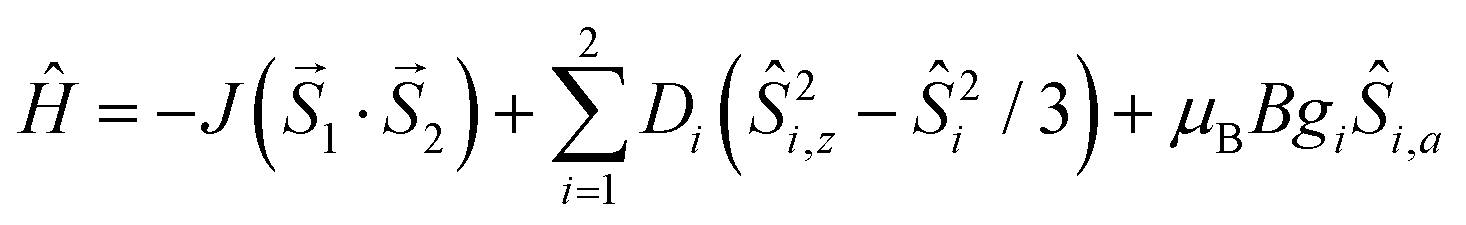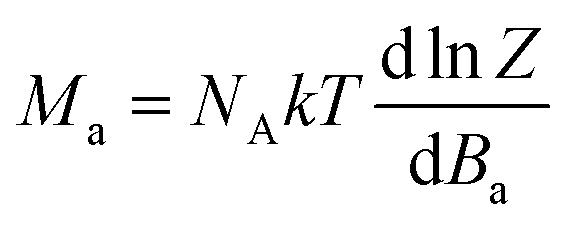DOI:
10.1039/C4DT03508A
(Paper)
Dalton Trans., 2015,
44, 2110-2121
Magnetic and structural properties of dinuclear singly bridged-phenoxido metal(II) complexes†
Received
15th November 2014
, Accepted 1st December 2014
First published on 3rd December 2014
Abstract
The reaction of a methanolic solution containing the bi-compartmental phenolic ligand 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) with MCl2·nH2O in the presence of NH4PF6 or NaClO4 afforded the dinuclear bridged-phenoxido dichlorido-metal(II) complexes [Co2(μ-LClO)(H2O)2Cl2][Co2(μ-LClO)(MeOH)2Cl2](PF6)2 (1), [Ni2(μ-LClO)(MeOH)2Cl2]PF6 (2), [Ni2(μ-LClO)(MeOH)(H2O)Cl2]ClO4·1.25H2O (3), [Cu2(μ-LClO)Cl2]PF6·1/2MeOH (4) and [Zn2(μ-LClO)Cl2]PF6·MeOH (5). The complexes were characterized by elemental microanalyses, conductivity measurements, IR and UV-Vis spectroscopy, mass spectrometry and single crystal X-ray crystallography. Each M(II) center within the dinuclear complex cations is octahedrally coordinated in complexes 1–3, and five-coordinated distorted square pyramidal in 4 and 5. Magnetic susceptibility measurements at variable temperature of the complexes 1–4 revealed weak to moderate antiferromagnetic coupling with |J| values = 8.38, 39.0, 30.2 and 0.79 cm−1, respectively. The results of DFT calculations correlate well with the experimentally determined antiferromagnetic coupling and show that the magnetic exchange coupling occurs mainly through the phenoxido bridge M–O–M. Implications of geometry around the central metal ion, M⋯M distance, M–O–M bond angle and overlapping of magnetic orbitals on the magnetic exchange coupling are discussed.
Introduction
The design of compartmental ligands capable of providing symmetrical and asymmetrical bimetallic cores is a growing topic. The metal complexes constructed from these ligands have been used as successful devices to mimic the active sites of a variety of metalloenzymes.1–13 With focus on compartmental ligands that are derived from phenolic containing compounds, these ligands have been launched to study the phosphodiester hydrolysis and DNA cleavage1–4 and to model purple acid phosphatases,5,6 Zn phosphesterases,7,8 Mn catalases,9 catecholase oxidases,10,11 metallo-β-lactamases (MβL)12 and hemocyanin.13 The use of these model compounds was very helpful to gain insight into biological systems and to elucidate some structural features about these systems. Recently, it has been reported that heteronuclear transition metal–lanthanide, 3d–4f metal complexes derived from multidentate Schiff bases containing phenolic and alkoxy groups have the capability of fixing atmospheric CO2 to produce carbonato-bridged polynuclear compounds.14–18 The resulting carbonato-bridged complexes showed interesting magnetic properties that open possibilities for their use as magnetic devices for single molecule magnets (SMM).14–18
Among many different types of binucleating compounds are phenol-based compartmental ligands, which possess two pendant chelating arms attached to the 2- and 6-positions of the phenol ring. These two arms, which can be symmetric or asymmetric, can accommodate two similar or dissimilar metal ions and hence produce dinuclear metal complexes bridged by the endogeneous deprotonated phenolic group and one or two exogeneous groups leading to unsaturated coordination environment around the central metal ions, or in some cases coordinate to weakly bound ligand(s).1–13,19–51 The distance between the two metal ions bridged via the phenoxido group is a crucial parameter in mediating the magnetic interaction between the two paramagnetic metallic centers. Also, their close proximity allows the cooperation between metal ions, where the distance between the two bridged metal ions are within the range of 2.9–4.0 Å, providing an excellent pathway for a strong antiferromagnetic coupling between 3d7–9 centers. In addition, the benzene ring present in these systems allows great synthetic flexibility, especially in tuning the solubility of the compounds.
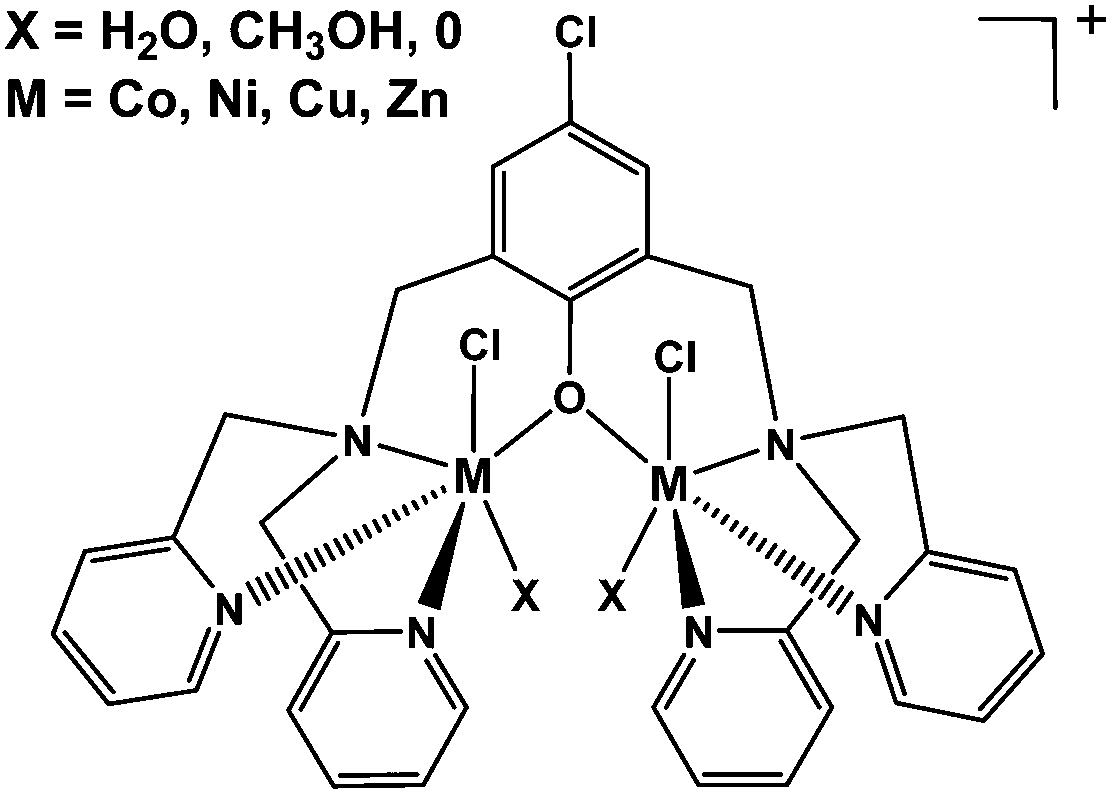
As it was indicated above, the phenol-based compounds with 2,6-pendant chelating coordinating arms have the tendency to bind two similar or dissimilar metal ions through the deprotonated phenolic group. In the presence of aliphatic and aromatic carboxylate compounds extra bridge(s) via the carboxylate moiety may be obtained.35–43 Similarly, bridged hydroxo-, methoxo- and phospho-ester compounds were also observed.22,44–48 However, it came to our attention that a few examples exist in literature for dinuclear bridged-phenoxido complexes with the formulas [M2(LRO)Cl2]Y and [M2(LMeO)(X)2]Y3, where X = H2O and CH3CN; Y = ClO4−, PF6− or BF4− and LROH = binucleating phenol.34,52–54 Therefore, this study was undertaken to explore the coordination properties of this class of compounds using bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) (Chart 1) with Co(II), Ni(II), Cu(II) and Zn(II) metal ions.
 |
| | Chart 1 Structure formulas of bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) and related compounds. | |
Results and discussion
Syntheses
The bi-compartmental phenolic ligand 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) was synthesized in about 80% by the reaction of 2,6-bis(2-chloromethyl)-4-chlorophenol with bis(2-pyridylmethyl)amine (DPA) in anhydrous CH3CN and in the presence of a slight excess of anhydrous K2CO3. The compound 2,6-bis(2-chloromethyl)-4-chlorophenol was obtained through the hydroxoformylation of 4-chlorophenol by 30% aqueous solution of formaldehyde in aqueous NaOH solution followed by acidification with acetic acid (pH < 6) to produce 2,6-bis(2-hydroxomethyl)-4-chlorophenol. The later compound was then converted to the corresponding 2,6-bis(2-chloromethyl)-4-chlorophenol via the reaction with concentrated hydrochloric acid. The ligand LCl-OH (Chart 1) was characterized by 1H and 13C NMR, ESI-MS and IR (see Experimental section).
The reaction of a methanolic solution of 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) with two equivalents of MCl2·nH2O (M = Co, n = 6; M = Ni, n = 6; M = Cu, n = 2; M = Zn, n = 0) in the presence of NH4PF6 or NaClO4 afforded the dinuclear dichloride–metal(II) complexes [Co2(μ-LClO)(H2O)2Cl2][Co2(μ-LClO)(MeOH)2Cl2](PF6)2 (1), [Ni2(μ-LClO)(MeOH)2Cl2]PF6 (2), [Ni2(μ-LClO)(MeOH)(H2O)Cl2]ClO4·1.25H2O (3), [Cu2(μ-LClO)Cl2]PF6·1/2MeOH (4) and [Zn2(μ-LClO)Cl2]PF6·MeOH (5) in moderate to high yields (60–90%) in which the ligand was deprotonated. The isolated complexes were characterized by elemental microanalyses, IR and UV-Vis spectroscopy, ESI-MS and single crystal X-ray crystallography. The magnetic properties of the complexes 1–4 were determined. The molar conductivity of the complexes as measured in CH3CN revealed their typical 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 electrolytic nature (ΛM = 132–140 Ω−1 cm2 mol−1).
:1 electrolytic nature (ΛM = 132–140 Ω−1 cm2 mol−1).
IR spectra of the complexes
The IR spectra of the complexes display some general characteristic features: (1) Medium broad absorption band in the 4430–4450 cm−1 region due to the stretching frequency ν(O–H) of the coordinated and/or solvent of crystallization CH3OH and H2O. (2) A series of strong to medium absorption bands over the 1610–1430 cm−1 region attributable to the pyridyl groups {ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and ν(CN)}. (3) The very sharp strong absorption band observed in complexes 1, 2, 4 and 5 around 840 cm−1 is assigned to the ν(P–F) of PF6− anion, whereas Ni(II) complex 3 displays two strong absorption bands at 1120 and 1097 cm−1 due to the stretching frequency ν(Cl–O) of the perchlorate counter anion. The split of the perchlorate band in [Ni2(μ-LClO)(MeOH)(H2O)Cl2]ClO4·1.25H2O (3) may be attributed to the strong involvement of the ClO4− ions in H-bonding with the aqua or MeOH molecules (see X-ray section). This interaction reduces the symmetry of the ClO4− ion from Td to C3υ or C2υ.
C) and ν(CN)}. (3) The very sharp strong absorption band observed in complexes 1, 2, 4 and 5 around 840 cm−1 is assigned to the ν(P–F) of PF6− anion, whereas Ni(II) complex 3 displays two strong absorption bands at 1120 and 1097 cm−1 due to the stretching frequency ν(Cl–O) of the perchlorate counter anion. The split of the perchlorate band in [Ni2(μ-LClO)(MeOH)(H2O)Cl2]ClO4·1.25H2O (3) may be attributed to the strong involvement of the ClO4− ions in H-bonding with the aqua or MeOH molecules (see X-ray section). This interaction reduces the symmetry of the ClO4− ion from Td to C3υ or C2υ.
UV-Vis spectra of the complexes
The UV-Vis spectra of the complexes 1–4 were measured in CH3CN. The spectrum of [Co2(μ-LClO)(H2O)2Cl2][Co2(μ-LClO)(MeOH)2Cl2](PF6)2 (1) displays three distinct bands at around 564, 500 and 463 nm. These bands are typical for six-coordinate high spin Co(II) complexes and they can be assigned to the spin-allowed transitions 4T2g(F) ← 4T1g(F), 4T1g(P) ← 4T1g(F) and 4A2g(F) ← 4T1g(F), respectively. Similarly, three bands were also detected for the complex cations [Ni2(μ-LClO)(MeOH)2Cl2]+ and [Ni2(μ-LClO)(MeOH)(H2O)Cl2]+ in complexes 2 and 3, respectively. These bands are located at 1050, 795 and 640 nm indicating octahedral geometry around 3d8 Ni(II) ion and result from the d–d electronic transitions 3T2g(F) ← 3A2g(F), 3T1g(F) ← 3A2g(F) and 3T1g(P) ← 3A2g(F), respectively.57
The spectrum of copper complex [Cu2(μ-LClO)Cl2]PF6·1/2MeOH (4) reveals the presence of two maxima at 457 and 695 nm. The broad single band at 695 nm suggests a distorted square pyramidal (SP) environment around the central Cu2+ ion.58 In general, the visible spectra of the five-coordinate SP Cu(II) complexes are most likely producing a broad band over the 550–700 nm range (dxz, dyz → dx2−y2) which occasionally may or may not be associated with a low-energy shoulder at λ > 800 nm, whereas the presence of a single d–d band at λ > 800 nm (dxy, dx2−y2 → dz2) with a high-energy shoulder is typical for trigonal bipyramidal (TBP) stereochemistry.58,59 Interestingly, the geometries of the complexes 1–4, as determined by UV-Vis spectroscopy in CH3CN solution, were in complete agreement with those obtained by single crystal X-ray crystallography.
Mass spectral characterization of complexes
ESI-Mass spectra of the five complexes were recorded in acetonitrile and all are shown in the ESI section (Fig. S1–S5†). The mass spectrum of [Co2(LClO)(H2O)2Cl2][Co2(LClO)(MeOH)2Cl2](PF6)2 (1) showed a peak at m/z = 757.04 that corresponds to [Co2(LClO)(H2O)Cl2]+ (calcd m/z = 756.87). In addition, another peak for a species was detected at m/z 825.04 which may result from additional coordination of H2O–MeOH to the Co(II) centers, [Co2(LClO)(H2O)3(MeOH)Cl2]+ (calcd m/z 824.94). Nickel(II) complexes 2 and 3 showed one major peak with m/z 757.08 which could be assigned to [Ni2(LClO)(H2O)Cl2]+ (calcd m/z = 756.40). The complexes [Cu2(LClO)Cl2]PF6·1/2MeOH (4) and [Zn2(LClO)Cl2]PF6·MeOH (5) revealed peaks at m/z = 767.03 and 771.07, respectively. These suggested the presence of protonated fragment [M2(LClOH)(H2O)Cl2]2+ (calcd m/z: M = Cu, 767.10; M = Zn, 770.80) that is related to those observed in complexes 1–3. The mass spectrum of Cu(II) complex 4 showed two more distinct fragments at m/z 903.03 and 835.03. The former fragment may arise from additional coordination of three acetonitrile molecules, [Cu2(LClO)(MeOH)(MeCN)3Cl2]+ (calcd m/z 903.28), whereas the second fragment may indicate the formation of [Cu2(LClOH)(MeOH)(H2O)3Cl2]2+ (calcd m/z 835.17). Species with additional coordination such as [Cu2(LClO)(MeOH)(MeCN)3Cl2]+, which observed above in complex 4 when CH3CN was used as a solvent in measuring mass spectra, have been recently reported in some dinuclear cobalt(II) complexes.2 Complexes 1–5 reveal also the presence of peaks at m/z 356.03, 356.04, 356.04, 356.01 and 278.17, respectively. Although we were unable to identify the origin of these species most likely they are attributed to ligand fragments. The acetonitrile mass spectrometry of [Cu2(LMe-O)2(OCH3)](ClO4)2 revealed a peak at m/z 357.1, which is located at about the same positions as for complexes 1–4 was incorrectly assigned for [Cu2(LMe-O)2(OH)]2+ ion.27 In addition to these peaks, the hexafluorophosphate complexes 1, 2, 4 and 5 displayed an m/z peak at 144.97 (100%) for PF6− (calcd m/z 144.97), whereas the corresponding perchlorate complex 3 showed a peak at m/z 98.95 (100%) (calcd ClO4−m/z 99.45).
Crystal structures of the complexes (1–5)
[Co2(μ-LClO)(H2O)2Cl2][Co2(μ-LClO)(MeOH)2Cl2](PF6)2 (1).
The crystal structure of 1 consists of two dinuclear complex cations [Co2(LClO)(X)2Cl2]+ (X = H2O [Co(1)/Co(2)] or MeOH [Co(3)/Co(4)] and PF6− counter ions. Perspective views of the complex cations together with partial atom numbering schemes are given in Fig. 1a and 1b, and selected bond parameters are summarized in Table S1.† Each Co(II) center within a dinuclear complex cation is octahedrally coordinated by three N-donor atoms of one bis(2-pyridylmethyl)aminomethyl group in a fac-arrangement, a terminal chloride ligands trans to N(amine) of bis(2-pyridylmethyl)aminomethyl group, the bridging O-atom of central 4-chlorophenolate moiety and an oxygen atom of aqua [Co(1)/Co(2)] or MeOH [Co(3)/Co(4)]. Both dinuclear subunits have a pseudo-2-fold axis passing through the O and Cl atoms of the central 4-chlorophenolate moiety. The Co–N bond distances are in the range 2.085(5)–2.187(5) Å, the Co–Cl bond lengths are 2.3700(18) and 2.4298(16) Å, and the Co–O bond lengths range from 2.087(4) to 2.178(5) Å. The Co(1)–O(1)–Co(2) and Co(3)–O(2)–Co(4) bond angles of the μ-phenoxido bridges are 134.2(2) and 133.65(19)°, respectively. The intra-dimeric Co(1)⋯Co(2) and Co(3)⋯Co(4) distances are 3.8840(12) and 3.9108(13) Å, respectively, and the shortest inter-dimer metal–metal separation is 6.3536(11) Å. Hydrogen bonds of type O–H⋯Cl are observed between aqua or MeOH donor ligands and chlorido acceptor ligands [O⋯Cl separations from 3.0145(5) to 3.262(4) Å] (Fig. S6†).
 |
| | Fig. 1 (a) Perspective views of Co1/Co2 and (b) Co3/Co4 of the dinuclear subunits of 1. | |
[Ni2(μ-LClO)(MeOH)2Cl2]PF6 (2) and [Ni2(μ-LClO)(MeOH)(H2O)Cl2]ClO4·1.25H2O (3).
The crystal structures of 2 and 3 consist of dinuclear complex cations [Ni2(μ-LClO)(X)2Cl2]+ (X = MeOH for 2, H2O and MeOH for 3), PF6− or ClO4− counter ions and additional disordered lattice water molecules in case of 3. Perspective views of the complex cations together with partial atom numbering schemes are given in Fig. 2 and 3, and selected bond parameters are summarized in Tables S2 and S3,† respectively. Each Ni(II) center within a dinuclear complex cation is octahedrally coordinated by three N donor atoms of one bis(2-pyridylmethyl)amino group in a fac-arrangement, a terminal chloride ligand anion trans to N(amine) of bis(2-pyridylmethyl)aminomethyl group, the bridging O atom of the central 4-chlorophenolate moiety and oxygen atom of a terminal MeOH, except for Ni(2) of 3, where MeOH is replaced by a aqua ligand. The dinuclear subunits also have a pseudo-2-fold axis passing through the O(1) and Cl(1) atoms of the central 4-chlorophenolate moiety. The Ni–N bond distances range from 2.042(3) to 2.111(3) Å, the Ni–Cl bond lengths from 2.3614(7) to 2.3826(9) Å, and the Ni–O bond lengths from 2.0690(17) to 2.174(2) Å. The Ni(1)–O(1)–Ni(2) bond angles of the μ-phenoxido bridges are 136.63(7) and 132.64(11)° for 2 and 3, respectively. The intra-dimeric Ni(1)⋯Ni(2) distances are 3.9682(5) and 3.9728(6) Å, and the shortest inter-dimer metal–metal separations are 7.6769(5) and 7.0198(7) Å, for 2 and 3, respectively. Hydrogen bonds of type O–H⋯Cl are observed between aqua or MeOH donor ligands and chlorido acceptor ligands [O⋯Cl separations range from 3.0145(5) to 3.262(4) Å]. Intra-dimer hydrogen bonds of type O–H⋯Cl were observed in both crystal structures [O⋯Cl separations from 2.9747(19) to 3.047(3) Å] (Fig. S7 and S8†). In case of 3, further hydrogen bonds of type O–H⋯O were observed [O(3)⋯O(5)(1/2 − x, 1/2 − y, 1/2 + z) = 2.957(4) Å].
 |
| | Fig. 2 Perspective view of 2. | |
 |
| | Fig. 3 Perspective view of 3. Disordered lattice water molecules are omitted. | |
[Cu2(μ-LClO)Cl2]PF6·1/2MeOH (4) and [Zn2(μ-LClO)Cl2]PF6·MeOH (5).
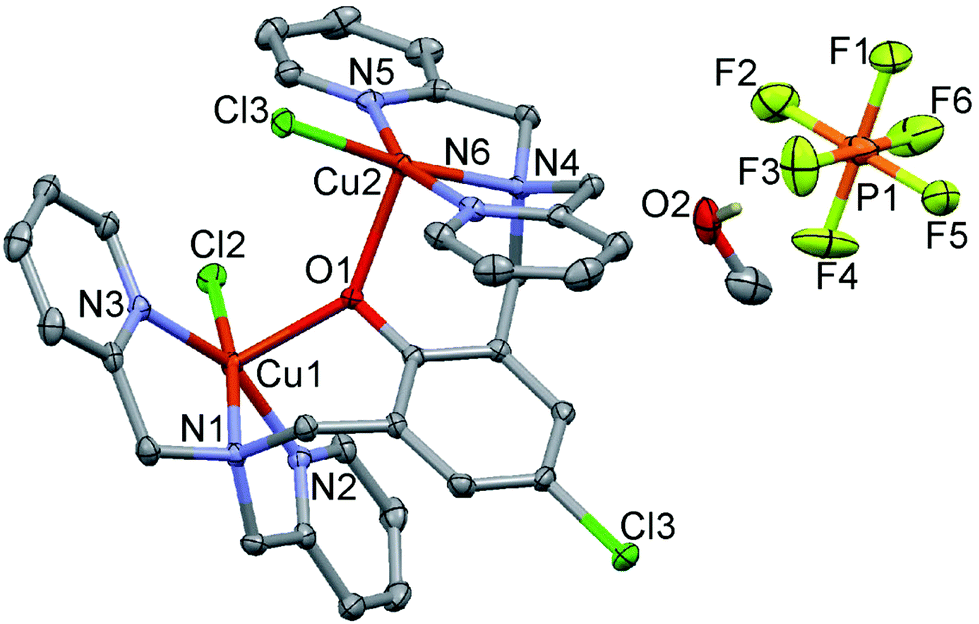
The crystal structures of 4 and 5 consist of dinuclear complex cations [M2(LClO)Cl2]+ (M = Cu for 4, and Zn for 5), PF6− counter ions and MeOH lattice solvent molecules. Perspective views of the crystal structures together with partial atom numbering schemes are depicted in Fig. 4 and 5, and selected bond parameters are presented in Tables S4 and S5,† respectively. The dinuclear subunits have also a pseudo-2-fold axis passing through O(1) and Cl(1) atoms of the central 4-chlorophenolate moiety. Each copper(II) or zinc(II) center within a dinuclear complex cation is penta-coordinated by three N-donor atoms of one bis(2-pyridylmethyl)aminomethyl group, a terminal chloride ligand in basal sites, and the bridging O(1) atom of central 4-chlorophenolate moiety, which occupies the axial position of the distorted square pyramids [τ-values: 0.20 and 0.19 for Cu(1)and Cu(2); 0.28 and 0.32 for Zn(1) and Zn(2), respectively.60 The axial Cu–O(1) and Zn–O(1) bond distances are 2.1915(11), 2.2089(11), 2.0518(11) and 2.0166(11) Å, respectively. The Cu/Zn–N bond distances are in the range from 1.9951(15) to 2.2730(14) Å, the Cu/Zn–Cl bond lengths vary from 2.2704(5) to 2.2995(4) Å. The metal centers deviate from their basal N3Cl plane by 0.217 Å for Cu(1), 0.192 Å for Cu(2), 0.328 Å for Zn(1), and 0.406 Å for Zn(2). The Cu(1)–O(1)–Cu(2) and Zn(1)–O(1)–Zn(2) bridging bond angles are 140.74(6) and 135.18(6)°, respectively. The corresponding intra-dimeric metal⋯metal distances are 4.1447(4) and 3.7612(4) Å, and the corresponding shortest inter-dimer metal–metal separations are 3.9446(4) and 4.1131(4) Å. Hydrogen bonds of type O–H⋯F are observed between O atoms of MeOH solvent molecules and F atoms of adjacent PF6− counter ions [O⋯F separations range from 2.886(4) to 3.029(2) Å] (Fig. S9 and S10†).
 |
| | Fig. 4 Perspective view of 4. | |
 |
| | Fig. 5 Perspective view of 5. | |
Magnetic properties of complexes 1–4
The experimental magnetic data of 1, depicted in Fig. 6, shows dominant antiferromagnetic exchange within the Co(II) dimer as evidenced by a decrease of the effective magnetic moment from 6.46μB (300 K) to 0.88μB (1.9 K) and also by the maximum of Mmolvs. T curve located at 21 K, which serves as a fingerprint for antiferromagnetically coupled homospin dimers.61 Furthermore, the significant zero-field splitting (ZFS) is expected in hexa-coordinate Co(II) complexes62 and therefore the following spin Hamiltonian was postulated| |  | (1) |
where the isotropic exchange (J), the zero-field splitting (D) and Zeeman term (g) are included. Then, the molar magnetization in a given direction of magnetic field Ba = B·(sinθcosφ, sinθsinφ, cosθ) was calculated as| |  | (2) |
Z is the partition function resulting from diagonalization of the spin Hamiltonian matrix. Finally, the integral (orientational) average of molar magnetization was calculated by eqn (3) in order to properly simulate experimental powder magnetization data.| |  | (3) |
 |
| | Fig. 6 The magnetic data for complex 1: Left: the temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: the isothermal magnetizations measured at T = 2, 5 and 10 K. Open circles represent the experimental data and solid lines represent the best fit using eqn (1) with J = −8.38 cm−1, D = 25.7 cm−1, g = 2.39, χTIP = 4.9 × 10−9 m3 mol−1, xPI = 0.79%. | |
Moreover, the small amount of monomeric paramagnetic impurity (PI) which accounts for an increase of molar magnetization (mean susceptibility) below 3 K was taken into consideration by eqn (4)
| | | Msample = (1 − xPI) Mmol + 2·xPIMPI | (4) |
where
MPI was calculated using the Brillouin function. Both temperature and field dependent magnetic data were included into fitting procedure which resulted in these parameters for
1:
J = −8.38 cm
−1,
D = 25.7 cm
−1,
g = 2.39,
χTIP = 4.9 × 10
−9 m
3 mol
−1,
xPI = 0.79% (
χTIP stands for the temperature-independent paramagnetism). The results confirmed the moderate antiferromagnetic exchange between Co(
II) atoms and the substantial role of magnetic anisotropy as deduced from the axial zero-field splitting parameter
D. The small discrepancies between experimental and calculated data are ascribed to the existence of two dimeric units within the asymmetric unit and also to the fact that one of the dimers forms supramolecular tetramers through O–H⋯Cl hydrogen bonds (Fig. S11
†). This probably creates more complex magnetic exchange pathways in the solid state.
The magnetic data for the dinuclear nickel(II) complexes 2 and 3 are plotted in Fig. 7, and Fig. S12,† respectively. In complex 2, the effective magnetic moment drops on cooling from 4.17μB (300 K) to 0.26μB (1.9 K) and the maximum of Mmolvs. T curve was found at 57 K, thus confirming strong antiferromagnetic exchange with S = 0 ground state. Such strong antiferromagnetic exchange means that excited molecular spin states S = 1 and S = 2, which bear information about magnetic anisotropy (D), are too high in energy, and therefore low temperature isothermal magnetization data (Mmol/NAμB < 0.1, Fig. 7) are non-informative concerning this issue. Therefore, only temperature data were used during the fitting procedure, which resulted in J = −39.0 cm−1, g = 2.19, χTIP = 2.2 × 10−9 m3 mol−1, xPI = 0.51%. Compound 3 shows very similar magnetic properties and the same magnetic analysis resulted in J = −30.2 cm−1, g = 2.24, χTIP = 3.4 × 10−9 m3 mol−1, xPI = 0.37% (Fig. S12†).
 |
| | Fig. 7 The magnetic data for 2: Left: the temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: the isothermal magnetizations measured at T = 2, 5 and 10 K. Open circles – experimental data, solid lines – calculated data using the eqn (1), with J = −39.0 cm−1, D = 0 cm−1 (fixed), g = 2.19, χTIP = 2.2 × 10−9 m3 mol−1, xPI = 0.51%. | |
In contrast to the above discussed results, the copper(II) complex 4 exhibited different magnetic behaviour (Fig. 8). The effective magnetic moment is almost constant in whole temperature range (μeff ≈ 2.57μB) and only below 10 K the small drop of μeff is observed (μeff = 2.32μB at T = 1.9 K). Moreover, there is no maximum on Mmolvs. T curve. Consequently, we can presume only a very weak antiferromagnetic exchange in 4. The magnetic analysis of both temperature and field dependent data resulted in J = −0.79 cm−1, g = 2.10, χTIP = 0.24 × 10−9 m3 mol−1, thus confirming a very weak magnetic exchange between Cu(II) atoms of the antiferromagnetic nature.
 |
| | Fig. 8 The magnetic data for 4: Left: the temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: the isothermal magnetizations measured at T = 2 and 5 K. Open circles represent the experimental data and solid lines represent the best fit using eqn (1) with J = −0.79 cm−1, g = 2.10, χTIP = 0.24 × 10−9 m3 mol−1. | |
Evaluation of magnetic properties using DFT calculations
In order to support the data of magnetic results which are showing large differences in the isotropic exchange mediated by the LCl-OH ligand for various metal atoms and possibly to get insight into the exchange mechanism, ab initio calculations based on DFT theory were used to calculate the J parameters in the dinuclear moieties [Co2(μ-LCl-O)Cl2(H2O)2]+ of 1, [Ni2(μ-LClO)(CH3OH)2Cl2]+ of 2, [Ni2(μ-LClO)(CH3OH)(H2O)Cl2]+ of 3, and [Cu2(μ-LClO)Cl2]+ of 4. The ORCA 3.0.1 computational package was used for all the calculations.63 Well established B3LYP functional64 and def2-TZVP(-f) basis set65 were used to calculate the energy difference Δ, between high spin (HS) and broken-symmetry (BS) spin states:
For the above mentioned structurally characterized dinuclear molecular fragments of 1–4 complexes, the following spin Hamiltonian for a dinuclear was used
| |  | (6) |
All the calculations utilized the RI approximation with the decontracted auxiliary def2-TZV/J Coulomb fitting basis set and the chain-of-spheres (RIJCOSX) approximation to exact exchange.66 Increased integration grids (Grid5 and GridX5in ORCA convention) and tight SCF convergence criteria were also used. The isotropic exchange J values were calculated by Ruiz's approach (eqn (7))67 and also by a more general Yamaguchi's approach (eqn (8))68:
| | | JRuiz = 2Δ/[(S1 + S2)(S1 + S2 + 1)] | (7) |
| | | JYam = 2Δ/[〈S2〉HS − 〈S2〉BS] | (8) |
The results of DFT calculations are summarized in Table 1. The J values calculated by DFT correlate well with the experimentally determined antiferromagnetic exchange in 1–4, and in the case of Co(II) and Ni(II) complexes the J's calculated by the Ruiz's approach are also very close to those found from magnetic analysis, whereas in Cu(II) compound, the experimentally determined J value is between JRuiz and JYam values.
Table 1 DFT-calculated net Mulliken spin densities (ρ), expected values <S2>, and isotropic exchange parameters (J) from high-spin (HS) and broken symmetry spin (BS) states of the dinuclear molecular fragments based on X-ray structures of 1–4
| |
|
HS |
BS |
<S2HS> |
<S2BS> |
Δ/cm−1 |
J
Ruiz/cm−1 |
J
Yam/cm−1 |
J
mag/cm−1 |
|
1
|
[Co2(LClO)Cl2(H2O)2]+ |
ρ(Co1) = 2.71 |
ρ(Co1) = −2.71 |
12.01 |
3.01 |
−56.728 |
−9.46 |
−12.6 |
−8.38 |
|
ρ(Co2) = 2.71 |
ρ(Co2) = 2.71 |
|
|
[Co2(LClO)Cl2(CH3OH)2]+ |
ρ(Co3) = 2.70 |
ρ(Co3) = −2.70 |
12.01 |
3.01 |
−52.939 |
−8.82 |
−11.76 |
|
ρ(Co4) = 2.71 |
ρ(Co4) = 2.70 |
|
2
|
[Ni2(LClO)(CH3OH)2Cl2]+ |
ρ(Ni1) = 1.63 |
ρ(Ni1) = −1.62 |
6.01 |
2.00 |
−112.338 |
−37.44 |
−56.06 |
−39.0 |
|
ρ(Ni2) = 1.63 |
ρ(Ni2) = 1.62 |
|
3
|
[Ni2(LClO)(CH3OH)(H2O)Cl2]+ |
ρ(Ni1) = 1.63 |
ρ(Ni1) = −1.62 |
6.01 |
2.00 |
−78.427 |
−26.14 |
−39.16 |
−30.2 |
|
ρ(Ni2) = 1.63 |
ρ(Ni2) = 1.63 |
|
4
|
[Cu2(LClO)Cl2]+ |
ρ(Cu1) = 0.58 |
ρ(Cu1) = −0.58 |
2.01 |
1.01 |
−0.494 |
−0.49 |
−0.99 |
−0.79 |
|
ρ(Cu2) = 0.58 |
ρ(Cu2) = 0.58 |
In case of [Co2(μ-LClO)(H2O)2Cl2]+ of 1, [Ni2(μ-LClO)(CH3OH)2Cl2]+ of 2 and [Cu2(μ-LClO)Cl2]+ of 4, the spin densities (Fig. 9) and also the non-orthogonal magnetic orbitals (Fig. 10) were visualized with the help of software Gabedit.69 Evidently, the unpaired electron of each Cu(II) atom is localized in the dx2−y2 orbital, which lies within the CuN3Cl plane. Therefore, there is a very weak overlap between magnetic orbitals of Cu(II) atoms, which results in such very weak antiferromagnetic exchange. On the contrary, the two unpaired electrons of Ni(II) atom are localized in the dx2−y2 and dz2 orbitals, leading to efficient overlap between magnetic orbitals through the bridged phenoxido Ni–O–Ni bond resulting in a strong antiferromagnetic exchange. Also, a consensus between DFT and magnetic analysis results in Ni(II) compounds 2 and 3 was acquired, showing that antiferromagnetic exchange is weaker in the case of 3. This trend can be related to the increase of Ni–O–Ni bond angle from 132.64° in 3 to 136.63° in 2, in such a way that overlap of magnetic orbitals between Ni(II) atoms is increasing from the right angle to straight angle. Thus, the larger Ni–O–Ni bond angle should result in a stronger antiferromagnetic exchange as it is actually observed. The situation concerning the Co(II) complex is similar to the Ni(II) complex meaning that super-exchange pathway is defined by Co–O–Co atoms and there is efficient overlap of magnetic orbitals leading to moderate antiferromagnetic exchange (Fig. 10). Also, there is non-negligible variation of DFT-derived J-values vs. Co–O–Co bond angle in 1, where larger Co–O–Co angle (134.15°) resulted in stronger antiferromagnetic exchange: JRuiz = −8.82 cm−1 for 133.66° angle in [Co2(μ-LClO)(CH3OH)2Cl2]+ and JRuiz = −9.46 cm−1 for 134.15° in [Co2(μ-LClO)(H2O)2Cl2]+ (Table 1). However, the effect of different solvent molecules (methanol or water) on magnetic exchange cannot be excluded.
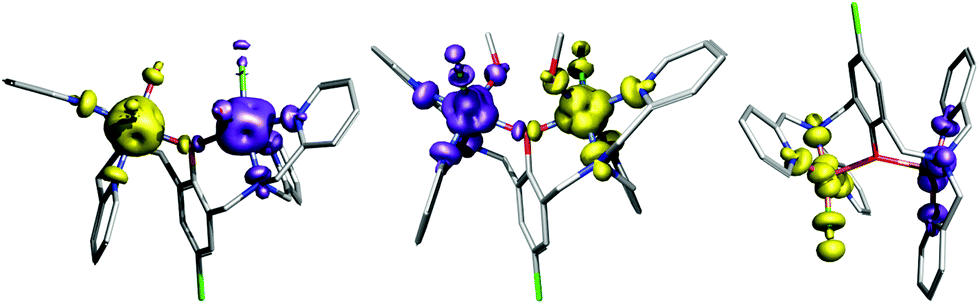
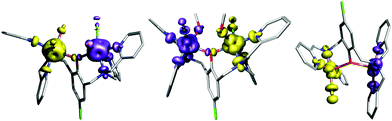 |
| | Fig. 9 The calculated the isodensity surfaces of the broken symmetry spin states using B3LYP/def2-TZVP(-f) for [Co2(μ-LClO)(H2O)2Cl2]+ of 1 (left), [Ni2(μ-LClO)(CH3OH)2Cl2]+ of 2 (middle) and [Cu2(μ-LClO)Cl2]+ of 4 (right). Positive and negative spin densities are represented by violet, and yellow surfaces, respectively, with the cutoff values of 0.005 e bohr−3. Hydrogen atoms were omitted for clarity. | |
 |
| | Fig. 10 The non-orthogonal magnetic orbitals of the broken-symmetry spin state visualized for [Co2(μ-LClO)(H2O)2Cl2]+ of 1 (A), [Ni2(μ-LClO)(CH3OH)2Cl2]+ of 2 (B) and [Cu2(μ-LClO)Cl2]+ of 4 (C). The values of overlap Sαβ between the corresponding orbitals are listed in right column. Hydrogen atoms were omitted for clarity. | |
Structure parameters and magnetic coupling
In general, bridged-phenoxido dinuclear metal(II) complexes, which are derived from ligands with pendant pyridyl and/or pyridyl derivative arms (Chart 1) exhibit weak to moderate antiferromagnetic coupling.8,37,53,54,70–73 Copper(II) complexes in which a singly bridged phenoxido group is the only bridge exhibit very weak antiferromagnetic coupling as this was the case in complex 4 (J = −0.79 cm−1). This has been observed in a number of related complexes such as [Cu2(μ-LMe-O)Cl2]ClO4 (J = 0 cm−1),53 [Cu2(μ-LMe-O)(OAc)2]ClO4·H2O (J = −0.6 cm−1)37 and [Cu2(μ-LMe-O)(H2O)Cl](ClO4)2 (J = −1.7 cm−1)73 and was attributed to the weak overlap between the phenolate apical oxygen orbitals and the unpaired electron at each Cu(II) center which is located in the equatorial plane (magnetic orbitals). Similar magnetic trends were reported with other 3d metal ions such as in [Ni2(μ-LMe-O)(μ-OAc)2]BF4·2MeOH (J = −2.3 cm−1) and [Mn2(μ-L′Me-O)(μ-OAc)2]ClO4 (J = −4.3 cm−1) (see Chart 1).2,8,70–72
Interestingly, nickel(II) complexes 2 and 3 revealed relatively high to moderate antiferromagnetic coupling with J values of −39.0 and −30.2 cm−1, respectively. As indicated above, this was attributed to the efficient overlap between the Ni(II) magnetic orbitals dx2−y2 and dz2 and the oxygen 2p orbitals of the phenolate oxygen atom and to an increase of the Ni–O–Ni bond angle. The paucity of the number of structurally and magnetically characterized Ni(II) compounds did not allow us to compare our results with literature data.
Although weak antiferromagnetic coupling was determined in Co(II) complex 1, [Co2(μ-LClO)(H2O)2Cl2][Co2(μ-LClO)(MeOH)2Cl2](PF6)2 (J = −8.38 cm−1), still this coupling is relatively stronger than those observed in related bridged phenoxido dinuclear Co(II) complexes based pyridyl and substituted pyridyl arms (J ranges from −0.1 to −5.4 cm−1).2,70,74 However, weak ferromagnetic coupling was also reported in some related systems: [Co2(μ-L1NO2-O)(μ-OAc)2]PF6 (J = +3.09 cm−1) and [Co2(μ-LBr-O)(μ-OAc)2]PF6 (J = +0.78 cm−1) (see Chart 1).75 No obvious correlation was found between the magnitude or sign of J and the nature of the substituents at the phenolic ring.75 In dicobalt(II) compounds, several factors were addressed to account for this weak interactions. These include the Co(II)–Co(II) and Co–O(phenoxido) bond distances as well as Co–O–Co bond angle,2,71,74,76,77 but unlike coupled dicopper and dinickel(II) complexes,78–80 the magneto–structural relationship for dicobalt(II) is not well resolved. The results of dicobalt(II) and dicopper(II) compounds clearly indicate that the μ-phenoxido bridge is a poor mediator for exchange magnetic interaction in this series of complexes.
Experimental
Materials and physical measurements
The compound bis(2-pyridylmethyl)amine (DPA) was purchased from TCI-America. All other chemicals were commercially available and used without further purification. Infrared spectra were recorded on a JASCO FTIR-480 plus spectrometer as KBr pellets. Electronic spectra were recorded using an Agilent 8453 HP diode array UV-Vis spectrophotometer. 1H and 13C NMR spectra were obtained at room temperature on a Varian 400 NMR spectrometer operating at 400 MHz (1H) and 100 MHz (13C). 1H and 13C NMR chemical shifts (δ) are reported in ppm and were referenced internally to residual solvent resonances (DMSO-d6: δH = 2.49, δC = 39.4 ppm). ESI-MS were measured on LC-MS Varian Saturn 2200 Spectrometer. The conductivity measurements were performed using Mettler Toledo Seven Easy conductivity meter and the cell constant was determined by the aid of 1413 μS cm−1 conductivity standard. The molar conductivity of the complexes were determined from ΛM = (1.0 × 103κ)/M, where κ = cell constant and M is the molar concentration of the complex. Elemental analyses were carried out by the Atlantic Microlaboratory, Norcross, Georgia U.S.A. Magnetic measurements of cobalt(II) (1) and nickel(II) compounds (2 and 3) were performed with a PPMS Dynacool VSM magnetometer (Quantum Design, Inc.) (T = 1.9–300 K at B = 1 T; B = 0–9 T at T = 2, 5 and 10 K), while the copper(II) complex 4 was measured on an MPMS XL7 SQUID magnetometer (Quantum Design, Inc.) (T = 1.9–300 K at B = 1 T; B = 0–5 T at T = 2 and 5 K). The magnetic data were corrected for diamagnetic susceptibilities and the signal of the sample holder.
Caution: Salts of perchlorate and their metal complexes are potentially explosive and should be handled with great care and in small quantities.
Synthesis of the ligand
The ligand 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH, Chart 1) was synthesized via 2,6-bis(chloromethyl)-4-chlorophenol, which in turn was obtained by the conversion of 4-chlorophenol to 2,6-bis(2-hydroxomethyl)-4-chlorophenol.
2,6-Bis(2-hydroxomethyl)-4-chlorophenol.
This compound was prepared based on published procedures,55,56 with modifications. An aqueous solution of NaOH (20.0 g, 0.5 mol in 20 mL of water) was added with stirring to a suspension of 4-chlorophenol (12.8 g, 0.1 mol) in 30 mL of 30% aqueous formaldehyde (0.3 mol). The mixture was kept at 30–40 °C for five days without further stirring, at which time the sodium salt of the target compound had precipitated. This was collected by filtration, washed with six 5 mL portions of saturated NaCl solution, and dissolved in 100 mL of boiling water. This solution was acidified by acetic acid until a pH of <6 had been reached and chilled in a refrigerator. The pale yellow product, which was collected by filtration was washed with 10 mL of H2O and then dried in air (yield: 18.6 g, 63%, based on 4-chlorophenol). Further recrystallization from ethyl acetate afforded off-white needles. Characterization: mp 166–167 °C (Lit.: 165 °C),561H NMR (Na salt, D2O): 4.22 (s, exchangeable proton), 4.73 (s, 4H; H2C–), 6.74 (s, 2H; protons at C3 and C5 of the phenyl group). 13C NMR (Na salt, D2O): 60.20 (CH2–), 116.33 (C4), 125.80 (C4 and C6), 130.94 (C3 and C5), 161.15 (C1). The compound was considered pure enough for further conversion to 2,6-bis(chloromethyl)-4-chlorophenol.
2,6-Bis(chloromethyl)-4-chlorophenol.
This compound was prepared by adapting a procedure for preparing 2,6-bis(chloromethyl)-4-methylphenol.81 A mixture of 2,6-bis(hydroxymethyl)-4-chlorophenol (5.3 g, 0.028 mol), and 60 mL of aqueous 36% HCl (0.7 mol) in CH2Cl2 (20 mL), charged into a 250 mL round bottom flask, was magnetically stirred overnight at room temperature. The resulting two phases were separated and the aqueous phase was extracted with CH2Cl2 (2 × 25 mL). The combined organic phases were vacuum evaporated and the crude light brown solid was collected and extracted with hot heptane (2 × 30 mL). Evaporation of heptane afforded the desired product as a white solid (yield, 4.2 g, 66%). Further recrystallization from heptane resulted in the formation of colorless needles. Characterization: mp. 92–93 °C (Lit. 92–92.5 °C).821H NMR (CDCl3): 4.56 (s, 4H; CH2–), 5.66 (s, 1H; exchangeable phenolic proton), 7.21 (s, 2H; phenyl protons at C3 and C5). 13C NMR (CDCl3): 41.60 (CH2–), 125.64 (C4), 126.31 (C1 and C6), 130.58 (C3 and C5), 151.74 (C1).
Synthesis of 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH).
To 2,6-bis(chloromethyl)-4-chlorophenol (1.12 g, 5 mmol) dissolved in anhydrous CH3CN (50 mL), bis(2-pyridylmethyl)amine (2.00 g, 10 mmol) was added. The mixture was treated with anhydrous K2CO3 (2.10 g, 15 mmol) and magnetically stirred under gentle reflux for 3 days, during which the color turned light yellow and a white precipitate was formed. This mixture was cooled in the refrigerator and then filtered off to remove KCl and unreacted K2CO3. The solvent was removed with a rotary evaporator under reduced pressure and the resulting dark brown liquid was solidified when stored over P2O5 in a desiccator under vacuum. Several crystallizations from Et2O with the aid of activated charcoal afforded yellow oil which was then solidified to produce the desired product as a pale yellow solid (yield: 2.2 g, 80%). Characterization: mp = 102–104 °C, Selected IR (KBr, cm−1): 3421 (mb) ν(O–H); 3060 (w), 3022 (w) (pyridyl/phenyl C–H stretching); 2925 (w), 2832 (w), (aliphatic C–H stretching), 1589 (vs), 1569 (s), 1475 (s), 1433 (s) (CC, CN pyridyl/phenyl ring stretching); 760 (vs) (C–H out of plane bending). ESI-MS in MeOH (100%) m/z = 551.232 (Calcd for [M + H]+ = 552.103), 573.214 (Calcd for [M + Na]+ = 574.095), 589.188 (Calcd for [M + K]+ = 590.193). 1H NMR (DMSO-d6, 400 MHz, δ in ppm): δ = 8.50, 8.49 (2H, phenyl protons), 7.73, 7.44 (m, 4H), 7.40, 7.24 (m, 4H) (pyridyl protons); 3.77 (s, 2H), 3.70 (s, 2H) (CH2-py); 3.33 (s, 2H, CH2-ph).13C NMR: (DMSO-d6, 100 MHz) δ = 158.4, 154.3, 148.7, 136.7, 128.0, 126.1, 122.7, 122.4, 121.6 (pyridyl and phenyl carbons); 58.9, 53.3 (CH2-ph and CH2-py).
Syntheses of metal(II) complexes (1–5)
A general method was used to synthesize the dinuclear dichloro metal(II) complexes (1–5). To a mixture containing MCl2·nH2O (M = Co, n = 6; M = Ni, n = 6; M = Cu, n = 2; M = Zn, n = 0) (0.40 mmol) and 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) (0.110 g, 0.20 mmol) dissolved in MeOH (20 mL), NH4PF6 (80 mg, 0.50 mmol) or NaClO4 (61 mg, 0.50 mmol) in case of complex 3 was added and the resulting solution was heated on a steam-bath for 5–10 min. The resulting solution was filtered while hot through celite and then allowed to crystallize at room temperature. The precipitate which was obtained over a period of 1–6 h was collected by filtration, washed with propan-2-ol and Et2O, and then dried at room temperature. Long needles (pink for Co, bluish green for Ni, light green for Cu and colorless for Zn) suitable for X-ray structure determination were obtained from dilute methanolic solutions of complexes 1, 3, 4 and 5. In case of 2, recrystallization of the complex from CH3CN afforded bluish green single crystals of X-ray quality.
[Co2(LCl-O)Cl2(H2O)2][Co2((LCl-O)Cl2(CH2OH)2](PF6)2 (1).
The complex was isolated as shiny pink long needles (overall yield: 163 mg, 87%). Characterization: Anal. Calcd for C66H72Cl6Co4F12N12O6P2 (MM = 1867.76 g mol−1): C, 42.44; H, 3.89; N, 9.00%. Found: C, 42.10; H, 3.91; N, 9.06%. Selected IR bands (cm−1): 3441 (s) ν(O–H), 1607 (vs) ν(CC); 1573 (m), 1482 (m), 1463 (m), 1447 (m) ν(CN); 844 (vs) ν(P–F). UV- Vis. {λmax, nm (ε per Co atom, M−1 cm−1)} in CH3CN: 463 (64.0), 500 (57.2), 543 (sh), 564 (83.4), ∼701 (17.6 b). ESI-MS (CH3CN) for positive ions: m/z = 825.04, [Co2(C32H30ClN6O)Cl2(H2O)3(CH3OH)]+; 757.04, [Co2(C32H30ClN6O)Cl2(H2O)]+; 356.03, and for negative ions: m/z = 144.97 (100%), PF6−. Molar conductivity in CH3CN, ΛM = 136 Ω−1 cm2 mol−1.
[Ni2((LCl-O)Cl2(CH3OH)2]PF6 (2).
This complex was isolated as bluish-green long needles (overall yield: 135 mg, 71%). Characterization: Anal. Calcd for C34H38Cl3F6Ni2N6O3P (MM = 947.43 g mol−1): C, 43.10; H, 4.04; N, 8.87%. Found: C, 43.25; H, 4.13; N, 9.10%. Selected IR bands (cm−1): 3430 (mb), 1607 (m) ν(CC); 1481 (w), 1463 (w), 1446 (m), 1435 (m) ν(CN); 842 (vs) ν(P–F). UV-Vis. {λmax, nm (ε per Ni atom, M−1 cm−1)} in CH3CN: 641 (11.7), 795 (5.3), 1050 (20.4, b). ESI-MS (CH3CN) for positive ions: m/z = 757.08, [Ni2(C32H30ClN6O)Cl2(H2O)]+; 356.04, and for negative ions: m/z = 144.97 (100%), PF6−. Molar conductivity in CH3CN, ΛM = 137 Ω−1 cm2 mol−1.
[Ni2((LCl-O)Cl2(H2O)(CH3OH)]ClO4·1.25H2O (3).
This was isolated as light bluish-green crystalline compound (overall yield: 168 mg, 71%). Characterization: Anal. Calcd for C33H38.5Cl4Ni2N6O8.25 (MM = 907.86 g mol−1): C, 42.66; H, 4.00; N, 9.26%. Found: C, 42.25; H, 4.23; N, 9.30%. Selected IR bands (cm−1): 3452 (mb), 1607 (s) ν(CC); 1573 (m), 1479 (m), 1450 (s), 1433 (s) ν(CN); 1120 (s), 1097 (vs) ν(Cl–O). UV- Vis. {λmax, nm (ε per Ni atom, M−1 cm−1)} in CH3CN: 640 (8.2), 795 (3.7), ∼1050 (14.3, b). ESI-MS (CH3CN) for positive ions: m/z = 757.08, [Ni2(C32H30ClN6O)Cl2(H2O)]+; 356.04, and for negative ions: m/z = 99.45 (100%), ClO4− = 99.45. Molar conductivity, ΛM (CH3CN) = 132 Ω−1 cm2 mol−1.
[Cu2(LCl-O)Cl2]PF6·1/2CH3OH (4).
The complex was isolated as light green crystalline compound (overall yield: 110 mg, 59%). Characterization: Anal. Calcd for C32.5H32Cl3Cu2F6N6O1.5P (MM = 909.06 g mol−1): C, 42.94; H, 3.55; N, 9.24%. Found: C, 42.45; H, 3.78; N, 9.06%. Selected IR bands (cm−1): 3442 (w, b) ν(O–H), 1611 (s) ν(CC); 1575 (w), 1482 (m), 1462 (m), 1440 (s) ν(CN); 844 (vs) ν(P–F). UV-Vis. {λmax, nm (ε per Cu atom, M−1 cm−1)} in CH3CN: 457 (103.8), 695 (128, b). ESI-MS (CH3CN) for positive ions: m/z = 903.03, [Cu2(C32H30ClN6O)Cl2(MeOH)(MeCN)3]+; 835.03, [Cu2(C32H31ClN6O)Cl2(MeOH)(H2O)3]2+; 767.03, [Cu2(C32H31ClN6O)Cl2(H2O)]+; 356.01, and for negative ions: m/z = 144.97 (100%), PF6−. Molar conductivity, ΛM (CH3CN) = 140 Ω−1 cm2 mol−1.
[Zn2(LCl-O)Cl2]PF6·CH3OH (5).
This complex was isolated as colorless long needles (overall yield: 140 mg, 75%). Characterization: Anal. Calcd for C33H34Cl3F6N6O2PZn2 (MM = 928.78 g mol−1): C, 42.67; H, 3.69; N, 9.05%. Found: C, 43.01; H, 3.71; N, 9.17%. Selected IR bands (cm−1): 3445 (m, b) ν(O–H), 1609 (vs) ν(CC); 1576 (m), 1468 (m), 1463 (s), 1443 ν(CN); 846 (vs) ν(P–F). ESI-MS (CH3CN) for positive ions: m/z = 771.07, [Zn2(C32H31ClN6O)Cl2(H2O)]+, 278.17, and for negative ions: m/z = 144.96 (100%), PF6−. Molar conductivity, ΛM (CH3CN) = 132 Ω−1 cm2 mol−1.
X-ray crystal structure analysis
The X-ray single-crystal data of compounds 1–5 were collected on a Bruker-AXS APEX CCD diffractometer at 100(2) K. The crystallographic data, conditions retained for the intensity data collection and some features of the structure refinements are listed in Table 2. The intensities were collected with Mo-Kα radiation (λ = 0.71073 Å). Data processing, Lorentz-polarization and absorption corrections were performed using APEX, and the SADABS computer programs.83 The structures were solved by direct methods and refined by full-matrix least-squares methods on F2, using the SHELXTL program package.84 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were located from difference Fourier maps, assigned with isotropic displacement factors and included in the final refinement cycles by use of HFIX (parent C atom) or DFIX (parent O atom) utility of the SHELXTL program. Molecular plots were performed with the Mercury program.85 In case of 1, data affected by twin components (8.6%) were excluded from refinement. In case of 3, Uij constraints were applied for disordered water oxygen molecules and their hydrogen atoms excluded from refinements.
Table 2 Crystallographic data and processing parameters for complexes 1–5a
| Compound |
1
|
2
|
3
|
| Empirical formula |
C66H72Cl6Co4F12N12O6P2 |
C34H38Cl3F6N6Ni2O3P |
C33H36Cl4N6Ni2O8.25 |
| Formula mass |
1867.72 |
947.40 |
907.86 |
| System |
Triclinic |
Monoclinic |
Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c |
P21/n |
|
a (Å) |
10.7772(10) |
13.4024(5) |
10.3560(5) |
|
b (Å) |
11.8843(11) |
18.0346(7) |
19.7894(9) |
|
c (Å) |
32.324(3) |
32.6751(12) |
19.0876(9) |
|
α (°) |
97.470(6) |
90 |
90 |
|
β (°) |
94.477(6) |
98.282(2) |
96.752(2) |
|
γ (°) |
106.833(5) |
90 |
90 |
|
V (Å3) |
3899.7(6) |
7815.4(5) |
3884.7(3) |
|
Z
|
2 |
8 |
4 |
|
T (K) |
100(2) |
100(2) |
100(2) |
|
μ (mm−1) |
1.168 |
1.282 |
1.301 |
|
D
calc (Mg m−3) |
1.519 |
1.610 |
1.552 |
| Crystal size (mm) |
0.33 × 0.26 × 0.21 |
0.26 × 0.22 × 0.15 |
0.33 × 0.27 × 0.22 |
|
θ max (°) |
25.50 |
27.00 |
29.04 |
| Data collected |
23532 |
64869 |
50341 |
| Unique refl./Rint |
23532/— |
8523/0.0304 |
10297/0.0392 |
| Parameters/Restraints |
994/6 |
504/2 |
515/27 |
| Goodness-of-fit on F2 |
1.047 |
0.993 |
1.046 |
|
R
1/wR2 (all data) |
0.0705/0.2168 |
0.0344/0.1086 |
0.0481/0.1381 |
| Residual extrema (e Å−3) |
1.41/−0.85 |
1.00/−0.62 |
1.62/−0.88 |
| Compound |
4
|
5
|
|
CCDC 1034090, 1034091, 1034092, 1034093 and 1034094 contain the crystallographic data and the CIF format for the complexes, 1, 2, 3, 4 and 5, respectively.
|
| Empirical formula |
C32.5H32Cl3Cu2F6N6O1.5P |
C33H34Cl3F6N6O2PZn2 |
| Formula mass |
909.06 |
928.76 |
| System |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
|
a (Å) |
12.0536(5) |
12.1205(4) |
|
b (Å) |
24.9462(11) |
24.9231(8) |
|
c (Å) |
12.0840(5) |
12.2116(3) |
|
α (°) |
90 |
90 |
|
β (°) |
103.631(2) |
103.081(2) |
|
γ (°) |
90 |
90 |
|
V (Å3) |
3531.2(3) |
3593.16(19) |
|
Z
|
4 |
4 |
|
T (K) |
100(2) |
100(2) |
|
μ (mm−1) |
1.549 |
1.676 |
|
D
calc (Mg m−3) |
1.710 |
1.717 |
| Crystal size (mm) |
0.30 × 0.25 × 0.18 |
0.26 × 0.22 × 0.15 |
|
θ max (°) |
29.090 |
30.010 |
| Data collected |
108528 |
122582 |
| Unique refl./Rint |
9419/0.0382 |
2090/0.0434 |
| Parameters/Restraints |
483/1 |
483/1 |
| Goodness-of-fit on F2 |
1.137 |
1.103 |
|
R
1/wR2 (all data) |
0.0322/0.0940 |
0.0276/0.0819 |
| Residual extrema (e Å−3) |
0.82/−0.47 |
0.58/−0.49 |
Conclusion
Five dinuclear bridged-phenoxido dichloro–metal(II) complexes [Co2(μ-LClO)(H2O)2Cl2][Co2(μ-LClO)(MeOH)2Cl2](PF6)2 (1), [Ni2(μ-LClO)(MeOH)2Cl2]PF6 (2), [Ni2(μ-LClO)(MeOH)(H2O)Cl2]ClO4·1.25H2O (3), [Cu2(μ-LClO)Cl2]PF6·1/2MeOH (4) and [Zn2(μ-LClO)Cl2]PF6·MeOH (5) have been synthesized based on 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenolate ion (LCl-O−) and structurally characterized. The complexes 1–4 exhibit weak to moderate antiferromagnetic coupling. Attempts were made to correlate the experimental coupling constants, J of the complexes to their structural parameters and to the metal 3d and intervening oxygen 2p orbitals. The dicopper(II) and dicobalt(II) complexes exhibit weak antiferromagnetic coupling, whereas the corresponding dinickel(II) compounds reveal moderate to relatively strong magnetic coupling.
Acknowledgements
This research was financially supported by the Department of Chemistry-University of Louisiana at Lafayette, and by the Ministry of Education, Youth and Sports of the Czech Republic (a project no. LO1305). FAM acknowledges support by NAWI Graz.
References
- D. Montagner, V. Gandin, C. Marzano and A. Erxleben, Eur. J. Inorg. Chem., 2014, 4084 CrossRef CAS.
- L. J. Daumann, P. Comba, J. A. Larrabee, G. Schenk, R. Stranger, G. Cavigliasso and L. R. Gahan, Inorg. Chem., 2013, 52, 2029 CrossRef CAS PubMed.
- P. Comba, L. R. Gahan, V. Mereacre, G. R. Hanson, A. K. Powell, G. Schenk and M. Zajaczkowski-Fischer, Inorg. Chem., 2012, 51, 7669 CrossRef PubMed.
- M. Jarenmark, M. Haukka, S. Demeshko, F. Tuczek, L. Zuppiroli, F. Meyer and E. Nordlander, Inorg. Chem., 2011, 50, 3866 CrossRef CAS PubMed.
- A. Neves, M. Lanzanaster, A. J. Bortoluzzi, R. A. Peralla, A. Cassellato, E. E. Castellano, P. Herrald, M. J. Riley and G. Schenk, J. Chem. Soc., 2007, 129, 7486 CrossRef CAS PubMed.
- M. Ghiladi, C. J. Mckenzie, A. Meler, A. K. Powell, J. Ulstrup and S. Wocadlo, J. Chem. Soc., Dalton Trans., 1997, 4011 RSC.
- B. Das, H. Daver, M. Pyrkosz-Bulska, E. Persch, S. K. Barman, R. Mukherjee, E. Gumienna-Kontecka, M. Jarenmark, F. Himo and E. Nordlander, J. Inorg. Biochem., 2014, 132, 6 CrossRef CAS PubMed.
- S. Bosch, P. Comba, L. R. Gahan and G. Schenk, Inorg. Chem., 2014, 53, 9036 CrossRef CAS PubMed.
- Y. Gultneh, Y. T. Tesema, T. B. Yisgedu, R. J. Butcher, G. Wang and G. T. Yee, Inorg. Chem., 2006, 45, 3023 CrossRef CAS PubMed.
- A. Biswas, L. K. Das and A. Ghosh, Polyhedron, 2013, 61, 253 CrossRef CAS PubMed.
- N. A. Rey, A. Neves, A. J. Bortoluzzi, C. T. Pich and H. Terenzi, Inorg. Chem., 2007, 46, 348 CrossRef CAS PubMed.
- L. J. Daumann, J. A. Larrabee, P. Comba, G. Schenk and L. R. Gahan, Eur. J. Inorg. Chem., 2013, 3082 CrossRef CAS.
- K. D. Karlin, Z. Tyeklár, A. Farooq, M. S. Haka, P. Ghosh, R. W. Cruse, Y. Gultneth, J. C. Hayes, P. J. Toscano and J. Zubieta, Inorg. Chem., 1992, 31, 1436 CrossRef CAS.
- S. Sakamoto, T. Fujinami, K. Nishi, N. Matsumoto, N. Mochida, T. Ishida, Y. Sunatsuki and N. Re, Inorg. Chem., 2013, 52, 7218 CrossRef CAS PubMed.
- K. Ehama, Y. Omichi, S. Sakamoto, T. Fujinami, N. Matsumoto, N. Mochida, T. Ishida, Y. Sunatsuki and M. Tsuchimoto, Inorg. Chem., 2013, 52, 12828 CrossRef CAS PubMed.
- S. Sakamoto, S. Yamauchi, H. Hagiwara, N. Matsumoto, Y. Sunatsuki and N. Re, Inorg. Chem. Commun., 2012, 26, 20 CrossRef CAS PubMed.
- E. Colacio, J. Ruiz, A. J. Mota, M. A. Palacios, E. Cremades, E. Ruiz, F. J. White and E. K. Brechin, Inorg. Chem., 2012, 51, 5857 CrossRef CAS PubMed.
- S. Titos-Padilla, J. Ruiz, J. M. Herrera, K. Euan, E. K. Brechin, W. Wersndorfer, F. Lloret and E. Colacio, Inorg. Chem., 2013, 52, 9620 CrossRef CAS PubMed.
- R. T. Paine, Y.-C. Tan and X.-M. Gan, Inorg. Chem., 2001, 40, 7009 CrossRef CAS PubMed.
- C. Huang, H. Gou, H. Zhu and W. Huang, Inorg. Chem., 2007, 46, 5537 CrossRef CAS PubMed.
- R. Lomoth, P. Huang, J. Zheng, L. Sun, L. Hammarström, B. Akermark and S. Styring, Eur. J. Inorg. Chem., 2002, 2965 CrossRef CAS.
- T. N. Sorrell, D. L. Jameson and C. J. O'Connor, Inorg. Chem., 1984, 23, 190 CrossRef CAS.
- R. K. Edgal, A. D. Bond and C. J. Mckenzie, Dalton Trans., 2009, 3833 Search PubMed.
- R. K. Edgal, F. B. Larsen, A. D. Bond and C. J. Mckenzie, Inorg. Chim. Acta, 2005, 358, 376 CrossRef PubMed.
- P. D. Southon, D. J. Price, P. K. Nielsen, C. J. Mckenzie and C. J. Kepert, J. Am. Chem. Soc., 2011, 133, 10885 CrossRef CAS PubMed.
- A. Banerjee, A. Guha, J. Adhikary, A. Khan, K. Manna, S. Dey, E. Zangrando and D. Das, Polyhedron, 2013, 60, 102 CrossRef CAS PubMed.
- P. Dalgaard, A. Hazell, C. J. Mckenzie, B. Moubaraki and K. S. Murray, Polyhedron, 2000, 19, 1909 CrossRef CAS.
- S. Sarkar, S. Majumder, S. Sasmal, L. Carrella, E. Rentschler and S. Mohanta, Polyhedron, 2013, 50, 370 Search PubMed.
- S. Uozumi, H. Furutachi, M. Ohba, H. Ōkawa, D. E. Fenton, K. Shindo, S. Murata and D. J. Kitko, Inorg. Chem., 1998, 37, 6281 CrossRef CAS.
- M. Thirumavalavan, P. Akilan, P. Amudha and M. Kandaswamy, Polyhedron, 2004, 23, 519 CrossRef CAS PubMed.
- G. Ambrosi, M. Formica, V. Fusti, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2008, 252, 1121 CrossRef CAS PubMed.
- R. K. Seidler-Edgal, F. B. Johansson, S. Veltze, E. M. Skou, A. D. Bond and C. J. McKenzie, Dalton Trans., 2011, 40, 3336 RSC.
- D. H. Lee, J. H. Im, S. U. Son, Y. K. Chung and J.-I. Hong, J. Am. Chem. Soc., 2003, 125, 7752 CrossRef CAS PubMed.
- F. Michel, S. Torelli, F. Thomas, C. Duboc, C. Philouze, C. Belle, S. Hamman, E. Saint-Aman and J. L. Pierre, Angew. Chem., Int. Ed., 2005, 44, 438 CrossRef CAS PubMed.
- A. S. Borovik and L. Que Junior, J. Am. Chem. Soc., 1988, 110, 2345 CrossRef CAS.
- T. Manago, S. Hayami, H. Oshio, S. Osaka, H. Hasuyama, R. H. Herber, K. J. Berry and Y. Maeda, J. Chem. Soc., Dalton Trans., 1999, 1001 RSC.
- S. J. Smith, C. J. Noble, R. C. Palmer, G. R. Hanson, G. Schenk, L. R. Gahan and M. J. Riley, J. Biol. Inorg. Chem., 2008, 13, 499 CrossRef CAS PubMed.
- H. Diril, H.-R. Chang, M. J. Nilges, X. Zhang, J. A. Potenza, H. J. Schugar, S. S. Isied and D. N. Hendrickson, J. Am. Chem. Soc., 1989, 111, 5102 CrossRef CAS.
- S. Svane, F. Kryuchkov, A. Lennartson, C. J. McKenzie and F. Kjeldsen, Angew. Chem., Int. Ed., 2012, 51, 3216 CrossRef CAS PubMed.
- S. Blanchard, G. Blain, E. Riviere, M. Nierlich and G. Blondin, Chem. – Eur. J., 2003, 9, 4260 CrossRef CAS PubMed.
- L. F. Taylor and O. P. Anderson, J. Am. Chem. Soc., 1988, 110, 1986 CrossRef.
- M. Suzuki, M. Mikuriya, S. Murata, A. Uehara and H. Oshio, Bull. Chem. Soc. Jpn., 1987, 60, 4305 CrossRef CAS.
- A. S. Borovik, M. P. Hendrich, T. R. Holman, E. Munck, V. Papaefthymiou and L. Que Junior, J. Am. Chem. Soc., 1990, 112, 6031 CrossRef CAS.
- K. Matsufuji, H. Shiraishi, Y. Miyasato, T. Shiga, M. Ohba, T. Yokoyama and H. Ōkawa, Bull. Chem. Soc. Jpn., 2005, 78, 851 CrossRef CAS.
- K. Selmeczi, C. Michel, A. Milet, I. Gautier-Luneau, C. Philouze, J.-L. Pierre, D. Schnieders, A. Rompel and C. Belle, Chem. – Eur. J., 2007, 13, 9093 CrossRef CAS PubMed.
- L. M. Berreau, A. Saha and A. M. Arif, Dalton Trans., 2006, 183 RSC.
- R. C. Holz and J. M. Brink, Inorg. Chem., 1994, 33, 4609 CrossRef CAS.
- J. J. Maloney, M. Glogowski, D. F. Rohrbach and F. L. Urbach, Inorg. Chim. Acta, 1987, 127, L33 CrossRef CAS.
- C. Huang, S. Gou, H. Zhu and W. Huang, Inorg. Chem., 2007, 46, 5537 CrossRef CAS PubMed.
- R. T. Paine, Y.-C. Tan and X.-M. Gan, Inorg. Chem., 2001, 40, 7009 CrossRef CAS PubMed.
-
(a) J.-L. Tian, W. Gu, S.-P. Yan, D.-Z. Liao and Z.-H. Jiang, Z. Anorg. Allg. Chem., 2008, 634, 1775 CrossRef CAS;
(b) S. Halder, S. Dey, C. Rizzoli and P. Roy, Polyhedron, 2014, 78, 85 CrossRef CAS PubMed.
- S. Torelli, C. Belle, I. Gautier-Luneau, J. L. Pierre, E. Saint-Aman, J. M. Latour, L. Le Pape and D. Luneau, Inorg. Chem., 2000, 39, 3526 CrossRef CAS.
- Y. Nishida, H. Shimo, H. Maehara and S. Kida, J. Chem. Soc., Dalton Trans., 1985, 1945 RSC.
- S. S. Massoud, T. Junk, M. Mikuriya, N. Naka and F. A. Mautner, Inorg. Chem. Commun., 2014, 50, 48 CrossRef CAS PubMed.
- A. K. Moshfegh, B. Mazandarani, A. Nahid and G. H. Hakimelahi, Helv. Chim. Acta, 1982, 65, 1229 CrossRef CAS.
- C. A. Lenz and M. Rychlik, Tetrahedron Lett., 2013, 54, 883 CrossRef CAS PubMed.
-
C. E. Housecroft and A. G. Sharpe, Inorg. Chem., 4th edn, Pearson, Harlow, England, 2012, p. 691 Search PubMed.
-
B. J. Hathaway, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, England, 1987, vol. 5, p. 533 Search PubMed.
-
(a) S. S. Massoud, F. R. Louka, Y. K. Obaid, R. Vicente, J. Ribas, R. C. Fischerc and F. A. Mautner, Dalton Trans., 2013, 42, 3968 RSC;
(b) S. S. Massoud, R. S. Perkins, K. D. Knierim, S. P. Comiskey, K. H. Otero, C. L. Michel, W. M. Juneau, J. H. Albering, F. A. Mautner and W. Xu, Inorg. Chim. Acta, 2013, 399, 177 CrossRef CAS PubMed;
(c) S. S. Massoud, L. Le Quan, K. Gatterer, J. H. Albering, R. C. Fischer and F. A. Mautner, Polyhedron, 2012, 31, 601 CrossRef CAS PubMed;
(d) F. A. Mautner, R. Vicente and S. S. Massoud, Polyhedron, 2006, 25, 1673–1680 CrossRef CAS PubMed;
(e) U. Mukhopadhyay, I. Bernal, S. S. Massoud and F. A. Mautner, Inorg. Chim. Acta, 2004, 357, 3673 CrossRef CAS PubMed.
- A. W. Addison, T. N. Rao, J. Reedijk, J. V. Rijin and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349 RSC.
-
R. Boča, A Handbook of Magnetochemical Formulae, Elsevier, Amsterdam, 2012 Search PubMed.
- R. Boča, Coord. Chem. Rev., 2004, 248, 757 CrossRef PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73 CrossRef CAS.
-
(a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS;
(b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS PubMed;
(c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed;
(d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
-
(a) A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS PubMed;
(b) A. Schafer, C. Huber and R. J. Ahlrichs, Chem. Phys., 1994, 100, 5829 Search PubMed;
(c) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98 CrossRef CAS PubMed.
-
(a) E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391 CrossRef CAS;
(b) E. Ruiz, A. Rodríguez-Fortea, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 2003, 24, 982 CrossRef CAS PubMed.
-
(a)
K. Yamaguchi, Y. Takahara and T. Fueno, in Applied Quantum Chemistry, ed. V. H. Smith, Reidel, Dordrecht, 1986, p. 155 Search PubMed;
(b) T. Soda, Y. Kitagawa, T. Onishi, Y. Takano, Y. Shigeta, H. Nagao, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 2000, 319, 223 CrossRef CAS.
- A. R. Allouche, J. Comput. Chem., 2011, 32, 174 CrossRef CAS PubMed.
- Z. Tomkowicz, S. Ostrovsky, S. Foro, V. Calvo-Perez and W. Haase, Inorg. Chem., 2012, 15, 6046 CrossRef PubMed.
- S. Ostrovsky, Z. Tomkowicz and W. Haase, Inorg. Chem., 2010, 49, 6942 CrossRef CAS PubMed.
- H. Sdams, D. Bradshaw and D. E. Fenton, Inorg. Chem. Acta, 2002, 332, 195 CrossRef.
- S. Turba, S. P. Foxon, A. Beitat, F. W. Heinemann, K. Petukhov, P. Müller, O. Walter, F. Lloret, M. Julve and S. Schindler, Inorg. Chem., 2012, 51, 88 CrossRef CAS PubMed.
- B. E. Schultz, B.-H. Ye, X.-Y. Li and S. I. Chan, Inorg. Chem., 1997, 36, 2617 CrossRef CAS.
- Y. Simon-Manso, J. Phys. Chem. A, 2005, 109, 2006 CrossRef CAS PubMed.
-
(a) O. Fabelo, L. Canadillas-Delgado, J. Pasan, F. S. Delgado, F. Lloret, J. Cano, M. Julve and C. Ruiz-Perez, Inorg. Chem., 2009, 48, 11342 CrossRef CAS PubMed;
(b) V. Tudor, G. Marin, F. Lloret, V. C. Kravtsov, Y. A. Siminov, M. Julve and M. Andruh, Inorg. Chim. Acta, 2008, 361, 3446 CrossRef CAS PubMed.
- F. B. Johansoon, A. D. Bond, U. G. Nielsen, B. Moubaraki, K. S. Murray, K. J. Berry, J. A. Larrabee and C. J. Mckenzie, Inorg. Chem., 2008, 47, 5079 CrossRef PubMed.
- X. H. Bu, M. Du, L. Zhang, D. Z. Liao, J. K. Tang, R. H. Zhang and M. Shionoya, J. Chem. Soc., Dalton Trans., 2001, 595 Search PubMed.
- A. Rodriguez-Fortea, P. Alemany, S. Alvarez and E. Ruiz, Chem. – Eur. J., 2001, 7, 626 CrossRef.
- V. H. Crawford, H. W. Richardson, J. R. Wasson, D. J. Hodgson and W. E. Hatfield, Inorg. Chem., 1976, 15, 2107 CrossRef CAS.
- Q. Liu and T. Rovis, J. Am. Chem. Soc., 2006, 128, 2552 CrossRef CAS PubMed.
- Y. Chen, J. Zhang, H. Zhao, N. Jiang and D. Wu, Youji Huaxue, 1989, 2, 132 Search PubMed.
-
(a)
Bruker, SAINT v. 7.23, Bruker AXS Inc., Madison, Wisconsin, USA, 2005 Search PubMed;
(b)
G. M. Sheldrick, SADABS v. 2, University of Goettingen, Germany, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, A64, 112 CrossRef PubMed.
- C. F. Macrae, P. R. Edington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, T. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The mass spectra of the complexes 1–5 are shown in Fig. S1–S5, respectively. Packing plots for crystal structures of compounds 1–5 are presented in Fig. S6–S10 and the corresponding selected bond parameters are summarized in Tables S1–S5, respectively. Fig. S11 and S12 represent the formation of the supramolecular tetramer of complex 1 and magnetic data for complex 3. CCDC 1034090–1034094 for complexes 1–5. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03508a |
| ‡ Present address: Department of Chemistry, Louisiana State University, Choppin Hall, Baton Rouge LA, 70803, USA. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Roland C.
Fischer
c,
Zdeněk
Trávníček
*b and
Franz A.
Mautner
*d
b,
Roland C.
Fischer
c,
Zdeněk
Trávníček
*b and
Franz A.
Mautner
*d