
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, characterization and cellular location of cytotoxic constitutional organometallic isomers of rhenium delivered on a cyanocobalmin scaffold†
Giuseppe
Santoro
a,
Theodora
Zlateva
b,
Albert
Ruggi
a,
Luca
Quaroni‡
b and
Fabio
Zobi
*a
aDepartment of Chemistry, University of Fribourg, Chemin du Musée 9, CH-1700 Fribourg, Switzerland. E-mail: fabio.zobi@unifr.ch; Fax: (+41) 044 635 6802; Tel: (+41) 044 635 4623
bPaul Scherrer Institute, Swiss Light Source, 5232, Villigen-PSI, Switzerland
First published on 9th March 2015
Abstract
Constitutional isomers of cyanocobalamin adducts based on a fluorescent rhenium tris-carbonyl diimine complex were prepared, characterized and tested against PC-3 cancer cells. The adducts differ only in the relative binding position of the organometallic species which is either bound at the cyano or the 5′-hydroxo group of vitamin B12. When tested for their cytotoxic potency, the species showed IC50 values in the low μM rage. Upon conjugation to the vitamin an energy transfer process causes an extremely low quantum yield of fluorescence emission, making the conjugates unsuitable for fluorescence imaging. However, by exploiting the vibrational signature of the fac-[Re(CO)3]+ core, their cellular distribution was evaluated via FTIR spectromicroscopy.
Introduction
The development of facial-rhenium(I) tris-carbonyl diimine complexes (where diimine = N^N chelating bipyridine or phenanthroline type ligand) as anticancer and cell imaging agents has received considerable attention in the last decade.1–12 The complexes are particularly attractive due to their ease of preparation, structural modification and appropriate emission wavelength for visible detection which may be tuned by e.g. lowering the LUMO energy. The cytotoxic properties of a number of fac-[Re(CO)3(N^N)L]n compounds have been tested on a variety of cancer cell lines with IC50 values averaging in the low μM range.13–16 Furthermore, the emissive properties of the complexes (generally emitting in the yellow/green region of the visible spectrum) has allowed a detailed picture of their cellular distribution.3,17–19Reported modifications of the axial or of the diimine ligands of the species, either directed at increasing the inhibitory potency of active compounds or at fine-tuning their emissive properties, tend to increase the hydrophobicity of the complexes.1,5 In general this is perceived as a limitation for the possible in vivo application of the compounds. Attempts to increase water solubility of the complexes have been made by e.g. introducing poly(ethyleneglycol) chains, sulfonates groups or peptides.20–22
An alternative approach to increase hydrophilicity of organometallic and inorganic anticancer agents has been presented by Alberto. The strategy entails direct coordination of the drug to the axially coordinated CN− group of vitamin B12 (cyanocobalamin).23–26 We find this approach elegant and effective in increasing the biocompatibility of metal-based drugs. Indeed, the use of vitamin B12 as a drug delivery system has attracted the interest of several researchers in the field of medicinal chemistry. The axial CN− group, however, is not the only anchoring site which may be exploited for congenial derivatization of the vitamin. The e-propionamide of the peripheral corrin ring27 and the 2′- and the 5′-hydroxyl group of the ribose sugar28,29 are also valid alternatives. While conjugation to cyanocobalamin increases the biocompatibility of organometallic drugs, the known quenching exhibited by vitamin B12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 30–34 limits its use in fluorescent imaging experiments intended at specifying the intracellular fate of the drug. In some cases, however, the right choice of ligand and spacer might partially overcome this limitation.35
30–34 limits its use in fluorescent imaging experiments intended at specifying the intracellular fate of the drug. In some cases, however, the right choice of ligand and spacer might partially overcome this limitation.35
In the last few years our research efforts have been directed at the development of biocompatible-site specific metal based carbon monoxide releasing molecules (CORMs) via conjugation of an active metal carbonyl complex to natural biomolecules.36–38 Within this context we focused our attention on vitamin B12 as a delivery scaffold for CORMs.39,40 Recently we have also been interested in developing FTIR spectromicroscopy as an imaging technique for organometallic species comprising ligands such as CN−, CNR−, CO and NO.41
In this contribution we describe the synthesis, characterization and cytotoxicity against PC-3 prostate cancer cells of two vitamin B12 adducts bearing a fac-[Re(CO)3(Me2-phen)]+ unit (where Me2-phen = 4,7-dimethyl-1,10-phenanthroline). The adducts (1 and 2 in Fig. 1) are constitutional isomers differing only in the relative position of the carbonyl complex which is either bound at cyano group or the 5′-hydroxyl group of the ribose sugar of the vitamin. Upon conjugation of the fac-[Re(CO)3(Me2-phen)]+ complex to the vitamin an energy transfer process causes an extremely low quantum yield of fluorescence emission, making such constructs unsuitable for fluorescence imaging. However, by exploiting the vibrational signature of the species, the uptake and cellular distribution of the adducts in 3T3 fibroblasts was studied via FTIR spectromicroscopy.
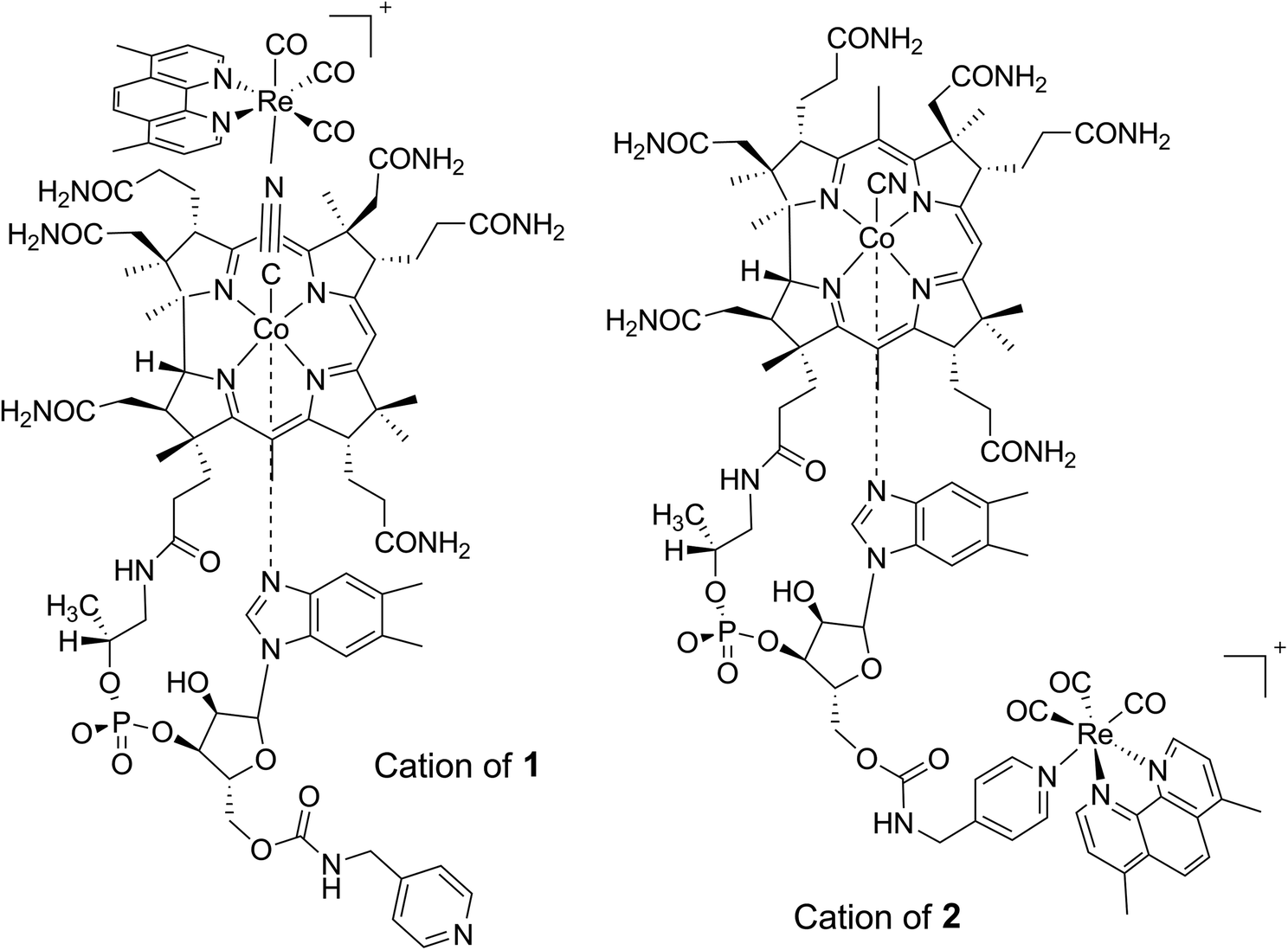 | ||
| Fig. 1 Structures of cations 1 and 2 used in the present study. The adducts are constitutional isomers differing only in the binding arrangement of the metal carbonyl unit. | ||
Results and discussion
Chemical characterization of cyanocobalamin conjugates
Vitamin B12 adducts 1 and 2 were synthesized in moderate to low yields according to the reaction depicted in Scheme 1. In a first step cyanocobalamin was derivatized with 4-picolylamine according to a published procedure.40 The isolated derivative was then reacted with one equivalent of fac-[Re(CO)3(Me2-phen)Br] activated via the removal of Br− with AgCO2CF3. HPLC-MS traces recorded during the course of the reaction revealed the formation numerous products of which the two major species were the complexes of interest. According to HPLC-peak area analysis of all vitamin B12-based species present in the reaction mixture (and assuming similar extinction coefficients for the Soret band), compounds 1 and 2 formed in a relative ratio of 1:1 in ca. 30% yield. However, in order to obtain analytically pure substances, several consecutive preparative HPLC runs had to be carried out. This procedure generally decreased the isolated yield to ca. 15%.
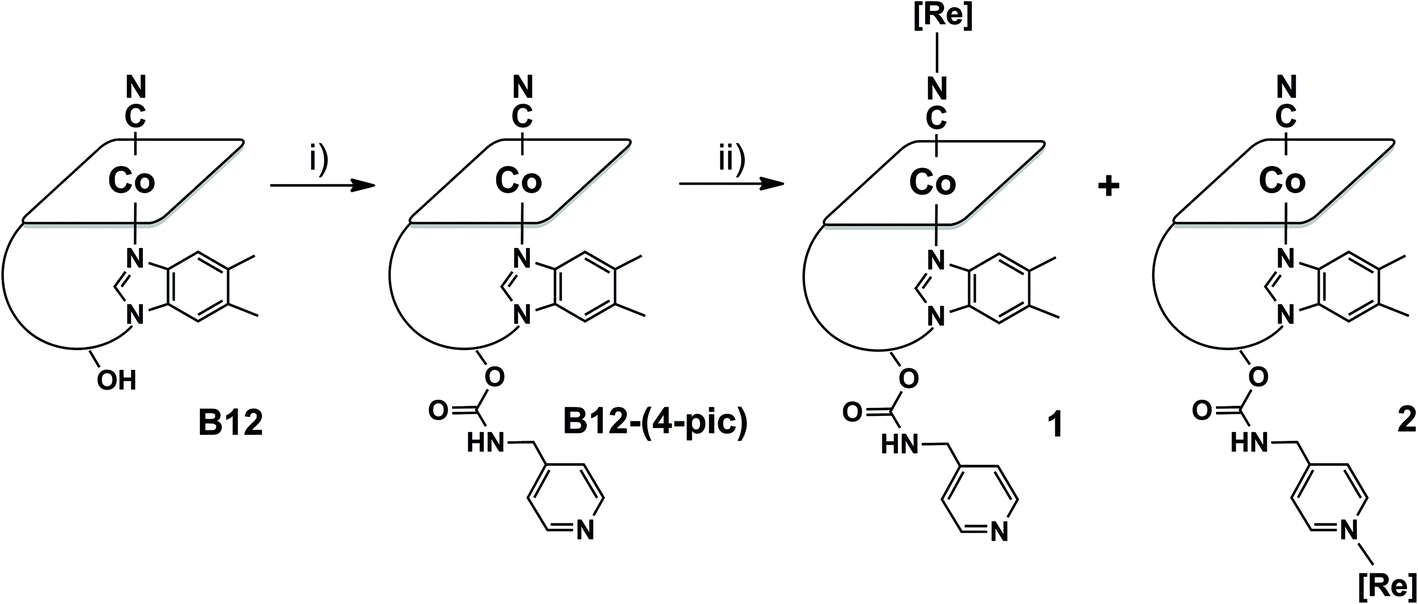 | ||
| Scheme 1 Synthetic scheme for the preparation of 1 and 2. Reagents and conditions: (i) CDT, DMSO, 12 h, RT followed by 4-picolylamine, 24 h, RT; (ii) MeOH and CH2Cl2 mixture 1:1, [Re(CO2CF3)(CO)3(Me2-phen)] 48 h, RT. [Re] = [Re(CO)3(Me2-phen)]+. | ||
The observed masses of the two main products matched exactly, at a value of 1966.7 m/z, and displayed a well-defined rhenium isotope pattern confirming binding of the metal tricarbonyl unit. The presence of two distinguishable species (HPLC retention times of 20.9 and 24.7 min for 1 and 2 respectively) was a clear indication to us that the fac-[Re(CO)3]+ core had addressed binding to both the pending pyridine and the axial cyano group. This conjecture was soon confirmed by the FTIR spectra of the isolated products (see ESI†). In KBr, the spectrum of 1 shows a νC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N band at 2175 cm−1 and the typical νCO stretching frequencies at 2033 and 1937 cm−1. Compound 2 shows a similar pattern with signals at 2134, 2030 and 1924 cm−1 for the νCN and νCO vibrational modes respectively. The hypsochromic νCN shift observed in 1 is characteristic of Co–CN–M bridges (M = transition metal) previously documented for several adducts of this type.24–26
N band at 2175 cm−1 and the typical νCO stretching frequencies at 2033 and 1937 cm−1. Compound 2 shows a similar pattern with signals at 2134, 2030 and 1924 cm−1 for the νCN and νCO vibrational modes respectively. The hypsochromic νCN shift observed in 1 is characteristic of Co–CN–M bridges (M = transition metal) previously documented for several adducts of this type.24–26
To further characterize constitutional isomers 1 and 2, their 1H-NMR spectra were recorded and compared to that of B12-(4-pic). Fig. 2 shows the aromatic region of the spectra. A number of interesting features are observed in the spectrum of 1. In isomer 1 (spectrum B) phenanthroline resonances (peak labeled as “phen#,#”; see ESI† for assignments) show a clear splitting. Assuming free rotation of the Re complex about the Co–CN–Re axis, one would in principle expect the respective hydrogens in the sets of signals to be equal and give rise to the same pattern observed in the NMR spectrum of 2 (spectrum C). As expected, in the latter the phenanthroline resonances appear as two doublets and a singlet.
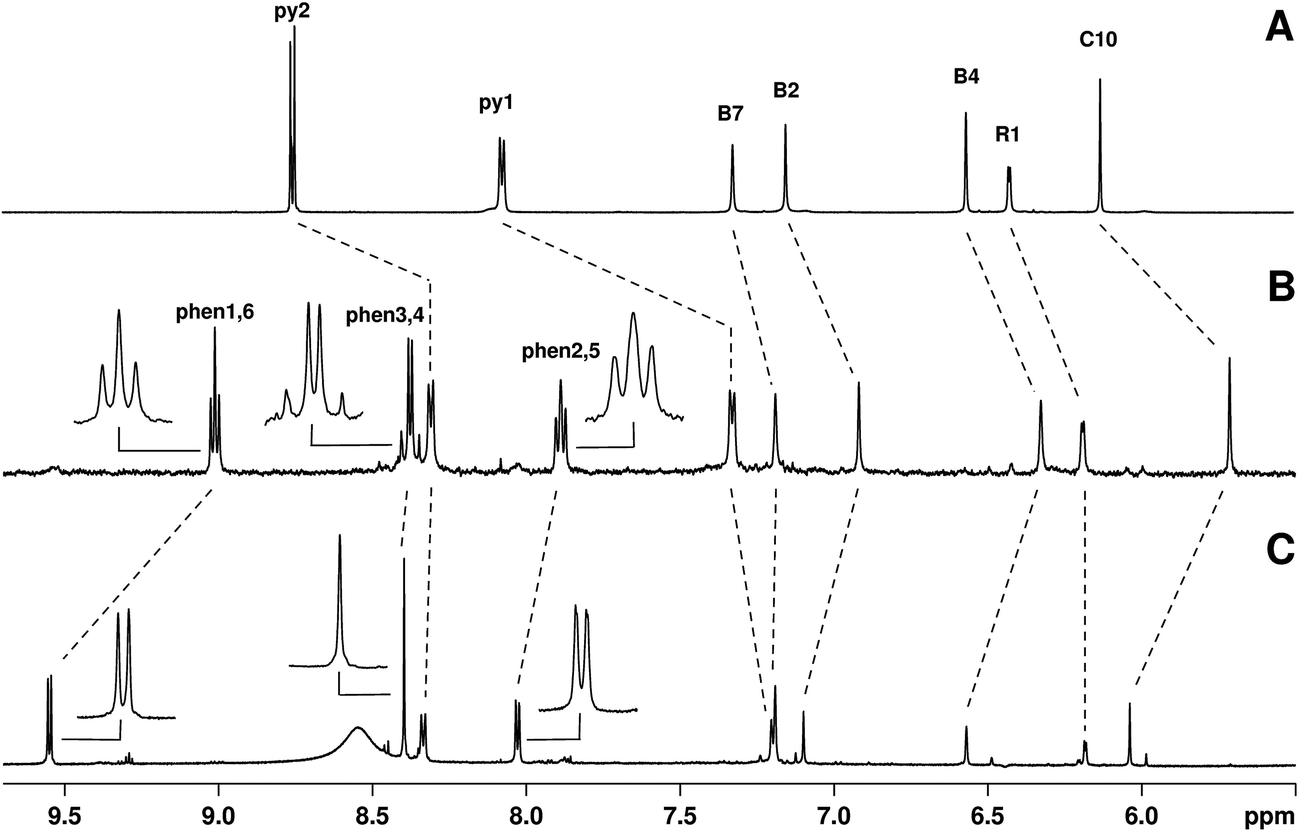 | ||
| Fig. 2 Comparison of aromatic region (5.5–10 ppm) of 1H-NMR spectra of (A to C respectively) B12-(4-pic), 1, and 2. Inserts in spectra B and C are meant to highlight the splitting of the Me2-phen signals in 1. Small peaks visible in spectrum C are ascribed to a minor conformer of 2. Resonances labeled py are those of the pending 4-picoline ligand, while phen are those of the 4,7-dimethyl-1,10-phenanthroline. Signals were all referenced to the residual HOD signal set at 4.80 ppm. The broad signal in C (ca. 8.5 ppm) is tentatively ascribed to resonance of the carbamate. | ||
The apparent loss of the magnetic homotopic nature of the phenanthroline protons in 1 points to either two equally favorable conformers of the metal complex, or to the fact that a single conformer is present in solution and that the nuclei experience different magnetic and chemical environments as a structural consequence. We rationalized this observation via two-dimensional J-resolved and COSY NMR spectroscopy and a theoretical conformational and rotational analysis of 1 (see ESI† for COSY). As shown in Fig. 3, J-resolved NMR spectroscopy indicates the presence of two distinct conformers in solutions. The analysis of the phen3,4 pattern further shows that resonance ascribed to these specific protons arises from a mixture of both an AX and AB system. Finally, the theoretical analysis confirms the presence of two minima in the rotational energy profile of 1 (Fig. 3, right) considered as a function of the rotation of the fac-[Re(CO)3(Me2-phen)]+ unit about the Co–CN–Re bridge.
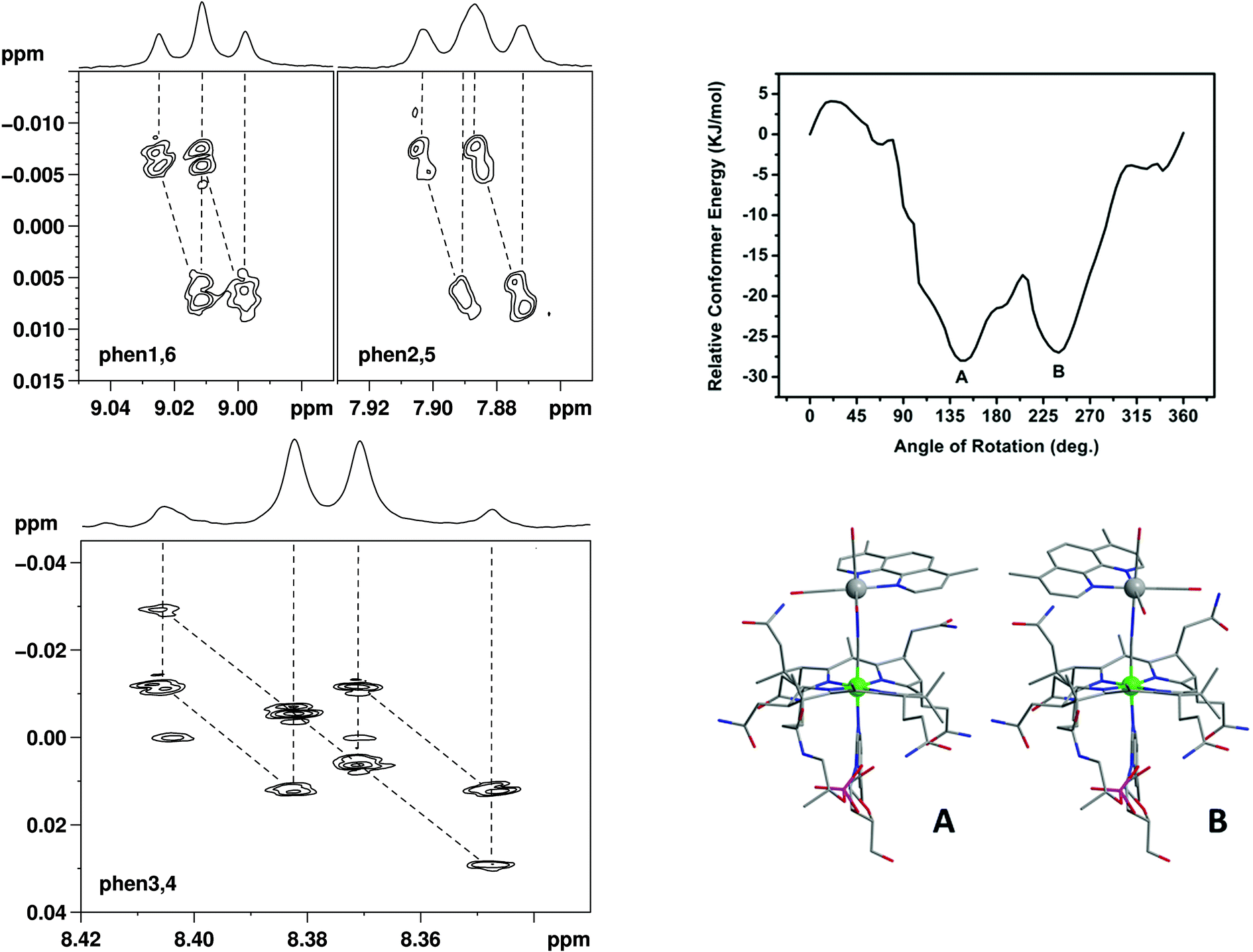 | ||
| Fig. 3 Left: two-dimensional J-resolved NMR spectra of the phenanthroline resonances in isomer 1. Top right: rotational energy profile of 1 as a function of the rotation of the fac-[Re(CO)3(Me2-phen)]+ unit about the Co–CN–Re axis. Bottom right: structure of minimum conformers A and B. | ||
No broadening of the resonances of the pending 4-picoline unit was observed in the NMR spectrum of either isomer. This indicates that gyration of the pyridine unit is principally unhindered in both molecules and that binding of [Re(CO)3(Me2-phen)]+ in 2 does not impose a severe steric strain. Finally, as evidenced from the small peaks visible in spectrum C, 2 is present in solution as a mixture of two conformers with a relative abundance of ca. 9:1. The relative ratio of the two species varied slightly over a 72 h monitoring period both in methanol and water. We made no attempts to elucidate structural differences between these major and minor adducts.
Photophysical characterization
Complexes 1 and 2 were analyzed by UV-Vis absorption spectroscopy and steady-state and time-resolved fluorescence spectroscopy, together with the pristine complex [Re(CO)3(Me2-phen)MeOH]+ in methanol solutions (Table 1). UV-Vis absorption and luminescence emission spectra of [Re(CO)3(Me2-phen)MeOH]+ are reported in Fig. 4. The typical absorption of Re(I) complexes is observed, with a strong ligand-centered (1LC) band centered at 269 nm and a weaker broad band centered at 369 nm, which can be assigned to a metal-to-ligand charge transfer (1MLCT). The pristine complex [Re(CO)3(Me2-phen)MeOH]+ shows a yellow emission, with a broad structureless band centered at 567 nm. The long lifetime displayed by this emission (61 ns) confirms the triplet nature of the excited state and the absence of a vibrational structure suggests that the excited state has a 3MLCT character.2,42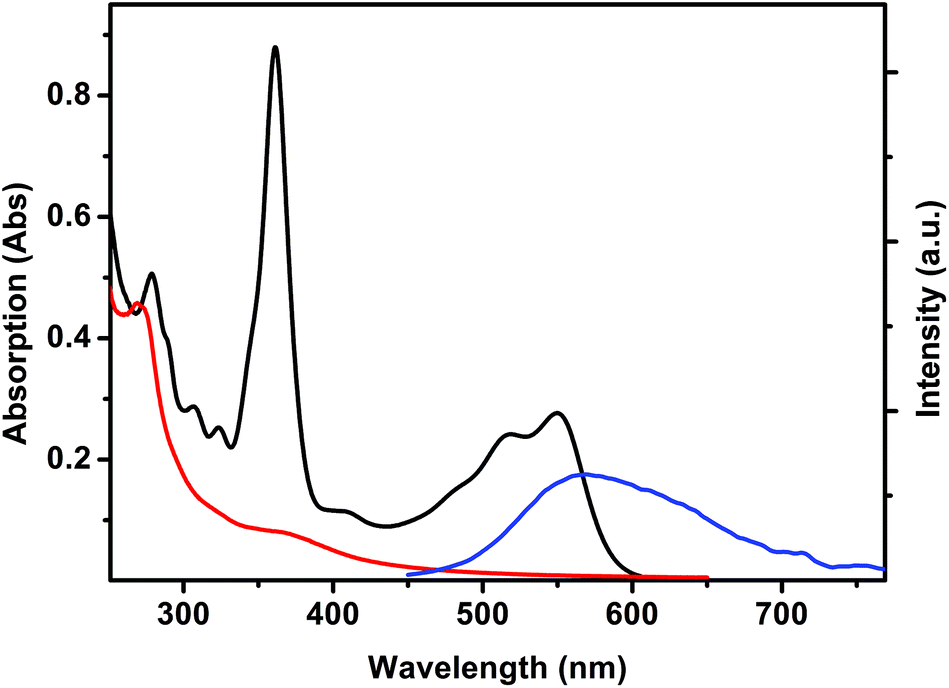 | ||
| Fig. 4 UV-Vis absorption spectra of [Re(CO)3(Me2-phen)MeOH]+ (0.63 mM, red), vitamin B12 (0.013 mM, black) and emission spectrum of [Re(CO)3(Me2-phen)MeOH]+ (λexc = 406 nm, blue) in MeOH solutions. | ||
| λ em (nm) | τ 298 K (ns) | τ 77 K (μs) | |
|---|---|---|---|
| [Re(CO) 3 (Me 2 -phen)MeOH] + | 567 | 61 | 5.1 |
| 1 | 540 (sh) | 25 | 9.5 |
| 596 | 51 | 8.6 | |
| 2 | 560 | 22 | 5.9 |
The emission of [Re(CO)3(Me2-phen)MeOH]+ is partially overlapped with the absorption band of vitamin B12 (Fig. 4). In fact, in the presence of vitamin B12, a pronounced quenching of the luminescence is observed. Since the luminescence lifetime of the rhenium complex remains unchanged, such a quenching can be interpreted as due to a trivial energy transfer.
The UV-Vis spectra of complexes 1 and 2 (ESI†) show a predominance of the vitamin B12 absorption in the region located between 320 and 650 nm. However, in the region below 320 nm, a strong absorption band centered at 274 nm is observed. Such a band is due to the 1LC transition of the [Re(CO)3(Me2-phen)]+ moiety. The emission of complexes 1 and 2 (ESI†) is red-shifted with respect to the pristine complex, however the two compounds show a different behavior in terms of emission spectra. Complex 2 shows a structureless emission band centered at 590 nm, with a monoexponential lifetime of 22 ns. Complex 1 shows a more complicated emission, with a maximum located at 596 nm and a prominent shoulder lying at 540 nm. Time-resolved spectroscopy of complex 1 provides further strong evidence of the presence of two different conformers in solution: by measuring the fluorescence lifetime on the shoulder and on the maximum, two different lifetimes of 25 ns and 51 ns respectively were observed. The presence of two conformers is in agreement with the data obtained from NMR spectroscopy (vide supra) and it is further confirmed by low temperature time-resolved spectroscopy. Both constitutional isomers 1 and 2 and the [Re(CO)3(Me2-phen)MeOH]+ complex show a remarkably long (in the range of microseconds) lifetime at 77 K (see Table 1), as expected for triplet emitting states. Analogously to what was observed at room temperature, complex 1 shows two different lifetimes (9.5 μs and 8.6 μs) at 540 nm and 596 nm, respectively, whilst complex 2 shows a monoexponential lifetime of 5.9 μs. The low lifetimes observed for all the molecules presented in this study, with respect to the values reported in literature for Re(I) phenanthroline complexes, are probably due to different effects.43 In the case of the [Re(CO)3(Me2-phen)MeOH]+ complex, the presence of MeOH as a ligand likely provides a non-radiative decay pathway constituted by the O–H vibrator. Conversely, in the case of complexes 1 and 2, the observed low lifetimes can be explained in terms of energy/electron transfer between the excited state of Re(I) and the vitamin B12 moiety. However, the low lifetimes observed at 77 K with respect to literature complexes having CN or pyridine substituents (which usually show low-temperature lifetimes in the orders of milliseconds)44 suggest the predominance of an energy transfer, in agreement with the observed good overlapping between Re(I) emission and vitamin B12 absorption. The energy transfer causes an extremely low quantum yield (<0.01) for complexes 1 and 2, making such molecules not suitable for fluorescence imaging.
The cytotoxicity of three compounds was also tested on 3T3 fibroblasts grown as an adherent phenotype on CaF2 windows (Table 2). This particular cell line and phenotype was selected in order to perform infrared spectromicroscopy studies aimed at imaging the distribution of the compounds in cells (vide infra). Similarly to what we found for PC-3 cancer cells, the rhenium complex and 1 were the most cytotoxic species with IC50 values in the 50 μM range. Interestingly 2 showed no toxicity against this cell line, up to a tested concentration of 200 μM. Our results do not permit a detailed biological discussion but it is possible that when bound the 5′-hydroxyl group the cytotoxic potency of the fac-[Re(CO)3(Me2-phen)]+ unit is shielded by vitamin B12.
Cellular uptake and localization
We studied the uptake and internalization of compounds 1 and 2 with live 3T3 fibroblasts via synchrotron infrared (IR) spectromicroscopy. As a bio-imaging technique IR spectromicroscopy is capable of providing chemical information at micrometers spatial resolution by exploiting both endogenous and exogenous vibrational frequencies of the measured sample. Organometallic species comprising ligands such as CN−, CNR−, CO and NO are particularly well suited for this technique as they show unique vibrational signatures in a region of the spectrum free of interference from the vibrations of most cellular molecules and thus amiable to rapid interpretation.46,47 In general spectra are calculated by measuring the reference in the medium, in the absence of cells, and moving to the location of the cell to measure the sample trace repeatedly. Therefore time dependent spectra effectively represent spectral changes in the cell location.As expected from the cytotoxicity studies, when 3T3 cells were treated with a solutions of [Re(CO)3(Me2-phen)(MeOH)]+ or 1 (100 μM concentration), they showed severe signs of toxicity. After addition of the adducts, the cells rapidly entered what appeared visually as a necrotic phenotype (typically after 10 min of incubation) characterized by cell enlargement and swelling and subsequent membrane rupture. In order to perform viable experiments we focused on 1 and reduced its concentration to 30 μM. As outlined in the previous section, when 1 is dissolved in the cellular medium the carbonyl complex de-coordinates from vitamin B12, thus it is the [Re(CO)3(Me2-phen)(X)]n complex (where X = solvent or other molecule) which is actually monitored. The above mentioned change in concentration allowed us to study the cell for ca. 30 min without any observable changes in the overall cellular structure. During this period of time, no accumulation of the detached [Re(CO)3(Me2-phen)]+ unit was observed. Spectra recorded by using the medium as the reference, did not show any increase in carbonyl bands over time. Measurement of cellular spectra relative to air (Fig. 5), shows that the compound is present in the cellular location at a concentration comparable to the external one. The results seem to suggest that the [Re(CO)3(Me2-phen)(X)]n complex passively penetrates the cell through the cellular membrane until the extracellular and intracellular concentrations are equal, but it is not actively taken up by the cell. It should be noted that under these experimental conditions, with 3T3 cells grown as an adherent phenotype on CaF2 windows, the first signs of necrosis became apparent within 1 h. IR mapping experiments were not performed.
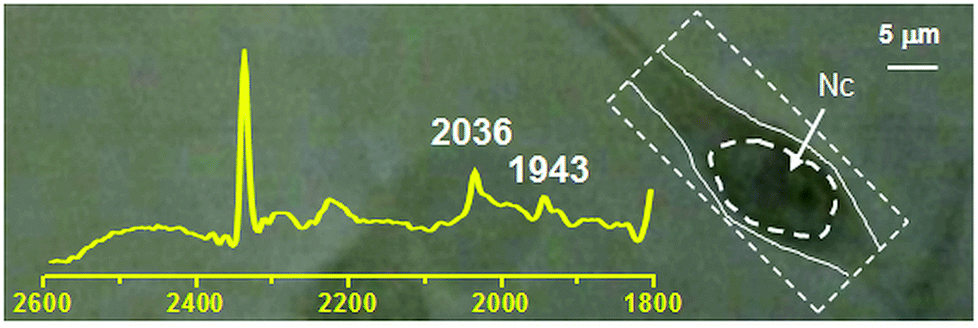 | ||
| Fig. 5 IR spectrum (2600–1800 cm−1 region) superimposed on optical image of a 3T3 fibroblast incubated with a 30 μM solution of 1. The spectrum refers to the measured area defined by the segmented box encompassing the cell to the right. The carbonyl stretching vibrations of 1 at frequencies of 2036 and 1943 cm−1 respectively are indicated. Nc = cell nuclear area identified as previously described.48,49 Intensity of absorbance of peaks at 2036 and 1943 cm−1 is 0.01245 and 0.0081 respectively. | ||
Similar experiments were performed by incubating 3T3 cells with 2 (100–300 μM concentration). In this case difference IR spectra showed evolving spectroscopic changes associated with a slow cellular uptake of the cobalamin adduct (ESI†). Weak bands associated to the νCO stretching frequencies were observed as soon as spectra on the cellular location started being measured, indicating that some uptake had already occurred.
The time course of the accumulation of the CO complex was characterized by an initial slow phase followed by a steep increase in rate about 60 min after exposure (ESI†). After 3.5 h of continuous monitoring the accumulation of 2 approached a plateau. The positions of the νCO stretching frequencies shifted during uptake. During the initial phase of slow uptake the bands peaked at 1915 cm−1 and 2025 cm−1. They progressively shifted to higher frequency as the rate of uptake increased and reached stable positions at 1922 cm−1 and 2028 cm−1 after about 60 min. These changes might be interpreted as the compound experiencing different cellular environments but it is also possible that the organometallic unit is completely detached from the carrier vitamin once in the cell.
After the compound had reached an appreciable intracellular concentration, we performed IR mapping at diffraction limited spatial resolution in order to obtain information on the localization of 2. The maps of Fig. 6 show the distribution of the A1 absorption band throughout the cell, revealing that 2 is present in the cytoplasmic region but is much more concentrated in the proximity of the nucleus. In order to highlight the cellular distribution of 2, the absorption at 2852 cm−1 was also mapped. This band arises from CH2 stretching modes from long alkyl chains and can be used to map the distribution of acyl lipids throughout the cell. This is a marker for the regions of the cell that are rich in membranes, such as mitochondria, the endoplasmic reticulum and the Golgi.50 As is clear from Fig. 6, lipid concentration is highest in the perinuclear environment, where the endoplasmatic reticulum is usually located,50 and shows a remarkably different distribution as compared to 2.
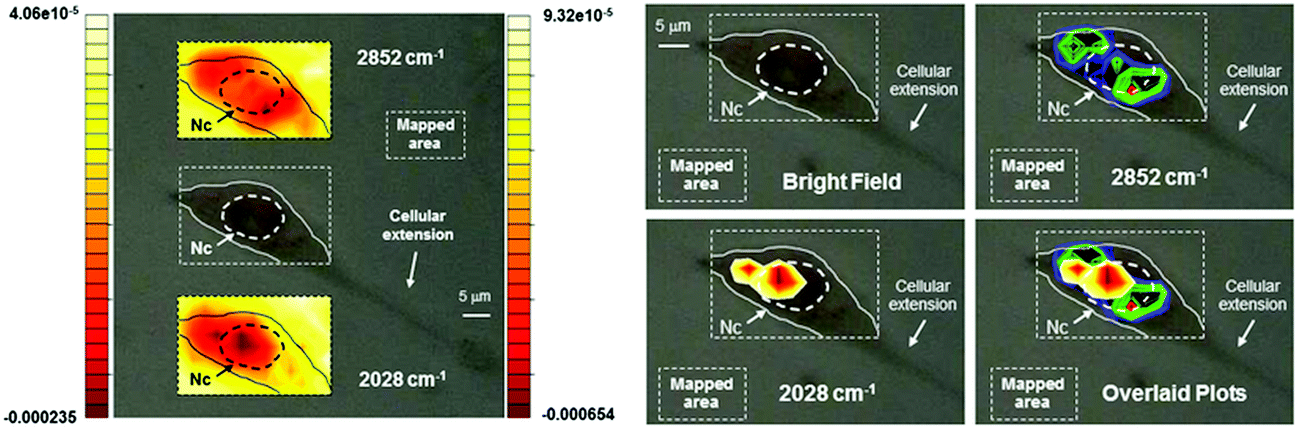 | ||
| Fig. 6 Optical images of a 3T3 fibroblast incubated with 2 (300 μM concentration). Left: top and bottom inserts show contour images reconstructed from mapping the intensities of the 2nd derivative of the lipid absorption at 2852 cm−1 and the A1 carbonyl stretching vibration at 2028 cm−1 respectively. The scales represent relative units of 2nd derivative of absorbance with the left scale referring to the top insert. Nc = cell nuclear area identified as previously described.48,49 Right: same cell with maxima of reconstructed integrated contour intensities (same scale as to the left with minima set to −9.5 × 10−5 and −0.00024 units for the 2852 cm−1 and the 2028 cm−1 vibration respectively) of the 2nd derivative of the same lipid absorption and the A1 stretching frequency finally superimposed in the bottom right panel to highlight the different chemical distribution of the molecular species. Pixel maps of the same images may be found in ESI.† | ||
Conclusions
Constitutional isomers of vitamin B12 adducts bearing a fac-[Re(CO)3(Me2-phen)]+ unit appended either at the cyano or the 5′-hydroxo group of the vitamin were prepared and characterized. The isomers were obtained in moderate yields and they show the presence of two distinct conformers in aqueous media where they are stable over a 72 h period. Compound 1, however, was found to be unstable in cell media. The inhibitory potency of 1, 2 and the [Re(CO)3(Me2-phen)MeOH][NO3] complex was tested on PC-3 prostate cancer cells and 3T3 fibroblasts. All three drugs showed IC50 values in the low μM rage with the pristine rhenium complex and 1 being the most active compounds. Steady-state and time-resolved fluorescence spectroscopy measurements indicate an energy transfer between the overlapping Re(I) emission and vitamin B12 absorption eventually causing an extremely low quantum yield, and thus making the molecules unsuitable for fluorescence imaging. However, synchrotron infrared spectromicroscopy provided evidence that 2 accumulated in live 3T3 fibroblasts with predominant signals concentrated in the proximity of the cell nucleus.Experimental section
Chemicals and solvents for synthesis were purchased from standard sources. All synthesis were performed in the dark under an inert N2 atmosphere unless otherwise noted. B12-4-picolinamine (B12-(4-pic))40 and [ReBr(CO)3(Me2-phen)] (where Me2-phen = 4,7-dimethyl-1,10-phenanthroline) were synthesized as previously described.51 The [Re(CO)3(Me2-phen)MeOH](NO3) was obtained by reacting [ReBr(CO)3(Me2-phen)] in methanol with AgNO3 overnight. Elemental analyses (EA) were performed on a Leco CHNS-932 elemental analyzer. For EA measurements, samples of vitamin B12 containing species were ground to a very fine powder and subjected to high vacuum for at least 3 days. This procedure is necessary in order to eliminate all residual solvent molecules and to obtain accurate results. Solid state IR spectra were recorded in a PerkinElmer Spectrum BX FT-IR spectrometer. UV/Vis spectra were taken on a Perkin Elmer Cary 50 spectrometer with a Peltier thermostat. Steady-state fluorescence spectra were taken with a Perkin Elmer LS50B and time-resolved emission spectra were taken with an Edinburgh Lifespec II, using a picosecond pulsed diode laser (406 nm). 77 K lifetime measurements were performed in liquid nitrogen. All absorption and emission measurements were performed by using spectrophotometric grade methanol (Aldrich). Mass spectra were recorded in the positive mode on an Esquire HCT from Bruker (Bremen, Germany) with electrospray ionization (ESI). High-resolution electrospray mass spectra were recorded on a Bruker maXis QTOF-MS instrument (Bruker Daltonics GmbH, Bremen, Germany). The samples were dissolved in a 1:1 mixture of MeOH and 0.1% formic acid and analyzed via continuous flow injection at 30 μL min−1. The mass spectrometer was operated in positive ion mode with a capillary voltage of 4 kV, an endplate offset of −500 V, nebulizer pressure of 5.8 psig, and a drying gas flow rate of 4 L min−1 at 180 °C. The instrument was calibrated with a sodium formate solution (500 μl H2O:500 μl iPrOH:20 μl HCOOH:20 μl 0.1 M NaOHaq.). The resolution was optimized at 30000 FWHM in the active focus mode. The accuracy was better than 2 ppm in a mass range between m/z 118 and 2000. All solvent used were purchased in best LC-MS qualities.
Analytical HPLC method
Instrument: MERCK HITACHI LaChrom with a D-7000 interface coupled with a Diode Array detector L-7455 and a pump L-7100 system. Column: Macherey-Nagel, EC250/3 Nucleosil 100-5 C18. Flow rate 0.5 mL min−1. Absorbance monitored at 250 nm. Solutions: A: 0.1% trifluoroacetic acid in water; B: methanol. Chromatographic method: 0–5 min: isocratic flow of 90% A – 10% B; 5–35 min: linear gradient to 100% B; 35–45 min: isocratic flow of 100% B; 45–50 min: linear gradient to 10% B; 50–55 min: isocratic flow of 90% A – 10% B.Synthesis of {B12-(4-pic)-[Re(CO)3(Me2-phen)]}(CO2CF3) species (1 for CN-bound Re and 2 py-bound Re)
A total of 18.7 mg of [ReBr(CO)3(Me2-phen)] (0.033 mmol) was dissolved in 3 mL of MeOH and 7.3 mg of AgCO2CF3 (0.033 mmol, 1 eq.) were added. After 12 h of stirring at RT, the mixture was filtered through celite and then dried under vacuum. The resulting yellow solid residue was dissolved with a mixture MeOH and CH2Cl2 1:1 and 50 mg of B12-(4-pic) (0.033 mmol, 1 eq.) were added to the solution. After stirring for 48 h the reaction was stopped and the mixture purified by HPLC. Two products were identified and separated. Yield 12 mg (18%) for 1 and 10 mg (15%) for 2. Analytical data for 1. HPLC: retention time 20.9 min. ESI-MS analysis (positive mode) gave a peak at m/z = 1966.7 [M+]. 1H NMR, 400 MHz (CD3OD, δ ppm): 0.43 (s, 3H); 1.08 (m, 2H); 1.24 (overlapping: s + d, 6H); 1.31 (m, 2H); 1.35 (s, 3H); 1.36 (s, 6H); 1.51 (m, 2H); 1.63 (m, 1H); 1.70 (s, 3H); 1.72 (d, 2H); 1.87 (m, 4H); 2.01 (m, 2H); 2.12 (d, 1H); 2.21 (s, 3H); 2.22 (s, 3H); 2.29 (m, 2H); 2.36 (overlapping: s + m, 8H); 2.45 (overlapping: s + m, 6H); 2.60 (overlapping: d + m, 8H); 2.82 (m, 4H); 2.99 (m, 2H); 3.01 (s, 3H); 3.02 (s, 3H); 3.07 (d, 1H); 3.40 (m, 1H); 3.65 (d, 1H); 3.90 (d, 1H); 4.15 (m, 3H); 4.33 (m, 3H); 4.71 (d, 1H); 4.92 (d, 1H); 5.71 (s, 1H); 6.19 (d, 1H); 6.33 (s, 1H); 6.92 (s, 1H); 7.19 (s, 1H); 7.33 (d, 2H); 7.89 (t, 2H); 8.31 (d, 2H); 8.37 (dd, 2H); 9.01 (t, 2H). I.r. (solid state, KBr, cm−1): νCO 2033, 1937; νCN 2175. Analytical data for 2. HPLC: retention time 24.7 min. ESI-MS analysis (positive mode) gave a peak at m/z = 1966.7 [M+]. 1H NMR, 400 MHz (CD3OD, δ ppm): 0.45 (s, 3H); 1.12 (m, 4H); 1.16 (overlapping: s + d, 6H); 1.31 (m, 2H); 1.36 (s, 3H); 1.38 (s, 3H); 1.44 (s, 3H); 1.74 (m, 1H); 1.81 (d, 1H); 1.88 (s, 3H); 2.05 (overlapping: d + m, 5H); 2.10 (m, 2H); 2.27 (s, 3H); 2.29 (overlapping: s + m, 4H); 2.35 (dt, 3H); 2.54 (overlapping: s + m, 12H); 2.58 (overlapping: s + m, 13H); 2.84 (m, 2H); 2.99 (overlapping: s + m, 8H); 3.65 (dd, 1H); 4.11 (overlapping: d + m, 5H); 4.23 (d, 2H); 4.29 (m, 1H); 4.50 (d, 1H); 4.70 (m, 1H); 6.18 (d, 1H), 6.57 (s, 1H); 7.10 (s, 1H); 7.19 (overlapping: d + s, 3H); 8.03 (d, 2H); 8.33 (d, 2H); 8.40 (s, 2H); 9.55 (d, 2H). I.r. (solid state, KBr, cm−1): νCO 2030, 1924; νCN 2134.
Fibroblast cell culture for FTIR spectromicroscopy measurements
NIH 3T3 Swiss Albino Mouse Fibroblast cells (ECACC Catalogue Number 85022108) were cultured directly on CaF2 windows (Crystran, Poole, UK) in Dulbecco's Modified Eagle Medium (DMEM) containing 5 mM glucose and 10% fetal bovine serum (FBS). Cells were grown for 16 to 24 hours, until the desired degree of coverage was obtained (about 20%–30%). The cell-coated window was transferred to a custom-built solution sample holder for FTIR microscopy, as previously described.52 All cell culture materials were purchased from Invitrogen (Invitrogen, Life Technologies, Paisley, UK).Fibroblast cell treatment
Cell treatment with isomers 1 and 2 was performed under dim light. Addition of the cobalamin adducts was performed by first removing bulk DMEM medium and covering the CaF2 windows with a DMEM solution of the species of 0.03–1 mM. The sample holder was then enclosed, kept at 37 °C and used for FTIR spectromicroscopy measurements.FTIR spectromicroscopy measurements
FTIR Spectromicroscopy experiments were performed at beamline X01DC of the Swiss Light Source, comprising a Bruker Vertex 70v interferometer (Bruker Optics) and a Bruker Hyperion 3000 microscope. For these measurements, a KBr-supported Ge-multilayer beamsplitter was used together with a liquid nitrogen-cooled MCT detector. The interferometer was scanned with an acquisition rate of 40 kHz to 4 cm−1 resolution. Measurements were performed in transmission. 250 scans were collected for each measurements, with a 60 s interval between measurements. Single channel spectra were obtained by performing a Fourier Transform of the interferogram after apodization with a Blackman-Harris 3-Term function, using a zero-filling factor of 2 and a Mertz phase correction.FTIR mapping measurements
The distribution of IR absorption bands was recorded by mapping the sample over a rectangular grid pattern. For the highest resolution maps, confocal blades were set to a square aperture of 5 × 5 μm2 and point spacing was set at 2.5 μm. Using a 36× objective with 0.5 NA, these conditions give a theoretical spatial resolution close to the diffraction limit at the wavelength of absorption of CO ligands. For low resolution maps, confocal blades were set to an aperture of 10 × 10 μm2 and point spacing was set at 5 or 10 μm.Data analysis
FTIR data analysis and assembly of IR maps was performed using the software package OPUS 6.5 (Bruker Optics). Spectral plots were constructed using Origin 8.0 (Originlab, Northampton, MA, USA).Computational details
Rotational energy profile of 1 was calculated by using Molecular Mechanics (MMFF force field, angle step 5°) starting from a DFT optimised structure of the species (B3LYP, 6-31G*; Co and Re were modeled by using a LANL2DZ basis set), whose bond lengths were found in good agreement with crystallographic data of vitamin B12 and related Re-vitamin B12 complexes, with the exception of a slightly elongated (ca. 0.2 Å) Co–CN bond.39,53 The rotational axis was defined by using the dihedral angle N(phen)–Re–Co–N(corrin). All calculations were performed using Spartan '14 software package.Acknowledgements
Financial support from the Swiss National Science Foundation (Grant# PP00P2_144700) is gratefully acknowledged. The authors are grateful to Felix Fehr (University of Fribourg) for NMR measurements.References
- K. K. W. Lo, M. W. Louie and K. Y. Zhang, Coord. Chem. Rev., 2010, 254, 2603 CrossRef CAS PubMed.
- K. K. W. Lo, K. Y. Zhang and S. P. Y. Li, Eur. J. Inorg. Chem., 2011, 3551 CrossRef CAS.
- R. G. Balasingham, M. P. Coogan and F. L. Thorp-Greenwood, Dalton Trans., 2011, 40, 11663 RSC.
- K. K. W. Lo, A. W. T. Choi and W. H. T. Law, Dalton Trans., 2012, 41, 6021 RSC.
- M. W. Louie, T. T. H. Fong and K. K. W. Lo, Inorg. Chem., 2011, 50, 9465 CrossRef CAS PubMed.
- E. Alberti, M. P. Coogan and D. Donghi, J. Biol. Inorg. Chem., 2014, 19, S824 Search PubMed.
- M. P. Coogan, R. P. Doyle, J. F. Valliant, J. W. Babich and J. Zubieta, J. Labelled Compd. Radiopharm., 2014, 57, 255 CrossRef CAS PubMed.
- M. P. Coogan and V. Fernandez-Moreira, Chem. Commun., 2014, 50, 384 RSC.
- K. E. Henry, R. G. Balasingham, A. R. Vortherms, J. A. Platts, J. F. Valliant, M. P. Coogan, J. Zubieta and R. P. Doyle, Chem. Sci., 2013, 4, 2490 RSC.
- M. P. Coogan, V. Fernandez-Moreira, B. M. Kariuki, S. J. A. Pope and F. L. Thorp-Greenwood, Angew. Chem., Int. Ed., 2009, 48, 4965 CrossRef CAS PubMed.
- L. A. Mullice, F. L. Thorp-Greenwood, R. H. Laye, M. P. Coogan, B. M. Kariuki and S. J. A. Pope, Dalton Trans., 2009, 6836 RSC.
- S. Clede and C. Policar, Chem. – Eur. J., 2015, 21, 942 CrossRef CAS PubMed.
- A. J. Amoroso, M. P. Coogan, J. E. Dunne, V. Fernandez-Moreira, J. B. Hess, A. J. Hayes, D. Lloyd, C. Millet, S. J. A. Pope and C. Williams, Chem. Commun., 2007, 3066 RSC.
- C. Redshaw, S. Watkins, S. M. Humphrey, P. C. B. Page, S. Ashby, Y. M. Chao, C. J. Herbert and A. Mueller, R. Soc. Chem. Adv., 2013, 3, 23963 CAS.
- H. N. Banerjee, D. Vaughan, J. Medley, G. Hyman, C. Krauss, C. Parson, S. Mandal, P. Olczak, M. Mbagu, D. Kebulu, S. Pramanik and F. Sarkar, Cancer Res., 2013, 73(8 suppl.), 4485 Search PubMed.
- C. Parson, V. Smith, C. Krauss, H. N. Banerjee, J. A. Krause, J. M. Wachira, D. Giri, A. Winstead and S. K. Mandal, J. Bioprocess. Biotech., 2013, 4, 1 Search PubMed.
- M. Bartholoma, J. Valliant, K. P. Maresca, J. Babich and J. Zubieta, Chem. Commun., 2009, 493 RSC.
- I. Kitanovic, S. Z. Can, H. Alborzinia, A. Kitanovic, V. Pierroz, A. Leonidova, A. Pinto, B. Spingler, S. Ferrari, R. Molteni, A. Steffen, N. Metzler-Nolte, S. Wolfl and G. Gasser, Chem. – Eur. J., 2014, 20, 2496 CrossRef CAS PubMed.
- S. Clede, F. Lambert, C. Sandt, Z. Gueroui, M. Refregiers, M. A. Plamont, P. Dumas, A. Vessieres and C. Policar, Chem. Commun., 2012, 48, 7729 RSC.
- M. W. Louie, H. W. Liu, M. H. C. Lam, Y. W. Lam and K. K. W. Lo, Chem. – Eur. J., 2011, 17, 8304 CrossRef CAS PubMed.
- A. W. T. Choi, M. W. Louie, S. P. Y. Li, H. W. Liu, B. T. N. Chan, T. C. Y. Lam, A. C. C. Lin, S. H. Cheng and K. K. W. Lo, Inorg. Chem., 2012, 51, 13289 CrossRef CAS PubMed.
- K. A. Stephenson, S. R. Banerjee, T. Besanger, O. O. Sogbein, M. K. Levadala, N. McFarlane, J. A. Lemon, D. R. Boreham, K. P. Maresca, J. D. Brennan, J. W. Babich, J. Zubieta and J. F. Valliant, J. Am. Chem. Soc., 2004, 126, 8598 CrossRef CAS PubMed.
- P. Ruiz-Sanchez, S. Mundwiler, B. Spingler, N. R. Buan, J. C. Escalante-Semerena and R. Alberto, J. Biol. Inorg. Chem., 2008, 13, 335 CrossRef CAS PubMed.
- P. Ruiz-Sanchez, S. Mundwiler, A. Medina-Molner, B. Spingler and R. Alberto, J. Organomet. Chem., 2007, 692, 1358 CrossRef CAS PubMed.
- S. Mundwiler, B. Spingler, P. Kurz, S. Kunze and R. Alberto, Chem. – Eur. J., 2005, 11, 4089 CrossRef CAS PubMed.
- S. Kunze, F. Zobi, P. Kurz, B. Spingler and R. Alberto, Angew. Chem., Int. Ed., 2004, 43, 5025 CrossRef CAS PubMed.
- J. Alsenz, G. J. Russell-Jones, S. Westwood, B. Levet-Trafit and P. C. de Smidt, Pharm. Res., 2000, 17, 825 CrossRef CAS.
- A. K. Petrus, A. R. Vortherms, T. J. Fairchild and R. P. Doyle, ChemMedChem, 2007, 2, 1717 CrossRef CAS PubMed.
- X. Y. Wang, L. H. Wei and L. P. Kotra, Bioorg. Med. Chem., 2007, 15, 1780 CrossRef CAS PubMed.
- D. J. Li, Y. M. Yang, X. X. Cao, C. Xu and B. M. Ji, J. Mol. Struct., 2012, 1007, 102 CrossRef CAS PubMed.
- B. S. Liu, Z. C. Liu and G. Jing, Anal. Lett., 2005, 38, 1367 CrossRef CAS PubMed.
- H. N. Hou, Z. D. Qi, Y. W. OuYang, F. L. Liao, Y. Zhang and Y. Liu, J. Pharm. Biomed. Anal., 2008, 47, 134 CrossRef CAS PubMed.
- Z. B. Shang, Y. J. Wen, X. Q. Yan, H. H. Sun, Y. Wang and W. J. Jin, Luminescence, 2014, 29, 598 CrossRef CAS PubMed.
- F. Samari, B. Hemmateenejad, Z. Rezaei and M. Shamsipur, Anal. Methods, 2012, 4, 4155 RSC.
- A. R. Vortherms, A. R. Kahkoska, A. E. Rabideau, J. Zubieta, L. L. Andersen, M. Madsen and R. P. Doyle, Chem. Commun., 2011, 47, 9792 RSC.
- F. Zobi, A. Degonda, M. C. Schaub and A. Y. Bogdanova, Inorg. Chem., 2010, 49, 7313 CrossRef CAS PubMed.
- F. Zobi and O. Blacque, Dalton Trans., 2011, 40, 4994 RSC.
- F. Zobi, Future Med. Chem., 2013, 5, 175 CrossRef CAS PubMed.
- F. Zobi, L. Quaroni, G. Santoro, T. Zlateva, O. Blacque, B. Sarafimov, M. C. Schaub and A. Y. Bogdanova, J. Med. Chem., 2013, 56, 6719 CrossRef CAS PubMed.
- F. Zobi, O. Blacque, R. A. Jacobs, M. C. Schaub and A. Y. Bogdanova, Dalton Trans., 2012, 41, 370 RSC.
- L. Quaroni, M. Obst, M. Nowak and F. Zobi, Angew. Chem., Int. Ed., 2015, 54, 318 CrossRef CAS PubMed.
- A. Kumar, S. S. Sun and A. J. Lees, Top. Organomet. Chem., 2010, 29, 1 CAS.
- B. Probst, M. Guttentag, A. Rodenberg, P. Hamm and R. Alberto, Inorg. Chem., 2011, 50, 3404 CrossRef CAS PubMed.
- L. A. Sacksteder, M. Lee, J. N. Demas and B. A. Degraff, J. Am. Chem. Soc., 1993, 115, 8230 CrossRef CAS.
- M. Cummings, K. Higginbottom, C. J. McGurk, O. G. W. Wong, B. Koberle, R. T. D. Oliver and J. R. Masters, Biochem. Pharmacol., 2006, 72, 166 CrossRef CAS PubMed.
- K. Meister, J. Niesel, U. Schatzschneider, N. Metzler-Nolte, D. A. Schmidt and M. Havenith, Angew. Chem., Int. Ed., 2010, 49, 3310 CrossRef CAS PubMed.
- C. Policar, J. B. Waern, M. A. Plamont, S. Clede, C. Mayet, R. Prazeres, J. M. Ortega, A. Vessieres and A. Dazzi, Angew. Chem., Int. Ed., 2011, 50, 860 CrossRef CAS PubMed.
- S. Clede, C. Policar and C. Sandt, Appl. Spectrosc., 2014, 68, 113 CrossRef CAS PubMed.
- C. Sandt, J. Frederick and P. Dumas, J. Biophotonics, 2013, 6, 60 CrossRef CAS PubMed.
- G. van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112 CrossRef CAS PubMed.
- P. Kurz, B. Probst, B. Spingler and R. Alberto, Eur. J. Inorg. Chem., 2006, 2966 CrossRef CAS.
- L. Quaroni and T. Zlateva, Analyst, 2011, 136, 3219 RSC.
- N. Marino, A. E. Rabideau and R. P. Doyle, Inorg. Chem., 2011, 50, 220 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt03598d |
| ‡ Current address, Functional Genomic Center, ETH/University of Zürich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. |
| This journal is © The Royal Society of Chemistry 2015 |