
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe relationship between the strength of hydrogen bonding and spin crossover behaviour in a series of iron(III) Schiff base complexes†
Ivan
Nemec
 ,
Radovan
Herchel
and
Zdeněk
Trávníček
*
,
Radovan
Herchel
and
Zdeněk
Trávníček
*
Regional Centre of Advanced Technologies and Materials, Department of Inorganic Chemistry, Faculty of Science, Palacký University, Tř. 17. listopadu 12, CZ-77146 Olomouc, Czech Republic. E-mail: zdenek.travnicek@upol.cz
First published on 19th January 2015
Abstract
X-ray crystal structures and magnetic properties of an isostructural series of iron(III) Schiff base complexes with the general formula [Fe(L5)(NCX)]·Solv (where H2L5 = N,N′-bis(2-hydroxy-naphthylidene)-1,6-diamino-4-azahexane, X = S, Solv = tetrahydrofuran, 1a; X = S, Solv = methanol and 0.5 pyrazine, 1b; X = S, Solv = butanone, 1c; Solv = N,N′-dimethylformamide, X = S (1d) or X = Se (1d′); X = S, Solv = dimethyl sulfoxide, 1e) are reported. In the crystals, the individual [Fe(L5)(NCX)] molecules are connected through weak C–H⋯O, C–H⋯π or C–H⋯S non-covalent contacts into 2D supramolecular networks, while the guest-solvent (Solv) molecules are trapped in the cavities between two adjacent layers, which are furthermore stabilized by N–H⋯O hydrogen bonds connecting the Solv oxygen atom with the amine group of the [Fe(L5)(NCX)] molecule, with the N⋯O distances varying from 2.921(6) Å (in 1d′) to 3.295(2) Å (in 1a). The magnetic properties of the complexes were tuned by the different Solv molecules and as a result of this, four new spin crossover (SCO) compounds with cooperative spin transitions are reported, which are accompanied by thermal hysteresis in two cases (1d and 1e): 1c, T1/2 = 84 K; 1d, T1/2↓ = 232 K, T1/2↑ = 235 K and 1e, T1/2↓ = 127 K, T1/2↑ = 138 K. The role of the N–H⋯O hydrogen bonding in the occurrence and tuning of SCO was also computationally studied using a topological analysis, and also by evaluation of non-covalent interaction (NCI) indexes. Both theoretical approaches showed a clear relationship between the strength of the N–H⋯O hydrogen bonds and T1/2, as already inferred from X-ray structural and magnetic data.
Introduction
The spin crossover (SCO) phenomenon, as a well-known example of molecular bistability, attracts the attention of material chemists due to its potential applications in molecular switches, memory, displays or hybrid materials.1 It represents the spin transition between the low-spin (LS, in the case of FeIII octahedral complexes, S = 1/2, 2T2g) and high-spin (HS) states (S = 5/2, 6A1g) upon external perturbations such as thermal and/or pressure changes or light irradiation.2 Such a dramatic change in the electronic structure is accompanied by changes in the magnetic and structural properties, which can be detected and monitored by basic experimental techniques such as magnetometry, various kinds of spectroscopy or X-ray structural analysis.2 The SCO phenomenon is very sensitive to the environment of the SCO molecule. In isolated systems, the spin state conversion follows the Maxwell–Boltzmann statistics in most cases,3 but when cooperative interactions (represented by non-covalent contacts such as hydrogen bonding and/or π–π stacking interactions) come into play, the transition might be very abrupt (within the interval of a few kelvins). Furthermore, if these interactions are able to propagate large changes in the crystal structure upon SCO, then thermal hysteresis may appear.4In general, the role of solvent molecules of crystallization (Solv) in the SCO process is still not clear, because there are several contrasting examples describing the influence of solvents. The presence of the guest molecules in the crystal lattice of the SCO compounds can dramatically affect their magnetic behaviour5 by changing the cooperative interactions with respect to the parent compound and therefore the lattice phonon distribution is affected.2 It can be also rationalized that non-covalent interactions, such as hydrogen bonds, may cause a charge redistribution on the ligands and consequently, a change in the ligand field strength can occur.6 The stabilization of the LS state is usually observed for the solvated samples and there are many examples of a downward shift of T1/2 (or just occurrence of SCO in the formerly LS-only solvated compounds) upon desolvation of the sample.7
In accord with these observations, the solvation of a sample usually tends to increase T1/2. Alternatively, upon guest molecule exchange, the variation in T1/2 and in steepness of the transition is observed, as has been reported recently in the remarkable single-crystal-to-single-crystal transformation studies.8 Rational tuning of the SCO behaviour by the lattice solvent exchange was reported first for the nanoporous metal–organic frameworks (MOFs) built by [Fe2(azpy)(NCS)4] molecules (azpy = trans-4,4′-azopyridine) in 2002 by Kepert et al.,9 and then, other similar studies have followed.10 The vapour and gas adsorption studies on {[Fe(PYZ)Ni(CN)4]} Hoffmann-type metal–organic frameworks (MOFs) revealed the relationship between the guest size and T1/2 (PYZ = pyrazine),11 and similar relationships were reported also for 0D supramolecular systems such as [Fe(4ditz)3](PF6)2·Solv (4ditz = 1,4-bis(tetrazole-1-yl)butane, Solv = CH3OH, CH3CH2OH) or [Fe(bdpt)2]·Solv (Hbdpt = 3-(5-bromo-2-pyridyl)-5-(4-pyridyl)-1,2,4-triazole), Solv = CH3OH, CH3CH2OH).12 Another possible Solv–SCO relationship was found in MOFs such as [Fe(bpbd)2(NCS)2]·Solv (bppd = 2,3-bis(4′-pyridyl)-2,3-butanediol, Solv = CH3CN, CH3OH, CH3CH2OH, CH3CH2OH (CH3)2CO), and it was shown that lower values of the Solv dielectric constants (εr) result in the LS state being more stabilized.13 The solvent–SCO relationship was documented also for Fe(III) SCO compounds several times but the systematic study is still missing.14
Recently, we have reported on the SCO phenomenon observed in a series of [Fe(L5)(L1)]·Solv compounds (Scheme 1, H2L5 = N,N′-bis(2-hydroxy-naphthylidene)-1,6-diamino-4-azahexane, L1 stands for pseudohalide ligands).15 The thiocyanate and selenocyanate complexes from the mentioned series, [Fe(L5)(NCX)]·CH3CN and [Fe(L5)(NCS)]·(CH3)2CO (X = S, Se), exhibited magnetic behaviour dependent on the presence of a solvent molecule incorporated into the crystal structure: the acetonitrile solvate showed SCO while the latter compound stayed in the HS state down to 2 K. In both cases, the solvent molecule is connected with the donor nitrogen amine atom from the L5 ligand via a hydrogen bond (the nitrogen/oxygen atom from the Solv molecules serves as an acceptor). In order to explore the influence of the co-crystallized Solv molecules on SCO in greater detail, we decided to investigate the properties of the aforementioned system utilizing various Solv molecules. In this work, we report on the crystal structures and magnetic properties of an isostructural (for bond lengths and selected structural parameters see Table 1, for crystallographic data see Table 2) series of [Fe(L5)(NCX)]·Solv complexes, where X = S, Solv = tetrahydrofuran (THF) (1a); X = S, Solv = methanol (MeOH) and 0.5 pyrazine (PYZ) (1b); X = S, Solv = butanone (MEK) (1c); X = S, Solv = N,N′-dimethylformamide (DMF) (1d); X = Se, Solv = DMF (1d′); X = S, Solv = dimethyl sulfoxide (DMSO) (1e). Moreover, previously published compounds 1f (X = S, Solv = 0.5 MeOH and 0.5 MEK) and 1g (X = S, Solv = acetone) were included in the discussion for comparative purposes.15,16 Apparently, the use of PYZ as a guest molecule deviates from the series because PYZ can potentially form a N–H⋯N hydrogen bond, while the other guests from the series are O-acceptors of hydrogen bonding. However, the use of this guest allowed us to study the influence of a methanol molecule on the magnetic behaviour of the [Fe(L5)(NCS)] molecule (vide infra), since the preparation of the pure methanol solvate is impossible by direct synthesis in methanol.17 The previously reported [Fe(L5)(NCX)]·CH3CN compounds were not included because they are not isostructural with compounds 1a–1g.
 | ||
| Scheme 1 Schematic representation of the H2L5 ligand. | ||
| Fe–Nam | Fe–NNCX | Fe–Nima | Fe–Oa | α/° | Σ/° | |
|---|---|---|---|---|---|---|
| a The average values calculated from two bond lengths. | ||||||
| 1a | 2.2137(14) | 2.0849(15) | 2.082 | 1.939 | 84.5 | 59.3 |
| 1b | 2.209(2) | 2.085(2) | 2.080 | 1.945 | 85.8 | 61.5 |
| 1c | 2.1937(14) | 2.0908(16) | 2.069 | 1.942 | 84.7 | 56.8 |
| 1d 298 K | 2.183(3) | 2.086(3) | 2.068 | 1.941 | 82.5 | 58.8 |
| 1d 150 K | 2.0087(16) | 1.9447(17) | 1.925 | 1.884 | 84.6 | 25.5 |
| 1d′ 308 K | 2.173(3) | 2.096(4) | 2.064 | 1.935 | 82.0 | 56.6 |
| 1d′ 150 K | 2.003(4) | 1.942(4) | 1.927 | 1.884 | 83.3 | 25.3 |
| 1e | 2.185(2) | 2.077(2) | 2.077 | 1.940 | 87.5 | 56.1 |
| 1f Fe1 | 2.218(3) | 2.094(3) | 2.080 | 1.940 | 84.3 | 65.7 |
| 1f Fe2 | 2.210(3) | 2.082(3) | 2.078 | 1.944 | 81.0 | 56.6 |
| 1g | 2.205(2) | 2.088(2) | 2.077 | 1.941 | 83.2 | 60.0 |
| 1a | 1b | 1c | 1d 150 K | 1d 298 K | 1d′ 150 K | 1d′ 308 K | 1e | |
|---|---|---|---|---|---|---|---|---|
| a R 1 = ∑(|Fo|−|Fc|)/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. | ||||||||
| Formula | C32H33Fe1N4O3S1 | C31H31Fe1N5O3S1 | C32H33Fe1N4O3S1 | C31H32Fe1N5O3S1 | C31H32Fe1N5O3S1 | C31H32Fe1N5O3Se1 | C31H32Fe1N5O3Se1 | C30H31Fe1N4O3S2 |
| M r/g mol−1 | 609.54 | 609.52 | 609.53 | 610.53 | 610.53 | 657.43 | 657.43 | 615.56 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P |
P |
P |
P |
P |
P |
| a | 10.0481(2) | 10.2159(4) | 10.2135(3) | 10.2652(3) | 10.2153(6) | 10.2722(7) | 10.2841(3) | 10.2094(3) |
| b | 10.8818(2) | 10.6166(5) | 10.9173(3) | 11.4487(3) | 11.1783(6) | 11.4128(6) | 11.2785(3) | 11.0128(4) |
| c | 13.1468(2) | 13.0796(5) | 13.0351(3) | 12.7914(4) | 13.2477(6) | 12.8071(9) | 13.2216(4) | 12.9469(4) |
| α | 98.4710(10) | 83.965(4) | 98.967(2) | 106.073(2) | 100.939(4) | 105.151(5) | 101.184(3) | 98.641(3) |
| β | 92.739(2) | 87.370(3) | 93.341(2) | 92.837(2) | 93.011(4) | 93.370(5) | 93.834(3) | 92.784(2) |
| γ | 91.3720(10) | 89.972(4) | 90.673(2) | 97.652(2) | 93.339(4) | 96.060(5) | 92.497(2) | 95.509(3) |
| V | 1419.48(4) | 1409.23(10) | 1432.94(7) | 1425.77(7) | 1479.62(14) | 1435.30(16) | 1498.57(7) | 1429.56(8) |
| T/K | 100(2) | 100(2) | 100(2) | 150(2) | 298(2) | 150(2) | 308(2) | 160(2) |
| ρ calc (g cm−1) | 1.426 | 1.436 | 1.413 | 1.422 | 1.370 | 1.521 | 1.457 | 1.430 |
| μ (mm−1) | 0.639 | 0.652 | 0.640 | 0.644 | 0.621 | 1.835 | 1.758 | 0.713 |
| F(000) | 638 | 636 | 638 | 638 | 638 | 674 | 674 | 642 |
| Goodness of fit | 1.066 | 0.954 | 1.054 | 0.947 | 1.030 | 0.909 | 0.913 | 1.075 |
| Data/restraints/param. | 4970/0/370 | 4943/0/371 | 5027/3/405 | 5015/0/370 | 5205/0/360 | 5026/0/372 | 5271/2/360 | 5009/0/363 |
| r int/rσ | 0.0161/0.0204 | 0.0315/0.0553 | 0.0179/0.0218 | 0.0240/0.0314 | 0.0262/0.0428 | 0.0362/0.0658 | 0.0324/0.0593 | 0.0232/0.0321 |
R
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a/wR2b (all data) a/wR2b (all data) |
0.0345/0.0792 | 0.0620/0.0976 | 0.0367/0.0814 | 0.0438/0.0895 | 0.0802/0.1500 | 0.0839/0.1588 | 0.0855/0.1201 | 0.0533/0.1195 |
|
R
1a/wR2b (I > 2σ(I)) |
0.0286/0.0776 | 0.0396/0.0929 | 0.0298/0.0789 | 0.0328/0.0931 | 0.0522/0.1416 | 0.0616/0.1523 | 0.0467/0.1127 | 0.0412/0.1156 |
| CCDC number | 943181 | 943182 | 943183 | 1030134 | 943184 | 1030135 | 943185 | 1030136 |
The presented series of the [Fe(L5)(NCX)]·Solv compounds is unique due to very similar structural frameworks of constitutive [Fe(L5)(NCX)] molecules (Fig. 1, see ESI, Fig. S1–S7†). Due to only very tiny structural differences (both at the molecular and supramolecular level) between the particular members of the series, the [Fe(L5)(NCX)] framework cannot be responsible for the differences in their magnetic behaviours, but the properties of the Solv molecules must be considered as the main perturbative term responsible for the (non)occurrence and characteristics of SCO.
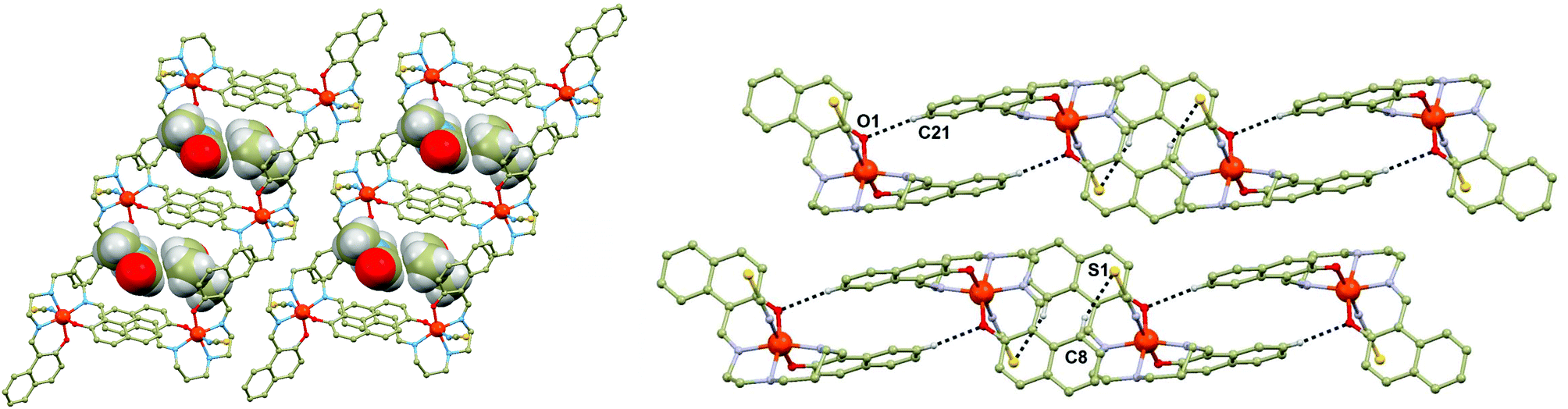 | ||
| Fig. 1 Perspective view of fragments of the crystal structures of the complexes 1a–d, with the guest molecules highlighted in the space-fill model (left). Parts of the crystal structures of the complexes 1a–d, showing selected non-covalent contacts (black dashed lines) (right). Selected hydrogen atoms of the complex molecules were omitted for clarity, except for those involved in the selected non-covalent contacts. The lengths of the non-covalent contacts (in Å): 1a, d(C21⋯O1) = 3.475(2), d(C8⋯S1) = 3.753(2); 1b = d(C21⋯O1) = 3.507(3), d(C8⋯S1) = 3.818(3); 1c, d(C21⋯O1) = 3.623(2), d(C8⋯S1) = 3.826(2); 1d 298 K, d(C21…O1) = 3.546(5), d(C8…S1) = 3.816(4); 1d 150 K, d(C21…O1) = 3.456(2), d(C8…S1) = 3.819(2); 1d′ 308 K, d(C21…O1) = 3.552(5), d(C8…Se1) = 3.914(4); 1d′ 150 K, d(C21…O1) = 3.468(6), d(C8…Se1) = 3.882(5); 1e, d(C21…O1) = 3.448(3), d(C8…S1) = 3.801(3); 1f, Fe1, d(C21…O1) = 3.475(2), d(C8…S1) = 3.754(2); 1f, Fe2, d(C48…O3) = 3.519(3), d(C8…S1) = 3.796(3); 1g, d(C21…O1) = 3.474(3), d(C8…S1) = 3.843(2). | ||
Results
Synthesis and infrared spectroscopy
The synthesis of compounds 1a–1e is uncomplicated and it can be performed in two consecutive steps: (1) the preparation of the [Fe(L5)Cl] precursor complex;15 (2) the reaction between the precursor and potassium pseudohalide in methanol in the presence of the Solv molecules.Infrared spectra of 1a–1e are very similar for all the compounds in the series. The only differences between them originate from the presence of the different guest molecules of crystallization (in cm−1, ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) = 1699 in 1c, 1663 in 1d, 1665 in 1d′, ν(O–H) = 3272 in 1b). The differences in the frequency of the ν(N–H) vibrations were also observed (between 3163 and 3242 cm−1). This should reflect different strengths of N–H⋯N hydrogen bonds within the present series (vide infra), but the obtained results are not very informative (see ESI, Fig. S8†), probably due to the broadness of the observed bands, which prevented us from performing a deeper study.
O) = 1699 in 1c, 1663 in 1d, 1665 in 1d′, ν(O–H) = 3272 in 1b). The differences in the frequency of the ν(N–H) vibrations were also observed (between 3163 and 3242 cm−1). This should reflect different strengths of N–H⋯N hydrogen bonds within the present series (vide infra), but the obtained results are not very informative (see ESI, Fig. S8†), probably due to the broadness of the observed bands, which prevented us from performing a deeper study.
General description of molecular and crystal structures
The molecular structure of the [Fe(L5)(NCX)]-type complexes was well-described in our previous work,15 and therefore it is discussed only briefly and the main attention is focused on the tiny differences in the molecular geometry between the particular members of the 1a–1g series, and in the case of the SCO compounds, on the LS vs. HS molecular structure distinctions. Doubly deprotonated pentadentate ligand L52− provides an N3O2 donor set to the iron(III) central atom and the remaining coordination site is occupied by the nitrogen atom from the anionic NCX− ligand. The pentadentate ligand coordinates to the iron(III) atom through the oxygen atoms in the cis position as is typical for this group of compounds with the propyl-ethyl aliphatic part of the pentadentate ligand.15,18,19 The chromophore bond lengths are listed in Table 1. The presented compounds (1a–e) exhibit very similar metal–ligand bond lengths. The longest bond in the HS state is between the iron atom and amine nitrogen atom (Nam) and the length of this bond adopts values ranging from 2.18 to 2.22 Å. The imino nitrogen atoms (Nim) as well as nitrogen atoms of the NCX ligand form bonds with the iron atom of the approximately same length: d(Fe–Nim), the average value calculated from two bond lengths = 2.07–2.08 Å, d(Fe–NNCX) = 2.08–2.09 Å.The Fe–O bond lengths are the shortest adopting values close to 1.94 Å. The LS structures were determined only for compounds 1d and 1d′. Both compounds possess very similar bond lengths and selected structural parameters (Table 1), which correspond well with the values found for the purely LS cyanido complexes reported previously.15,19,20
The crystal structures of 1a–1g are very similar. They are composed of the [Fe(L5)(NCX)] and Solv molecules which form a three-dimensional framework via C–H⋯O and C–H⋯π weak interactions. The essential part of the framework consists of a centrosymmetric dimer of two adjacent [Fe(L5)(NCX)] molecules interconnected by a rather weak C–H⋯O non-covalent contact between the CH group of the naphthalene ring and the phenolic oxygen atom (Fig. 1). Further small stabilization within the dimer is provided by an offset ring–ring stacking interaction of the naphthalene rings. The interconnections between the adjacent dimeric units, lying in the same layer, are provided by very weak C–H⋯π and C–H⋯S non-covalent contacts. The cavities with the Solv molecules are placed between the dimeric units (Fig. 1).
Each cavity is occupied by two same guest molecules in most of the cases, but two exceptions can be found within the present series: two CH3OH and one PYZ molecules in 1b, MEK and CH3OH in 1f. The Solv guests from the cavity are hydrogen bonded to the amine groups of the [Fe(L5)(NCX)] molecules from the upper, or lower {[Fe(L5)(NCX)]2}n layer. The cavities are of similar size with the volumes ranging from 224.4 Å3 in 1b to 295 Å3 in 1d′. The contraction of the framework induced by SCO is documented also by a change in the cavity size by approximately 13% (1d, 247.3 at 150 K vs. 283.3 Å3 at 298 K) and 16% (1d′, 247.0 Å3 at 150 K vs. 294.8 Å3 at 308 K). Additional stabilization of the Solv molecule is provided by weak contacts such as C–H⋯O and C–H⋯π interactions. In particular, the C–H⋯O contact between the CH group from Solv and the phenolic oxygen atom from the [Fe(L5)(NCX)] molecule is of importance, because together with the N–H⋯O hydrogen bond, these two contacts form ring synthons (Fig. 2): R22(6) in 1b and 1f (CH3OH molecules), R22(8) in 1c, 1f (MEK molecule), 1e and 1g, R22(9) in 1d and 1d′. It must be noted that the aliphatic chain (in most of the cases the C–H⋯O contact provided by the carbon atom C12, Fig. 2) from the L5 ligand also interacts with the solvent oxygen atom. Besides these dominant contacts other weak non-covalent C–H⋯O, C–H⋯S and C–H⋯π interactions can be found in the crystal structures of 1a–g.
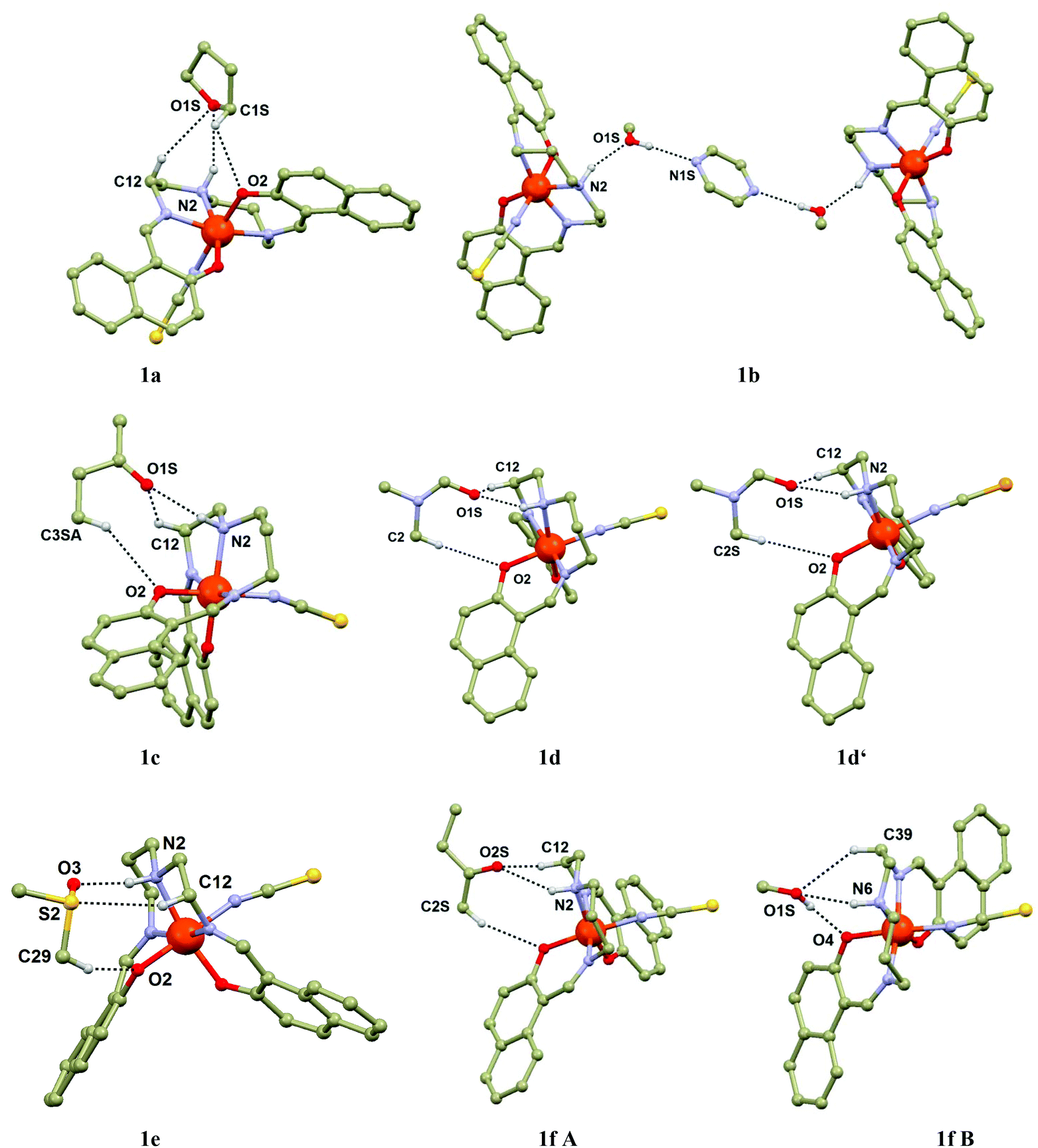 | ||
| Fig. 2 Perspective view of non-covalent interactions of the Solv molecule with [Fe(L5)(NCS)] in 1a–f. Most of hydrogen atoms were omitted for clarity, except for those involved in hydrogen bonding and non-covalent contacts (black dashed lines). The lengths of selected non-covalent contacts (in Å): 1a, d(N2⋯O1S) = 3.295(2), d(C1S⋯O2) = 3.120(2), d(C12⋯O1S) = 3.591(2); 1b, d(N2⋯O1S) = 3.110(3), d(O1S⋯N1S) = 2.839(4); 1c, d(N2⋯O1S) = 2.989(2), d(C3SA⋯O2) = 3.705(6), d(C12⋯O1S) = 3.448(3); 1d 298 K, d(N2⋯O1S) = 2.944(5), d(C2S⋯O2) = 3.965(8), d(C12⋯O1S) = 3.512(6); 1d 150 K, d(N2⋯O1S) = 2.941(2), d(C2S⋯O2) = 3.586(3), d(C12⋯O1S) = 3.576(3); 1d′ 308 K, d(N2⋯O1S) = 2.935(6), d(C3SA⋯O2) = 3.972(9), d(C12⋯O1S) = 3.524(6); 1d′ 150 K, d(N2⋯O1S) = 2.921(6), d(C3SA⋯O2) = 3.623(7), d(C12⋯O1S) = 3.571(6); 1e, d(N2⋯O3) = 3.004(3), d(C29⋯O2) = 3.499(4), d(C12⋯S2) = 3.797(3); 1f, d(N2⋯O2S) = 3.152(4) Å, d(C2S⋯O2) = 3.526(5) Å, d(C12⋯O2S) = 3.357(4) Å, d(O1S⋯O4) = 2.771(3) Å, d(N6⋯O1S) = 3.264(4) Å, d(C39⋯O1S) = 3.493(4) Å. | ||
Magnetic properties
The compounds of the presented series can be divided into two subgroups according to their magnetic behaviour, i.e. purely HS compounds and SCO compounds.The magnetic data for the purely HS compounds, 1a–c, 1f, are shown in ESI, Fig. S9–S11 and Table S1.† The magnetic behaviours observed for 1a–c, 1f and 1g are essentially similar. The effective magnetic moment at room temperature adopts slightly higher values (μeff ≈ 6.0–6.1μB) than the spin-only value calculated for S = 5/2 and g = 2.0 (5.92μB). The μeff values stay almost constant down to 20 K, where the zero-field splitting (ZFS) and/or weak magnetic interactions mediated through the non-covalent interactions start to dominate the magnetic behaviour and this is observed as an abrupt drop in the μeff values to ca. 5μB. The magnetic data were analysed using the following eqn (1),
| Ĥ = D(Ŝz2 − Ŝ2/3) − zj〈Sa〉Ŝa + μBBgiŜi,a | (1) |
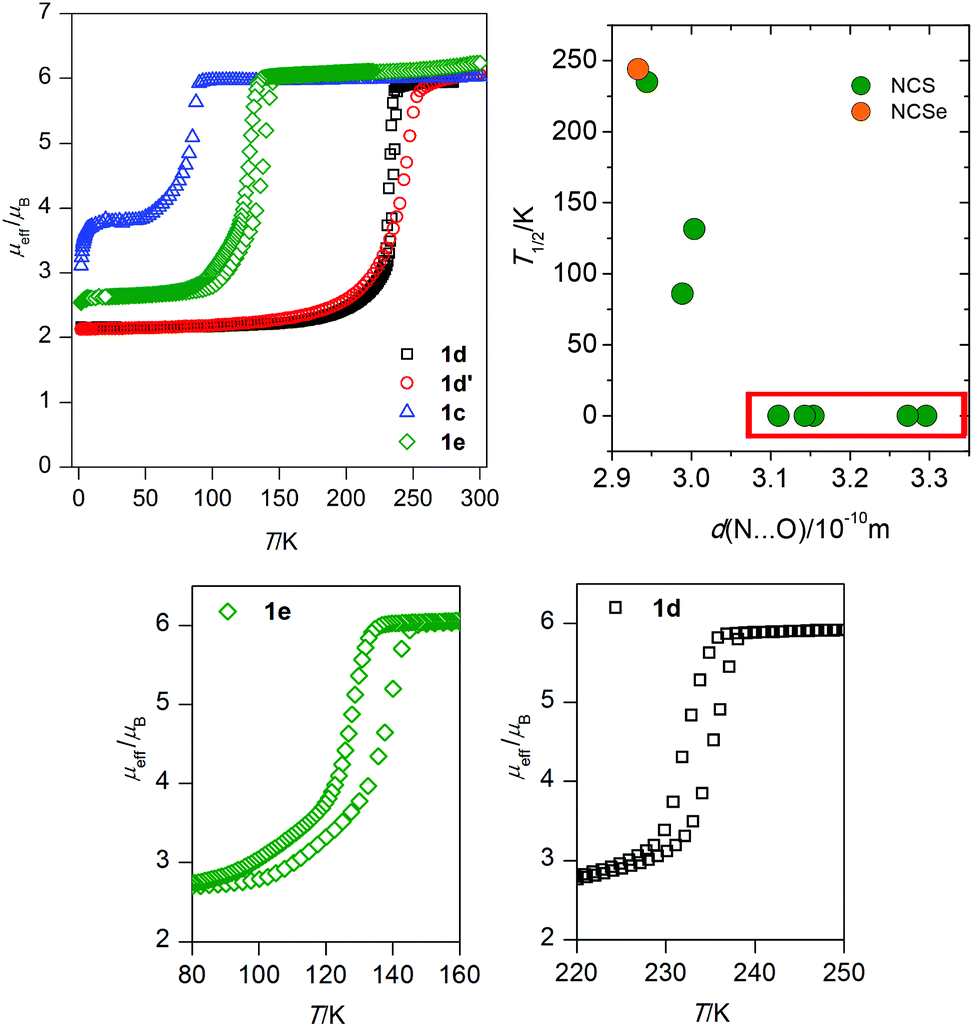 | ||
| Fig. 3 Temperature dependence of effective magnetic moment for compounds 1c, 1d, 1d′ and 1e (top left). Plot of possible T1/2 dependence on the length of the hydrogen bond between the amine group and the acceptor atom from the guest solvent molecule (top left). The T1/2 values for the purely HS compounds were set to zero and highlighted by a red rectangle. Detailed view of the thermal hysteresis in 1e and 1d (below). | ||
Discussion
Magnetostructural relationship
When looking for the reason for such a striking difference in the magnetic behaviours found in this isostructural series, attention must be focused on the role of the guest solvent molecules, which are the major variable within the present series. If one inspects the relationship between the length of the N–H⋯O contact and occurrence of SCO, or even more, its correlation with T1/2, a possible association can be found. The donor–acceptor (D–A) distances of the N–H–O hydrogen bonds are summarized in Table 3, together with the values of T1/2 of the SCO compounds and several basic properties of the guest molecules. It is apparent that SCO occurs only in the compounds with the D–A distance shorter than ca. 3.0 Å and moreover, the compounds with the shortest D–A distance have the highest T1/2 (1d, 1d′). Comparison between the crystal structure and magnetic properties of the compounds 1c and 1f might serve as a good example of such a relationship. Both compounds have the MEK guest molecules and 1f has additional CH3OH molecules in the cavities. In 1f (purely HS) the D–A distance is 3.154(4) Å, while this distance is significantly shorter in 1c (SCO, Fig. 3): 2.988(2) Å. From the magnetic and structural data presented for compounds 1a–1g, the relationship between T1/2 and hydrogen bonding might seem to be straightforward. However, as is well established, the T1/2 value is thermodynamically defined as the ratio of the enthalpy and entropy of SCO. In particular, the entropy originates in the molecular and lattice vibrations22 which cannot be purposely modulated but can be easily influenced by chemical modifications (such as ligand substitution, different lattice solvent etc.).2 Therefore, the rational tuning of the SCO phenomenon is always a challenging task which involves simultaneous changes of entropy and enthalpy.| d(N⋯O)/Å | T 1/2/K |
V
solva/Å3 |
ε
rb |
|
|---|---|---|---|---|
| a Molecular volumes calculated by the Molinspiration program predictions.28 b According to ref. 29. | ||||
| 1a | 3.295(2) | HS | 78.0 | 7.5 |
| 1b | 3.110(3) | HS | 37.2 | 33.0 |
| 1c | 2.988(2) | 84 | 81.5 | 18.6 |
| 1d 308 K | 2.941(2) | 232↓ 235↑ | 77.5 | 38.3 |
| 1d 150 K | 2.944(4) | |||
| 1d′ 308 K | 2.935(5) | 244 | 77.5 | 38.3 |
| 1d′ 150 K | 2.921(6) | |||
| 1e | 3.004(3) | 127↓ 138↑ | 71.4 | 47.2 |
| 1f Fe1 | 3.272(4) CH3OH | HS | 37.2 | 33.0 |
| 1f Fe2 | 3.154(4) MEK | 81.5 | 18.6 | |
| 1g | 3.143(3) | HS | 64.7 | 21.0 |
For example, any prediction fails in the case of the isostructural series, where the SCO complexes [Fe(pa)3]Cl2·Solv (pa = 2-picolylamine)5c have very similar surroundings of the [Fe(pa)3]2+ cation consisting of relatively strong N–H⋯Cl hydrogen bonds, and different Solv guests are involved in the supramolecular system only by weak non-covalent contacts. However, the difference between the aforementioned system and the present series must be emphasized. In contrast to the [Fe(pa)3]Cl2·Solv series, the 1a–1g compounds have a complex framework built by weak non-covalent contacts and the strongest contact is formed between the solvent molecule and the amine group, which is directly involved in the coordination of the metal centre. Therefore, a dominant role of this contact in influencing the ligand field strength might be expectable due to the charge transfer of the electron density from the acceptor to hydrogen bonding donor upon formation of the hydrogen bond.23
Finally, it should be pointed out that each solvent molecule has different intrinsic properties such as electron density distribution, vibrational states, basicity, polarity etc., which might affect the SCO behaviour or Solv⋯SCO complex interaction. Therefore, the accurate T1/2 prediction based only on the D⋯A distance is not expectable and this can be also documented within the 1a–g series in which 1e has a longer D–A distance but a higher T1/2 than 1c (Table 3).
Abruptness of SCO
We proposed previously,15 inspired by findings reported by Halcrow et al.,24 that the dihedral angle between the least-square planes of the aromatic rings (α)25 could be a valuable structural parameter to characterize the cooperativeness of the spin transition in this group of compounds. By comparison of the LS and HS values, one can expect larger differences (Δα) for the compounds with more abrupt transitions. In the case of 1d and 1d′, a larger α(LS) – α(HS) difference is found for 1d (Δα = 2.1°) than for 1d′ (Δα = 1.3°) and from Fig. 3 it is apparent that the spin transition is indeed more abrupt for 1d. Another parameter (δxyz) which might be useful in the characterization of the structural changes observed upon spin transition can be defined as a sum of the absolute values of LS and HS coordinate differences (xi, yi, zi, only non-hydrogen atoms) divided by the number of the atoms (N) involved in the calculation: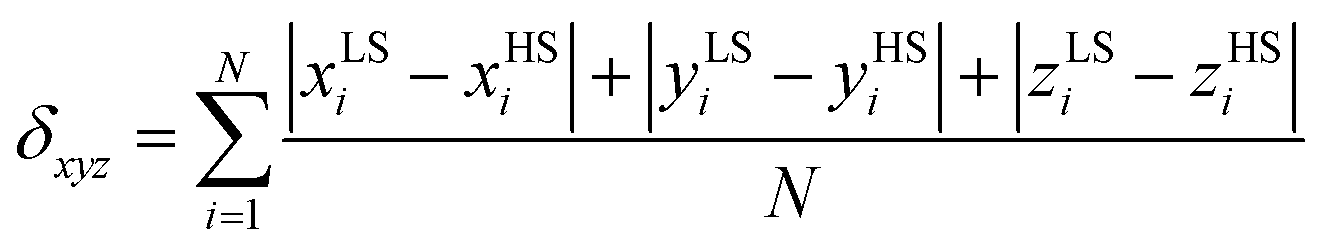 | (2) |
This parameter literally measures the difference between the LS and HS crystal structure of the SCO compound. It could be expected that the compounds exhibiting cooperative SCO behaviours might have changes in the crystal structure that are more pronounced (and the δxyz values higher) than those exhibiting gradual transitions. This can be understood on the basis of the elastic interaction model developed by Spiering et al.26 where the cooperativity is defined as the interaction between the LS and HS species in the SCO solids, which are expected to be stronger for molecules capable of forming a “larger” point defect in the crystal lattice upon spin transition. Furthermore, it can be expected that the use of this parameter should be valid for the compounds consisting of the SCO molecules with a similar second coordination sphere, i.e. with non-covalent interactions of similar strength, which is, as mentioned above, the case of the herein studied series of compounds. The LS and HS structures are available for two compounds in this work: 1d and 1d′. The δxyz values calculated for these compounds differ significantly: 0.0526 (1d) and 0.0463 (1d′). Only one SCO compound with determined LS and HS structures belonging to the group of [Fe(L5)(NCX)] compounds was reported previously: [Fe(L5)(NCSe)]·CH3CN.15 The δxyz value calculated for this compound is equal to 0.0300. Again, as in the case of the α parameter, the largest δxyz value is found for the most abrupt transition in 1d. In order to compare these values properly, it is necessary to define the abruptness of SCO. This can be done by utilizing a modification of the previously published SCO smoothness equation:27TS = T(xHS = 0.9) − T(xHS = 0.1): TS(1d) = 24 K, TS(1d′) = 45 K and TS([Fe(L5)(NCSe)]·CH3CN) = 76 K. The Ts values found for 1d, 1d′ and [Fe(L5)(NCSe)]·CH3CN agree with expected changes of abruptness in above mentioned compounds.
Theoretical insight into intermolecular interactions
With the aim of quantitatively analysing the impact of the N–H⋯O contact on the SCO behaviour, or more precisely, on critical temperature T1/2, we analysed the bonding properties between [Fe(L5)(NCX)] and the solvent molecule in {[Fe(L5)(NCX)]⋯Solv} moieties. All the calculations were based on geometries following from the experimental X-ray structures, but all the hydrogen atom positions were optimized using the B3LYP functional30 together with the atom-pairwise dispersion correction to the DFT energy with Becke–Johnson damping (D3BJ)31 using ORCA 3.0.1.32 The polarized triple-ζ-quality basis set, def2-TZVP(-f), was used for iron, nitrogen, sulphur and selenium atoms, while the def2-SVP basis set was used for carbon and hydrogen atoms.33 The calculations also utilized the RI approximation with the decontracted auxiliary def2-TZV/J and def2-SVP/J Coulomb fitting basis sets34 and the chain-of-spheres (RIJCOSX) approximation to exact exchange35 as implemented in ORCA.First, the non-covalent interaction (NCI) index was utilized to visualize both attractive (hydrogen bonding, van der Waals) and repulsive (steric) interactions based on the properties of the electron density using program NCIPLOT.36 The method is based on the analysis of the reduced gradient of density s, defined as
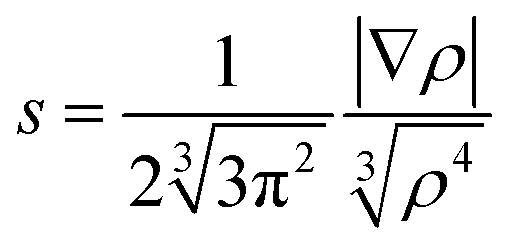 | (3) |
In order to accomplish our intention, the NCIPLOT was used to calculate the NCI index for all available {[Fe(L5)(NCX)]⋯Solv} moieties 1a–g, utilizing their HS experimental single-crystal X-ray structures with optimized hydrogen atom positions. The keyword LIGAND was used to address only NCI between the complex and the solvent requiring the use of promolecular densities. The results are divided into two groups, purely HS compounds (1a, 1b, 1f, 1g) and SCO compounds (1c–e) – Fig. 4. As an example of NCI calculation, the molecular structure of {[Fe(L5)(NCS)]⋯DMF} (1d) is shown in Fig. 4c, together with NCI isosurfaces coloured according to the nature and strength of these interactions showing the N–H⋯O contact (orange-red colour), C–H⋯O and C–H⋯π contacts (green-blue colour) and O-lone-pair⋯O-lone-pair contact (blue colour). This 3D plot of NCI then can be transformed into a 2D plot, in which the above mentioned NCIs are represented by troughs (Fig. 4a and b). The troughs found approximately in the region of −0.015 < sign(λ2)ρ < +0.015 represents weak interactions. In the group of SCO compounds, the strongest and attractive interactions belonging to the N–H⋯O contact are found in the region of −0.027 < sign(λ2)ρ < −0.024 (Fig. 4d). Furthermore, the sign(λ2)ρ values of these troughs, corresponding to the strength of the N–H⋯O contact, nicely correlate with T1/2 (Fig. 5).
 | ||
| Fig. 4 Left: (a) non-covalent interaction (NCI) analysis of SCO compounds (1c–e). (b) NCI analysis of HS compounds (1a, 1b, 1f, 1g). Right: (c) Plot of the NCI isosurface (s = 0.3) coloured according to the RGB scheme over the range of −0.03 < sign(λ2)ρ < 0.01 for {[Fe(L5)(NCS)]⋯DMF} (1d). Red indicates strong attraction; green indicates very weak interaction and blue indicates weak repulsion. (d) NCI analysis of SCO compounds (1c–e) zoomed to the N–H⋯O contact region. | ||
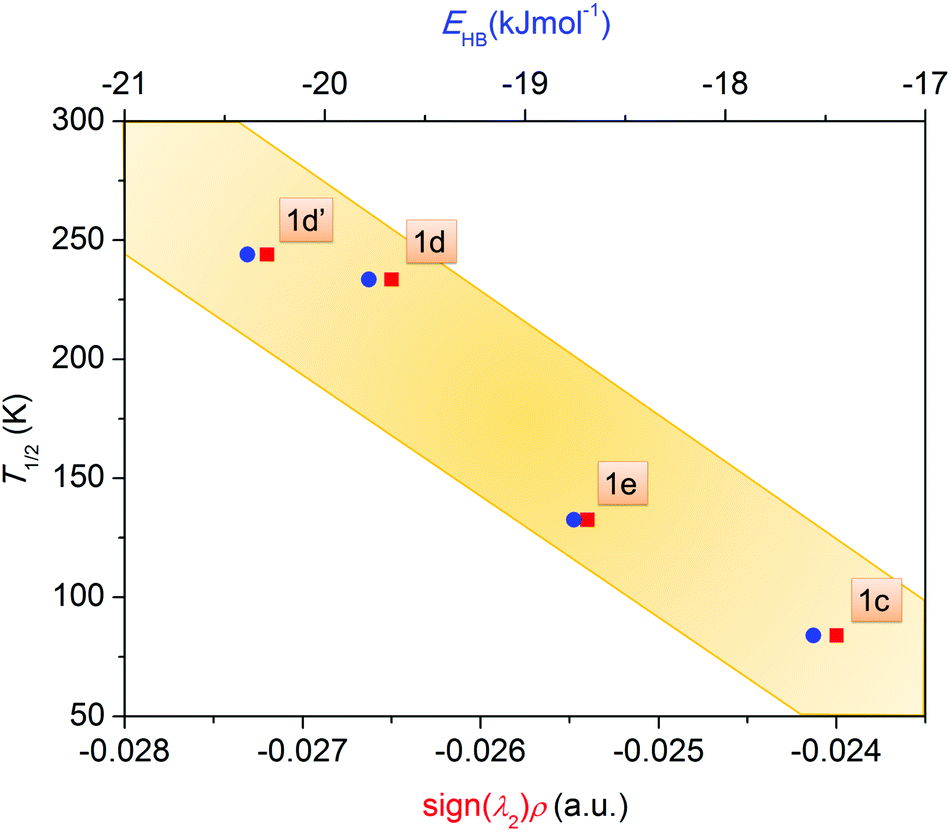 | ||
| Fig. 5 The plot of spin transition temperature T1/2 of SCO compounds (1c–e) as a function of strength of N–H⋯O contacts quantified either by sign(λ2)ρ through NCI analysis (red squares) or by the potential energy density V(r) calculated at BCP points (EHB = V(r)/2) (blue circles). | ||
The indispensable role of this N–H⋯O contact is further demonstrated in Fig. 4b for purely HS compounds, in which there are no troughs in the region of −0.027 < sign(λ2)ρ < −0.024, which means that there are no N–H⋯O contacts that are strong enough. We can only observe the strong and attractive O–H⋯O contact in 1f between MeOH and phenolic oxygen atoms. This contact should act in an opposite direction (than the N–H⋯O contact) in charge transfer due to hydrogen bonding because the solvent molecule acts as a hydrogen bond donor. The strongest N–H⋯O contact is found in 1b, sign(λ2)ρ = −0.020, but evidently the strength of this contact is not sufficient to induce SCO behaviour.
To push further our effort to substantiate the role of the N–H⋯O contact in SCO behaviour within the presented series of complexes, we used another approach based on topological analysis using the total molecular electronic density ρ(r) and the Laplacian of ρ(r) (∇2ρ(r)) based on atoms in molecule (AIM) calculations.37 The single point energy DFT calculations on {[Fe(L5)(NCX)]⋯Solv} moieties resulted in the geometry-basis-wavefunction (GBW) files, which were then transformed to the MOLDEN format and analysed using a program Multiwfn 3.3.5 (A Multifunctional Wavefunction Analyzer).38 The so-called bond critical points (BCP) of the type (3,−1) were located in the N–H⋯O contacts and in these points, the potential energy density V(r) was calculated, because it was shown that the energy of hydrogen bonds (EHB) can be approximated as EHB = V(r)/2.39 The results depicted in Fig. 5 (right) proved that the energy of the N–H⋯O hydrogen bonds within the series of SCO compounds correlates with T1/2, showing that the stronger the N–H⋯O hydrogen bond in the HS X-ray structure is formed, the higher T1/2 is observed.
To summarize, both theoretical approaches based on ab initio calculations identified the N–H⋯O hydrogen bond strength (energy) as the key driving force for observing SCO in the isostructural series of the [Fe(L5)(NCX)]·Solv complexes.
Conclusions
In this article the crystal structures and magnetic properties of the compounds belonging to an isostructural series of iron(III) Schiff base complexes were reported. From the magnetic and structural data, a possible relationship between the occurrence of SCO (and its critical temperature) and the strength of the hydrogen bonding between the guest solvent molecules and the amine group from the [Fe(L5)(NCX)] molecules was outlined. Remarkably, there is no apparent correlation of the SCO behaviour with the other parameters such as the size of the host cavity, the guest volume or its dielectric constant (Table 3), and the guest molecule substitution affects T1/2 much more significantly than the substitution in the monodentate NCX ligand, as was documented on compounds 1d and 1d′. The N–H⋯O hydrogen bonding in 1a–f compounds was studied by combined DFT and Bader charge analysis calculations in greater detail, and these calculations identified a correlation between the strength of the N–H⋯O hydrogen bond and T1/2 in the SCO compounds of this series. More concretely, the stronger N–H⋯O hydrogen bond (i.e. the shorter distance) implies a higher value of T1/2. This hypothesis can be supported also by previous work where a similar relationship was observed for solution studies of anion binding Fe(II) SCO cations.40 However, further research involving a preparation of the [Fe(L5)(NCX)]·Solv compounds with a different type of the guest Solv molecules is inevitable in order to prove this hypothesis.Furthermore, the group of [Fe(L5)(NCX)]·Solv compounds is interesting not only for the above mentioned relationship in the host–guest system. These compounds are promising also due to the capability of the host [Fe(L5)(NCX)] framework to propagate cooperative interactions resulting in the occurrence of abrupt SCO with thermal hysteresis (1d, ΔT = 3 K, 1e, ΔT = 11 K). This, along with the possibility of exploiting the Solv guests as T1/2 tuners, gives an opportunity to prepare cooperative Fe(III) SCO compounds with T1/2 at ambient temperature.
Experimental
Synthesis
All reagents and solvents were purchased from commercial sources (Sigma Aldrich, Acros Organics) and used as received.The complex 1a was prepared by the reaction of [Fe(L5)Cl]17 (100 mg, 0.186 mmol) with KNCS (20 mg, 0.206 mmol) in tetrahydrofuran (10 cm3) and methanol (20 cm3). This solution was stirred and heated for 10 minutes. Then it was filtered through a paper filter and left to crystallize for several days. Yield (based on [Fe(L5)Cl]) = 74%.
The complex 1b was prepared by the reaction of [Fe(L5)Cl] (100 mg) with KNCS (20 mg, 0.206 mmol) in methanol (25 cm3) and 4 g of pyrazine was added afterwards. This solution was stirred and heated for 10 minutes. Then it was filtered through a paper filter and left to crystallize for several days. Yield (based on [Fe(L5)Cl]) = 12%.
The complexes 1c, 1d, 1d′ were prepared in the very same way. The precursor complex [Fe(L5)Cl] (100 mg, 0.186 mmol) was reacted with KNCS/Se (20 (S) or 30 (Se) mg, 0.206 mmol) in 20 cm3 of methanol and X ml of Solv (X = 10 cm3 MEK (1c), X = 3 cm3 DMF (1d, 1d′) and the resulting mixture was stirred and heated for 10 minutes. Then it was filtered through a paper filter and left to crystallize for several days. In the case of 1d and 1d′ the suitable single-crystals were obtained by slow diffusion of diethyl ether to the solution. Yields (based on [Fe(L5)Cl]) = 67% for 1c, 35% for 1d, 24% for 1d′.
The complex 1e was prepared by the reaction of [Fe(L5)Cl] (100 mg, 0.186 mmol) and KNCS (20 mg, 0.206 mmol) in the mixture of CH3OH and DMSO (5 cm3). This solution was stirred and heated for 10 minutes. Then it was filtered through a paper filter and the solution volume was reduced by nitrogen gas flow. When the first microcrystals appeared, the solution was left to crystallize for several days at −18 °C. Yield (based on [Fe(L5)Cl]) = 25%.
All prepared samples are of dark violet colour when ground. In liquid nitrogen, the samples exhibiting SCO turned green as reported previously for this group of compounds.41 Elemental analysis: 1a, Mr = 609.54, C32H33Fe1N4O3S1, Found: C, 62.9; H, 5.5; N, 9.1, requires C, 63.1; H, 5.5; N, 9.2%, IR mid (in cm−1): ν(N–H) = 3240 (w), ν(C–H)aromatic = 3039 (w), ν(C–H)aliphatic = 2974, 2926, 2871 (m), ν(NCS) = 2061 (vs), ν(CN) and ν(CC) = 1603, 1536, 1506 (vs); 1b, Mr = 609.52, C31H31Fe1N5O3S1, Found: C, 61.3; H, 5.3; N, 11.4, requires C, 61.1; H, 5.1; N, 11.5%, IR mid (in cm−1): ν(O–H) = 3272 (w), ν(N–H) = 3204 (w), ν(C–H)aromatic = 3049 (w), ν(C–H)aliphatic = 2973, 2926, 2865 (m), ν(NCS) = 2053 (vs), ν(CN) and ν(CC) = 1602, 1538, 1506 (vs); 1c, Mr = 609.53, C32H33Fe1N4O3S1, Found: C, 62.9; H, 5.4; N, 9.2, requires C, 63.1; H, 5.5; N, 9.2%, IR mid (in cm−1): ν(N–H) = 3242 (w), ν(C–H)aromatic = 3050 (w), ν(C–H)aliphatic = 2973, 2924, 2871 (m), ν(NCS) = 2057 (vs), ν(CO) = 1699, ν(CN) and ν(CC) = 1603, 1537, 1505 (vs); 1d, Mr = 610.53, C31H32Fe1N5O3S1, Found: C, 61.1; H, 5.2; N, 11.4, requires C, 61.0; H, 5.3; N, 11.5%, IR mid (in cm−1): ν(N–H) = 3166 (w), ν(C–H)aromatic = 3049 (w), ν(C–H)aliphatic = 2967, 2924, 2861 (m), ν(NCS) = 2058 (vs), ν(CO) = 1663, ν(CN) and ν(CC) = 1605, 1539, 1506 (vs); 1d′Mr = 657.42, C31H32Fe1N5O3Se1, Found: C, 56.7; H, 5.0; N, 10.5, requires C, 56.6; H, 4.9; N, 10.7%, IR mid (in cm−1): ν(N–H) = 3163 (w), ν(C–H)aromatic = 3046 (w), ν(C–H)aliphatic = 2967, 2926, 2859 (m), ν(NCSe) = 2060 (vs), ν(CO) = 1665, ν(CN) and ν(CC) = 1602, 1536, 1506 (vs); 1e, Mr = 615.57, C30H31Fe1N4O3S2, Found: C, 58.6; H, 5.1; N, 9.3, requires C, 58.5; H, 5.1; N, 9.1%, IR mid (in cm−1): ν(N–H) = 3167 (w), ν(C–H)aromatic = 3049 (w), ν(C–H)aliphatic = 2969, 2925, 2867 (m), ν(NCS) = 2060 (vs), ν(CN) and ν(CC) = 1603, 1537, 1506 (vs).
Equipment, measurements and software
Elemental analysis (CHN) was performed on a FLASH 2000 CHNS Analyzer (ThermoFisher Scientific). Magnetic data were measured on powdered samples pressed into the pellets using an MPMS XL-7 Quantum Design SQUID magnetometer. The experimental data were corrected for the diamagnetism of the constituent atoms using Pascal's constants.Magnetic susceptibility and magnetization measurements were done using a SQUID magnetometer (MPMS, Quantum Design) from T = 2 K at B = 0.1 T. The magnetization data were taken at T = 2.0 and 4.6 K. The effective magnetic moment was calculated as usual: μeff/μB = 798(χ′T)1/2 when SI units are employed. Analysis of magnetic data was done with the package POLYMAGNET.42 The visualization of non-covalent interactions based on NCIPLOT calculation was done with the help of a VMD program.43
Single crystal X-ray diffraction data were collected using an Oxford diffraction Xcalibur2 CCD diffractometer with a Sapphire CCD detector installed in a fine-focus sealed tube (Mo-Kα radiation, λ = 0.71073 Å) and equipped with Oxford Cryosystems nitrogen gas-flow apparatus. All structures were solved by direct methods using SHELXS9744 and SIR-9245 incorporated into the WinGX program package.46 For each structure, its space group was checked by the ADSYMM procedure of the PLATON47 software. All structures were refined using full-matrix least-squares on Fo2 − Fc2 with SHELXTL-9744 with anisotropic displacement parameters for non-hydrogen atoms. All the hydrogen atoms were found in differential Fourier maps and their parameters were refined using a riding model with Uiso(H) = 1.2 or 1.5 Ueq (atom of attachment). All the crystal structures were visualized using the Mercury software.48
Non-routine aspects of the structure refinement are as follows: some parts (carbon atoms) of the solvent molecules in the compounds 1c, 1d and 1d′ are disordered over two positions: 1c, C1SA/B, C2SA/B, C3SA/B, C4SA/B (occupancy factors, A:B = 0.51:0.49).
Acknowledgements
We acknowledge the financial support from the Grant Agency of the Czech Republic (GAČR P207/11/0841) and the National Programme of Sustainability I (LO1305) of the Ministry of Education, Youth and Sports of the Czech Republic. We also would like to thank Mr Pavel Zoufalý for the help with the preparation of compound 1e.Notes and references
- (a) J. F. Letard, P. Guionneau and L. Goux-Capes, Spin Crossover in Transition Metal Compounds III, 2004, vol. 235, pp. 221–251 Search PubMed; (b) Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Ltd Search PubMed.
- P. Gutlich and H. A. Goodwin, Spin Crossover in Transition Metal Compounds I, 2004, vol. 233, pp. 1–47 Search PubMed.
- W. Kosaka, H. Tokoro, T. Matsuda, K. Hashimoto and S. Ohkoshi, J. Phys. Chem. C, 2009, 113, 15751–15755 CAS.
- (a) L. Salmon, A. Bousseksou, B. Donnadieu and J.-P. Tuchagues, Inorg. Chem., 2005, 44, 1763–1773 CrossRef CAS PubMed; (b) M. Kepenekian, B. Le Guennic and V. Robert, J. Am. Chem. Soc., 2009, 131, 11498–11502 CrossRef CAS PubMed; (c) B. Weber, E. S. Kaps, J. Obel, K. Achterhold and F. G. Parak, Inorg. Chem., 2008, 47, 10779–10787 CrossRef CAS PubMed; (d) A. Tissot, R. Bertoni, E. Collet, L. Toupet and M.-L. Boillot, J. Mater. Chem., 2011, 21, 18347–18353 RSC.
- (a) J. M. Holland, J. A. McAllister, C. A. Kilner, M. Thornton-Pett, A. J. Bridgeman and M. A. Halcrow, Dalton Trans., 2002, 548–554 RSC; (b) F. J. Munoz Lara, A. B. Gaspar, D. Aravena, E. Ruiz, M. C. Munoz, M. Ohba, R. Ohtani, S. Kitagawa and J. A. Real, Chem. Commun., 2012, 48, 4686–4688 RSC; (c) M. Hostettler, K. W. Tornroos, D. Chernyshov, B. Vangdal and H. B. Burgi, Angew. Chem., Int. Ed., 2004, 43, 4589–4594 CrossRef CAS PubMed.
- M. Clemente-León, E. Coronado, M. C. Giménez-López and F. M. Romero, Inorg. Chem., 2007, 46, 11266–11276 CrossRef PubMed.
- (a) I. Šalitroš, J. Pavlík, R. Boča, O. Fuhr, C. Rajadurai and M. Ruben, CrystEngComm, 2010, 12, 2361–2368 RSC; (b) T. D. Roberts, F. Tuna, T. L. Malkin, C. A. Kilner and M. A. Halcrow, Chem. Sci., 2012, 3, 349–354 RSC; (c) A. Bhattacharjee, V. Ksenofontov, K. H. Sugiyarto, H. A. Goodwin and P. Gutlich, Adv. Funct. Mater., 2003, 13, 877–882 CrossRef CAS.
- (a) R. J. Wei, J. Tao, R. B. Huang and L. S. Zheng, Inorg. Chem., 2011, 50, 8553–8564 CrossRef CAS PubMed; (b) B. Li, R. J. Wei, J. Tao, R. B. Huang, L. S. Zheng and Z. P. Zheng, J. Am. Chem. Soc., 2010, 132, 1558–1566 CrossRef CAS PubMed; (c) N. Wannarit, N. Nassirinia, S. Amani, N. Masciocchi, S. Youngme, O. Roubeau, S. J. Teat and P. Gamez, Inorg. Chem., 2014, 53, 9827–9836 CrossRef CAS PubMed; (d) J. S. Costa, S. Rodríguez-Jiménez, G. A. Craig, B. Barth, C. M. Beavers, S. J. Teat and G. Aromí, J. Am. Chem. Soc., 2014, 136, 3869–3874 CrossRef CAS PubMed.
- G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762–1765 CrossRef CAS PubMed.
- see e.g.: (a) Y.-C. Chuang, C.-T. Liu, C.-F. Sheu, W.-L. Ho, G.-H. Lee, C.-C. Wang and Y. Wang, Inorg. Chem., 2012, 51, 4663–4671 CrossRef CAS PubMed; (b) V. Niel, A. L. Thompson, M. C. Muñoz, A. Galet, A. E. Goeta and J. A. Real, Angew. Chem., Int. Ed., 2003, 42, 3760–3763 CrossRef CAS PubMed; (c) X.-Y. Chen, R.-B. Huang, L.-S. Zheng and J. Tao, Inorg. Chem., 2014, 53, 5246–5252 CrossRef CAS PubMed.
- P. D. Southon, L. Liu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 CrossRef CAS PubMed.
- (a) M. Bartel, A. Absmeier, G. N. L. Jameson, F. Werner, K. Kato, M. Takata, R. Boča, M. Hasegawa, K. Mereiter, A. Caneschi and W. Linert, Inorg. Chem., 2007, 46, 4220–4229 CrossRef CAS PubMed; (b) J.-B. Lin, W. Xue, B.-Y. Wang, J. Tao, W.-X. Zhang, J.-P. Zhang and X.-M. Chen, Inorg. Chem., 2012, 51, 9423–9430 CrossRef CAS PubMed.
- S. M. Neville, G. J. Halder, K. W. Chapman, M. B. Duriska, B. Moubaraki, K. S. Murray and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 12106–12108 CrossRef CAS PubMed.
- see e.g. (a) M. D. Timken, C. E. Strouse, M. S. Soltis, S. A. Daverio, D. N. Hendrickson, A. M. Abdel-Mawgoud and S. R. Wilson, J. Am. Chem. Soc., 1986, 108, 395 CrossRef CAS PubMed; (b) D. J. Harding, D. Sertphon, P. Harding, K. S. Murray, B. Moubaraki, J. D. Cashion and H. Adams, Chem. – Eur. J., 2013, 19, 1082–1090 CrossRef CAS PubMed; (c) J. C. Dias, B. Vieira, I. C. Santos, L. C. J. Pereira and V. da Gama, Inorg. Chim. Acta, 2009, 362, 2076–2079 CrossRef CAS; (d) Z.-Y. Li, J.-W. Dai, Y. Shiota, K. Yoshizawa, S. Kanegawa and O. Sato, Chem. – Eur. J., 2013, 19, 12948–12952 CrossRef CAS PubMed; (e) S. Hayami, Z.-Z. Gu, H. Yoshiki, A. Fujishima and O. Sato, J. Am. Chem. Soc., 2001, 123, 11644–11650 CrossRef CAS PubMed; (f) E. W. T. Yemeli, G. R. Blake, A. P. Douvalis, T. Bakas, G. O. R. Alberda van Ekenstein and P. J. van Koningsbruggen, Chem. – Eur. J., 2010 DOI:10.1002/chem.201002100; (g) S. Hayami, K. Hiki, T. Kawahara, Y. Maeda, D. Urakami, K. Inoue, M. Ohama, S. Kawata and O. Sato, Chem. – Eur. J., 2009, 15, 3497–3508 CrossRef CAS PubMed.
- I. Nemec, R. Herchel, R. Boča, Z. Trávníček, I. Svoboda, H. Fuess and W. Linert, Dalton Trans., 2011, 40, 10090–10099 RSC.
- I. Nemec and Z. Trávníček, Private communication to the Cambridge Structural Database, 2014, deposition number CCDC: 943180 Search PubMed.
- P. Masárová, P. Zoufalý, J. Moncol, I. Nemec, J. Pavlik, M. Gembický, Z. Trávníček, R. Boča and I. Šalitroš, New J. Chem., 2015, 39, 508–519 RSC.
- I. Nemec, R. Boča, M. Gembický, L. Dlháň, R. Herchel and F. Renz, Inorg. Chim. Acta, 2009, 362, 4754–4759 CrossRef CAS.
- C. Krüger, P. Augustín, I. Nemec, Z. Trávníček, H. Oshio, R. Boča and F. Renz, Eur. J. Inorg. Chem., 2013, 902–915 CrossRef.
- I. Šalitroš, R. Boča, L. Dlháň, M. Gembický, J. Kozisek, J. Linares, J. Moncol, I. Nemec, L. Perašinová, F. Renz, I. Svoboda and H. Fuess, Eur. J. Inorg. Chem., 2009, 3141–3154 CrossRef.
- (a) R. Boča, Theoretical Foundations of Molecular Magnetism, Elsevier, Amsterdam, 1999 Search PubMed; (b) R. Boča, Coord. Chem. Rev., 2004, 248, 757–815 CrossRef.
- M. Sorai and S. Seki, J. Phys. Chem. Solids, 1974, 35, 555–570 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford university Press, 1997 Search PubMed.
- M. A. Halcrow, Structure:Function Relationships in Molecular Spin-Crossover Materials 147 in Spin-Crossover Materials: Properties and Applications, ed. M. A. Halcrow, John Wiley & Sons, Ltd., 1st edn, 2013 Search PubMed.
- R. Pritchard, S. A. Barrett, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2008, 3159–3168 RSC.
- N. Willenbacher and H. Spiering, J. Phys. C: Solid State Phys., 1988, 21, 1423–1439 CrossRef.
- P. Guionneau, M. Marchivie, G. Bravic, J. F. Letard and D. Chasseau, Spin Crossover in Transition Metal Compounds II, 2004, vol. 234, pp. 97–128 Search PubMed.
- Molinspiration program predictions, Molinspiration Cheminformatics, 2012.
- D. R. Lide, CRC Handbook of Chemistry and Physics, CRC Press, 85th edn, 2004 Search PubMed.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS; (b) A. D. Becke, Chem. Phys., 1993, 98, 1372–1377 CAS; (c) A. D. Becke, Chem. Phys., 1993, 98, 5648–5652 CAS; (d) P. J. Stephens, F. K. Devlin, C. F. Chabalowski and M. J. Frisch, Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- (a) S. Grimme, J. Antony, S. Ehrlich and H. J. Krieg, Chem. Phys., 2010, 132, 154104 Search PubMed; (b) S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- (a) A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef; (b) A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef; (c) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- (a) K. Eichkorn, O. Treutler, H. Ohm, M. Haser and R. Ahlrichs, Chem. Phys. Lett., 1995, 240, 283–290 CrossRef CAS; (b) K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- (a) E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed; (b) J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- C. F. Matta, R. J. Boyd and A. Becke, The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design, Wiley-VCH, Weinheim, 2007 Search PubMed.
- T. Lu and F. J. Chen, Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 285, 170–173 CrossRef CAS.
- (a) S. A. Barrett and M. A. Halcrow, RSC Adv., 2014, 4, 11240–11243 RSC; (b) Z. Ni, S. R. Fiedler and M. P. Shores, Dalton Trans., 2011, 40, 944–950 RSC; (c) Z. Ni, A. M. McDaniel and M. P. Shores, Chem. Sci., 2010, 1, 615–621 RSC.
- I. Nemec, R. Boča, R. Herchel, Z. Trávníček, M. Gembický and W. Linert, Monatsh. Chem., 2009, 140, 815–828 CrossRef CAS.
- R. Boča, Program POLYMAGNET, STU Bratislava, 2008 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- (a) A. L. Speck, “PLATON, a multipurpose crystallographic tool”, Utrecht University, Utrecht, The Netherlands, 2001 Search PubMed; (b) Y. Le Page, J. Appl. Crystallogr., 1988, 21, 983–984 CrossRef.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Results of magnetic data analysis for the HS compounds, detailed crystal structure information for compounds 1a–1e. CCDC 943182–943185, 1030134–1030136. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03400g |
| This journal is © The Royal Society of Chemistry 2015 |