in 50 mM Tris–HCl buffer (pH 8.0) containing 1% DMF] was treated with hydrogen peroxide (0.5 mM) in the dark for 1 h at 37 °C. Lanes 1–9: 1, 2.5, 5.0, 7.5, 10, 50, 75, 100 and 300 μM of 1–4. | |
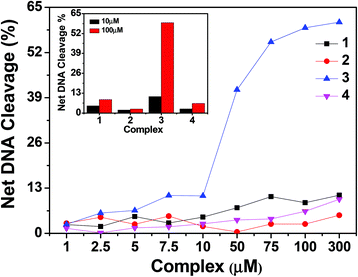 |
| | Fig. 13 Concentration dependent chemical nuclease activity by 1–4; 300 ng of SC pUC19 DNA at different concentrations of the complexes [1–300 μM in 50 mM Tris–HCl buffer (pH 8.0) containing 1% DMF] was treated with hydrogen peroxide (0.5 mM) in the dark for 1 h at 37 °C. The net DNA cleavage percent was calculated using eqn (4). The inset shows a bar diagram representation of the net DNA cleavage of different complexes at 10 and 100 μM. | |
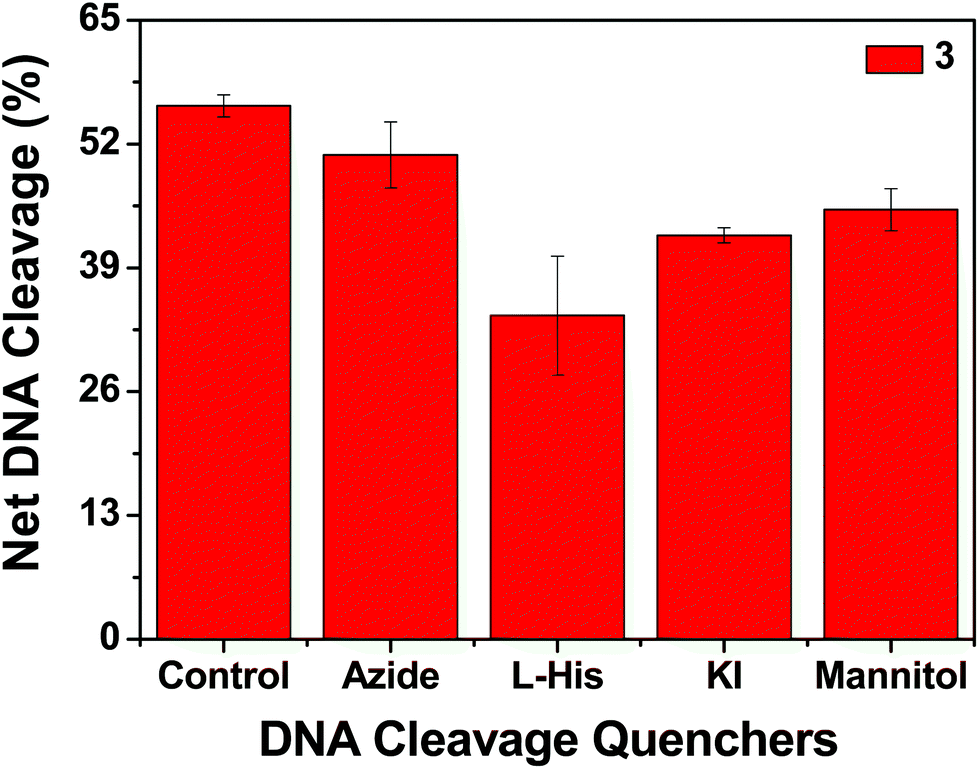
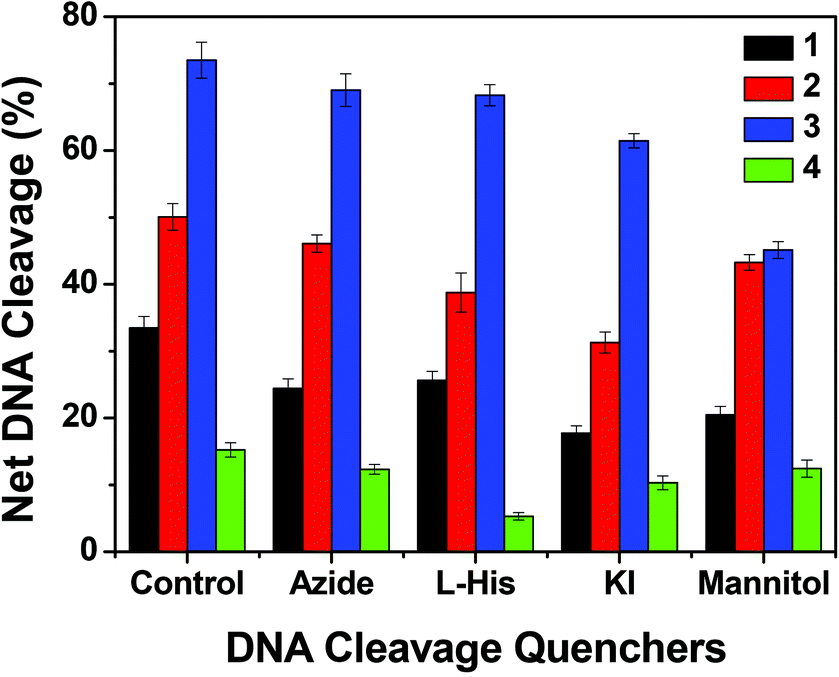
In order to elucidate the probable mechanistic aspects of the chemical-induced DNA cleavage activity by these complexes various inhibitors were used. The chemical-induced DNA cleavage reactions may involve reactive oxygen species (ROS) such as singlet oxygen (1O2) and hydroxyl radicals (˙OH). Therefore, NaN3 and L-histidine were used as singlet oxygen quenchers, while KI and D-mannitol were employed as hydroxyl radical quenchers. Complexes 1, 2 and 4 did not show any appreciable inhibition in the chemical-induced DNA cleavage activity in the presence of the various additives, which may be due to the diminished chemical nuclease activity of these complexes (ESI† Fig. S11). On the other hand, addition of singlet oxygen quenchers like NaN3 and L-histidine inhibited the DNA cleavage activity of complex 3 by ∼6% and ∼22% respectively. Similarly, in the presence of the hydroxyl radical scavengers KI and D-mannitol, the chemical nuclease activity of complex 3 was reduced by ∼14% and ∼11% respectively (Fig. 14). These results suggest that among all the copper complexes, 3 exhibits chemical-induced DNA cleavage activity probably via both singlet oxygen and hydroxyl radical pathways.
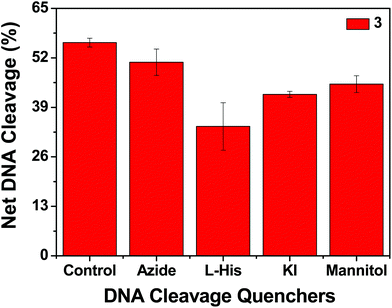 |
| | Fig. 14 Chemical nuclease activity of SC pUC19 DNA by 3 in the presence of various additives in 50 mM Tris–HCl buffer (pH 8.0) containing 1% DMF. SC pUC19 DNA (300 ng) in the presence of various additives was treated with hydrogen peroxide (0.5 mM) in the dark for 1 h at 37 °C with 3 (100 μM). The additive concentrations were: sodium azide (0.5 mM), L-histidine (0.5 mM), KI (0.5 mM) and D-mannitol (0.5 mM). | |
Photo-induced DNA cleavage.
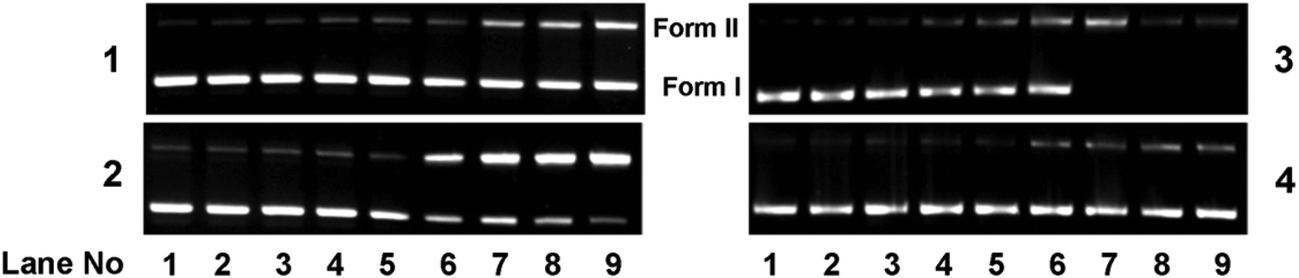
To investigate if the chemical nuclease activity of the complexes was also associated with photonuclease activity, photo-induced DNA cleavage was carried out with 300 ng pUC19 DNA in the presence and absence of complexes 1–4 (Fig. 15). The extent of DNA cleavage by the complexes was monitored in a concentration dependent manner as shown in Fig. 16. All the complexes (except 4) showed ∼10% or more photo-induced DNA cleavage activity at a complex concentration of 10 μM, which ultimately was saturated at a complex concentration of 100 μM. Among the copper(I) complexes, 2 exhibited greater (∼55%) photo-induced DNA cleavage activity than 1 (∼40%). On the other hand, in copper(II) complexes, 3 showed a significantly higher photo-induced DNA cleavage activity of ∼80%, whereas 4 exhibited a minimal DNA cleavage activity of ∼18% (Fig. 16). The higher DNA cleavage activity of 3 may be attributed to its higher binding affinity to DNA as shown in binding studies and may also be because of its solubility in aqueous medium and the presence of a quinonoidal group in the thiosemicarbazone ligand coordinated to the metal. Control experiments suggest that neither DMF (1%) nor the ligands showed any photo-induced DNA cleavage activity, which implies that the ligands or DMF alone are cleavage inactive under similar conditions (ESI† Fig. S12).
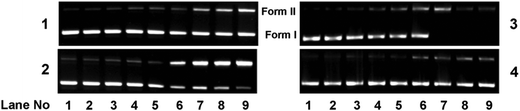 |
| | Fig. 15 Gel diagram showing the concentration dependent DNA cleavage by 1–4; 300 ng of SC pUC19 DNA at different concentrations of the complexes [1–300 μM in 50 mM Tris–HCl buffer (pH 8.0) containing 1% DMF] was photo-irradiated with UVA at 350 nm for 1 h. Lanes 1–9: 1, 2.5, 5.0, 7.5, 10, 50, 75, 100 and 300 μM of 1–4. | |
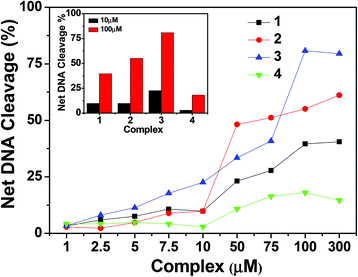 |
| | Fig. 16 Concentration dependent DNA cleavage by 1–4; 300 ng of SC pUC19 DNA at different concentrations of the complexes [1–300 μM in 50 mM Tris–HCl buffer (pH 8.0) containing 1% DMF] was photo-irradiated with UVA at 350 nm for 1 h. The net DNA cleavage percent was calculated using eqn (4). The inset shows a bar diagram representation of the net DNA cleavage of different complexes at 10 and 100 μM. | |
To understand the mechanistic aspects of the photonuclease activity of these complexes, we used the same additives as those used in exploring the mechanism of chemical nuclease activity. The DNA cleavage reaction involving molecular oxygen can proceed by two mechanistic pathways, namely, by a type-II process involving singlet oxygen species (1O2) or by a photo-redox pathway involving reactive hydroxyl radicals (˙OH).72 In the case of copper(I) complexes, the singlet oxygen quenchers, such as NaN3 and L-histidine, showed a reduced photonuclease activity of complex 1 by ∼9% and ∼8% and of complex 2 by ∼4% and ∼12% respectively. Similarly the hydroxyl radical scavengers, KI and D-mannitol, exhibited a significant inhibition of photo-induced DNA cleavage activity of complex 1 by ∼26% and ∼13% and of complex 2 by ∼19% and ∼7% respectively (Fig. 17 and ESI† Fig. S13), while in the case of copper(II) complexes, the presence of singlet oxygen quenchers, NaN3 and L-histidine, decreased the photonuclease activity of complex 3 by ∼4% and ∼5% and complex 4 by ∼3% and ∼10% respectively. Similarly, KI and D-mannitol (hydroxyl radical quenchers) showed an inhibition of DNA cleavage activity by ∼12% and ∼28% for complex 3 and ∼5% and ∼3% for complex 4 (Fig. 17 and ESI† Fig. S12). These results suggest that 1 and 2 exhibit photo-induced DNA cleavage activity possibly via both singlet oxygen and hydroxyl radical pathways while the mechanistic pathway for 3 and 4 cannot be stated with a degree of certainty. Of the two pathways, the hydroxyl radical dominates over the singlet oxygen pathway as the hydroxyl radical scavengers showed higher inhibitory effect than the singlet oxygen quenchers.
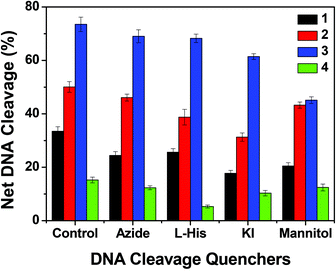 |
| | Fig. 17 DNA cleavage of SC pUC19 DNA by 1–4 in the presence of various additives in 50 mM Tris–HCl buffer (pH 8.0) containing 1% DMF. SC pUC19 DNA (300 ng) in the presence of various additives was photo-irradiated at 350 nm for 1 h with 1–4 (100 μM). The additive concentrations were: sodium azide (0.5 mM), L-histidine (0.5 mM), KI (0.5 mM) and D-mannitol (0.5 mM). | |
Anticancer activity
Inhibition of cancer cell viability.
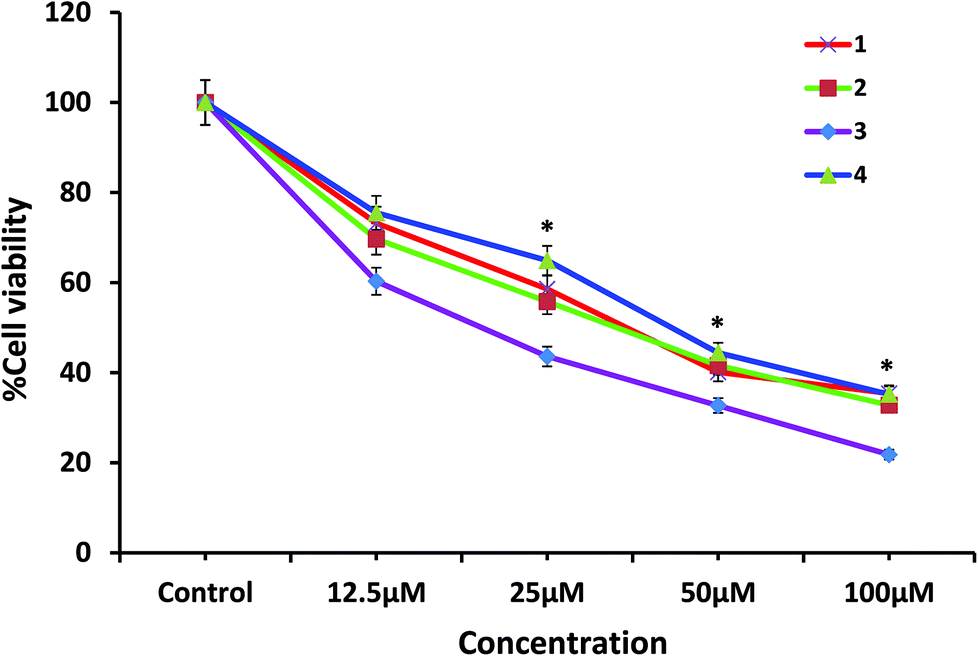
In the present study, the antiproliferative efficacy of 1–4 was assayed by determining the viability of HeLa cells using the MTT assay. The ligands (HL1, HL2 and H2L4) and metal precursors (CuBr, CuCl and CuCl2) gave IC50 values of >200 μM but the other ligand (HL3) gave IC50 values of 98 μM, whereas corresponding complexes 1–4 gave values in the range 20–36 μM. The significant decrease in inhibitory activity for the ligand compared to the metal complex clearly indicates that incorporation of copper into the ligand environment has a marked effect on cytotoxicity. A possible explanation is that by coordination the polarity of the ligand and of the central metal ion are reduced through the charge equilibration, which favors permeation of the complexes through the lipid layer of the cell membrane.73 The present results are consistent with the observation that metal complexes can exhibit greater biological activities than the free ligand.36
Comparing the activity of four complexes, the cytotoxic activity follows the order 3 > 2 > 1 > 4, which is reflected by the IC50 values with the dose dependence illustrated in Table 7 and Fig. 18. It is remarkable that 3, having a quinonoidal group in the thiosemicarbazone ligand coordinated to the metal, is most active. This is in correlation with the fact that the derivatives of quinoline are found to show good biological activities such as antioxidation, antiproliferation, and anti-inflammation.74,75
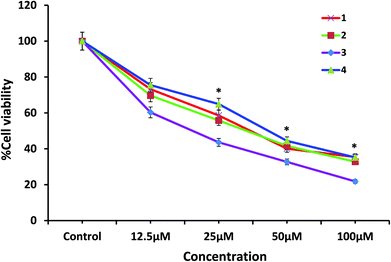 |
| | Fig. 18 Effect of 1, 2, 3 and 4 on cancer cell viability and growth: HeLa cells were treated with different concentrations of the test compounds for 72 h and then cell viability was measured by the MTT assay. Data are reported as the mean ± S.D. for n = 6 and compared against the control using a Student's t-test. (* denotes significance compared to the control.) | |
Table 7 Cytotoxic scores in HeLa cancer cells for 1–4
| Compounds |
IC50 (μM) |
|
1
|
33.5 ± 4.67 |
|
2
|
31.5 ± 5.72 |
|
3
|
19.8 ± 3.54 |
|
4
|
36 ± 6.74 |
A possible single shot drug for cancer cure has been elusive so far, due to their multiple occurrences in more than a hundred forms, and several cases of recurrence of cancer post chemotherapy and surgery are well known. Interestingly, equating the efficacy of our synthesized novel copper compounds against the presently available common chemodrugs sold to patients, we found that Cisplatin, Gefitinib, Gemcitabine, 5-Florouracil, and Vinorelbine had an IC50 of 13 μM, 20 μM, 35 μM, 40 μM and 48 μM respectively on HeLa cells under conditions similar to our experiment.76 These findings give a positive revelation about the potential of our copper compounds as future neoplastic precursor drug candidates.
Nuclear staining assay.
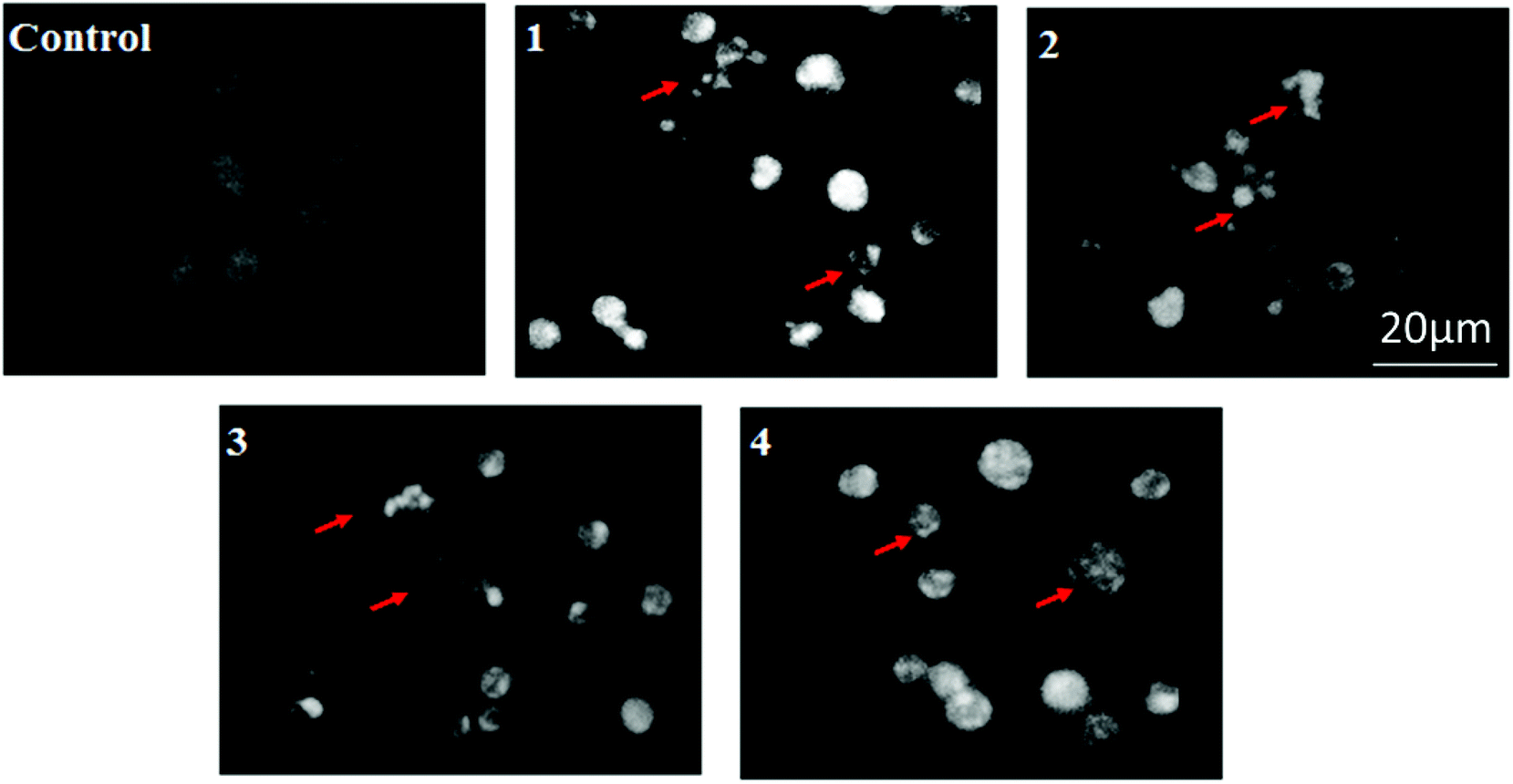
To investigate the apoptotic potential of test compounds in HeLa cells, DAPI staining was performed. Chromatin condensation during the process of apoptosis (type I programmed cell death) is a characterizing marker of nuclear alteration. HeLa cells were treated with 30 μM, 25 μM, 15 μM and 30 μM of 1, 2, 3 and 4 respectively. All the doses were given below the calculated IC50 and the cells were incubated for 24 h before the DAPI nuclear staining assay. Control cells hardly showed any sort of condensation in comparison to the test compound's activity (as shown in Fig. 19) when the cells were examined under a fluorescent microscope, with a DAPI filter. All images clearly demonstrate the brightly condensed chromatin bodies and the nuclear blebbings, marked by arrows in the figure. The drug treated groups, besides showing nuclear changes, also revealed a shrinking morphology, which is another important hallmark of apoptosis.
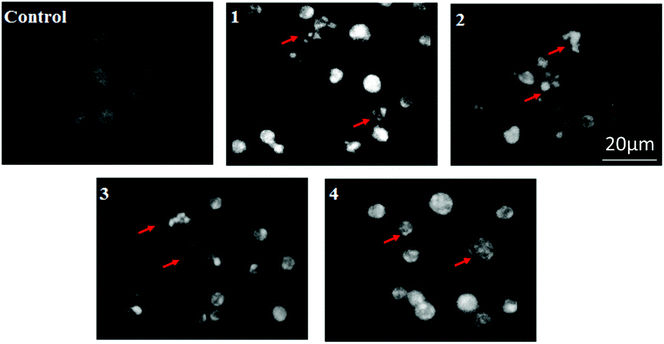 |
| | Fig. 19 Study of apoptosis by morphological changes in nuclei of HeLa cells: after treatment, HeLa cells from the control and the treated group were fixed with 3.7% formaldehyde for 15 min, permeabilized with 0.1% Triton X-100 and stained with 1 μg ml−1 DAPI for 5 min at 37 °C. The cells were then washed with PBS and examined by fluorescence microscopy (Olympus IX 71) (200×). | |
Conclusion
The following are the salient observations and findings of this work:
(a) Two Cu(I) complexes 1 [Cu(HL1)(PPh3)2Br]·CH3CN and 2 [Cu(HL2)(PPh3)2Cl]·DMSO and two Cu(II) complexes 3 [(Cu2L32Cl)2(μ-Cl)2]·2H2O and 4 [Cu(L4)(Py)] of thiosemicarbazone ligands were synthesized and characterized by structural, analytical, and spectral methods.
(b) The copper complexes 1–4 showed good DNA binding propensity. Their DNA binding activities were determined using UV-Vis absorption titration, competitive DNA binding fluorescence experiments, thermal denaturation studies and circular dichroism spectroscopy. The experimental results show that the complexes interact with CT-DNA probably by groove binding mode, with binding constants ranging from 104 to 105 M−1. The competitive DNA binding fluorescence experiments suggest that among all the complexes, 3 showed the highest quenching constant (KSV) and Kapp values.
(c) Among all the complexes, 3 displayed a significant chemical nuclease activity in the presence of hydrogen peroxide of ∼60%. All the complexes showed good photo-induced cleavage of pUC19 supercoiled plasmid DNA with complex 3 showing the highest photo-induced DNA cleavage activity of ∼80%.
(d) The results from the mechanistic study suggested that the chemical nuclease activity of complex 3 and the photonuclease activity of complexes 1–2 proceed probably by both singlet oxygen and hydroxyl radical pathways.
(e) In addition, the in vitro antiproliferative activity of complexes 1–4 against the HeLa cell line was assayed. The cytotoxicity of the complexes is affected by various functional groups attached to the thiosemicarbazone derivative whereby 3 was particularly potent against the cells tested.
(f) The results on pharmacological activity of the copper complexes reported in this paper reveal that compound 3 shows the highest activity, which may be due to its solubility in aqueous medium and the presence of a quinonoidal group in the thiosemicarbazone ligand coordinated to the metal.
(g) The results obtained from the present copper complexes are of importance for the development of metal-based agents for anti-cancer applications. Further work is in progress to better identify the mechanism of action and to prepare more potent related compounds for the treatment of cancer.
Acknowledgements
The authors thank the reviewers for their comments and suggestions, which were helpful in preparing the revised version of the manuscript. Funding for this research was provided by the Department of Science and Technology, Govt. of India [grant no. SR/FT/CS-016/2008 (R.D.) and grant no. SR/FT/CS-010/2009 (R.A.)] and the Council of Scientific and Industrial Research, India [grant 37(1535)/12/EMR-II) (A.B.)]. R.D. thanks Prof. S.K. Chattopadhyay, Indian Institute of Engineering Science and Technology, Shibpur, for electrochemical studies and fruitful discussions. S.B. gratefully acknowledges the Council of Scientific and Industrial Research, New Delhi, India, for the award of a senior research fellowship [grant 9/983(0012)2k13-EMR-I]. A.K.P. acknowledges the ICMR, India (grant 45/25/2012-Bio/BMS) for providing a fellowship.
Notes and references
- T. Boulikas and M. Vougiouka, Oncol. Rep., 2003, 10, 1663 CAS.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451 CrossRef CAS PubMed.
- M. Galanski, V. B. Arion, M. A. Jakupec and B. K. Keppler, Curr. Pharm. Des., 2003, 9, 2078 CrossRef CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307 CrossRef CAS PubMed.
- A. M. Angeles-Boza, P. M. Bradley, P. K. L. Fu, S. E. Wicke, J. Bacsa, K. M. Dunbar and C. Turro, Inorg. Chem., 2004, 43, 8510 CrossRef CAS PubMed.
- B. Rosenberg, L. VamCamp, J. E. Trosko and V. H. Mansour, Nature, 1969, 222, 385 CrossRef CAS.
- Z. Wu, Q. Liu, X. Liang, X. Yang, N. Wang, X. Wang, H. Sun, Y. Lu and Z. Guo, J. Biol. Inorg. Chem., 2009, 14, 1313 CrossRef CAS PubMed.
- D. S. Raja, N. S. P. Bhuvanesh and K. Natarajan, Dalton Trans., 2012, 41, 4365 RSC.
- P. J. Bednarski, F. S. Mackay and P. J. Sadler, Anti-Cancer Agents Med. Chem., 2007, 7, 75 CrossRef CAS.
- S. Sharma, S. K. Singh, M. Chandra and D. S. Pandey, J. Inorg. Biochem., 2005, 99, 458 CrossRef CAS PubMed.
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215 RSC.
- C. M. Chang, V. J. Klema, B. J. Johnson, M. Mure, J. P. Klinman and C. M. Wilmot, Biochemistry, 2010, 49, 2540 CrossRef CAS PubMed.
- E. I. Solomon, P. Chen, M. Metz, S.-K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570 CrossRef CAS.
- P. Wojciech and M. D. J. D. Nicholas, BBA-Protein Struct. Mol. Enzymol., 1985, 828, 130 CrossRef.
- J. A. Tainer, E. D. Getzoff, J. S. Richardson and D. C. Richardson, Nature, 1983, 306, 284 CrossRef CAS.
- Y. Matoba, T. Kumagai, A. Yamamoto, H. Yoshitsu and M. Sugiyama, J. Biol. Chem., 2006, 281, 8981 CrossRef CAS PubMed.
-
J. K. Barton, in Bioinorganic Chemistry, ed. I. Bertini, H. B. Grey, S. J. Lippard and J. S. Valentine, University Science Book, Mill Valley, 1994, p. 455 Search PubMed.
-
(a) C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815 CrossRef CAS PubMed;
(b) A. N. Kate, A. A. Kumbhar, A. A. Khan, P. V. Joshi and V. G. Puranik, Bioconjugate Chem., 2014, 25, 102 CrossRef CAS PubMed;
(c) D. S. Raja, N. S. P. Bhuvanesh and K. Natarajan, Inorg. Chem., 2011, 50, 12852 CrossRef CAS PubMed;
(d) M. A. Cater, H. B. Pearson, K. Wolyniec, P. Klaver, M. Bilandzic, B. M. Paterson, A. I. Bush, P. O. Humbert, S. L. Fontaine, P. S. Donnelly and Y. Haupt, ACS Chem. Biol., 2013, 8, 1621 CrossRef CAS PubMed.
-
(a) J. Easmon, G. Purstinger, G. Heinisch, T. Roth, H. H. Fiebig, W. Holzer, W. Jager, M. Jenny and J. Hofmann, J. Med. Chem., 2001, 44, 2164 CrossRef CAS PubMed;
(b) P. Jutten, W. Schumann, A. Hartl, H. M. Dahse and U. Grafe, J. Med. Chem., 2007, 50, 3661 CrossRef PubMed;
(c) D. C. Greenbaum, Z. Mackey, E. Hansell, P. Doyle, J. Gut, C. R. Caffrey, J. Lehrman, P. J. Rosenthal, J. H. McKerrow and K. Chibale, J. Med. Chem., 2004, 47, 3212 CrossRef CAS PubMed.
- R. W. Brockman, J. R. Thomson, M. J. Bell and H. E. Skipper, Cancer Res., 1956, 16, 167 CAS.
- M. B. Ferrari, F. Bisceglie, C. Casoli, S. Durot, I. M. Badarau, G. Pelosi, E. Pilotti, S. Pinelli and P. Tarasconi, J. Med. Chem., 2005, 48, 1671 CrossRef PubMed.
- D. R. Richardson, P. C. Sharpe, D. B. Lovejoy, D. Senaratne, D. S. Kalinowski, M. Islam and P. V. Bernhardt, J. Med. Chem., 2006, 49, 6510 CrossRef CAS PubMed.
- J. A. Ludwig, G. Szakacs, S. E. Martin, B. F. Chu, C. Cardarelli, Z. E. Sauna, N. J. Caplen, H. M. Fales, S. V. Ambudkar, J. N. Weinstein and M. M. Gottesman, Cancer Res., 2006, 66, 4808 CrossRef CAS PubMed.
- D. S. Kalinowski, Y. Yu, P. C. Sharpe, M. Islam, Y. T. Liao, D. B. Lovejoy, N. Kumar, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2007, 50, 3716 CrossRef CAS PubMed.
- J. Chen, Y. W. Huang, G. Liu, Z. Afrasiabi, E. Sinn, S. Padhye and Y. Ma, Toxicol. Appl. Pharmacol., 2004, 197, 40 CrossRef CAS PubMed.
-
(a) D. S. Raja, G. Paramaguru, N. S. P. Bhuvanesh, J. H. Reibenspies, R. Renganathan and K. Natarajan, Dalton Trans., 2011, 40, 4548 RSC;
(b) P. J. Jansson, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2010, 53, 5759 CrossRef CAS PubMed;
(c) S. Adsule, V. Barve, D. Chen, F. Ahmed, Q. P. Dou, S. Padhye and F. H. Sarkar, J. Med. Chem., 2006, 49, 7242 CrossRef CAS PubMed;
(d) H. Huang, Q. Chen, X. Ku, L. Meng, L. Lin, X. Wang, C. Zhu, Y. Wang, Z. Chen, M. Li, H. Jiang, K. Chen, J. Ding and H. Liu, J. Med. Chem., 2010, 53, 3048 CrossRef CAS PubMed.
- D. X. West, I. H. Hall, K. G. Rajendran and A. E. Liberta, Anti-Cancer Drugs, 1993, 4, 231 CrossRef PubMed.
- A. I. Matesanz, C. Joie and P. Souza, Dalton Trans., 2010, 39, 7059 RSC.
-
(a) S. Padhye, Z. Afrasiabi, E. Sinn, J. Fok, K. Mehta and N. Rath, Inorg. Chem., 2005, 44, 1154 CrossRef CAS PubMed;
(b) Z. Afrasiabi, E. Sinn, W. Lin, Y. Ma, C. Campana and S. Padhye, J. Inorg. Biochem., 2005, 99, 1526 CrossRef CAS PubMed.
-
(a) P. Kalaivani, R. Prabhakaran, P. Poornima, F. Dallemer, K. Vijayalakshmi, V. V. Padma and K. Natarajan, Organometallics, 2012, 31, 8323 CrossRef CAS;
(b) L. Otero, M. Vieites, L. Boiani, A. Denicola, C. Rigol, L. Opazo, C. Olea-Azar, J. D. Maya, A. Morello, R. L. K. Siegel, O. E. Piro, E. Castellano, M. González, D. Gambino and H. Cerecetto, J. Med. Chem., 2006, 49, 3322 CrossRef CAS PubMed;
(c) M. Baldini, M. B. Ferrari, F. Bisceglie, P. P. Dall'Aglio, G. Pelosi, S. Pinelli and P. Tarasconi, Inorg. Chem., 2004, 43, 7170 CrossRef CAS PubMed;
(d) M. Baldini, M. B. Ferrari, F. Bisceglie, G. Pelosi, S. Pinelli and P. Tarasconi, Inorg. Chem., 2003, 42, 2049 CrossRef CAS PubMed;
(e) P. Kalaivani, R. Prabhakaran, E. Ramachandran, F. Dallemer, G. Paramaguru, R. Renganathan, P. Poornima, V. V. Padma and K. Natarajan, Dalton Trans., 2012, 41, 2486 RSC;
(f) K. Sampath, S. Sathiyaraj and C. Jayabalakrishnan, Med. Chem. Res., 2014, 23, 958 CrossRef CAS;
(g) J. G. D. Silva, A. A. R. Despaigne, S. R. W. Louro, C. C. Bandeira, E. M. Souza-Fagundes and H. Beraldo, Eur. J. Med. Chem., 2013, 65, 415 CrossRef PubMed;
(h) E. Ramachandran, S. P. Thomas, P. Poornima, P. Kalaivani, R. Prabhakaran, V. V. Padma and K. Natarajan, Eur. J. Med. Chem., 2012, 50, 405 CrossRef CAS PubMed;
(i) J. Lu, H. Guo, X. Zeng, Y. Zhang, P. Zhao, J. Jiang and L. Zang, J. Inorg. Biochem., 2012, 112, 39 CrossRef CAS PubMed.
- Z. Afrasiabi, E. Sinn, S. Padhye, S. Dutta, S. Padhye, C. Newton, C. E. Anson and A. K. Powell, J. Inorg. Biochem., 2003, 95, 306 CrossRef CAS.
- E. M. Jouad, G. Larcher, M. Allain, A. Riou, G. M. Bouet, M. A. Khan and X. D. Thanh, J. Inorg. Biochem., 2001, 86, 565 CrossRef CAS.
- S. Sharma, F. Athar, M. R. Maurya, F. Naqvi and A. Azam, Eur. J. Med. Chem., 2005, 40, 557 CrossRef CAS PubMed.
- T. Wang and Z. Guo, Curr. Med. Chem., 2006, 13, 525 CrossRef CAS.
- A. Gaál, G. Orgován, Z. Polgári, A. Réti, V. G. Mihucz, S. Bősze, N. Szoboszlai and C. Streli, J. Inorg. Biochem., 2014, 130, 52 CrossRef PubMed.
- T. Rosu, E. Pahontu, S. Pasculescu, R. Georgescu, N. Stanica, A. Curaj, A. Popescu and M. Leabu, Eur. J. Med. Chem., 2010, 45, 1627 CrossRef CAS PubMed.
- D. Palanimuthu, S. V. Shinde, K. Somasundaram and A. G. Samuelson, J. Med. Chem., 2013, 56, 722 CrossRef CAS PubMed.
- D. S. Raja, N. S. P. Bhuvanesh and K. Natarajan, Eur. J. Med. Chem., 2011, 46, 4584 CrossRef CAS PubMed.
- F. Bisceglie, M. Baldini, M. B. Ferrari, E. Buluggiu, M. Careri, G. Pelosi, S. Pinelli and P. Tarasconi, Eur. J. Med. Chem., 2007, 42, 627 CrossRef CAS PubMed.
- Z. C. Liu, B. D. Wang, Z. Y. Yang, Y. Li, D. D. Qin and T. R. Li, Eur. J. Med. Chem., 2009, 44, 4477 CrossRef CAS PubMed.
- Z. Afrasiabi, E. Sinn, P. P. Kulkarni, V. Ambike, S. Padhye, D. Deobagakar, M. Heron, C. Gabbutt, C. E. Anson and A. K. Powell, Inorg. Chim. Acta, 2005, 358, 2023 CrossRef CAS PubMed.
- A. R. Cowley, J. R. Dilworth, P. S. Donnelly and J. M. White, Inorg. Chem., 2006, 45, 496 CrossRef CAS PubMed.
- M. C. Rodriguez-Arguelles, L.-S. Ee, J. Sanmartin, P. Pelagatti and F. Zami, J. Inorg. Biochem., 2005, 99, 2231 CrossRef PubMed.
- L. J. Ashfield, A. R. Cowley, J. R. Dilworth and P. S. Donnelly, Inorg. Chem., 2004, 43, 4121 CrossRef CAS PubMed.
- T. S. Lobana, S. Khanna, R. J. Butcher, A. D. Hunter and M. Zeller, Polyhedron, 2006, 25, 2755 CrossRef CAS PubMed.
- P. M. Krishna and K. H. Reddy, Inorg. Chim. Acta, 2009, 362, 4185 CrossRef CAS PubMed.
-
(a) S. P. Dash, S. Pasayat, Saswati, H. R. Dash, S. Das, R. J. Butcher and R. Dinda, Polyhedron, 2012, 31, 524 CrossRef CAS PubMed;
(b) S. Pasayat, S. P. Dash, Saswati, P. K. Majhi, Y. P. Patil, M. Nethaji, H. R. Dash, S. Das and R. Dinda, Polyhedron, 2012, 38, 198 CrossRef CAS PubMed;
(c) Saswati, R. Dinda, C. S. Schmiesing, E. Sinn, Y. P. Patil, M. Nethaji, H. Stoeckli-Evans and R. Acharyya, Polyhedron, 2013, 50, 354 CrossRef CAS PubMed;
(d) S. P. Dash, S. Pasayat, S. Bhakat, S. Roy, R. Dinda, E. R. T. Tiekink, S. Mukhopadhyay, S. K. Bhutia, M. R. Hardikar, B. N. Joshi, Y. P. Patil and M. Nethaji, Inorg. Chem., 2013, 52, 14096 CrossRef CAS PubMed;
(e) S. P. Dash, A. K. Panda, S. Pasayat, R. Dinda, A. Biswas, E. R. T. Tiekink, Y. P. Patil, M. Nethaji, W. Kaminsky, S. Mukhopadhyay and S. K. Bhutia, Dalton Trans., 2014, 43, 10139 RSC.
- Part 1: S. Ghosh and S. Purohit, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1987, 26A, 131 CAS.
- T. S. Lobana, Rekha, R. J. Butcher, A. Castineiras, E. Bermejo and P. V. Bharatam, Inorg. Chem., 2006, 45, 1535 CrossRef CAS PubMed.
- E. Blanc, D. Schwarzenbach and H. D. Flack, J. Appl. Crystallogr., 1991, 24, 1035 CrossRef CAS.
-
Bruker, SADABS, SAINT, SHELXTL and SMART, Bruker AXS Inc., Madison, Wisconsin, SA, 2003 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Eur. J. Med. Chem., 2011, 46, 3376 CrossRef CAS PubMed.
- W. M. Dai, K. W. Lai, A. Wu, W. Hamaguchi, M. Y. Lee, L. Zhou, A. Ishii and S. Nishimoto, J. Med. Chem., 2002, 45, 758 CrossRef CAS PubMed.
- S. Mukhopadhyay, P. K. Panda, B. Behera, C. K. Das, M. K. Hassan, D. N. Das, N. Sinha, A. Bissoyi, K. Pramanik, T. K. Maiti and S. K. Bhutia, Food Chem. Toxicol., 2014, 64, 369 CrossRef CAS PubMed.
- S. Mukhopadhyay, P. K. Panda, D. N. Das, N. Sinha, B. Behera, T. K. Maiti and S. K. Bhutia, Acta Pharmacol. Sin., 2014, 35, 814 CrossRef CAS PubMed.
- T. S. Lobana, P. K. Bhatia and E. R. T. Tiekink, J. Chem. Soc., Dalton Trans., 1989, 749 RSC.
- V. Philip, V. Suni, M. R. P. Kurup and M. Nethaji, Polyhedron, 2006, 25, 1931 CrossRef CAS PubMed.
- A. Sreekanth and M. R. P. Kurup, Polyhedron, 2003, 22, 3321 CrossRef CAS PubMed.
- T. S. Lobana, P. Kumari, G. Hundal and R. J. Butcher, Polyhedron, 2010, 29, 1130 CrossRef CAS PubMed.
- S. Datta, D. K. Seth, R. J. Butcher and S. Bhattacharya, Inorg. Chim. Acta, 2011, 377, 120 CrossRef CAS PubMed.
- V. M. Leovac, G. A. Bogdanović, S. Jovanović, L. Joksović, V. Marković, M. D. Joksović, S. M. Denčić, A. Isaković, I. Marković, F. W. Heinemann, S. Trifunović and I. Đalović, J. Inorg. Biochem., 2011, 105, 1413 CrossRef CAS PubMed.
- M. Nirmala, R. Manikandan, G. Prakash and P. Viswanathamurthi, Appl. Organomet. Chem., 2014, 28, 18 CrossRef CAS.
- P. Chakraborty, J. Adhikary, B. Ghosh, R. Sanyal, S. K. Chattopadhyay, A. Bauzá, A. Frontera, E. Zangrando and D. Das, Inorg. Chem., 2014, 53, 8257 CrossRef CAS PubMed.
-
(a) C. R. Kowol, P. Heffeter, W. Miklos, L. Gille, R. Trondl, L. Cappellacci, W. Berger and B. K. Keppler, J. Biol. Inorg. Chem., 2012, 17, 409 CrossRef CAS PubMed;
(b) Y. H. Zhou, J. Tao, D. L. Sun, L. Q. Chen, W. G. Jia and Y. Cheng, Polyhedron, 2015, 85, 849–856 CrossRef CAS PubMed;
(c) S. Naskar, S. Naskar, H. M. Figge, W. S. Sheldrick, M. Corbella, J. Tercero and S. K. Chattopadhyay, Polyhedron, 2012, 35, 77–86 CrossRef CAS PubMed.
-
(a) D. Mishra, S. Naskar, M. G. B. Drew and S. K. Chattopadhyay, Inorg. Chim. Acta, 2006, 359, 585 CrossRef CAS PubMed;
(b) P. Paul, D. K. Seth, M. G. Richmond and S. Bhattacharya, RSC Adv., 2014, 4, 1432 RSC.
- P. Kumar, S. Gorai, M. K. Santra, B. Mondal and D. Manna, Dalton Trans., 2012, 41, 7573 RSC.
- A. K. Patra, T. Bhowmick, S. Ramakumar, M. Nethaji and A. R. Chakravarty, Dalton Trans., 2008, 6966 RSC.
- Y. An, S. D. Liu, S. Y. Deng, L. N. Ji and Z. W. Mao, J. Inorg. Biochem., 2006, 100, 1586 CrossRef CAS PubMed.
- S. Banerjee, A. Hussain, P. Prasad, I. Khan, B. Banik, P. Kondaiah and A. R. Chakravarty, Eur. J. Inorg. Chem., 2012, 3899 CrossRef CAS.
- L. Li, Q. Guo, J. Dong, T. Xu and J. Li, J. Photochem. Photobiol., B, 2013, 125, 56 CrossRef CAS PubMed.
- P. K. Sasmal, S. Saha, R. Majumdar, S. De, R. R. Dighe and A. R. Chakravarty, Dalton Trans., 2010, 39, 2147 RSC.
- P. G. Avaji, C. H. V. Kumar, S. A. Patil, K. N. Shivananda and C. Nagaraju, Eur. J. Med. Chem., 2009, 44, 3552 CrossRef CAS PubMed.
- I. R. Canelón and P. J. Sadler, Inorg. Chem., 2013, 52, 12276 CrossRef PubMed.
- J. S. Casas, M. S. G. Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197 CrossRef CAS.
- M. Ahmed and K. Jamil, Biol. Med., 2011, 3, 60 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1 and Fig. S1–S13. CCDC 1002348–1002351 for 1–4. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03764b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a