
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Complexation of Cm(III) with the recombinant N-lobe of human serum transferrin studied by time-resolved laser fluorescence spectroscopy (TRLFS)
N.
Bauer
*ab,
V. C.
Smith
c,
R. T. A.
MacGillivray
c and
P. J.
Panak
ab
aKarlsruhe Institute of Technology (KIT), Campus North, Institute for Nuclear Waste Disposal (INE), P.O. Box 3640, 76021 Karlsruhe, Germany. E-mail: Nicole.bauer@kit.edu; Fax: (+)49 721 608 23927; Tel: (+)49 721 608 24652
bUniversity of Heidelberg, Institute of Physical Chemistry, Im Neuenheimer Feld 253, 69120 Heidelberg, Germany
cUniversity of British Columbia, Department of Biochemistry and Molecular Biology and Centre for Blood Research, 2350 Health Sciences Mall, Vancouver, British Columbia V6 T 1Z3, Canada
First published on 8th December 2014
Abstract
The complexation of Cm(III) with the recombinant N-lobe of human serum transferrin (hTf/2N) is investigated in the pH range from 4.0 to 11.0 using TRLFS. At pH ≥ 7.4 a Cm(III) hTf/2N species is formed with Cm(III) bound at the Fe(III) binding site. The results are compared with Cm(III) transferrin interaction at the C-lobe and indicate the similarity of the coordination environment of the C- and N-terminal binding sites with four amino acid residues of the protein, two H2O molecules and three additional ligands (e.g. synergistic anions such as carbonate) in the first coordination sphere. Measurements at c(carbonate)tot = 0.23 mM (ambient carbonate concentration) and c(carbonate)tot = 25 mM (physiological carbonate concentration) show that an increase of the total carbonate concentration suppresses the formation of the Cm(III) hTf/2N species significantly. Additionally, the three Cm(III) carbonate species Cm(CO3)+, Cm(CO3)2− and Cm(CO3)33− are formed successively with increasing pH. In general, carbonate complexation is a competing reaction for both Cm(III) complexation with transferrin and hTf/2N but the effect is significantly higher for the half molecule. At c(carbonate)tot = 0.23 mM the complexation of Cm(III) with transferrin and hTf/2N starts at pH ≥ 7.4. At physiological carbonate concentration the Cm(III) transferrin species II forms at pH ≥ 7.0 whereas the Cm(III) hTf/2N species is not formed until pH > 10.0. Hence, our results reveal significant differences in the complexation behavior of the C-terminal site of transferrin and the recombinant N-lobe (hTf/2N) towards trivalent actinides.
Introduction
If radionuclides are accidentally released into the environment, they (and in particular actinides) can cause a serious health risk upon incorporation into bodily tissues. Since they have no essential function in the human body little is known about the biochemistry of actinides in man. To allow the development of potential decontamination therapies, a detailed understanding of the mechanisms of relevant biochemical reactions is required.1One potential reaction that absorbed actinides might undergo is the coordination to human serum transferrin. Transferrin is an iron carrier protein in blood with a molecular mass of 79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 570 Da consisting of a glycopolypeptide of 679 amino acid residues.2–4 The ternary structure of transferrin is characterized by folding into two similar lobes which are joined by a short peptide chain.5 Each lobe consists of two domains separated by a cleft housing the metal binding site for Fe(III). In both lobes Fe(III) is coordinated by two tyrosines, one aspartate, one histidine and the synergistic anion carbonate in a distorted octahedral geometry.6,7 Coordination of Fe(III) and carbonate at the binding site leads to a conformational change of transferrin from an open to a closed form. This structural change stabilizes the transferrin metal ion complex and is important for the recognition of the metal ion transferrin complex by the receptor.2,8
570 Da consisting of a glycopolypeptide of 679 amino acid residues.2–4 The ternary structure of transferrin is characterized by folding into two similar lobes which are joined by a short peptide chain.5 Each lobe consists of two domains separated by a cleft housing the metal binding site for Fe(III). In both lobes Fe(III) is coordinated by two tyrosines, one aspartate, one histidine and the synergistic anion carbonate in a distorted octahedral geometry.6,7 Coordination of Fe(III) and carbonate at the binding site leads to a conformational change of transferrin from an open to a closed form. This structural change stabilizes the transferrin metal ion complex and is important for the recognition of the metal ion transferrin complex by the receptor.2,8
In blood, transferrin is about 30% saturated with iron. Consequently, non-saturated transferrin is available for the complexation of other metal ions. Besides ferric iron about 30 other metal ions have been identified to bind to transferrin, among them actinides such as Th(IV), Pa(V), U(VI), Np(IV), Np(V), Pu(IV), Am(III) and Cm(III).9–20 In our previous study, the complexation of Cm(III) with transferrin was studied in the pH range from 3.5 to 11.0 at ambient carbonate concentration.15 At pH ≥ 7.4 Cm(III) is bound at the Fe(III) binding site of transferrin (Cm(III) transferrin species II) whereas at lower pH a nonspecific partially bound Cm(III) transferrin species is formed (Cm(III) transferrin species I).15 The fluorescence spectrum of the Cm(III) transferrin species II is in very good agreement to the spectrum of Cm2Tf (Cm(III) bound at the C- and N-terminal binding sites) and CmCTf (Cm(III) bound only at the C-terminal binding site) observed by Sturzbecher-Hoehne et al. at pH 8.6.16 Furthermore, the influence of carbonate on the complexation of Cm(III) with transferrin was investigated.21 An increase of the total carbonate concentration favors the formation of the Cm(III) transferrin species II with Cm(III) bound at the Fe(III) binding site of transferrin at significantly lower pH values which proves directly that carbonate acts as synergistic anion for Cm(III) complexation with transferrin. On the other hand, at high carbonate concentration (25 mM) carbonate complexation is an important competition reaction which suppresses Cm(III) transferrin complex formation significantly.21
Although the binding ligands are conserved in the C- and N-terminal binding sites of transferrin, the sites are not kinetically or thermodynamically equivalent. The stability constants for the N- and C-lobe of Fe2Tf differ significantly (logKC = 21.4 and logKN = 20.3),22 and the difference is almost two orders of magnitude for Cm2Tf (logKC = 8.8 ± 0.3 and logKN = 7.0 ± 0.1 at pH 8.6).16 Therefor a high excess of transferrin was used in the previous studies to ensure exclusive coordination of Cm(III) at the C-terminal binding site and the formation of a Cm(III) transferrin complex with 1:1 stoichiometry.15,21 Only transferrin in the closed conformation with two metal ions bound at the Fe(III) binding sites can be brought into the cells via receptor mediated endocytosis. Hence, it is necessary to study the complexation of Cm(III) with transferrin at both the C- and N-lobes to achieve detailed information on the complexation properties of the N-terminal binding site compared to the C-terminal site. Since the stability constant of Cm(III) coordinated at the N-lobe of transferrin is significantly lower compared to the stability constant of the C-lobe it is not possible to achieve the formation of CmNTf with exclusive coordination of Cm(III) at the N-terminal binding site. Therefor the recombinant N-lobe of human serum transferrin (hTf/2N) which features only the N-terminal binding site is used as a model for the N-lobe of transferrin.
In the present study we investigate the complexation of Cm(III) with hTf/2N. The Cm(III) hTf/2N complexation is studied in the pH range from 4.0 to 11.0 at ambient and physiological carbonate concentration and at room temperature as well as at physiological temperature using time-resolved laser fluorescence spectroscopy (TRLFS). This is a very sensitive method to study complexation reactions of lanthanides and actinides in the submicromolar concentration range.23 Cm(III) is used as a representative for trivalent actinides because of its excellent fluorescence properties.24 Spectroscopic parameters like shape and position of the emission bands as well as the fluorescence lifetime provide information on the coordination environment of the metal ion and its complex geometry.
Experimental section
Chemicals and sample preparation
All protein samples were prepared in Tris-HCl (tris(hydroxymethyl)aminomethane) buffered solutions (10 mM, pH 7.4) with a physiological sodium chloride concentration of 150 mM NaCl using ultrapure water (Millipore, Billerica, MA, USA; 18.2 MΩ cm). Fe(III) hTF/2N was expressed in baby hamster kidney cells and purified from the tissue culture medium as described previously.25 The purity of the hTF/2N was confirmed by SDS-polyacrylamide gel electrophoresis and electrospray mass spectrometry. To remove the Fe(III), Fe(III) hTF/2N was added to an excess of 0.5 M sodium citrate, 1 mM nitrilotriacetic acid (NTA) and 1 mM EDTA (ethylenediaminetetraacetate) (pH 4.9) and incubated at room temperature for 10 minutes. The resulting apo-hTF/2N was purified using 22 filtration cycles with Amicon Centrifugal Filter Units, 10 kDa (6500 rpm). The protein concentration of the apo-hTf/2N stock solution was determined by UV/Vis spectroscopy at λ = 280 nm using an extinction coefficient of ε = 38500 M−1 cm−1.26
The Cm(III) stock solution used for the TRLFS studies (c(Cm) = 3.33 × 10−6 M in H2O) had an isotopic mass distribution of 89.7% Cm-248, 9.4% Cm-246, ≤1% Cm-243, Cm-244, Cm-245 and Cm-247. The Cm(III) concentration of the TRLFS samples was fixed at 1.00 × 10−7 M by adding 30 μl of the Cm(III) stock solution to 970 μl of a Tris-HCl buffered hTf/2N solution with a concentration of 4.85 × 10−6 M resulting in a final hTf/2N concentration of 5.00 × 10−6 M. Complexation studies were carried out at varying pH values between 4.0 and 11.0 using NaOH and HCl solutions of different concentrations (1.0 M, 0.5 M, 0.1 M, and 0.01 M). TRLFS measurements were performed at room temperature (296 K) and physiological temperature (310 K). For the measurements at physiological carbonate concentration the samples were prepared with Tris buffer containing 25 mM NaHCO3.
Time-resolved laser fluorescence spectroscopy (TRLFS)
TRLFS was performed using a Nd-YAG (Continuum Surelite Laser) pumped dye laser system (NARROWscan D-R Dye Laser) with a repetition rate of 10 Hz. For excitation of Cm(III) a wavelength of 396.6 nm was used. Emission spectra were recorded after a delay of 1 μs to discriminate short-lived fluorescence of organic compounds. The pulse width was set to 1 ms. After spectral decomposition by a spectrograph (Shamrock 303i) with a 1199 lines mm−1 grating the spectra were recorded with an ICCD camera (iStar Gen III, ANDOR) containing an integrated delay controller. For better comparison all spectra are normalized to the same peak area.For lifetime measurements, the delay time between the laser pulse and the detection of the fluorescence emission was increased continuously with time intervals of Δt = 20 μs. The lifetime τ is obtained by fitting the fluorescence intensity I as a function of the delay time τ according to
| I(λ) = I0(λ)·e−t/τ | (1) |
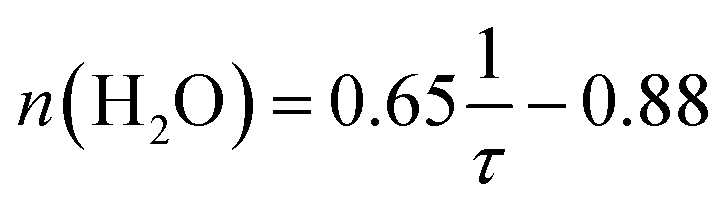 | (2) |
Results and discussion
Complexation of Cm(III) with hTf/2N at room temperature and ambient carbonate concentration
Complexation of Cm(III) with hTf/2N as a function of pH was investigated at ambient carbonate concentration (c(carbonate)tot = 0.23 mM) and room temperature (296 K) using two samples with an initial pH of 7.4 corresponding to physiological blood conditions. The pH of the samples was adjusted stepwise with HCl or NaOH, respectively. At each pH step, a fluorescence spectrum was recorded (Fig. 1, top). The spectra display a strong pH dependency of the complexation reaction. Up to pH 7.4 the system is dominated by the Cm(III) aquo ion, displaying an emission band at λmax = 593.8 nm.29–31 With increasing pH, an emission band at λmax = 598.7 nm increases which is attributed to the first Cm(III) hydrolysis species Cm(OH)2+.32 Above pH 7.7 a Cm(III) hTf/2N complex species is formed and becomes the dominant species for pH ≥ 8.3. The spectra display a sharp emission band with an emission maximum at λmax = 620.3 nm and a hot band at λmax = 603.1 nm. Furthermore, the spectra show the presence of a small amount of a Cm(III) EDTA species displaying an emission maximum at λmax = 603.8 nm.31 The EDTA impurity has its origin in the purification process of apo-hTf/2N.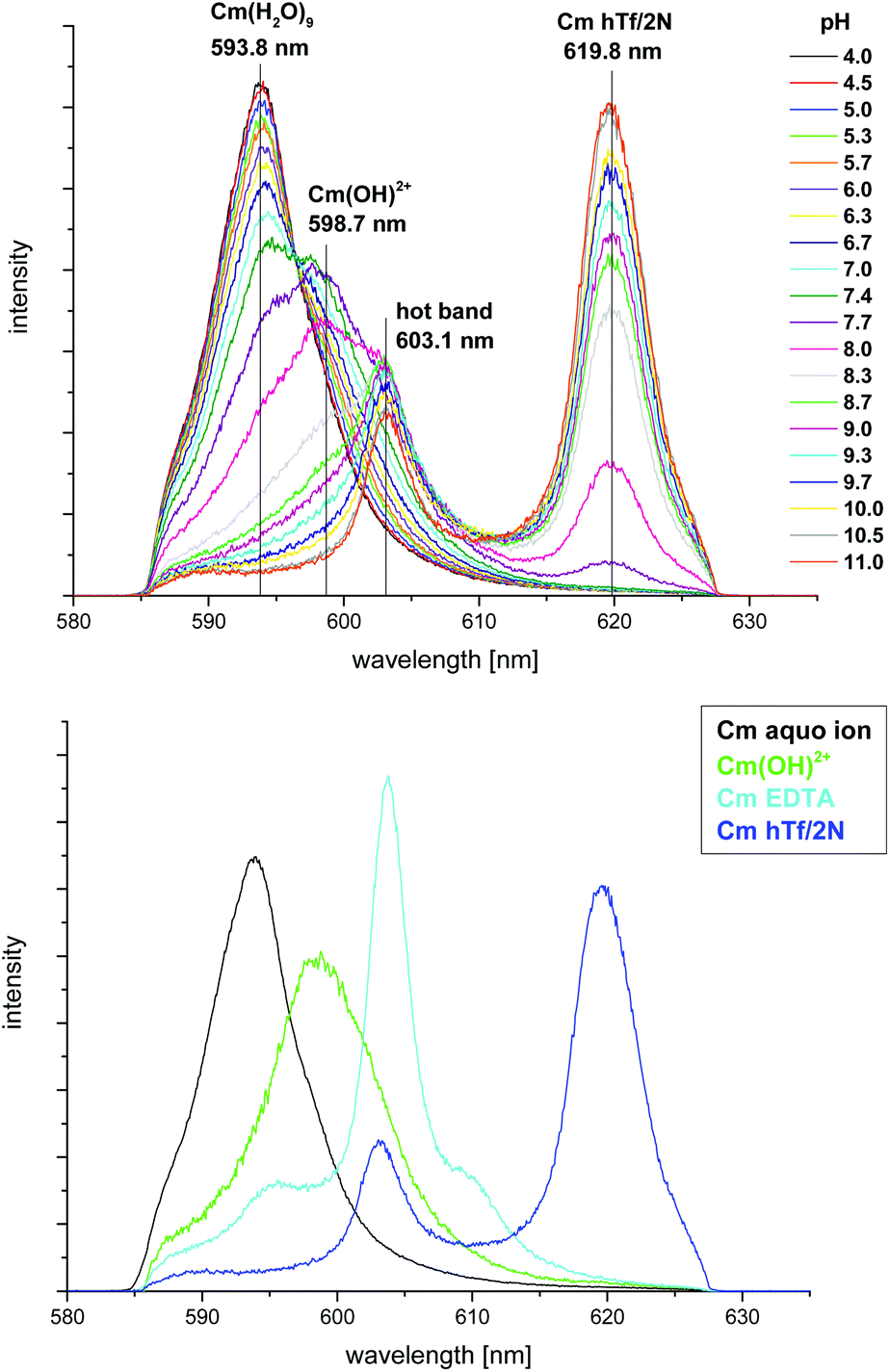 | ||
| Fig. 1 Top: normalized fluorescence spectra of the Cm(III) hTf/2N complexation at ambient carbonate concentration and room temperature in the pH range between 4.0 and 11.0; bottom: normalized fluorescence spectra of the Cm(III) aquo ion, the first Cm(III) hydrolysis species, Cm(III) EDTA and the Cm(III) hTf/2N species; c(Cm) = 1.0 × 10−7 M, c(hTf/2N) = 5.1 × 10−6 M, 10 mM Tris, T = 296 K. | ||
The emission band of the Cm(III) hTf/2N complex species is identical to that of the Cm(III) transferrin species II with Cm(III) bound at the C-terminal binding site observed in previous studies.15,16,21 This species is characterized by very sharp emission bands at λmax = 619.9 nm and λmax = 602.9 nm (hot band). The bathochromic shift of the emission band of 26 nm relative to the emission band of the Cm(III) aquo ion is extraordinary and confirms complexation of Cm(III) at the Fe(III) binding site of the protein.15 The fluorescence lifetime of the emission band of the Cm(III) hTf/2N species is determined to be τ = 217 ± 5 μs which is in very good agreement with the value of the Cm(III) transferrin species II (τ = 221 ± 5 μs) and corresponds to two quenching water molecules in the first coordination sphere.15,16,27,28 Assuming an overall coordination number of nine for Cm(III), the TRLFS results indicate that the first coordination sphere of Cm(III) complexed with transferrin as well as with hTf/2N is composed of four amino acid ligands from the protein, two H2O molecules and three additional ligands (e.g. synergistic anions like carbonate).15,16 These results show that the coordination environment of Cm(III) complexed at the C-terminal Fe(III) binding site of transferrin is identical to that of Cm(III) in the hTf/2N complex. The experimental conditions of our previous measurements with transferrin ensure exclusive complexation of Cm(III) at the binding site of the C-lobe whereas complexation occurs at the N-terminal binding site with hTf/2N. Hence, our results confirm the similarity of the coordination environment of the binding sites in the N- and C-lobe of the protein.
In addition to the Cm(III) transferrin species II, the studies on the complexation of Cm(III) with transferrin reveal the presence of an unspecific and only partially bound Cm(III) transferrin species with five water molecules and four additional ligands in the first coordination sphere (Cm(III) transferrin species I) which is formed in the pH range from 3.5 to 9.7.15 In contrast to the measurements with transferrin, no unspecific Cm(III) species is formed with hTf/2N. Instead the first Cm(III) hydrolysis species Cm(OH)2+ is formed in this pH range. This indicates that the binding of Cm(III) either occurs on the C-lobe or amino acids from both lobes are involved in the formation of the Cm(III) transferrin species I.
The fluorescence spectra of the pure components (the Cm(III) aquo ion, Cm(OH)2+, Cm(III) EDTA and the Cm(III) hTf/2N species, Fig. 1 bottom) were determined from the pH dependent fluorescence spectra. They were used to determine the fractions of the four species at various pH values by peak deconvolution of the emission spectra. The fluorescence intensity factors (fi factors) which describe the decrease or increase of the fluorescence intensity relative to a reference species, in this case the Cm(III) aquo ion, have been determined for all Cm(III) species to be around 1 ± 0.5. This means that there is no significant decrease or increase of the fluorescence intensity upon complexation. Nevertheless, the fluorescence intensity factors are taken into account for the calculation of the concentration ratios but the speciation in dependence of pH (species distribution, Fig. 2) shows no significant change upon consideration of the fi factors. The Cm(III) aquo ion is present up to pH 8.3, its concentration decreases with increasing pH, whereas the fraction of the first Cm(III) hydrolysis species Cm(OH)2+ increases. It is the dominating species in the pH range from 6.5 to 8.2. At pH ≥ 7.4, the Cm(III) hTf/2N species is formed. The formation of this species corresponds to the formation of the Cm(III) transferrin species II.15 Both species are formed at pH values above 7.4 which underlines the similarity of the C- and N-terminal binding sites. Since the maximum ratio of the Cm(III) EDTA species is about 5%, it has no significant impact on the speciation and can be neglected.
 | ||
| Fig. 2 Speciation of Cm(III) with hTf/2N as a function of pH at ambient carbonate concentration and room temperature (the curves do not represent mathematical fits but are for guidance only); c(Cm) = 1.0 × 10−7 M and c(hTf/2N) = 5.1 × 10−6 M, 10 mM Tris, T = 296 K. | ||
Kinetics of the complexation reaction and stability of the Cm(III) hTf/2N complex
Many chelating ligands (such as DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid)) show slow complexation kinetics.33–36 Since the effective ionic radius of Cm(III) is about two times larger than that of Fe(III), it is expected not to fit exactly into the C- and N-terminal Fe(III) binding sites of transferrin which might result in slow complexation kinetics.37 Therefore, the kinetics of the complexation of Cm(III) with hTf/2N was analyzed at different pH values in the time range from 0 to 189 hours. Fig. 3 depicts the normalized fluorescence spectra of two Cm(III) hTf/2N samples at pH 8.0 (top) and 10.0 (bottom) with time. Up to a period of 24 hours, the spectra do not change significantly with time which indicates fast kinetics of the complex formation. At both pH values, equilibrium was reached after five minutes. This is in good agreement with the data on the kinetics of the complexation of Cm(III) at the C-terminal binding site of transferrin at pH 7.4 and 9.015 and confirms fast complexation of Cm(III) at the C- and the N-terminal Fe(III) binding sites. Therefore, measurements without long-lasting equilibration periods are legitimate for all studies on the interaction of Cm(III) with transferrin and hTf/2N. | ||
| Fig. 3 Normalized fluorescence spectra of the Cm(III) hTf/2N complexation at room temperature and ambient carbonate concentration at pH 8.0 (top) and pH 10.0 (bottom) as a function of time; c(Cm) = 1.0 × 10−7 M and c(hTf/2N) = 5.0 × 10−6 M, 10 mM Tris, time range 0–189 h. | ||
To investigate the stability of the Cm(III) hTf/2N complex species, fluorescence spectra were recorded after long equilibration times. The spectra of the Cm(III) transferrin species I and II at pH 7.4 and 9.0 observed in our previous study are unaltered up to 96 hours which indicates the formation of very stable complexes.15 In contrast, the spectra of Cm(III) with hTf/2N at pH 8.0 and 10.0 start to change after one day. The emission band of the Cm(III) hTf/2N species at λmax = 620.3 nm decreases significantly, in particular at pH 10.0, which indicates decomplexation of Cm(III) in the time range from 24–189 hours. The results show that the Cm(III) complex formed with the intact transferrin is more stable than that formed with the half molecule. Although the same amino acid residues are present at both binding sites the primary structure of the protein strain is not identical for the C- and N-lobe4 and the stability constants, which are a measure for the complex stability, differ significantly for complexation of Cm(III) at the C- and N-terminal binding sites of transferrin (logKC = 8.8 ± 0.3 and logKN = 7.0 ± 0.1 at pH 8.6).16 This is also reflected in the increased stability of Cm(III) bound at the C-terminal binding site of transferrin in comparison with the Cm(III) hTf/2N species. The absence of the second lobe seems to have a significant influence on the stability of the protein itself. Decomplexation occurs in the whole pH range in which the Cm(III) hTf/2N species is present in solution, but the effect is significantly higher when the pH is increased. Hence, the deprotonation of amino acid residues which might change the structure of hTf/2N seems to lead to a further decrease of the stability of the half molecule.
Complexation of Cm(III) with hTf/2N at physiological temperature
The complexation of Cm(III) with hTf/2N as a function of pH was investigated at ambient carbonate concentration and physiological temperature (310 K). The normalized fluorescence spectra in the pH range from 4.0 to 11.0 (data not shown) are comparable to those observed at room temperature. They display the emission bands of the Cm(III) aquo ion (λmax = 593.9 nm), the first Cm(III) hydrolysis species (λmax = 598.7 nm), the Cm(III) EDTA species (λmax = 603.5 nm) and the Cm(III) hTf/2N species (λmax = 619.9 nm, hot band at λmax = 603.2 nm) which are formed successively with increasing pH.The species concentrations in dependence of pH (species distribution, Fig. 4) have been determined using the fractions of the different Cm(III) species derived from peak deconvolution and the fluorescence intensity factors which are around 1 ± 0.5. The increase of the temperature from 296 K to 310 K has a significant impact on the formation of the different Cm(III) species. They are formed at pH values that are about 0.5 units lower than the respective pH values at room temperature (Fig. 2). At physiological temperature, the Cm(III) hTf/2N species is formed above pH 7.0 whereas the formation starts at pH ≥ 7.4 at room temperature conditions. This is comparable to the difference in the formation of the Cm(III) transferrin species II at these two temperatures.15 At 310 K, complex formation also occurs at pH values that are 0.5 units lower than the respective pH values at room temperature. The analogy of the temperature dependency of the Cm(III) complexation with hTf/2N and transferrin again underlines the similarity of the C- and N-terminal binding sites.
 | ||
| Fig. 4 Speciation of Cm(III) with hTf/2N as a function of pH at ambient carbonate concentration and physiological temperature (the curves do not represent mathematical fits but are for guidance only); c(Cm) = 1.0 × 10−7 M and c(hTf/2N) = 5.1 × 10−6 M, 10 mM Tris, T = 310 K. | ||
Complexation of Cm(III) with hTf/2N at physiological carbonate concentration
In order to study the influence of carbonate on the complexation of Cm(III) with hTf/2N, the complex formation was studied as a function of pH at physiological carbonate concentration (c(carbonate)tot = 25 mM). The term physiological carbonate concentration describes a total concentration of the species CO32−, HCO3− and CO2(aq.) of c(carbonate)tot = 25 mM with the speciation of CO32−, HCO3− and CO2(aq.) depending on the pH.21Fig. 5 (top) shows the normalized fluorescence spectra in the pH range from 4.0 to 11.0. At low pH the spectra are dominated by the emission band of the Cm(III) aquo ion (λmax = 593.9 nm). In contrast to the measurements at ambient carbonate concentration, the emission band of the first Cm(III) hydrolysis species is not observed under physiological carbonate conditions. Instead, three Cm(III) carbonate species Cm(CO3)+, Cm(CO3)2− and Cm(CO3)33− with emission maxima at λmax = 598.8 nm, 603.2 nm and 605.8 nm are formed.32 Comparison with blank solutions (without hTf/2N) in the same pH range proves that these emission bands result from Cm(III) carbonate interactions and do not represent additional transferrin species. The Cm(III) hTf/2N species with λmax = 619.7 nm is formed at pH ≥ 10.5 which indicates a distinct competition between carbonate and hTf/2N. Fig. 5 (bottom) shows the fluorescence spectra of the pure components (Cm(III) aquo ion, Cm(CO3)+, Cm(CO3)2−, Cm(CO3)33− and Cm(III) hTf/2N species). As shown in Fig. 6 (top), the fraction of the Cm(III) aquo ion decreases with increasing pH whereas up to 90% of the Cm(III) mono-, di- and tri-carbonate complexes are formed successively. The Cm(CO3)+ and Cm(CO3)2− species dominate the speciation in the pH range from 5.8 to 7.2 and from 7.2 to 8.9, respectively. Additionally, the Cm(CO3)33− complex is formed in the pH range from 7.7 to 11.0. The Cm(III) hTf/2N species forms above pH 10.0 at physiological carbonate conditions. In comparison, at ambient carbonate concentration, the complex formation starts at a significantly lower pH value (pH 7.4). Hence, an increase of the carbonate concentration suppresses the formation of the Cm(III) hTf/2N species significantly. This indicates that carbonate complexation is an important competition reaction to the formation of the Cm(III) hTf/2N species which has to be considered when complexation studies with hTf/2N are performed at physiological conditions.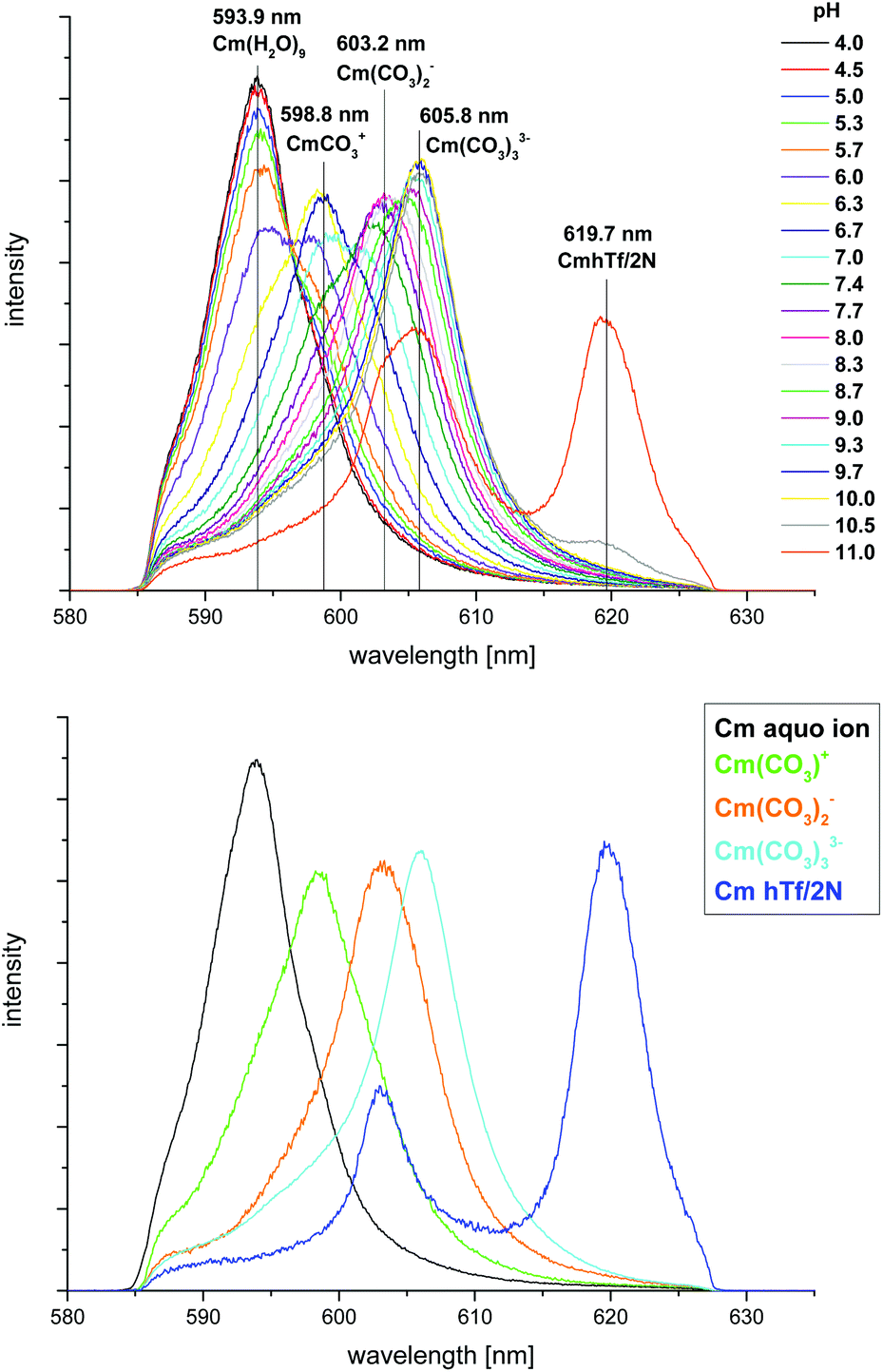 | ||
| Fig. 5 Top: normalized fluorescence spectra of the Cm(III) hTf/2N complexation at physiological carbonate concentration and room temperature in the pH range from 4.0 to 11.0; bottom: normalized fluorescence spectra of the Cm(III) aquo ion, the Cm(III) carbonate species and the Cm(III) hTf/2N species; c(Cm) = 1.0 × 10−7 M, c(hTf/2N) = 5.1 × 10−6 M and c(carbonate)tot = 2.5 × 10−2 M, 10 mM Tris, T = 296 K. | ||
 | ||
| Fig. 6 Speciation of Cm(III) with hTf/2N as a function of pH at physiological carbonate concentration and room temperature (top) and physiological temperature (bottom); the curves do not represent mathematical fits but are for guidance only; c(Cm) = 1.0 × 10−7 M, c(hTf/2N) = 5.1 × 10−6 M, c(carbonate)tot = 2.5 × 10−2 M, 10 mM Tris, T = 296 K (top) and T = 310 K (bottom). | ||
In addition to the measurements at room temperature, the complexation of Cm(III) with hTf/2N as a function of pH was also studied at physiological carbonate concentration (c(carbonate)tot = 25 mM) and physiological temperature (310 K). The spectra are very similar to those observed at room temperature conditions. They depict the emission bands of the Cm(III) aquo ion, the three Cm(III) carbonate species Cm(CO3)+, Cm(CO3)2− and Cm(CO3)33− and the Cm(III) hTf/2N species which are formed successively with increasing pH. The species distribution (Fig. 6, bottom) shows that the Cm(III) aquo ion dominates the speciation up to pH 5.7. With increasing pH the Cm(CO3)+, Cm(CO3)2− and Cm(CO3)33− species are formed successively. Compared to room temperature conditions the formation of the Cm(III) carbonate species starts at slightly lower pH values. Above pH 9.3 the concentration of the Cm(III) hTf/2N species increases continuously. In contrast, the formation of the Cm(III) hTf/2N species at room temperature does not occur until pH 10.0. Hence, an increase of the temperature favors the complexation of Cm(III) with hTf/2N at lower pH values.
The speciation of Cm(III) hTf/2N complexation was compared to the measurements of Cm(III) transferrin at the same conditions.21 While the spectroscopic data on the complexation of Cm(III) with hTf/2N and at the C-terminal binding site of transferrin agree very well at ambient carbonate concentration (complex formation starts at pH 7.4), the species distributions reveal significant differences at physiological carbonate concentration: at room temperature, the Cm(III) transferrin species II is formed at pH ≥ 7.0.21 This indicates that an increase of carbonate favors the formation of the Cm(III) transferrin species II at lower pH values, with carbonate acting as a synergistic anion for Cm(III) complexation at the C-terminal binding site of transferrin. At higher pH (pH ≥ 7.4), the competing effect of carbonate plays a distinct role resulting in a decrease of the fractions of the Cm(III) transferrin species II.21 In the case of hTf/2N, carbonate complexation suppresses the formation of the Cm(III) hTf/2N species significantly, even at low pH conditions. For that reason, this species is not formed until pH > 10.0 at physiological carbonate concentration. In general, carbonate complexation is a competing reaction for both Cm(III) complexation with hTf/2N and at the C-terminal binding site of transferrin but the effect is significantly higher for the half molecule.
There is only deficient knowledge concerning the competitive effect of carbonate on the complexation of metal ions to transferrin. Nevertheless, the competition between carbonate and transferrin has been observed in previous studies for some lanthanide and actinide ions.38–41 Based on complexation studies of Gd(III) with transferrin in vitro, Zak and Aisen suggest that transferrin may not be an important carrier protein for Gd(III) in vivo because of the formation of Gd(III) carbonate complexes.41 Taylor et al. reported that only about 20% of Eu(III), Yb(III), Am(III) and Cm(III) are bound to transferrin in vivo which is also attributed to the competition with carbonate.39,40
Although the species distributions reveal significant differences in the complexation behavior of Cm(III) with the C-lobe of transferrin and hTf/2N at physiological carbonate concentration, the temperature dependency of the Cm(III) hTf/2N complexation reaction is comparable to the data on Cm(III) hTf/2N and Cm(III) transferrin interaction at ambient carbonate concentration. In general, at physiological temperature (310 K) the Cm(III) complexes with hTf/2N and the C-terminal binding site of transferrin are formed at pH values that are 0.5 units lower than the respective pH values at room temperature (valid for ambient and physiological carbonate conditions). Hence, an increase of the temperature favors the complexation of Cm(III) with hTf/2N as well as at the C-terminal binding site of transferrin at lower pH values. This indicates that Cm(III) transferrin and Cm(III) hTf/2N complexation are endothermic and entropy-driven reactions.
Because of the potential transport of actinides into cells, the complexation of Cm(III) with hTf/2N (N-lobe) and transferrin (C-lobe) at physiological conditions (pH 7.4, T = 310 K, c(carbonate)tot = 25 mM) is of particular interest. In the case of hTf/2N, only the Cm(CO3)+ and Cm(CO3)2− species are formed under physiological conditions whereas the formation of the Cm(III) hTf/2N species is completely suppressed. In contrast, results from our previous study show the formation of about 29% Cm(CO3)+, 38% Cm(CO3)2−, 18% Cm(CO3)33− and 15% of the Cm(III) transferrin species II with Cm(III) bound at the C-terminal binding site under the same conditions. Though the fraction of the Cm(III) transferrin species II is low compared to the fraction of the Cm(III) carbonate species, a complex with Cm(III) bound at the C-terminal binding site of transferrin might be recognized by the receptor followed by endocytosis which could be a possible pathway for the distribution of Cm(III) in the human body. In general, carbonate is a competitive ligand for both hTf/2N and transferrin but the impact on the Cm(III) hTf/2N species is significantly higher compared to Cm(III) complexation at the C-terminal binding site of transferrin. The different complexation behavior of hTf/2N and the C-lobe of transferrin at physiological carbonate concentration might either result from the thermodynamic difference of the binding sites (logKC = 8.8 ± 0.3 and logKN = 7.0 ± 0.1 at pH 8.6),16 or from the fact that the presence or absence of the second lobe might have a significant impact on the stability of the protein itself as well as on the regarding Cm(III) complexes. Nevertheless, the differences in the complexation behavior of Cm(III) with hTf/2N and transferrin underline the inequality of the C- and N-terminal binding sites.
Conclusions
In the present study, the complexation of Cm(III) with the recombinant N-lobe of human serum transferrin (hTf/2N) was investigated using TRLFS. The results were compared to the measurements on Cm(III) complexation at the C-terminal binding site of transferrin from previous studies resulting in detailed information on the complexation properties of the C- and N-terminal binding sites.15,21 The spectroscopic characteristics of the emission bands show that the Cm(III) hTf/2N species is comparable to the Cm(III) transferrin species II (Cm(III) complexed at the C-terminal site) which indicates the similarity of the coordination environment of the C- and N-terminal binding sites. In both complexes Cm(III) is coordinated by four amino acid residues of the protein, two H2O molecules and three additional ligands (e.g. synergistic anions like carbonate).15Measurements at physiological carbonate concentration reveal that the synergistic anion carbonate is a competitive ligand for complexation of Cm(III) with hTf/2N. This is comparable to the influence of carbonate on Cm(III) transferrin but the impact on the Cm(III) hTf/2N species (N-lobe) is significantly higher compared to Cm(III) complexation at the C-terminal binding site of transferrin. Hence, Cm(III) forms more stable complexes with the C-terminal binding site of transferrin compared to hTf/2N which is in good agreement with the thermodynamic difference of the binding sites.16 Furthermore, the presence of a second lobe seems to improve the stability of the protein itself and the Cm(III) complexes significantly.
Notes and references
- A. E. V. Gorden, J. D. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207 CrossRef CAS PubMed.
- H. Z. Sun, H. Y. Li and P. J. Sadler, Chem. Rev., 1999, 99, 2817 CrossRef CAS PubMed.
- R. T. A. MacGillivray, E. Mendez, J. G. Shewale, S. K. Sinha, J. Linebackzins and K. Brew, J. Biol. Chem., 1983, 258, 3543 CAS.
- R. T. A. MacGillivray, E. Mendez, S. K. Sinha, M. R. Sutton, J. Linebackzins and K. Brew, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 2504 CrossRef CAS.
- J. Wally, P. J. Halbrooks, C. Vonrhein, M. A. Rould, S. J. Everse, A. B. Mason and S. K. Buchanan, J. Biol. Chem., 2006, 281, 24934 CrossRef CAS PubMed.
- R. T. A. MacGillivray, S. A. Moore, J. Chen, B. F. Anderson, H. Baker, Y. G. Luo, M. Bewley, C. A. Smith, M. E. P. Murphy, Y. Wang, A. B. Mason, R. C. Woodworth, G. D. Brayer and E. N. Baker, Biochemistry, 1998, 37, 7919 CrossRef CAS PubMed.
- S. Bailey, R. W. Evans, R. C. Garratt, B. Gorinsky, S. Hasnain, C. Horsburgh, H. Jhoti, P. F. Lindley, A. Mydin, R. Sarra and J. L. Watson, Biochemistry, 1988, 27, 5804 CrossRef CAS.
- P. D. Jeffrey, M. C. Bewley, R. T. A. MacGillivray, A. B. Mason, R. C. Woodworth and E. N. Baker, Biochemistry, 1998, 37, 13978 CrossRef CAS PubMed.
- W. R. Harris, C. J. Carrano, V. L. Pecoraro and K. N. Raymond, J. Am. Chem. Soc., 1981, 103, 2231 CrossRef CAS.
- S. Scapolan, E. Ansoborlo, C. Moulin and C. Madic, Radiat. Prot. Dosim., 1998, 79, 505 CrossRef CAS.
- I. Llorens, C. Den Auwer, P. Moisy, E. Ansoborlo, C. Vidaud and H. Funke, FEBS J., 2005, 272, 1739 CrossRef CAS PubMed.
- C. Den Auwer, I. Llorens, P. Moisy, C. Vidaud, F. Goudard, C. Barbot, P. L. Solari and H. Funke, Radiochim. Acta, 2005, 93, 699 CrossRef CAS.
- A. Jeanson, M. Ferrand, H. Funke, C. Hennig, P. Moisy, P. L. Solari, C. Vidaud and C. Den Auwer, Chem. – Eur. J., 2010, 16, 1378 CrossRef CAS PubMed.
- E. Ansoborlo, O. Prat, P. Moisy, C. Den Auwer, P. Guilbaud, M. Carriere, B. Gouget, J. Duffield, D. Doizi, T. Vercouter, C. Moulin and V. Moulin, Biochimie, 2006, 88, 1605 CrossRef CAS PubMed.
- N. Bauer, D. R. Fröhlich and P. J. Panak, Dalton Trans., 2014, 43, 6689 RSC.
- M. Sturzbecher-Hoehne, C. Goujon, G. J. P. Deblonde, A. B. Mason and R. J. Abergel, J. Am. Chem. Soc., 2013, 135, 2676 CrossRef CAS PubMed.
- R. Racine, P. Moisy, F. Paquet, H. Metivier and C. Madic, Radiochim. Acta, 2003, 91, 115 CrossRef CAS.
- J. Michon, S. Frelon, C. Garnier and F. Coppin, J. Fluoresc., 2010, 20, 581 CrossRef CAS PubMed.
- D. M. Taylor and L. C. Farrow, Nucl. Med. Biol., 1987, 14, 27 CAS.
- D. M. Taylor, J. Alloys Compd., 1998, 271, 6 CrossRef.
- N. Bauer and P. J. Panak, New J. Chem., 2014 Search PubMed , submitted.
- P. Aisen, A. Leibman and J. Zweier, J. Biol. Chem., 1978, 253, 1930 CAS.
- N. M. Edelstein, R. Klenze, T. Fanghänel and S. Hubert, Coord. Chem. Rev., 2006, 250, 948 CrossRef CAS PubMed.
- I. J. Kim, R. Klenze and H. Wimmer, Eur. J. Solid State Inorg. Chem., 1991, 28, 347 Search PubMed.
- A. B. Mason, W. D. Funk, R. T. A. MacGillivray and R. C. Woodworth, Protein Expression Purif., 1991, 2, 214 CrossRef CAS.
- Q. Y. He, A. B. Mason, B. A. Lyons, B. M. Tam, V. Nguyen, R. T. A. MacGillivray and R. C. Woodworth, Biochem. J., 2001, 354, 423 CrossRef CAS.
- T. Kimura and G. R. Choppin, J. Alloys Compd., 1994, 213/214, 313 CrossRef CAS.
- T. Kimura, G. R. Choppin, Y. Kato and Z. Yoshida, Radiochim. Acta, 1996, 72, 61 CrossRef CAS.
- J. V. Beitz, Radiochim. Acta, 1991, 52–3, 35 Search PubMed.
- J. V. Beitz and J. P. Hessler, Nucl. Technol., 1980, 51, 169 CAS.
- R. Klenze, J. I. Kim and H. Wimmer, Radiochim. Acta, 1991, 52–3, 97 Search PubMed.
- T. Fanghanel, T. Konnecke, H. Weger, P. Paviet-Hartmann, V. Neck and J. I. Kim, J. Solution Chem., 1999, 28, 447 CrossRef CAS.
- E. Toth, E. Brucher, I. Lazar and I. Toth, Inorg. Chem., 1994, 33, 4070 CrossRef CAS.
- S. L. Wu and W. D. Horrocks, J. Chem. Soc., Dalton Trans., 1997, 1497 RSC.
- V. Kubicek, J. Havlickova, J. Kotek, T. Gyula, P. Hermann, E. Toth and I. Lukes, Inorg. Chem., 2010, 49, 10960 CrossRef CAS PubMed.
- A. Bremer, A. Geist and P. J. Panak, Dalton Trans., 2012, 41, 7582 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751 CrossRef.
- W. R. Harris, Inorg. Chem., 1986, 25, 2041 CrossRef CAS.
- J. R. Cooper and H. S. Gowing, Int. J. Radiat. Biol., 1981, 40, 569 CrossRef CAS.
- D. M. Taylor, J. R. Duffield, D. R. Williams, L. Yule, P. W. Gaskin and P. Unalkat, Eur. J. Solid State Inorg. Chem., 1991, 28, 271 CAS.
- O. Zak and P. Aisen, Biochemistry, 1988, 27, 1075 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |