
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetraguanidino-functionalized phenazine and fluorene dyes: synthesis, optical properties and metal coordination†
Elvira
Bindewald
a,
Roxana
Lorenz
a,
Olaf
Hübner
a,
Dominik
Brox
b,
Dirk-Peter
Herten
b,
Elisabeth
Kaifer
a and
Hans-Jörg
Himmel
*a
aAnorganisch-Chemisches Institut, Im Neuenheimer Feld 270. and Ruprecht-Karls-Universität Heidelberg, 69120 Heidelberg, Germany. E-mail: hans-jorg.himmel@aci.uni-heidelberg.de; Fax: +49-6221-545707
bCellnetworks Cluster and Inst. for Physical Chemistry, Im Neuenheimer Feld 267 and Ruprecht-Karls-Universität Heidelberg, 69120 Heidelberg, Germany
First published on 21st January 2015
Abstract
In this work the first phenazine derivatives with guanidino substituents were prepared and their structural and electronic properties studied in detail. The guanidino groups decrease the HOMO–LUMO gap, massively increase the quantum yield for fluorescence and offer sites for metal coordination. The yellow-orange colored 2,3,7,8-tetraguanidino-substituted phenazine shows intense fluorescence. The wavelength of the fluorescence signal is strongly solvent dependent, covering a region from 515 nm in Et2O solution (with a record quantum yield of 0.39 in Et2O) to 640 nm in water. 2,3-Bisguanidino-substituted phenazine is less fluorescent (maximum quantum yield of 0.17 in THF), but exhibits extremely large Stokes shifts. In contrast, guanidino-functionalized fluorenes emit only very weakly. Subsequently, the influence of coordination on the electronic properties and especially the fluorescence of the phenazine system was analysed. Coordination first takes place at the guanidino groups, and leads to a blue shift of the luminescence signal as well as a massive decrease of the luminescence lifetime. Luminescence is almost quenched completely upon CuI coordination. On the other hand, in the case of ZnII coordination the fluorescence signal remains strong (quantum yield of 0.36 in CH3CN). In the case of strong zinc Lewis acids, an excess of metal compound leads to additional coordination at the phenazine N atoms. This is accompanied by significant red-shifts of the lowest-energy transition in the absorption and fluorescence spectra. Pentanuclear complexes with two phenazine units were isolated and structurally characterized, and further aggregation leads to chain polymers.
Introduction
Amino-substituted phenazines and fluorenes are important dyes. Hence the phenazine derivative mauveine (Perkin's dye, which actually was shown to be a mixture of several compounds which differ in the number of methyl groups) was the first commercialized synthetic dye.1 Although its last commercial use (coloring of British stamps) ended in 1901, mauveine was of great importance for the development of the modern chemical industry.2 Other known phenazine dyes include safranin and Neutral red. All these compounds feature amino substituents. The amino functions increase the electron density on the aromatic system and the quantum yield of fluorescence by suppressing nonradiative relaxation processes. There are also other applications that make phenazines attractive for modern chemistry, spanning from organic electronics (phenazines have been integrated in solar cell3 and organic light emitting diode (OLED)4 devices) to sensors (e.g. pH5 or metal sensing6,7) and pharmaceutical purposes (especially DNA intercalation). Fluorenes are attractive building blocks in polymers, and polyfluorenes are electrically conductive and electroluminescent.8 Hence fluorenes and polymers including fluorene units are intensively applied in OLED devices.9 Fluorene and triphenylene derivatives have been integrated into porous covalent organic frameworks with distinct optical and electronic properties.10,11The influence of metal coordination on the optical properties of fluorophores was intensively studied in the past.12,13 Photoluminescent sensors based on the photo-induced electron transfer (PET) mechanism were built by linking fluorophors (through a spacer) to ionophores, which are responsible for metal coordination.14,15 In its bound state, the ionophore usually forms a chelating complex with the metal ion. For example, for zinc sensing,13 the ionophore often contains pyridyl groups.16 In classical PET probes, the HOMO energy of the free (uncoordinated) ionophore (e.g. “formed” by N donor atom lone pairs) is slightly higher than the relevant orbital energy of the fluorophore, so that intramolecular electron transfer (through the spacer) upon photoexcitation of the fluorophore leads to fluorescence quenching. In the bound (metal coordinated) ionophore the HOMO energy is then drawn down below the fluorophore orbital energy, leading to strong fluorescence. This is termed CHEF (chelation enhanced fluorescence) effect. On the other hand, coordination of transition metals with unpaired electrons such as Cu2+ (like coordination of heavy metal ions) generally leads to fluorescence quenching.17
In this work we report the first phenazine and fluorene derivatives with guanidino substituents, namely 2,3,7,8-tetrakis(tetramethylguanidino)-phenazine (1) and 2,3,6,7-tetrakis(tetramethylguanidino)-fluorene (2). The guanidino groups, which outperform in several aspects the amino groups, fulfil several tasks. They massively increase the quantum yield for fluorescence in the phenazine derivative. In addition, they increase the solubility in organic solvents and also in aqueous solutions, and prohibit, at least to some extent, aggregation processes which could lead to quenching of the fluorescence. Many previous experiments with guanidino-substituted aromatic compounds by our group showed that the guanidino groups increase significantly the HOMO energy and decrease the HOMO–LUMO gap. Last but not least, they offer sites for metal coordination. In this work, the coordination chemistry and optical properties (absorption and emission spectra) will be addressed in particular.
Neutral guanidines as well as guanidinates were shown to be excellent ligands, which were applied in coordination chemistry for manifold purposes.18 CuI complexes of tripodal tris(2-guanidinylethyl)amine ligands were used for the preparation of end-on bonded superoxo-complexes which exhibit a rich chemistry.19 Other guanidine ligands were used as catalysts, e.g. in lactide polymerization20 or Heck-type reactions.21 Generally only the imino-N atom of the guanidine binds to the metal, since the lone-pairs on the amino N atoms are engaged in π-interactions. However, in exceptional cases the amino group also binds to the metal, leading to additional hemilabile metal–ligand bonding which is of interest for catalytic applications.22 A detailed analysis of the guanidine-metal bonding on the basis of a experimental charge-density study revealed σ- as well as π-bonding contributions, in line with the observed elongation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N imino bond upon coordination.23 Our group recently established guanidino-functionalized aromatic compounds (GFAs) as a new class of strong organic electron donors.24,25 The Lewis-structures of two representatives, namely 1,2,4,5-tetrakis(tetramethylguanidino)benzene, 3,26 and 2,3,5,6-tetrakis(tetramethylguanidino)pyridine, 4,27 are shown in Scheme 1. According to cyclic voltammetric (CV) experiments, both compounds exhibit a similar electron-donor capacity with an E1/2 value (in CH2Cl2) of −0.76 V versus Fc/Fc+ (Fc = ferrocene) for two-electron oxidation. On the other hand, 4 but not 3 could be used in photochemical reductive C–C coupling reactions.28 We have analysed in some detail the effect of alkylation of the pyridine N atom on the electronic properties of 4. Alkylation leads to a red-shift of the lowest-energy electronic transition (LUMO ← HOMO excitation). We show in this work that coordination at the N atoms of the phenazine core of compound 1 also changes the electronic properties.
N imino bond upon coordination.23 Our group recently established guanidino-functionalized aromatic compounds (GFAs) as a new class of strong organic electron donors.24,25 The Lewis-structures of two representatives, namely 1,2,4,5-tetrakis(tetramethylguanidino)benzene, 3,26 and 2,3,5,6-tetrakis(tetramethylguanidino)pyridine, 4,27 are shown in Scheme 1. According to cyclic voltammetric (CV) experiments, both compounds exhibit a similar electron-donor capacity with an E1/2 value (in CH2Cl2) of −0.76 V versus Fc/Fc+ (Fc = ferrocene) for two-electron oxidation. On the other hand, 4 but not 3 could be used in photochemical reductive C–C coupling reactions.28 We have analysed in some detail the effect of alkylation of the pyridine N atom on the electronic properties of 4. Alkylation leads to a red-shift of the lowest-energy electronic transition (LUMO ← HOMO excitation). We show in this work that coordination at the N atoms of the phenazine core of compound 1 also changes the electronic properties.
 | ||
| Scheme 1 The new GFA compounds 1 and 2 and the previously reported GFAs 3 and 4. | ||
Results and discussion
Synthesis and structural characterization
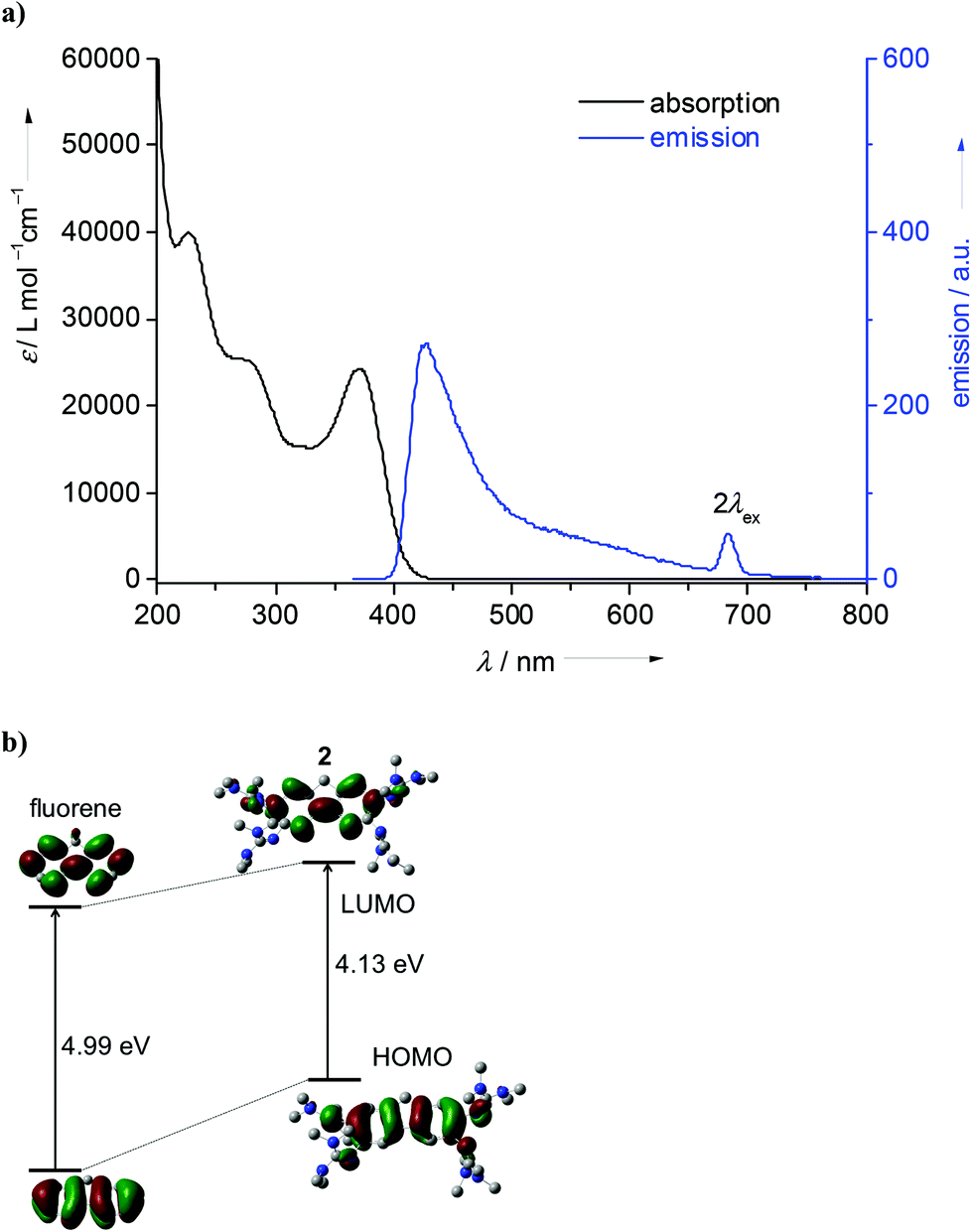
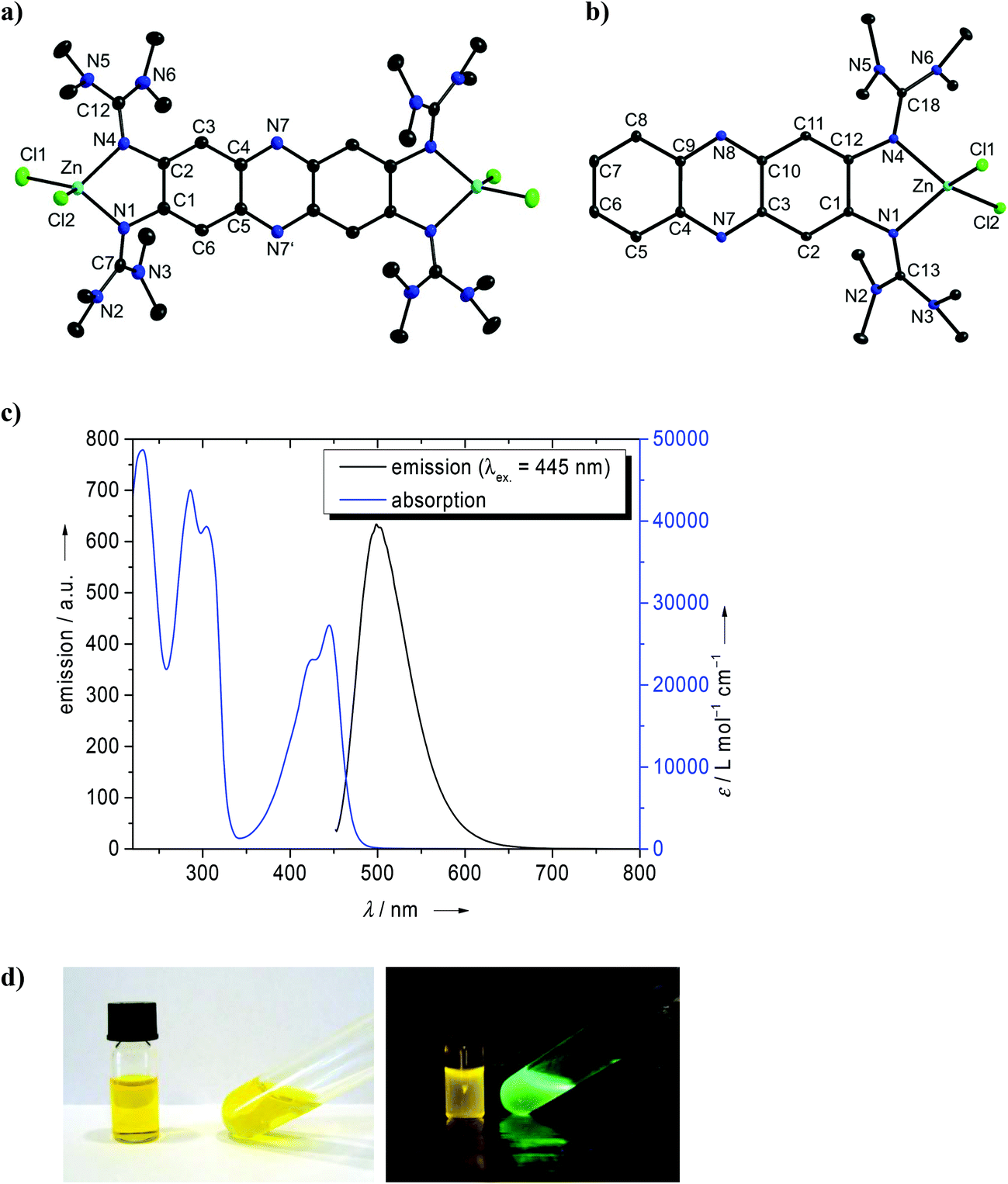
The new GFA 1 was synthesized in a three step reaction starting with 1,2,4,5-tetraamino-benzene. Heating an aqueous solution of 1,2,4,5-tetraamino-benzene·4HCl in the presence of NaOAc (sodium acetate) and under a stream of compressed air gave deep purple-colored 2,3,7,8-tetraamino-phenazine-trihydrochloride, a reaction which was already previously described.29 Then this compound was reacted with activated tetramethyl-urea (the “Vilsmeier” salt 2-chloro-1,1,3,3-tetramethylformamidinium chloride, [ClC(NMe2)2]Cl) to yield 1 in the form of an orange-colored solid. Deeply orange-colored crystals suitable for a single-crystal X-ray diffraction analysis were obtained from Et2O solutions. We also synthesized the corresponding bisguanidine 1,2-bis(tetramethylguanidino)-phenazine, 5 (see Scheme 3), from 1,2-diamino-phenazine (also an orange-colored solid at ambient conditions). The amino precursor to compound 2, namely 2,3,6,7-tetraminofluorene-tetrahydrochloride, was synthesized according to the literature (with minor modifications, see Experimental details) starting with 2,7-dinitrofluorene (see Scheme 2).30 Guanidinylation was again achieved with activated tetramethyl-urea in the presence of NEt3. The structures of compounds 1, 2 and 5, as derived from single-crystal X-ray diffraction, are visualized in Fig. 1. The imino CN bond distances adopt characteristic average values of 1.300 Å in 1, 1.293 Å in 2 and 1.317 Å in 5. These values generally increase upon metal coordination (see below). According to 1H NMR studies the guanidino groups are highly flexible,31 so that at room temperature all 48 methyl protons in 1 and 2 and all 24 methyl protons of 5 exhibit the same chemical shift.
 | ||
| Scheme 2 Synthesis of the new compounds 1 and 2. | ||
 | ||
| Scheme 3 Lewis structure of the new bisguanidine 5. | ||
 | ||
| Fig. 1 Molecular structures of (a) the phenazine derivative 1 and (b) the corresponding bisguandine 5, as well as (c) the fluorene derivative 2. Vibrational ellipsoids drawn at the 50% probability level. Hydrogen atoms omitted for sake of clarity. Selected structural parameters (bond distances in Å, bond angles in °): For 1: N1–C1 1.395(4), N1–C7 1.302(4), N2–C7 1.390(4), N3–C7 1.363(4), N4–C2 1.403(4), N4–C12 1.298(4), N5–C12 1.382(4), N6–C12 1.390(4), N7–C4 1.346(4), N7′–C5 1.354(4), C1–C2 1.457(5), C1–C6 1.382(4), C2–C3 1.371(5), C3–C4 1.426(4), C4–C5 1.435(4), C5–C6 1.417(4), C1–N1–C7 121.4(3), C2–N4–C12 119.0(3). For 5: N1–C1 1.382(4), N1–C13 1.321(4), N2–C13 1.361(4), N3–C13 1.357(4), N4–C12 1.389(4), N4–C18 1.312(4), N5–C18 1.372(4), N6–C18 1.379(4), N1⋯HO1 2.039(4), N7⋯HO2 2.059(3), C1–N1–C13 121.6(3), C12–N4–C18 122.5(3). For 2: N1–C1 1.412(3), N1–C14 1.293(3), N2–C14 1.381(3), N3–C14 1.382(3), N4–C2 1.406(3), N4–C19 1.299(3), N5–C19 1.391(3), N6–C19 1.367(3), N7–C7 1.408(3), N7–C24 1.287(3), N8–C24 1.375(3), N9–C24 1.390(3), N10–C8 1.412(3), N10–C29 1.291(3), N11–C29 1.381(3), N12–C29 1.394(3), C1–N1–C14 119.85(18), C2–N4–C19 119.79(17), C7–N7–C24 123.12(17), C8–N10–C29 121.27(17). | ||
We also measured CV curves to study the redox activity of the new compounds. Fig. S1 (see ESI†) displays the CV curve for compound 1. It contains four oxidation waves, centered at −0.12, 0.08, 0.38 and 0.63 V vs. Fc/Fc+ (Fc = ferrocene), showing that the compound could be oxidized at a lower potential than ferrocene. Three reduction waves of different intensity appear at −0.39, −0.23 and 0.24 V. The curve shows that the guanidino groups indeed turn phenazine into an electron donor. On the other hand, they clearly indicate a non-reversible redox behaviour, in contrast to the fully reversible oxidation processes for compounds 3 and 4.26,27 On the basis of this result, the further discussion focuses on the optical properties rather than the redox-chemistry.
Optical properties
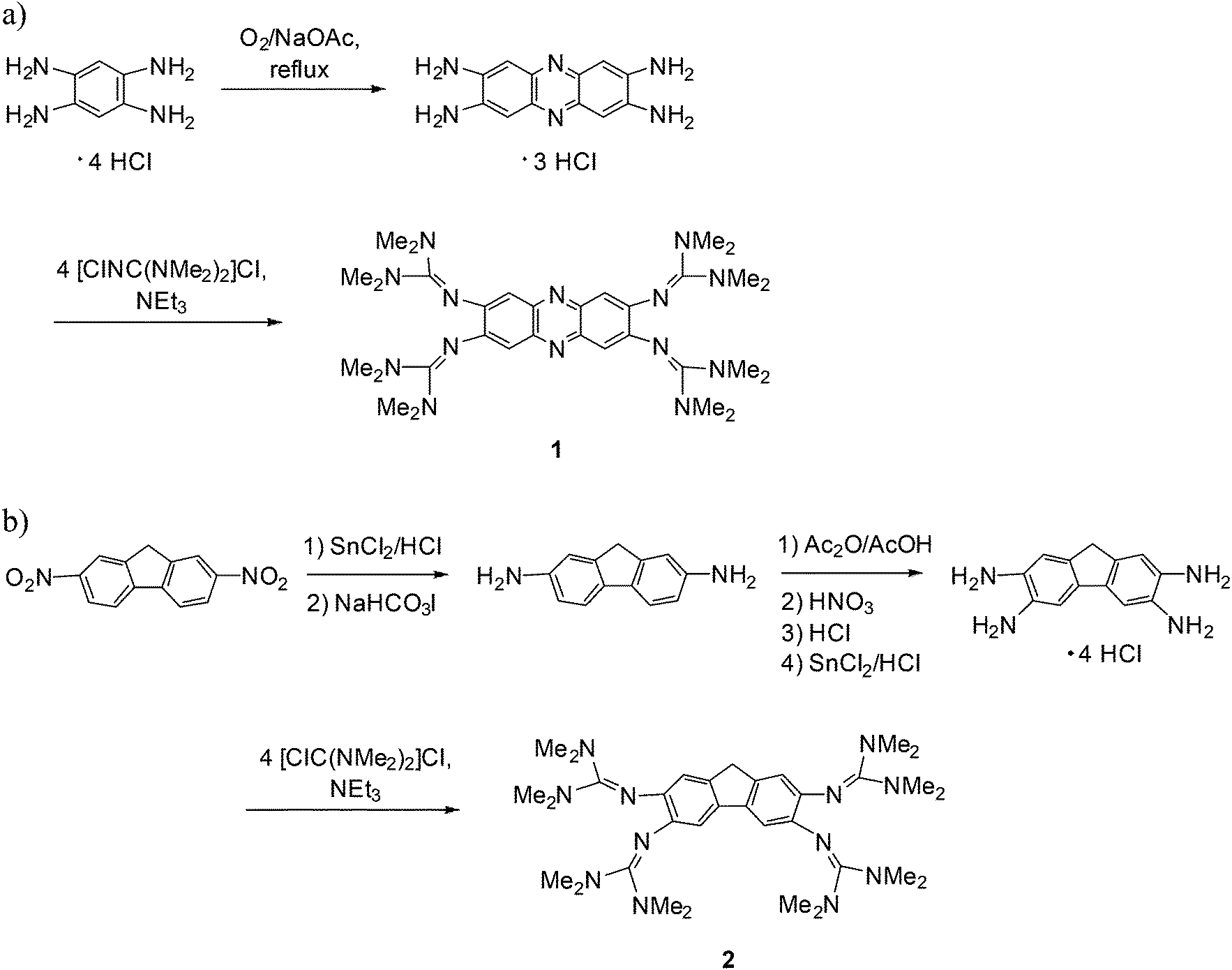
Next we studied the optical properties of the two new GFA compounds 1 and 2 and of bisguanidine 5. Compound 1 turned out to be soluble in several organic solvents and also water, yielding yellow-orange colored solutions. The electronic absorption spectrum of an aqueous solution of 1 is shown in Fig. 2. It contains strong bands at 229, 300 (with a shoulder at 311), 432 and 484 nm. Due to the high basicity of guanidines, a large portion of the molecules is protonated. Addition of HCl to the solution leads to a decrease of the 484 nm band, and an increase of the 432 nm band. Upon addition of 12 equivalents of HCl the 484 nm band is extinguished (see Fig. 2a). On the other hand, addition of NaOH leads to the increase of the band at higher wavelength (the maximum shifts slightly from 484 nm to 493 nm) at the expense of the 432 nm band (Fig. 2b). On the basis of these results we assign the 484 nm band to the unprotonated compound, and the band at 432 nm to the protonated species. The corresponding emission spectra are shown in Fig. 3. Without acid or base addition, the fluorescence quantum yield in water is 0.02. HCl addition leads to fluorescence quenching, and NaOH addition to a massive increase of the fluorescence signal. Hence only the unprotonated, but not the protonated species shows fluorescence. The emission maximum after addition of 10 eq. of HCl is reached at λmax = 634 nm, leading to a substantial Stokes shift of 4510 cm−1.32 Since the compound offers two different sites for protonation (the N atoms in the phenazine heterocycle and the imino N atom of the guanidino group), we studied protonation in some more detail. Addition of an ammonium salt (NH4)X (X = PF6 or BF4) led to twofold protonation. Crystals of the salt [1H2](BF4)2 were grown and the structural elucidation showed protonation exclusively at the guanidino groups (see Fig. S2 in the ESI†). On the other hand, quantum chemical calculations (B3LYP/6-311G**) predict a preference for the proton to bind to the N atom of the heterocycle. Hence the Gibbs free energy change (at 298 K, 1 bar) for tautomerization of 1H+ in the gas-phase according to Scheme 4 was calculated to be −23 kJ mol−1. Similar results were previously obtained for 2,3,5,6-tetrakis(tetramethylguanidino)pyridine, for which protonation at the pyridine N atom is favoured by −24 kJ mol−1 (ΔG value at 298 K, 1 bar).27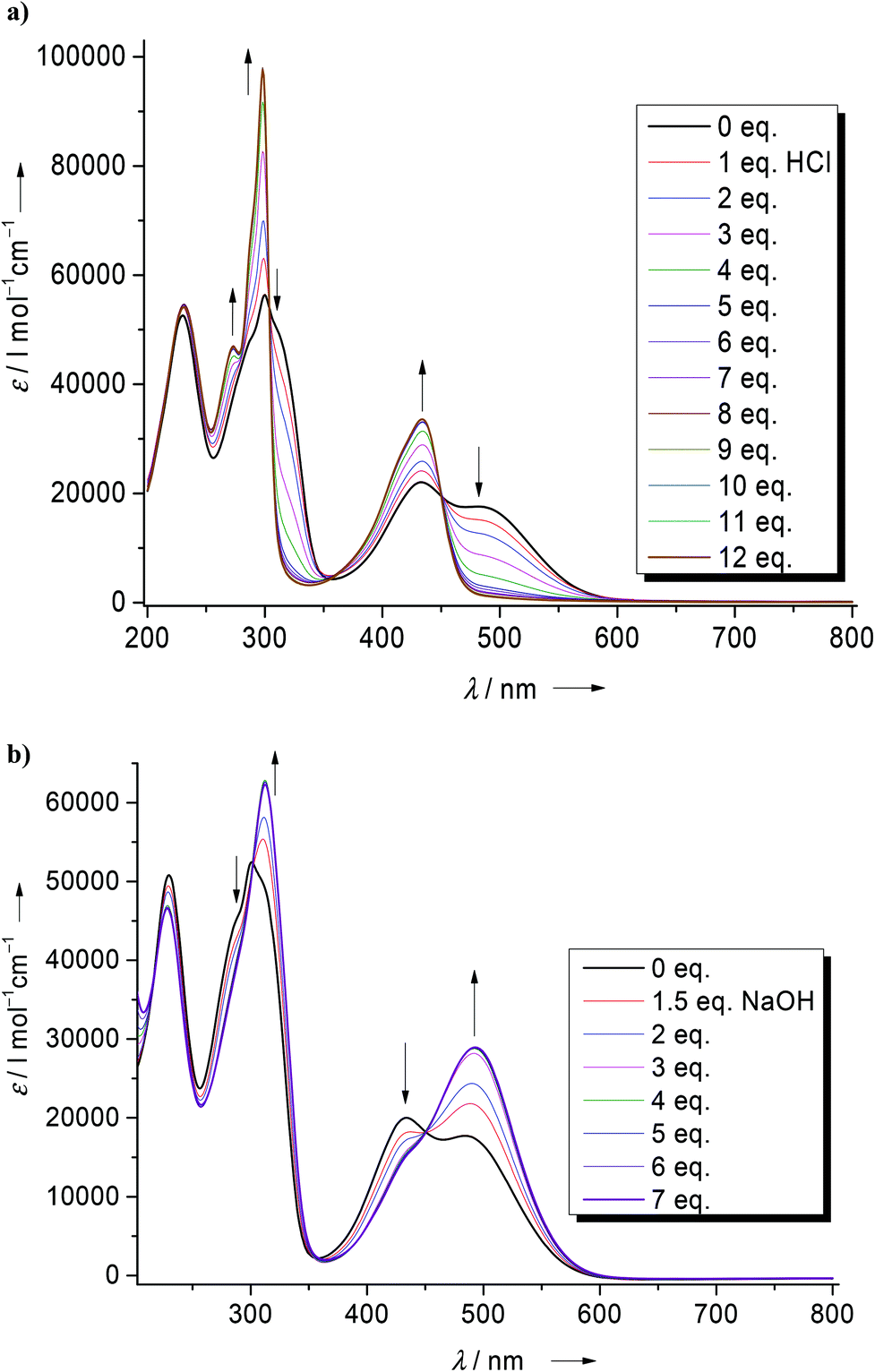 | ||
| Fig. 2 Effect of added HCl (a) and NaOH (b) on the electronic absorption spectra of an aqueous solution of compound 1. | ||
 | ||
| Fig. 3 Effect of added HCl (a) and NaOH (b) on the electronic emission spectra of an aqueous solution of compound 1. | ||
 | ||
| Scheme 4 The two tautomeric forms of 1H+ (which presumably are in equilibrium in solution). The structure on the right is more stable in the gas-phase according to quantum chemical calculations, but protonation experiments gave only evidence for protonation at the guanidino groups. | ||
Subsequently we studied the influence of different solvents on the absorption and emission spectra. In Fig. 4a, the light emitted from several solutions, excited with UV light, is visualized. It can be seen that the emission intensity and also the wavelength vary. The intensity is particularly high in Et2O, for which we determined a quantum yield of fluorescence of 0.39. To the best of our knowledge, this is the highest quantum yield measured for a molecular simple phenazine derivative. For comparison, Safranine O exhibits a quantum yield of 0.06 in H2O and 0.29 in EtOH,33 and dipyrido[3,2-a:2′,3′-c]phenazine of 0.009 in CH2Cl2.34Fig. 4b shows the absorption and emission spectra (excited at 480 nm) of 1 in CH3CN and Et2O. The emission maximum is reached at 515 nm, being thus strongly blue-shifted by more than 100 nm with respect to aqueous solution. The absorption maximum for the corresponding electronic transition is located at 484 nm, at the same position than in H2O, and thus the Stokes shift in Et2O solution (2130 cm−1) is much smaller than in aqueous solutions of the compound. For comparison, Fig. 4b also includes the UV/Vis spectrum of unsubstituted phenazine in CH3CN. The decrease of the HOMO–LUMO gap by the guanidino groups manifests itself in a massive red-shift of the lowest-energy band (located around 363 nm in phenazine, but 484 nm in 1). Table 1 gives an overview over the wavenumbers for absorption and emission and the Stokes shifts in several solvents.
 | ||
| Fig. 4 (a) Photos showing the emission of solutions of compound 1 in different organic solvents. (b) Electronic absorption and emission spectra for solutions of compound 1 in CH3CN and in Et2O solution. (c) Calculated HOMO and LUMO energies (B3LYP/6-311G**) of 1 relative to unsubstituted phenazine. | ||
| Compound | Solvent | λ max, abs | λ max, em | Stokes shift32 |
|---|---|---|---|---|
| a Only the lowest-energy band is listed. | ||||
| 1 | Et2O | 302, 444s, 464 | 515 | 2130 |
| CH2Cl2 | 311, 487 | 546 | 2220 | |
| CH3CN | 221, 309, 484 | 568 | 3060 | |
| H2O | 229, 300, 311s, 432, 484 | 640 | 5040 | |
| pH ≫ 7 | 493a | 634 | 4510 | |
| pH ≪ 7 | 432a | 583 | 6000 | |
| [1(ZnCl2)2] | CH3CN | 228, 316, 458, 484 | 506 | 898 |
| CH2Cl2 | 228, 319, 459, 486 | 512 | 1050 | |
| [(1)2(ZnCl2)5] | CH2Cl2 | 581a | 599 | 517 |
| 5 | Et2O | 277, 433 | 568 | 5490 |
| CH3CN | 233, 276, 393 | 599 | 8750 | |
| THF | 227, 278, 435 | 588 | 5980 | |
| [5(ZnCl2)2] | CH3CN | 231, 283, 302, 424, 442 | 514 | 3170 |
| THF | 231, 286, 304, 425, 445 | 498 | 2390 | |
At this stage, it appeared to us attractive to compare the optical properties of compound 1 bearing four guanidino groups with that of compound 5 bearing only two guanidino groups. Fig. S3 in the ESI† shows photos of the emission from solutions of 5 in different solvents, excited with UV light, and some representative absorption and emission spectra. Especially intense emission signals were observed in Et2O and THF. However, the quantum yields of 0.11 determined in Et2O and 0.17 in THF are smaller than for 1. The lowest-energetic band in the absorption spectrum of 5 (at 393 nm in CH3CN, 433 nm in Et2O and 435 nm in THF) is blue-shifted with respect to 1, but still red-shifted with respect to unsubstituted phenazine (363 nm in CH3CN). This fits with the expectations of a significantly higher HOMO energy in 1 compared with 5. The wavenumber of the emission maximum (568 nm in Et2O, 599 nm in CH3CN and 588 nm in THF) is much larger for 5. This leads to large Stokes shifts of 5490 cm−1 in Et2O, 8751 cm−1 in CH3CN and 5982 cm−1 in THF. Compound 1 has an average fluorescence lifetime of 4.6(2) ns in CH3CN, compared to 7.6(6) ns of compound 5 (see Table 2 and Fig. S4†). Interestingly, the average lifetimes are nearly independent of the excitation wavelength in CH3CN.
| Compound | Φ | Stokes shift/cm−1 | Lifetime/ns |
|---|---|---|---|
| 1 | 0.22 | 3060 | 4.6(2) |
| [1(ZnCl2)2] | 0.36 | 898 | 1.8(4) |
| 5 | 0.15 | 8750 | 7.6(6) |
| [5(ZnCl2)] | 0.08 | 3170 | 1.3(5) |
Finally, we studied the absorption and emission spectra of compound 2. As example, Fig. 5 shows the spectra recorded for a CH3CN solution. The lowest-energetic electronic transition occurred at 371 nm. The fluorescence signal was only weak (quantum yield below 0.01), with the maximum of emission at 428 nm and a long tail towards higher energies.
 | ||
| Fig. 5 (a) Electronic absorption and emission spectra for 2 in CH3CN solution. (b) Calculated HOMO and LUMO energies (B3LYP/6-311G**) of 2 relative to unsubstituted fluorene. | ||
Quantum chemical calculations were carried out to analyse in more detail the influence of the guanidino groups on the shape and energies of the HOMO and LUMO orbitals of phenazine and fluorene. The isodensity surfaces for these orbitals are visualized in Fig. 4c and 5b. It can be seen that the guanidino groups contribute to the HOMO, especially in the case of 1. As expected, they cause a significant increase of the HOMO energies and a less strong increase of the LUMO energy. Consequently, the HOMO–LUMO gap shrinks from 3.66 eV in phenazine to 3.24 eV in 1 and from 4.99 eV in fluorene to 4.13 eV in 2. The decrease of the HOMO–LUMO gap is in line with the observed colour changes and the difference in the lowest-energetic transitions detected in the absorption spectra (484 nm for 1 and 371 nm for 2 in CH3CN solution). It will be shown below that this band can indeed be assigned to the HOMO–LUMO excitation.
Coordination chemistry
Next we analysed the effect of metal coordination (especially d10 metal ions) on the optical properties of the new phenazine derivatives. We started our work by comparing the luminescence of a solution of 1 upon addition of various metal salts, and observed quenching of the luminescence signal upon addition of CuI salts, but a blue-shift of the signal in the presence of AgI or ZnII salts. To find out more about the coordination of these metals, we carried out more directed experiments. The reaction between compound 1 and 2 eq. of CuI yielded the complex [1(CuI)2] in a moderate yield of ca. 50%. The lowest energy band in the absorption spectrum (see Fig. S5 of the ESI†) occurred at 493 nm in CH3CN. This band has a shoulder at 465 nm, which we assign to excitation into an excited vibronic level (due to the difference of 1222 cm−1 and the similar intensity ratios in all experiments). In CH2Cl2, the band maximum shifts only slightly to 501 nm. An extremely weak luminescence signal (too weak to determine its quantum yield) was observed at 533 nm in CH3CN and 519 nm in CH2Cl2. Hence the coordination of two CuI units caused a sharp decrease of the luminescence signal of compound 1 to almost vanishing point. This is surprising since CuI is a d10 metal ion (like ZnII and AgI), which normally does not lead to luminescence quenching. Often luminescence quenching by CuI is explained by disproportionation of CuI in solution into CuII and Cu. However, our results clearly show coordination of CuI. We strictly avoided air contact, so that oxidation of CuI to CuII could be excluded. In this case, we could not exclude the participation of a triplet state.Reaction of 1 with two equivalents of ZnCl2 gave a product which could be identified as the dinuclear complex [1(ZnCl2)2]. Fig. 6a displays its structure from single-crystal X-ray diffraction experiments. Again, the absorption spectrum of this complex showed little difference to that of free 1, with the lowest-energetic band measured at 484 nm in CH3CN and 486 nm in CH2Cl2. However, in sharp contrast to the copper complex, the fluorescence signal remained strong. A quantum yield for fluorescence of 0.36 was measured in CH3CN solution. The quantum yield of free 1 in CH3CN solution amounts to only 0.22 (see Table 2). The increase of the quantum yield upon zinc coordination might point to the presence of a PET mechanism in free 1, which is removed upon coordination (see the discussion in the Introduction). The emission maximum of [1(ZnCl2)2] was reached at 506 nm in CH3CN and 512 nm in CH2Cl2, being significantly blue-shifted with respect to free 1 (568 nm). The Stokes shift decreases upon coordination in CH3CN from 3060 cm−1 (free 1) to 898 cm−1. At the same time, the average fluorescence lifetime of [1(ZnCl2)2] in acetonitrile (1.8(4) ns) is drastically shorter than for 1 (4.6(2) ns) underlining the strong influence of coordination on the optical properties (see Table 2).
 | ||
| Fig. 6 Molecular structures of (a) [1(ZnCl2)2] and (b) [5(ZnCl2)]. Vibrational ellipsoids drawn at the 50% probability level. Hydrogen atoms omitted for sake of clarity. Selected structural parameters (bond distances in Å, bond angles in °): For [1(ZnCl2)2]: Zn–Cl1 2.2238(9), Zn–Cl2 2.2582(8), Zn–N1 2.035(2), Zn–N4 2.039(2), N1–C1 1.408(3), N1–C7 1.342(3), N2–C7 1.338(4), N3–C7 1.356(4), N4–C2 1.409(3), N4–C12 1.334(3), N5–C12 1.344(3), N6–C12 1.359(3), N7–C4 1.351(3), N7′–C5 1.349(3), C1–C2 1.448(4), C1–C6 1.377(4), C2–C3 1.372(4), C3–C4 1.422(4), C4–C5 1.432(4), C5–C6 1.422(4). For [5(ZnCl2)]: Zn–Cl1 2.2306(8), Zn–Cl2 2.2473(7), Zn–N1 2.031(2), Zn–N4 2.034(2), N1–C1 1.397(3), N1–C13 1.345(3), N2–C13 1.355(3), N3–C13 1.338(3), N4–C12 1.398(3), N4–C18 1.351(3), N5–C18 1.352(3), N6–C18 1.337(3), Cl1–Zn–Cl2 113.49(3), N1–Zn–N4 81.74(8). (c) Absorption and emission spectra measured for [5(ZnCl2)] in THF solutions. (d) Photos showing the color arising from light absorption and that arising from light emission for [5(ZnCl2)] solutions in THF. For comparison, each photo also shows the uncoordinated ligand 5. It could be seen that coordination only slightly influences the absorption of light, while the emission is strongly changed. | ||
The corresponding mononuclear complex [5(ZnCl2)] was also synthesized (see Fig. 6b). As for [1(ZnCl2)2], coordination leads to an increase of the imino CN bond lengths (1.321(4)/1.312(4) Å in 5 and 1.345(3)/1.351(3) Å in [5(ZnCl2)]). The metal atom is in plane with the phenazine core. Also in this case, the absorption spectrum is similar to that of the free ligand 5, and the lowest-energy band showed at 442 nm in CH3CN and 445 nm (with a shoulder at 425 nm) in THF solutions. On the other hand, the emission spectrum clearly differs from that of the free ligand. Hence the maximum of emission is reached at 498 nm in THF and 514 nm in CH3CN solutions (see Fig. 6c). The large Stokes shift observed for free 5 (8750 cm−1 in CH3CN) is greatly reduced upon zinc coordination (3169 cm−1 for CH3CN) (see also the data in Table 1). To highlight this difference, Fig. 6d compares the absorption and emission colours. The average fluorescence lifetime of [5(ZnCl2)2] in CH3CN (1.3(5) ns) is also significantly reduced with respect to free 5 (7.6(6) ns).
When further ZnCl2 was added to a solution of [1(ZnCl2)2] in CH2Cl2, the colour of the solution gradually changed from yellow to purple. In Fig. 7a, the UV/Vis spectra recorded for an experiment in which a concentrated solution of ZnCl2 in CH3CN was step-wise added to a CH2Cl2 solution of [1(ZnCl2)2] (c = 1.28 × 10−5 mol l−1) is displayed, and the colour changes upon ZnCl2 addition are shown underneath (Fig. 7b). The yellow-colored solution turned to intense purple upon addition of 2 eq. of ZnCl2. In the UV/Vis spectra, the band at 486 nm with a shoulder at 459 nm decreased and gave way for an intense absorption at 581 nm and another one at 543 nm. The relative intensities of the two new absorptions varied and therefore they should belong to (at least) two different species in solution. In the case of solutions with low concentrations of [1(ZnCl2)2] (c = 0.64 × 10−5 mol l−1, see Fig. S6 in the ESI†), more equivalents of ZnCl2 were necessary to decrease the 486/459 nm band, pointing to an aggregation equilibrium. The obvious inference is that the excess ZnCl2 binds to the N atoms of the phenazine core and connects the [1(ZnCl2)2] complex units to oligomeric chains or rings (see Scheme 5). Aggregation-induced changes in the optical properties of metal complexes were also observed in other cases.35 Indeed we were able to synthesize the complex [(1)2(ZnCl2)5] by reaction of 1 with 2.5 eq. of ZnCl2. Purple-colored crystals of this compound were grown from a CH2Cl2–Et2O solvent mixture. Fig. 8 shows the molecular structure of [(1)2(ZnCl2)5] as derived from single-crystal X-ray diffraction. As expected, two [1(ZnCl2)2] complex units are linked via a ZnCl2 unit. The Zn–N bond distances within the [1(ZnCl2)2] units (in average 2.034 Å long) are shorter than the Zn–N bonds which link the [1(ZnCl2)2] units together (2.099 Å in average). This result indicates that the latter bonds are weaker, in line with the observed equilibrium (see Scheme 5).
 | ||
| Fig. 7 (a) Absorption spectra for addition of a ZnCl2 solution in CH3CN (1 μL = 0.2 eq.) to a 1.2811 × 10−5 mol l−1 solution (3 mL) of [1(ZnCl2)2] in CH2Cl2. (b) Photos showing the color changes upon ZnCl2 addition. | ||
 | ||
| Scheme 5 Formation and aggregation of the [1(ZnCl2)2] units via ZnCl2. | ||
 | ||
| Fig. 8 Molecular structure of [(1)2(ZnCl2)5]. Vibrational ellipsoids drawn at the 50% probability level. Hydrogen atoms omitted for sake of clarity. Selected structural parameters (bond distances in Å, bond angles in °): Zn1–N1 2.036(4), Zn1–N4 2.037(3), Zn1–Cl1 2.2079(15), Zn1–Cl2 2.2466(13), Zn2–N10 2.025(5), Zn2–N7 2.037(4), Zn2–Cl3 2.2268(16), Zn2–Cl4 2.2411(17), Zn5–N63 2.093(4), Zn5–N13 2.104(4), Zn5–Cl10 2.2375(13), Zn5–Cl9 2.2462(14), N1–C13 1.340(6), N4–C18 1.343(6), N7–C23 1.343(7), N10–C128 1.134(17), N1–Zn1–N4 82.74(14), N1–Zn1–Cl1 116.34(12), N4–Zn1–Cl1 107.03(10), N1–Zn1–Cl2 111.30(11), N4–Zn1–Cl2 116.89(11), Cl1–Zn1–Cl2 117.63(6), N10–Zn2–Cl4 126.36(15), N7–Zn2–Cl4 105.17(13), Cl3–Zn2–Cl4 113.89(6), N10–Zn2–N7 82.64(18), N10–Zn2–Cl3 103.87(17), N7–Zn2–Cl3 122.87(11), N63–Zn5–N13 107.68(15), N63–Zn5–Cl10 118.86(11), N13–Zn5–Cl10 97.03(11), N63–Zn5–Cl9 98.59(11), N13–Zn5–Cl9 120.09(10), Cl10–Zn5–Cl9 115.69(6). | ||
Quantum chemical calculations (B3LYP/def2-SV(P)) were carried out to obtain some preliminary information about the Gibbs free energy changes associated with the coordination of ZnCl2 to compound 1. For the first step, reaction of 1 with two equivalents ZnCl2 to give the dinuclear complex [1(ZnCl2)2], a ΔG value (at 298 K, 1 bar) of −365 kJ mol−1 was calculated. For the second step, reaction of two [1(ZnCl2)2] complex units with ZnCl2 to give the pentanuclear complex [(1)2(ZnCl2)5], we calculated a gas-phase Gibbs free energy change of −162 kJ mol−1. The calculated ΔG values neglect solvent effects, which are likely to be significant, especially in the case of ZnCl2. On the other hand, the difference in the Gibbs free energy changes between the first and the second step is in full agreement with the experimental results. According to Scheme 5, further aggregation might lead to a chain oligomer or polymer of the composition [1(ZnCl2)3]n. Indeed, we observed the formation of a solid material for addition of a large excess of ZnCl2 (the precipitation started upon addition of four equivalents for a CH2Cl2 solution of [1(ZnCl2)2] with c = 1.28 × 10−5 mol l−1, and of ten equivalents for the lower concentration (c = 0.64 × 10−5 mol l−1). In preliminary experiments, we recorded a diffuse reflectance measurement of this solid embedded in a BaSO4 matrix. This spectrum (see Fig. S7 in the ESI†) displays electronic absorptions around 305, 455 and 545 nm (the latter as a shoulder and with a tail towards larger wavenumbers). The position of the 545 nm band fits quite well with the absorption at 543 nm measured in the solution spectra. In summary we explain the spectra shown in Fig. 7a as follows: the 486 nm band clearly belongs to [1(ZnCl2)2], the 581 nm should belong to [(1)2(ZnCl2)5] and the 543 nm band is tentatively assigned to higher aggregates [1(ZnCl2)3]n.
When the experiment was repeated with ZnBr2 in place for ZnCl2, we observed a similar reaction pattern. On the other hand, with ZnMe2 the reaction stopped at the stage of the dinuclear complex; aggregation was not observed. The Lewis acidity of ZnMe2 is obviously not high enough for additional coordination at the phenazine N atoms. We also carried out some quantum chemical calculations (again B3LYP/def2-SV(P)), which are fully consistent with the experimental results. Hence reaction between 1 and two eq. of ZnMe2 to give [1(ZnMe2)2] is still mildly exergonic by −28 kJ mol−1, but further reaction of two eq. of [1(ZnMe2)2] with ZnMe2 is endergonic by +41 kJ mol−1 (at 298 K, 1 bar). Hence only strong Lewis acidic metals are capable to form the aggregates.
In Fig. 9a, the absorption spectra of free 1, [1(ZnCl2)2] and of a mixture of [1(ZnCl2)2] together with 2 eq. of ZnCl2, all dissolved in CH2Cl2, are compared. In Fig. 9b and 9c the TD-DFT spectra calculated with B3LYP/def2-SV(P) and BP/def2-SV(P), respectively, for the three species 1, [1(ZnCl2)2] and [(1)2(ZnCl2)5] are shown. The calculations (see Tables S1 and S2 and Fig. S8–S10 in the ESI† for details) show that the lowest-energetic transition of 1 (observed at 487 nm in CH2Cl2 and calculated at 441 nm (B3LYP)/503 nm (BP)) and of [1(ZnCl2)2] (observed at 486 nm and calculated at 462 nm (B3LYP)/540 nm (BP)) can be assigned to more or less pure HOMO–LUMO excitations. The calculated spectra also predict correctly a red-shift of the lowest-energetic electronic transition upon formation of [(1)2(ZnCl2)5]. In the case of [(1)2(ZnCl2)5], a number of orbitals contribute to the lower energy electronic transitions (see Table S1, Fig. S8 and S9 in the ESI†). The experimentally observed red-shift of 95 nm from [1(ZnCl2)2] to [(1)2(ZnCl2)5] compares with a calculated one of ca. 40 nm (B3LYP)/48 nm (BP). In summary, the calculations fully support our assignment.
 | ||
| Fig. 9 (a) Comparison between the experimentally obtained absorption spectra of 1, [1(ZnCl2)2] and a mixture of [1(ZnCl2)2] with a large excess of ZnCl2. (b) and (c) Simulations of the absortion spectra of 1, [1(ZnCl2)2] and [(1)2(ZnCl2)5] on the basis of TD-DFT calculations with (b) B3LYP/def2-SV(P) and (c) BP/def2-SV(P). | ||
A number of additional measurements were made to obtain further information about the aggregation equilibrium. Due to the distinct color changes upon aggregation, we predominantly relied on UV/Vis spectroscopy as analytical technique. However, it should be noted that aggregation is also visible with other techniques. For example, in the aromatic region in the 1H NMR spectrum of CD2Cl2 solutions, a signal at δ (ppm) = 6.89 can be assigned to [1(ZnCl2)2] and two signals at δ (ppm) = 7.04 and 6.71 to [(1)2(ZnCl2)5]. Higher aggregates are responsible for signals at δ (ppm) = 8.65 and 6.11. 2D HSQC experiments (HSQC = Heteronuclear Single Quantum Coherence) proved all these signals to arise from C–H protons, but a more detailed assignment was not possible.
The complex [(1)2(ZnCl2)5]) was dissolved in different solvents. The colour of the solution varied between yellow (in THF) and pinc (in CHCl3) (see Fig. 10b). The reason for the different colors can be found in differences in the degree of aggregation. Hence THF could compete successfully with [1(ZnCl2)2] in coordinating to ZnCl2, and consequently the solution mainly consist of [1(ZnCl2)2]. We observed that an initially pink-colored solution in THF in minutes changed colour to yellow, showing that THF not only prohibits aggregation, but also could break up the aggregates. On the other hand, CHCl3 solutions mainly consist of aggregates; the band due to [1(ZnCl2)2] at 486/459 nm is relatively small. Additional experiments were carried out in which the concentrations were varied for all three solvents (see Fig. S11 in the ESI†). These experiments show that the percentage of aggregates increases with increasing concentration (as already discussed for CH2Cl2), in line with the presence of an equilibrium. The effect of temperature on the relative band intensities turned out to be much weaker, and only slight variations were observed in the temperature region −60–(+20) °C in CH2Cl2 solution.
 | ||
Fig. 10 (a) Photo showing the color of solutions of [(1)2(ZnCl2)5] in CH3CN, THF, CH2Cl2 and CHCl3. (b) Comparison between the UV/Vis spectra of [(1)2(ZnCl2)5] (a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of [1(ZnCl2)2] and ZnCl2) in three different solvents (THF, CH2Cl2 and CHCl3), showing the equilibrium between [1(ZnCl2)2] and the aggregates. :1 mixture of [1(ZnCl2)2] and ZnCl2) in three different solvents (THF, CH2Cl2 and CHCl3), showing the equilibrium between [1(ZnCl2)2] and the aggregates. | ||
Finally, we inspected the fluorescence properties of aggregate mixtures. Fig. 11 displays the fluorescence signals measured for a solution of [(1)2(ZnCl2)5] in CH2Cl2. Two fluorescence signals were detected, one at 506 nm, which belongs to the dinuclear complex [1(ZnCl2)2], and another one at 599 nm. Measurements with different excitation wavelengths (see Fig. 11) clearly showed that the 581 nm absorption is the corresponding band of this species in the UV/Vis spectrum, and therefore the fluorescence signal at 599 nm could be assigned to the complex [(1)2(ZnCl2)5] (Stokes shift of 517 cm−1). We could qualitatively state that coordination at the phenazine N atoms greatly reduces the fluorescence quantum yield, although the absolute quantum yield for [(1)2(ZnCl2)5] cannot be determined due to the equilibrium. The oligomeric or polymeric [1(ZnCl2)3]n is fluorescence silent, and therefore the 543 nm band in the absorption spectra has no equivalent in the emission spectra. Our results show that the electronic and optical properties of the new fluorescent dye 1 could be greatly influenced by coordination.
 | ||
| Fig. 11 Fluorescence spectra for [(1)2(ZnCl2)5] in CH2Cl2 solution, excited with light of different wavelengths. | ||
Conclusions
Guanidino groups could be effectively used to tune the optical properties of aromatic compounds. They are a valuable alternative to the traditionally employed amino groups, which they outperform in several aspects. The guanidino groups decrease the HOMO–LUMO gap, increase the fluorescence quantum yield, and offer sites for metal coordination. In this work we show that substitution of phenazines with guanidino groups leads to highly fluorescent dyes. The absorption and emission spectra were studied in detail in several solvents. The Stokes shifts vary with the solvent and shifts of 2130–8750 cm−1 were measured. Especially remarkable are the Stokes shifts of 5040 cm−1 for 1 in H2O and of 8750 cm−1 for 5 in CH3CN. The quantum yield for fluorescence reaches a record value of 0.39 for 1 dissolved in Et2O. In contrast to the highly-fluorescent guanidino-functionalized phenazines, the guanidino-substituted fluorene 2, which was also synthesized in this work, shows little fluorescence. The bisguanidino-phenazine 5 also is a strongly fluorescent dye, but the fluorescence quantum yield is lower than for 1. We note that the extremely large Stokes shifts measured for 5 (up to 8750 cm−1) together with the relatively low wavenumber of the lowest-energetic transition in the UV/Vis spectrum might qualify the compound for applications in electroluminescent devices (see the discussion in ref. 34).The influence of coordination at the guanidino groups of the phenazine systems on the optical properties was subsequently analyzed. CuI coordination leads to luminescence quenching. By contrast, coordination with ZnCl2 leads to intensively fluorescent metal complexes. Both the dinuclear complex [1(ZnCl2)2] and the mononuclear complex [5(ZnCl2)] were structurally characterized. While little changes were observed in the absorption spectra upon coordination, the Stokes shifts decrease significantly. Also, the fluorescence lifetimes of 1 and 5 in CH3CN are dramatically reduced by coordination of ZnCl2. The measured lifetimes are independent of the excitation wavelength which makes these compounds highly interesting for further spectroscopic studies. For the strong Lewis basic compound 1, an excess of ZnCl2 initiates aggregation in the course of which the dinuclear [1(ZnCl2)2] complex units are connected by ZnCl2. The degree of aggregation is strongly solvent dependent. Aggregation is accompanied by a color change from yellow to purple, and the UV/Vis spectra displays lower-energetic bands. We were able to structurally characterize the compound [(1)2(ZnCl2)5], in which two [1(ZnCl2)2] units are connected via a ZnCl2 bridge. In ongoing work we are extending our studies to other guanidino-substituted fluorescent dyes.
Experimental details
If not stated otherwise, the reactions were carried out under Ar atmosphere using Schlenk techniques. Solvents were dried prior to use by standard methods, and preserved over 4 Å molecular sieves. NMR spectra were measured with Bruker Avance II 400 or Bruker Avance DPX AC200 NMR machine. Elemental analyses were carried out at the Microanalytical Laboratory of the University of Heidelberg. IR spectra were recorded with a Biorad Excalibur FTS 3000 spectrometer. UV/Vis measurements were recorded with a Varian Cary 5000 UV/Vis/NIR spectrophotometer. Fluorescence measurements were recorded with a Varian Cary Eclipse machine. Fluorescence quantum yields were obtained by using flouresceine in 0.1 M NaOH as standard (Φ = 0.82).36 Fluorescence lifetimes were determined with a PicoQuant FluoTime 100 Compact Fluorescence Lifetime spectrometer. Fluorescence excitation was performed with a 280 nm pulsed LED at 10 MHz repetition rate. Electrochemical studies were carried out at r.t. using an EG&G model 273A potentiostat/galvanostat. The auxiliary electrode was a platinum wire and the working electrode was a glassy carbon disk. The reference electrode was an aqueous Ag/AgCl in 3 M KCl electrode. A Bruker ApexQe FT-ICR (ESI) machine was used for mass spectrometry.2,3,7,8-Tetrakis(tetramethylguanidino)phenazine, 1 (in three steps)
1. Step (2,3,6,7-tetraamino-phenazine): this compound was prepared as described in the literature.29 Briefly, 1,2,4,5-tetraamino-benzene-tetrahydrochloride (1.338 g, 4.712 mmol) were dissolved in 30 mL destilled water. Then 3.091 g sodium acetate (37.69 mmol) were added. The reaction mixture was heated to reflux, while passing compressed air though the solution. After 5h, the reaction mixture was cooled to room-temperature, during which time a deep purple colored solid precipitated. The solid was separated by filtration, washed with water and dried under vacuum. Yield: 0.733 g (2.097 mmol, 89%). Elemental analysis for 2·(2,3,6,7-tetraamino-phenazine)·3HCl, C24H27Cl3N12 (349.65): calcd C 48.86, H 4.61, N 28.49; found C 48.27, H 4.63, N 27.71. 1H NMR (200 MHz, d6-DMSO): δ (ppm) = 5.47 (s, 8 H, NH2), 6.78 (s, 4 H, Har). MS (ESI+): m/z = 241.4 u ([M + H]+, 100%).2. Step (2-chloro-1,1,3,3-tetramethylformamidinium chloride, [ClC(NMe2)2]Cl): oxalylchloride (0.965 ml, 11.25 mmol, 1.428 g) was added drop-wise to a solution of 0.270 mL (2.250 mmol, 0.261 g) 1,1,3,3-tetramethyl-urea in 10 mL CHCl3. The pale yellow reaction mixture was heated to reflux for a period of 16h, and then allowed to cool to room temperature. Subsequently the solvent was removed under vacuum and the solid residue washed three times with 19 mL portions of Et2O. Finally the activated urea was dried under vacuum and directly used for the subsequent guanidinylation reaction.
3. Step (2,3,7,8-tetrakis(tetramethylguanidino)phenazine, 1): 0.745 ml (5.400 mmol) NEt3 were added to a solution of 0.133 g (0.45 mmol) 2,3,7,8-tetramino-phenazine·1.5HCl in 5 mL CH3CN. To this suspension, a solution of 0.385 g (2.250 mmol) 2-chloro-1,1,3,3-tetramethylformamidinium chloride in 10 mL CH3CN was added. Subsequently the reaction mixture was stirred for a period of 5 h at a temperature of 60 °C, leading to the precipitation of a brown-colored solid. After removal of the solvent under vacuum, the pale-brown solid was dissolved in destilled water. Upon addition of 25% NaOH solution, an orange-brown solid precipitated, which was extracted with CHCl3. The CHCl3 solution was dried over K2CO3 before the solvent was removed under vacuum. Yield of crude product: 0.199–0.227 g (0.314–0.359 mmol, 70–80%). Recrystallization from toluene at −18 °C afforded the orange-colored solid product in a yield of up to 74%. Deep-orange colored crystals were obtained from Et2O solutions at room temperature. Solutions of the compound are yellow-orange colored. Elemental analysis for C32H52N14 (632.85): calcd C 60.73, H 8.28, N 30.99; found C 60.12, H 7.63, N 30.33. 1H NMR (600.13 MHz, CD3CN): δ (ppm) = 6.78 (s, 4 H, Har), 2.72 (s, 48 H, Me). 13C NMR (150.92 MHz, CD3CN): δ (ppm) = 160.34 (Cguanidin), 150.25 (Car), 141.34 (Car), 114.05 (Car-H), 39.97 (Me). MS (ESI+): m/z = 633.3 u ([1 + H]+, 100.0%), 317.3 u ([1 + 2H]2+, 99.24%), 272.4 u ([1 − 2NMe2]2+, 14.77%). MS (FAB+): m/z = 633.5 u ([1 + H]+. UV/Vis (CH3CN, c = 1.58 × 10−5 mol l−1, d = 0.5 cm): λmax (ε, in L mol−1cm−1) = 222 (3.6228 × 104), 309 (5.6556 × 104), 480 (3.5612 × 104) nm. UV/Vis (H2O, c = 1.1503 × 10−5 mol l−1, d = 1 cm): λmax (ε, in L mol−1 cm−1) = 229 (5.7353 × 104), 300 (5.6211 × 104), 432 (2.0927 × 104), 484 (1.6961 × 104) nm. Fluorescence (CH3CN, λex = 480 nm): λmax,em = 568 nm. Fluorescence (H2O, λex = 484 nm): λmax,em = 640 nm. Quantum yield: Φ (H2O) = 0.02, Φ (CH3CN) = 0.22 and Φ (Et2O) = 0.39. IR (CsI): ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) = 3042w, 3001w, 2991w, 2930m, 2884m, 2794w, 1571s, 1508m, 1478m, 1433s, 1422s, 1379s, 1358m, 1333m, 1314w, 1266m, 1234m, 1189s, 1142s, 1106w, 1060m, 1019s, 932w, 923w, 907m, 867m, 855m, 794w, 777m, 741m, 718m, 676w, 669w, 654w, 597m, 583m, 530w, 457w cm−1. Crystal data for 1, C32H52N14: Mr = 632.88, 0.40 × 0.25 × 0.20 mm3, monoclinic, space group P2(1)/c, a = 774.7(2), b = 15.440(3), c = 15.074(3) Å, α = 90.00°, β = 96.68(3)°, γ = 90.00°, V = 1790.80 Å3, Z = 2, dcalc = 1.174 Mg m−3, Mo Kα radiation (graphite monochromated, λ = 0.71073 Å), T = 100 K, θrange 2.65 to 28.5°. Reflections measd. 8641, indep. 4477, Rint = 0.0445. Final R indices [I > 2σ(I)]: R1 = 0.059, wR2 = 0.153.
= 3042w, 3001w, 2991w, 2930m, 2884m, 2794w, 1571s, 1508m, 1478m, 1433s, 1422s, 1379s, 1358m, 1333m, 1314w, 1266m, 1234m, 1189s, 1142s, 1106w, 1060m, 1019s, 932w, 923w, 907m, 867m, 855m, 794w, 777m, 741m, 718m, 676w, 669w, 654w, 597m, 583m, 530w, 457w cm−1. Crystal data for 1, C32H52N14: Mr = 632.88, 0.40 × 0.25 × 0.20 mm3, monoclinic, space group P2(1)/c, a = 774.7(2), b = 15.440(3), c = 15.074(3) Å, α = 90.00°, β = 96.68(3)°, γ = 90.00°, V = 1790.80 Å3, Z = 2, dcalc = 1.174 Mg m−3, Mo Kα radiation (graphite monochromated, λ = 0.71073 Å), T = 100 K, θrange 2.65 to 28.5°. Reflections measd. 8641, indep. 4477, Rint = 0.0445. Final R indices [I > 2σ(I)]: R1 = 0.059, wR2 = 0.153.
2,3,6,7-Tetrakis(tetramethylguanidino)fluorene, 2 (in five steps)
1. Step (2,7-diamino-fluorene): 2.000 g (7.806 mmol) of 2,7-dinitrofluorene were added to a suspension of 14.11 g (74.40 mmol) SnCl2 in 48 mL Et2O. After addition of 30 mL concentrated HCl solution the suspension was heated under stirring for a period of 4 h to reflux. 2,7-diamino-fluorene·2HCl precipitated in the form of a pale-yellow colored solid, which was separated from the solution by filtration and subsequently washed three times with 10 mL portions of concentrated HCl solution. 1H NMR (200 MHz, d6-DMSO): δ (ppm) = 7.92 (d, 2 H, 1J = 8.2 Hz, H4/5ar), 7.47 (s, 2 H, H1/8ar), 7.28 (d, 2 H, 1J = 8.2 Hz, H3/6ar), 4.00 (s, 2 H, CH2). 13C NMR (150 MHz, d6-DMSO): δ (ppm) = 121.95 (C4/5ar), 121.08 (C1/8ar), 120.02 (C3/6ar), 36.36 (CH2). MS (FAB+): m/z = 196.5 u ([M]+, 100%). MS (ESI+): m/z = 197.2 u ([M + H]+, 100%). The 2,7-diamino-fluorene·2HCl was dissolved in H2O and a saturated NaHCO3 solution was added until a pH value of 8 was reached. The yellow precipitate of 2,7-diamino-fluorene was separated by filtration, dried under vacuum and used in the next step without further purification. 1H NMR (200 MHz, d6-DMSO): δ (ppm) = 7.25 (d, 2 H, 1J = 8.0 Hz, H4/5ar), 6.68 (s, 2 H, H1/8ar), 6.49 (d, 2 H, 1J = 8.0 Hz, H3/6ar), 4.87 (br s, 4 H, NH2), 3.57 ppm (s, 2 H, CH2).2. Step (2,7-diacetamidofluorene): 1.532 g (7.806 mmol) of 2,7-diaminofluorene were suspended in 135 mL glacial acetic acid. Then 45 mL acetic anhydride were added. Under an Ar atmosphere the solution was heated to reflux for a period of 30 min, then cooled back to room temperature and poured onto ice. The milky white precipitate of 2,7-diacetamidofluorene was separated by filtration and dried under vacuum. A yield of 75% referring to 2,7-dinitrofluorene was obtained. 1H NMR (200 MHz, d6-DMSO): δ (ppm) = 9.94 (s, 2 H, NH), 7.86 (s, 2 H, H1/8ar), 7.68 (d, 2 H, 1J = 8.2 Hz, H3/6ar), 7.48 (d, 2 H, 1J = 8.2 Hz, H4/5ar), 3.85 (s, 2 H, CH2), 2.06 (s, 6 H, CH3).
3. Step (3,6-dinitro-2,7-diaminofluorene): 1.703 g (6.073 mmol) 2,7-diacetamidofluorene were suspended in 40 mL CH2Cl2. After addition of 35 mL glacial acetic acid the mixture was cooled to −10 °C, and a −10 °C cold solution of 17.5 mL fuming nitric acid in 50 mL CH2Cl2 drop-wise slowly added, keeping the temperature below −5 °C. The color of the reaction mixture changes first from white to deep-green. After complete addition of the HNO3–CH2Cl2 solution it slowly bleached and an orange-colored precipitate formed. After stirring for an additional 10 min the solution was poured on ice. The yellow-colored precipitate was separated by filtration and washed with some H2O, yielding 67% of 3,6-dinitro-2,7-diacetamidofluorene. 1H NMR (200 MHz, d6-DMSO): δ (ppm) = 10.30 (s, 2 H, NH), 8.73 (s, 2 H, H4/5ar), 7.89 (s, 2 H, H1/8ar), 4.21 (s, 2 H, CH2), 2.11 (s, 6 H, CH3). MS (EI+): m/z = 370.1 u ([M]+, 52%), 279.1 u ([2,7-diacetamido-fluorene – H]+, 100%). Then 0.500 g (1.351 mmol) of 3,6-dinitro-2,7-diacetamidofluorene were suspended in 150 mL acetone. After addition of 30 mL concentrated HCl solution, the mixture was heated for 7 h to a temperature of 75 °C, leading to a color change from orange to deep-red. The reaction mixture was allowed to cool back to room temperature and then poured on ice. The deep-red colored precipitate was separated by filtration and dried under vacuum, yielding 78% of 3,6-dinitro-2,7-diaminofluorene. 1H NMR (200 MHz, d6-DMSO): δ (ppm) = 8.41 (s, 2 H, Har), 7.54 (br s, 4 H, NH2), 7.09 (s, 2 H, Har), 3.96s, 2 H, CH2).
4. Step (2,3,6,7-tetraminofluorene): 4.5 mL concentrated HCl solution was added to a suspension of 0.148 g (0.516 mmol) of 3,6-dinitro-2,7-diaminofluorene in 15 mL EtOH. The pale-yellow colored precipitate of 2,3,6,7-tetramino-fluorene·4HCl was separated by filtration, washed three times with 4 mL portions of concentrated HCl solution and dried under vacuum to obtain 2,3,6,7-tetramino-fluorene·4HCl in a yield of 58%. 1H NMR (200 MHz, d3-CD3CN): δ (ppm) = 7.90 (s, 2 H, H4/5ar), 7.62 (s, 2 H, H3/6ar), 4.08 (s, 2 H, CH2). MS (HR-ESI): m/z = 226.12128 u ([M]+, 100%).
5. Step (2,3,6,7-Tetrakis(N,N,N′,N′-tetramethylguanidino)fluorene, 2): 0.546 g (5.400 mmol) NEt3 were added to a solution of 0.102 g (0.450 mmol) 2,3,6,7-tetraminofluorene-tetrahydrochloride in 5 mL CH3CN. This suspension was cooled at 0 °C. To this suspension, a solution of 0.385 g (2.250 mmol) of 2-chloro-1,1,3,3-tetramethylformamidinium chloride in 8 mL CH3CN was added. The reaction mixture was stirred for a period of 2 h at a temperature of 0 °C, leading to the precipitation of a brown-colored solid. After removal of the solvent under vacuum, the light-brown solid was dissolved in 10% HCl solution. Upon the addition of 25% NaOH solution, the aqueous solution was extracted with Et2O (4 × 15 mL), and the organic phases were dried over KCO3. The solvent was removed under vacuum, and the crude product as yellow-brown solid was dried under vacuum. The purification was carried out by sublimation at 210–220 °C and the white solid was obtained. Yield: 0.162 g (0.261 mmol, 58%). Elemental analysis for C33H54N12 (618.86): calcd C 64.05, H 8.79, N 27.16; found C 63.57, H 8.89, N 26.25. 1H NMR (600 MHz, CD3CN): δ (ppm) = 6.63 (s, 2 H, H4ar), 6.51 (s, 2 H, H1ar), 3.55 (s, 2 H, CH2), 2.64 (s, 58 H, CH3). 13C NMR (150 MHz, CD3CN): δ (ppm) = 159.01 (s, Cguanidin), 143.88 (s, C2/3ar), 143.13 (s, C2/3ar), 136.47 (s, C5/6ar), 136.30 ppm (s, C5/6ar), 118.57 ppm (s, C1ar), 111.98 ppm (s, C4ar), 36.32 ppm (s, CH2), 39.76 ppm (s, CH3). MS (HR-ESI): m/z = 1237.92446 u ([2M + H]+, 4.90%), 619.46635 u ([M + H]+, 100%), 310.23695 u ([M+2H]2+, 32.0%). UV/Vis (CH3CN, c = 1.0341 × 10−5 mol l−1, d = 1 cm): λmax (ε, in L mol−1 cm−1) = 371 (2.431 × 104), 319 (1.529 × 104), 269 (2.546 × 104, shoulder), 226 (3.990 × 104) nm. Fluorescence (CH3CN, λex = 342 nm): λmax,em = 428 nm. IR (CsI): = 2999w, 2924m, 2886m, 2803w, 1612s, 1601s, 1587s, 1549m (shoulder), 1497m, 1476w, 1460m, 1437w, 1423m, 1406w, 1371s, 1333w, 1325w, 1277m, 1261m, 1233m, 1215m, 1190w, 1173w, 1165m, 1138s, 1107w, 1092w, 1063m, 1018s, 1001s, 955w, 939w, 920m, 858m, 833w, 800m, 789m, 735w, 671w, 637m, 579w, 505w cm−1. Crystal data for 2·CH3CN, C35H57N13: Mr = 659.94, 0.35 × 0.20 × 0.15 mm3, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 7.8850(16), b = 13.540(3), c = 18.573(4) Å, α = 103.13(3)°, β = 90.59(3)°, γ = 92.60(3)°, V = 1928.6(7) Å3, Z = 2, dcalc = 1.136 Mg m−3, Mo Kα radiation (graphite monochromated, λ = 0.71073 Å), T = 100 K, θrange 2.25 to 27.71°. Reflections measd. 31289, indep. 8875, Rint = 0.0820. Final R indices [I > 2σ(I)]: R1 = 0.0673, wR2 = 0.1721.
, a = 7.8850(16), b = 13.540(3), c = 18.573(4) Å, α = 103.13(3)°, β = 90.59(3)°, γ = 92.60(3)°, V = 1928.6(7) Å3, Z = 2, dcalc = 1.136 Mg m−3, Mo Kα radiation (graphite monochromated, λ = 0.71073 Å), T = 100 K, θrange 2.25 to 27.71°. Reflections measd. 31289, indep. 8875, Rint = 0.0820. Final R indices [I > 2σ(I)]: R1 = 0.0673, wR2 = 0.1721.
2,3-Bis(tetramethylguanidino)phenazine, 5
722 mg (7.14 mmol) of NEt3 was added to a suspension of 300 mg of 2,3-diaminophenazine (1.43 mmol) in 4 mL CH3CN. Then a solution of 1.22 g (7.14 mmol) of 2-chloro-1,1,3,3-tetramethylformamidinium chloride in 8 mL CH3CN was slowly drop-wise added over a period of 10 min. The reaction mixture, which turns deep-brown, was stirred for a period of 3 h at a temperature of 50 °C. After the mixture has cooled back to room-temperature, the solvent was removed in vacuum and the remaining deep-brown colored solid re-dissolved in water. The product was then precipitated by addition of 25% NaOH solution, and subsequently extracted 5 times with 15 mL portions of CHCl3. The organic phase was dried over K2CO3, and then the solvent removed to yield 337 mg (58%) crude product as brown-colored solid. The crude product was re-dissolved in CH3CN and the yellow-colored insoluble side product of the reaction was removed by filtration, before the solvent was removed from the filtrate under vacuum. In this way one obtains 121 mg (0.30 mmol, total yield 21%) orange-colored clean product. Elemental analysis for C22H30N8 (406.53): calcd C 65.00, H 7.44, N 27.56; found C 64.72, H 7.15, N 27.49. 1H NMR (400 MHz, CD3CN): δ (ppm) = 2.75 (s, 25 H, CH3), 6.84 (s, 2 H, CH5ar), 7.60 (q, J = 3.39 Hz, 2 H, Har), 7.96 (q, J = 3.39 Hz, 2 H, Har). 13C NMR (400 MHz, CD3CN): δ (ppm) = 40.15 (CH3), 113.05 (CH5ar), 128.13 (CHar), 129.48 (CHar). Four quaternary C atoms were not detected. IR (CsI): = 2927 w (ν(C–H)), 1577 vs. (ν(CC)), 1510m (C–H def), 1437 vs. (C–H def), 1383m (C–H def), 1233 w, 1133m, 1019m, 891 w, 757 w (Car–H def), 595 w, 505 w cm−1. MS (DART): m/z = 407.26683 ([M + H]+, 100.0%). [C22H31N8]+, calcd 407.26662, [M + H]+ (100.0%), found 407.26683, diff. −0.2 mmu. UV/Vis (CH3CN, c = 1.28 × 10−5 mol l−1, d = 1 cm): λmax (ε, in L mol−1 cm−1) = 233 (3.63 × 105), 276 (5.32 × 105), 393 (1.58 × 105) nm. UV/Vis (Et2O, c = 1.18 × 10−5 mol l−1, d = 1 cm): λmax (ε, in L mol−1 cm−1) = 277 (3.91 × 105), 433 (1.77 × 105) nm. Fluorescence (CH3CN, λex = 233 nm): λmax,em = 599 nm; Fluorescence (THF, λex = 447 nm): λmax,em = 588 nm. Quantum yield: Φ (THF) = 0.17, Φ (CH3CN) = 0.15, Φ (Et2O) = 0.11. Crystal data for 5·2H2O, C22H34N8O2: Mr = 442.57, 0.50 × 0.50 × 0.45 mm3, monoclinic, space group P2/c, a = 15.900(3), b = 9.2100(18), c = 16.379(3) Å, β = 94.82(3)°, V = 2390.0(8) Å3, Z = 4, dcalc = 1.230 Mg m−3, Mo Kα radiation (graphite monochromated, λ = 0.71073 Å), T = 100 K, θrange 2.21 to 30.04°. Reflections measd. 13359, indep. 6949, Rint = 0.0323. Final R indices [I > 2σ(I)]: R1 = 0.0910, wR2 = 0.2642.
[1(CuI)2]: Compound 1 (0.0669 g, 0.1058 mmol) were dissolved in 8 mL CH2Cl2. Then 2 equivalents of CuI (0.0403 g, 0.2116 mmol) were added. The dark red reaction mixture was stirred for a period of 1 h at room temperature. Subsequently the solvent was removed under vacuum. The dark red precipitate was washed with Et2O (3 × 5 mL), and dried under vacuum. The solid was re-dissolved in CH2Cl2 and upon addition of Et2O precipitated as a clean product. The product was filtrated and dried under vacuum. Yield: 0.055 g (0.0539 mmol, 51%). Elemental analysis for C32H52Cu2I2N14 (1013.75): calcd C 37.91, H 5.17, N 19.34; found C 37.65, H 5.06, N 19.18. MS (HR-ESI+): m/z = 823.29278 u ([1 + CuI + H+]+, 20.5%), 950.129723 u ([1 + CuI + I− + H+]+, 14.7%). MS (FAB+): m/z = 588.4 ([1 − NMe2]+, 21.2%), 633.5 u ([1 + H+]+, 9.6%), 696.1 u ([1 + Cu+]+, 17.3%), 824.0 u ([1 + CuI+2H+]+, 13.5%), 1012.7 u ([M+]+, 9.6%). UV/Vis (CH3CN, c = 0.9081 × 10−5 mol l−1, d = 1 cm): λmax (ε, in L mol−1 cm−1) = 226 (6.2067 × 104), 243 (5.7190 × 104), 317 (7.4615 × 104), ca. 465 (3.4082 × 104, shoulder), 493 nm (5.9110 × 104) nm. UV/Vis (CH2Cl2, c = 1.3024 × 10−5 mol l−1, d = 1 cm): λmax (ε, in L mol−1 cm−1) = 321 (8.8200 × 104), ca. 474 (4.2945 × 104, shoulder), 501 (7.2754 × 104) nm. Fluorescence signals almost extinguished by CuI coordination. Fluorescence (CH2Cl2, λex = 460 nm): λmax,em = ca. 519 nm. Fluorescence (CH3CN, λex = 465 nm): λmax,em = ca. 533 nm. IR (CsI): = 3047w, 3005w, 2936m, 2890m, 2793w, 1574s, 1515s, 1451s, 1417s, 1401s, 1325m, 1263m, 1232w, 1199m, 1157m, 1142m, 1108w, 1064w, 1026m, 958w, 917w, 905w, 855w, 810w, 796w, 785w, 740w, 733w, 714w, 696w, 668w, 610m, 502w cm−1.
[
1
(ZnCl2)2]: 0.0256 g (2 eq., 0.1878 mmol) of ZnCl2 was dissolved in 2 mL CH3CN and added dropwise to a solution of 0.0594 g (0.0939 mmol) of 1 in 2 mL CH3CN. The reaction mixture was stirred for a period of 2 h at room temperature. The solvent was removed to obtain an orange solid that was re-dissolved in 1.5 mL CH2Cl2 and filtered to remove the insoluble rest. Upon addition of Et2O the product precipitates as a crystalline orange solid. The product was filtered, washed three times with 1 mL portions of Et2O and dried under vacuum. Upon recrystallization from CH2Cl2 crystals of [1(ZnCl2)2] were obtained. Yield 0.0450 mg (0.0498 mmol, 53%) product. Elemental analysis for C32H52Cl4N14Zn2 (905.42): calcd C 42.45, H 5.79, N 21.66; found C 42.16, H 5.66, N 21.26. 1H NMR (600 MHz, CD3CN): δ (ppm) = 6.89 (s, 4 H, Har), 3.03 (s, 24 H, Me), 2.84 (s, 24 H, Me). 13C NMR (150 MHz, CD3CN): δ (ppm) = 166.23 (s, Cguan), 145.49 (s, Cq,ar), 141.51 (s, Cq,ar), 113.02 (s, Car–H), 41.21 (s, Me), 40.52 (s, Me). 1H NMR (600 MHz, CD2Cl2): δ (ppm) = 6.88 (s, 4 H, Har), 3.06 (s, 24 H, Me), 2.90 (s, 24 H, Me). 13C NMR (150 MHz, CD2Cl2): δ (ppm) = 165.74 (s, Cguan), 144.61 (s, Cq,ar), 141.09 (s, Cq,ar), 112.55 (s, Car–H), 41.17 (s, Me), 40.38 (s, Me). UV/Vis (CH3CN, c = 1.1486 × 10−5 mol l−1, d = 1 cm): λmax (ε, in L mol−1 cm−1) = 228 (5.8829 × 104), 316 (7.6583 × 104), 458 (3.7518 × 104), 484 (5.7725 × 104) nm. UV/Vis (CH2Cl2, c = 1.4137 × 10−5 mol l−1, d = 0.5 cm): λmax (ε, in L mol−1 cm−1) = 228 (5.3921 × 104), 319 (6.9684 × 104), 459 (3.5629 × 104), 486 (5.4647 × 104) nm. Fluorescence (CH3CN, λex = 484 nm): λmax,em = 506 nm. Fluorescence (CH2Cl2, λex = 486 nm): λmax,em = 512 nm. Quantum yield: Φ(CH3CN) = 0.36. IR (CsI): = 3049w, 3009w, 2935m, 2895w, 2799w, ca. 1615m shoulder, 1565s, 1522s, 1452s, 1420s, 1408s, 1400s, 1329m, 1261m, 1234w, 1193m, 1162m, 1144w, 1110w, 1065w, 1035m, 1027m, 957w, 919w, 907w, 861w, 810w, 789w, 740w, 735w, 715w, 707w, 611w, 575w, 561w, 506w, 503w, 492w, 473w cm−1. Crystal data for [1(ZnCl2)2]·2CH2Cl2, C36H60Cl12N14Zn2: Mr = 1245.12, 0.30 × 0.30 × 0.20 mm3, monoclinic, space group P2(1)/c, a = 13.162(3), b = 13.499(3), c = 16.472(3) Å, β = 108.30(3)°, V = 2778.63 Å3, Z = 2, dcalc = 1.488 Mg m−3, Mo Kα radiation (graphite monochromated, λ = 0.71073 Å), T = 100 K, θrange 4.4 to 60.1°. Reflections measd. 16059, indep. 8111, Rint = 0.0482. Final R indices [I > 2σ(I)]: R1 = 0.045, wR2 = 0.128.
[
5
(ZnCl2)]. 0.010 g (1 eq., 0.0738 mmol) of ZnCl2 was added to a solution of 0.030 g (0.0738 mmol) of 5 in 5 mL CH3CN. The reaction mixture was stirred for a period of 4 h at room temperature. The solvent was partially removed under vacuum before the solution was stored at +4 °C for several days. The needle shaped crystals which were formed in the solution were filtered off and washed three times with Et2O and n-hexane. The remaining yellow solid was dried under vacuum. Upon recrystallization from CH3CN crystals of [5(ZnCl2)] were obtained. Elemental analysis for C22H30Cl2N8Zn, (542.81): calcd C 48.68, H 5.57, N 20.64; found C 48.36, H 5.50, N 21.16. 1H NMR (400 MHz, CD3CN): δ (ppm) = 2.88 (br. s., 12 H), 3.06 (br. s., 12 H), 6.99 (s, 2 H), 7.70 (q, J = 3.39 Hz, 2 H), 8.04 (q, J = 3.64 Hz, 2 H). 13C NMR (400 MHz, CD3CN): δ (ppm) = 112.57 (CH5ar), 129.42 (CHar), 129.77 (CHar), 143.02 (Cq), 143.65 (Cq), 147.78 (Cq). IR (CsI): = 2932 w (ν(C–H)), 1571m (CC val), 1530m (C–H def), 1453s (C–H def), 1405s (C–H def), 1340m, 1232m, 1163m, 1036m, 897w, 816w, 762w (Car–H def), 603w, 503w cm−1. MS (HR-FAB): m/z = 540.1262 ([M]+, 80.0%). [C22H30N8ZnCl2]+ calcd 540.1262, [M]+ (80.0%); found 540.1262, Diff.: 0.0 mmu. UV/Vis (CH3CN, c = 1.18 × 10−5 mol l−1, d = 1 cm): λmax (ε, in L mol−1 cm−1) = 231 (3.34 × 105), 283 (3.79 × 105), 302 (3.40 × 105), 424 (2.02 × 105), 442 (2.31 × 105) nm. Fluorescence (THF, λex = 445 nm): λmax,em = 498 nm. Fluorescence (CH3CN, λex = 302 nm): λmax,em = 514 nm. Quantum yield: Φ(CH3CN) = 0.08. Crystal data for [5(ZnCl2)]·CH3CN, C24H33Cl2N9Zn: Mr = 583.86, 0.50 × 0.50 × 0.50 mm3, monoclinic, space group P2(1), a = 10.558(2), b = 23.182(5), c = 22.678(5) Å, β = 90.44(3)°, V = 5550.40(8) Å3, Z = 8, dcalc = 1.397 Mg m−3, Mo Kα radiation (graphite monochromated, λ = 0.71073 Å), T = 100 K, θrange 1.8 to 60.2°. Reflections measd. 32157, indep. 32131, Final R indices [I > 2σ(I)]: R1 = 0.039, wR2 = 0.101.
[(
1
)2(ZnCl2)5]: To 0.0362 g (2.5 eq., 0.2656 mmol) of ZnCl2 in CH2Cl2 was added a solution of 0.0672 g (0.1062 mmol) of 1 in 8 mL CH2Cl2. The reaction mixture was stirred for a period of 2 h at a temperature of 45 °C until it became dark red. After the mixture had cooled back to room temperature, the solvent was partially removed. To this concentrated solution was added 8 mL Et2O. The mixture was stored at 0 °C to get a crystalline red precipitate. The solid was filtrated, washed with Et2O and n-hexane and dried under vacuum. Purple crystals were obtained from a CH2Cl2–Et2O mixture. Yield: 0.0289 g (0.0149 mmol, 28%). Elemental analysis for C64H104Cl10N28Zn5 (1947.13): calcd C 39.48, H 5.38, N 20.14; found C 38.72, H 5.91, N 20.30. Crystal data for [(1)2(ZnCl2)5]·4.7CH2Cl2, C68.70H113.40Cl19.40N28Zn5: Mr = 2346.25, 0.35 × 0.30 × 0.30 mm3, triclinic, space group P, a = 17.629(4), b = 18.298(4), c = 18.587(4) Å, α = 90.87(3)°, β = 104.98(3)°, γ = 108.66(3)°, V = 5455.9(19) Å3, Z = 2, dcalc = 1.428 Mg m−3, Mo Kα radiation (graphite monochromated, λ = 0.71073 Å), T = 100 K, θrange 2.48 to 27.52°. Reflections measd. 45548, indep. 24658, Rint = 0.0342. Final R indices [I > 2σ(I)]: R1 = 0.0633, wR2 = 0.1829.
X-ray crystallographic study
Suitable crystals were taken directly out of the mother liquor, immersed in perfluorinated polyether oil and fixed on top of a cryo loop. Measurements were made with a Nonius-Kappa CCD diffractometer with low-temperature unit using graphite-monochromated Mo Kα radiation. The temperature was set to 100 K. The data collected were processed using the standard Nonius software.37 All calculations were performed using the SHELXT-PLUS software package. Structures were solved by direct methods with the SHELXS-97 program and refined with the SHELXL-97 program.38,39 Graphical handing of the structural data during solution and refinement was performed with XPMA.40 Atomic coordinates and anisotropic thermal parameters of non-hydrogen atoms were refined by full-matrix least-squares calculations. CCDC-1016464 (1), 1016465 (2), 1034621 ([1(ZnCl2)2]), 1016466 ([5(ZnCl2)]), 1016467 (5·2H2O), 1016468 ([1H2](BF4)2), and 1016832 ([(1)2(ZnCl2)5]) contain the supplementary crystallographic data for this paper..Details of the quantum chemical calculations
The density functional calculations on the uncoordinated compounds and protonated 1 were carried out with the B3LYP functional41 and the 6-311G** basis set.42 The GAUSSIAN43 program package was used. To speed up the investigation for the different complexes, the calculations, including the TD-DFT calculations, were performed with the smaller def2-SV(P) basis set44 in combination with the BP8641a,45 and B3LYP functionals. The structures were optimized with the BP86 functional, using the resolution of the identity (RI) approximation and the appropriate auxiliary SV(P) basis set.46 Then final energies were obtained with the B3LYP functional using thermodynamic contributions from the BP86 results. These calculations used the TURBOMOLE47 program package.A comparison of the structures (Table S2 in the ESI†) and the HOMO–LUMO gap of compound 1 shows that in the case of the structure the usage of a different basis set and functional and in the case of the HOMO–LUMO gap the usage of a different basis set has only a minor impact on the calculated quantities. Within the B3LYP/6-311G** calculations the HOMO–LUMO gap amounts to 3.24 eV, whereas within the B3LYP/def2-SV(P) calculations it amounts to 3.16 eV.
Acknowledgements
The authors thank the Deutsche Forschungsgemeinschaft (DFG) for continuous financial support.References
- (a) W. H. Perkin, J. Chem. Soc., 1896, 69, 596–637 RSC; (b) O. Meth-Cohn and M. Smith, J. Chem. Soc., Perkin Trans., 1994, 5–7 RSC; (c) J. S. de Melo, S. Takato, M. Sousa, M. J. Melo and A. J. Parola, Chem. Commun., 2007, 2624–2626 RSC.
- K. Hübner, Chem. Unserer Zeit, 2006, 40, 274–275 CrossRef.
- A. K. Jana, J. Photochem. Photobiol., A, 2000, 132, 1–17 CrossRef CAS.
- N. J. Lundin, A. G. Blackman, K. C. Gordon and D. L. Officer, Angew. Chem., 2006, 118, 2644–2646 ( Angew. Chem., Int. Ed. , 2006 , 45 , 2582–2584 ) CrossRef.
- P. Hashemi and R. A. Zarjani, Sens. Actuators, B, 2008, 135, 112–115 CrossRef CAS PubMed.
- J. J. Bryant, Y. Zhang, B. D. Lindner, E. A. Davey, A. L. Appleton, X. Qian and U. H. F. Bunz, J. Org. Chem., 2012, 77, 7479–7486 CrossRef CAS PubMed.
- S. Miao, C. G. Bangcuyo, M. D. Smith and U. H. F. Bunz, Angew. Chem., 2006, 118, 677–681 ( Angew. Chem., Int. Ed. , 2006 , 45 , 661–665 ) CrossRef.
- G. Hughes and M. R. Bryce, J. Mater. Chem., 2005, 15, 94–107 RSC.
- For some recent examples, see: (a) J. Huang, R. Tang, T. Zhang, Q. Li, G. Yu, S. Xie, Y. Liu, S. Ye, J. Qin and Z. Li, Chem. – Eur. J., 2014, 20, 1–11 CrossRef; (b) K. Zhang, C. Zhong, S. Liu, A. Liang, S. Dong and F. Huang, J. Mater. Chem. C, 2014, 2, 3270–3277 RSC; (c) X. J. Feng, S. F. Chen, Y. Ni, M. S. Wong, M. M. K. Lam, K. W. Cheah and G. Q. Lai, Org. Electron., 2014, 15, 57–64 CrossRef CAS PubMed; (d) Z. H. Li, M. S. Wong, H. Fukutani and Y. Tao, Org. Lett., 2006, 8, 4271–4274 CrossRef CAS PubMed.
- (a) Y. Li, J. Ding, M. Day, Y. Tao, J. Lu and M. D'Iorio, Chem. Mater., 2003, 15, 4936–4943 CrossRef CAS; (b) H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- M. Mastalerz, Angew. Chem., 2008, 120, 453–455 ( Angew. Chem., Int. Ed. , 2008 , 47 , 445–447 ) CrossRef.
- (a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed; (b) B. Valeur and I. Leray, Coord. Chem. Rev., 2000, 205, 3–40 CrossRef CAS; (c) C. Lodeiro, J. L. Capelo, J. C. Mejuto, E. Oliveira, H. M. Santos, B. Pedras and C. Nuñez, Chem. Soc. Rev., 2010, 39, 2948–2976 RSC; (d) H. N. Kim, W. X. Ren, J. S. Kim and J. Yoon, Chem. Soc. Rev., 2012, 41, 3210–3244 RSC.
- Z. Xu, J. Yoon and D. R. Spring, Chem. Soc. Rev., 2010, 39, 1996–2006 RSC.
- A. P. de Silva, T. S. Moody and G. D. Wright, Analyst, 2009, 134, 2385–2393 RSC.
- Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568–1600 RSC.
- R. D. Hancock, Chem. Soc. Rev., 2013, 42, 1500–1524 RSC.
- (a) D. Brox, A. Kiel, S. J. Wörner, M. Pernpointner, P. Comba, B. Martin and D. P. Herten, PLoS One, 2013, 8, e58049 CAS; (b) A. Sprödefeld, A. Kiel, D. P. Herten and R. Krämer, Z. Anorg. Allg. Chem., 2013, 639, 1636–1639 CrossRef; (c) A. Kiel, J. Kovacs, A. Mokhir, R. Krämer and D. P. Herten, Angew. Chem., 2007, 119, 3645–3649 ( Angew. Chem., Int. Ed. , 2007 , 46 , 3363–3366 ) CrossRef.
- (a) F. T. Edelmann, Adv. Organomet. Chem., 2008, 57, 183–352 CrossRef CAS; (b) F. T. Edelmann, Coord. Chem. Rev., 1994, 137, 403–481 CrossRef; (c) J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219–300 CrossRef CAS; (d) P. J. Bailey and S. Price, Coord. Chem. Rev., 2001, 214, 91–141 CrossRef CAS; (e) S. Herres-Pawlis, Nat. Chem., 2009, 57, 20–23 CAS; (f) M. P. Coles, Dalton Trans., 2006, 985–1001 RSC; (g) M. P. Coles, Chem. Commun., 2009, 3659–3676 RSC; (h) H.-J. Himmel, in Modeling of Molecular Properties, ed. P. Comba, Wiley-VCH, 2011 Search PubMed; (i) C. Jones, Coord. Chem. Rev., 2010, 254, 1273–1289 CrossRef CAS PubMed; (j) F. T. Edelmann, Adv. Organomet. Chem., 2013, 61, 55–374 CrossRef CAS.
- (a) C. Würtele, E. Gaoutchenova, K. Harms, M. C. Holthausen, J. Sundermeyer and S. Schindler, Angew. Chem., 2006, 118, 3951–3954 ( Angew. Chem., Int. Ed. , 2006 , 45 , 3867–3869 ) CrossRef; (b) D. Maiti, D.-H. Lee, K. Gaoutchenova, C. Würtele, M. C. Hothausen, A. A. N. Sarjeant, J. Sundermeyer, S. Schindler and K. D. Karlin, Angew. Chem., 2007, 120, 88–91 ( Angew. Chem., Int. Ed. , 2007 , 47 , 82–85 ) CrossRef; (c) M. P. Lanci, V. V. Smirnov, C. J. Cramer, E. V. Gauchenova, J. Sundermeyer and J. P. Roth, J. Am. Chem. Soc., 2007, 129, 14697–14709 CrossRef CAS PubMed; (d) D. Maiti, D.-H. Lee, K. Gaoutchenova, C. Würtele, M. C. Holthausen, A. A. N. Sarjeant, J. Sundermeyer, S. Schindler and K. D. Karlin, Angew. Chem., 2008, 120, 88–91 ( Angew. Chem., Int. Ed. , 2008 , 47 , 82–85 ) CrossRef; (e) R. L. Peterson, J. W. Ginsbach, R. E. Cowley, M. F. Qayyum, R. A. Himes, M. A. Siegler, C. D. Moore, B. Hedman, K. O. Hodgson, S. Fukuzumi, E. I. Solomon and K. D. Karlin, J. Am. Chem. Soc., 2013, 135, 16454–16467 CrossRef CAS PubMed; (f) C. Saracini, D. G. Liakos, J. E. Zapata Rivera, F. Neese, G. J. Meyer and K. D. Karlin, J. Am. Chem. Soc., 2014, 136, 1260–1263 CrossRef CAS PubMed.
- (a) J. Börner, U. Flörke, K. Huber, A. Döring, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2009, 15, 2362–2376 CrossRef PubMed; (b) J. Börner, I. dos Santos Vieira, A. Pawlis, A. Doering, D. Kuckling and S. Herres-Pawlis, Chem. – Eur. J., 2011, 17, 4507–4512 CrossRef PubMed; (c) J. Börner, I. dos Santos Vieira, M. D. Jones, A. Döring, D. Kuckling, U. Flörke and S. Herres-Pawlis, Eur. J. Inorg. Chem., 2011, 4441–4456 CrossRef; (d) I. dos Santos Vieira and S. Herres-Pawlis, Eur. J. Inorg. Chem., 2012, 765–774 CrossRef CAS; (e) I. dos Santos Vieira and S. Herres-Pawlis, Z. Naturforsch., B: Chem. Sci., 2012, 67, 320–330 CrossRef CAS PubMed.
- A. Maronna, O. Hübner, M. Enders, E. Kaifer and H.-J. Himmel, Chem. – Eur. J., 2013, 19, 8958–8977 CrossRef CAS PubMed.
- M. Reinmuth, C. Neuhäuser, P. Walter, M. Enders, E. Kaifer and H.-J. Himmel, Eur. J. Inorg. Chem., 2011, 83–90 CrossRef CAS.
- P. Roquette, A. Maronna, A. Peters, E. Kaifer, H.-J. Himmel, Ch. Hauf, V. Herz, E.-W. Scheidt and W. Scherer, Chem. – Eur. J., 2010, 16, 1336–1350 CrossRef CAS PubMed.
- H.-J. Himmel, Z. Anorg. Allg. Chem., 2013, 639, 1940–1952 CrossRef CAS.
- The strong electron donor capacity of GFA compounds makes their chemistry very different to that of previously synthesized aromatic compounds with guanidino substituents, such as 1,2-bis(tetramethylguanidino)benzene, which was first reported by the Ishikawa group: M. Kawahata, K. Yamaguchi, T. Ito and T. Ishikawa, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o3301–o3302 CAS.
- (a) A. Peters, E. Kaifer and H.-J. Himmel, Eur. J. Org. Chem., 2008, 5907–5914 CrossRef CAS; (b) A. Peters, C. Trumm, M. Reinmuth, D. Emeljanenko, E. Kaifer and H.-J. Himmel, Eur. J. Inorg. Chem., 2009, 3791–3800 CrossRef CAS.
- S. Stang, A. Lebkücher, P. Walter, E. Kaifer and H.-J. Himmel, Eur. J. Inorg. Chem., 2012, 4833–4845 CrossRef CAS.
- S. Stang, E. Kaifer and H.-J. Himmel, Chem. – Eur. J., 2014, 20, 5288–5297 CrossRef CAS PubMed.
- H. M. Gajiwala and R. Zand, Polymer, 2000, 41, 2009–2015 CrossRef CAS.
- (a) M. J. Namkung and T. L. Fletcher, J. Org. Chem., 1959, 25, 740–744 CrossRef; (b) X. Li, Y. Xiao and X. Qian, Org. Lett., 2008, 10, 2885–2888 CrossRef CAS PubMed.
- M. Reinmuth, C. Neuhäuser, P. Walter, M. Enders, E. Kaifer and H.-J. Himmel, Eur. J. Inorg. Chem., 2011, 83–90 CrossRef CAS.
- The term “Stokes shift”, as used in this article, also includes possible solvent effects or excited-state reactions (of the S1 electronic state), as long as internal conversion to a triplet state is not involved. In this we follow the explanation of J. R. Lakowicz in his seminal book “Principles of Fluorescence Spectroscopy” (Springer Science + Business Media, third edition, 2006, p. 6).
- M. L. Gómez, C. M. Previtali and H. A. Montejano, Spectrochim. Acta, Part A, 2004, 60, 2433–2439 CrossRef PubMed.
- (a) G. David, P. J. Walsh and K. C. Gordon, Chem. Phys. Lett., 2004, 383, 292–296 CrossRef CAS PubMed; (b) N. J. Lundin, A. G. Blackman, K. C. Gordon and D. L. Officer, Angew. Chem., 2006, 118, 2644–2646 ( Angew. Chem. Int. Ed. , 2006 , 45 , 2582–2584 ) CrossRef.
- See, for example: C. T. L. Ma and M. J. MacLachlan, Angew. Chem., 2005, 117, 4250–4254 ( Angew. Chem., Int. Ed. , 2005 , 44 , 4178–4182 ) CrossRef.
- A. T. R. Williams, S. A. Winfield and J. N. Miller, Analyst, 1983, 108, 1067–1071 RSC.
- DENZO-SMN, Data processing software, Nonius, 1998, http:// www.nonius.nl Search PubMed.
- (a) G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, Germany, 1997, http://shelx.uni-ac.gwdg.de/SHELX/index.html Search PubMed; (b) G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, http://shelx.uni-ac.gwdg.de/SHELX/index.html Search PubMed.
- International Tables for X-Ray Crystallography, vol. 4, Kynoch Press, Birmingham, 1974 Search PubMed.
- L. Zsolnai and G. Huttner, XPMA, University of Heidelberg, Germany, 1994, http://www.uni-heidelberg.de/institute/fak12/AC/huttner/soft-ware/software.html Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS; (c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- K. Raghavachari, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- (a) R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS; (b) O. Treutler and R. Ahlrichs, J. Chem. Phys., 1995, 102, 346–354 CrossRef CAS PubMed; (c) K. Eichkorn, O. Treutler, H. Öhm, M. Häser and R. Ahlrichs, Chem. Phys. Lett., 1995, 242, 652–660 CrossRef CAS; (d) R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1016464–1016468, 1016832, 1034621. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03572k |
| This journal is © The Royal Society of Chemistry 2015 |