
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iridium(III) N-heterocyclic carbene complexes: an experimental and theoretical study of structural, spectroscopic, electrochemical and electrogenerated chemiluminescence properties†
Gregory J.
Barbante
a,
Egan H.
Doeven
a,
Paul S.
Francis
a,
Bradley D.
Stringer
b,
Conor F.
Hogan
*b,
Peyman R.
Kheradmand
b,
David J. D.
Wilson
b and
Peter J.
Barnard
*b
aCentre for Chemistry and Biotechnology, School of Life and Environmental Sciences, Faculty of Science, Engineering and Built Environment, Deakin University, Geelong, Victoria 3216, Australia
bDepartment of Chemistry, La Trobe Institute for Molecular Science, La Trobe University, Melbourne, Victoria 3086, Australia. E-mail: p.barnard@latrobe.edu.au; c.hogan@latrobe.edu.au
First published on 5th February 2015
Abstract
Four cationic heteroleptic iridium(III) complexes have been prepared from methyl- or benzyl-substituted chelating imidazolylidene or benzimidazolylidene ligands using a Ag(I) transmetallation protocol. The synthesised iridium(III) complexes were characterised by elemental analysis, 1H and 13C NMR spectroscopy and the molecular structures for three complexes were determined by single crystal X-ray diffraction. A combined theoretical and experimental investigation into the spectroscopic and electrochemical properties of the series was performed in order to gain understanding into the factors influencing photoluminescence and electrochemiluminescence efficiency for these complexes, with the results compared with those of similar NHC complexes of iridium and ruthenium. The N^C coordination mode in these complexes is thought to stabilise thermally accessible non-emissive states relative to the case with analogous complexes with C^C coordinated NHC ligands, resulting in low quantum yields. As a result of this and the instability of the oxidised and reduced forms of the complexes, the electrogenerated chemiluminescence intensities for the compounds are also low, despite favourable energetics. These studies provide valuable insights into the factors that must be considered when designing new NHC-based luminescent complexes.
Introduction
Providing a versatile synthetic framework and fine control of electronic properties, N-heterocyclic carbenes (NHC) are emerging as a useful option for the development of novel luminescent materials.1–4 In recent years, luminescent complexes of metals such as Cu(I),5,6 Ag(I),7,8 Au(I),7,9–12 Ni(II),13 Pd(II),13,14 Pt(II),13–17 Ir(III),18–21 Ru(II),22–27 Os(II),28 Re(I),29,30 Zr(IV),31 and Hf(IV),31 incorporating NHC ligands have been devised for a wide range of fundamental investigations, photoluminescence applications and light-emitting devices. Similarly, NHC ligands have recently been exploited to manipulate the electrochemical and spectroscopic properties of metal complexes for electrogenerated chemiluminescence (also known as electrochemiluminescence or ECL).26,32,33 In a typical ECL reaction, the complex is electrochemically oxidised before receiving an electron from a strong reductant to generate its electronically excited state.34–42ECL can be separated into two operational modes that are distinguished by the source of the reductant; in annihilation ECL it is derived from electrochemical reduction of the metal complex itself, whereas in co-reactant ECL it is formed by the concurrent oxidation of a sacrificial reagent such as tri-n-propylamine (TPA). While the latter is important for the development highly sensitive analytical applications, the importance of the former lies in the insights that can be gained into the design and operation of light emitting devices (LEDs), in particular light emitting electrochemical cells (LECs). Control of the electronic properties of each ligand of a complex allows tuning of not only the energetics of the reactions required to generate the excited species, but also the emission colour. This is of great interest for the development of mixed metal-complex ECL systems43–46 for multiplexed analysis in the case of co-reactant ECL; or for colour tuneable LEDs in the case of annihilation ECL.
We have previously examined the electrochemical, photoluminescence and ECL properties of four Ru(II) complexes that each contain two bipyridine (bpy) ligands and a pyridine-functionalised imidazoylidene- or benzimidazolylidene-NHC ligand (5–8 in Scheme 1a).32 Compared to the benchmark ECL emitter [Ru(bpy)3]2+, annihilation ECL intensities of the four Ru(II) complexes ranged between 7% and 95%. Moreover, the imidazoylidene-NHC analogues exhibited a bathochromic shift (8–13 nm) due to destabilisation of the metal-based highest occupied molecular orbital (HOMO) with only minor destabilisation of the bpy ligand-based lowest unoccupied molecular orbital (LUMO). In contrast, the complexes containing a benzimidazolylidene-NHC were hypsochromically shifted (7–12 nm) due to HOMO stabilisation (with little or no effect on the LUMO), in which case it was concluded that π-back bonding mitigated the typically strong σ-donation of NHCs. Park and co-workers26 reported a wider spread of emissions (from greenish-yellow to red) in the annihilation and co-reactant ECL of three Ru(II) complexes bearing tridentate NHC ligands (9–11, Scheme 1b), but their ECL intensities were not compared to [Ru(bpy)3]2+.
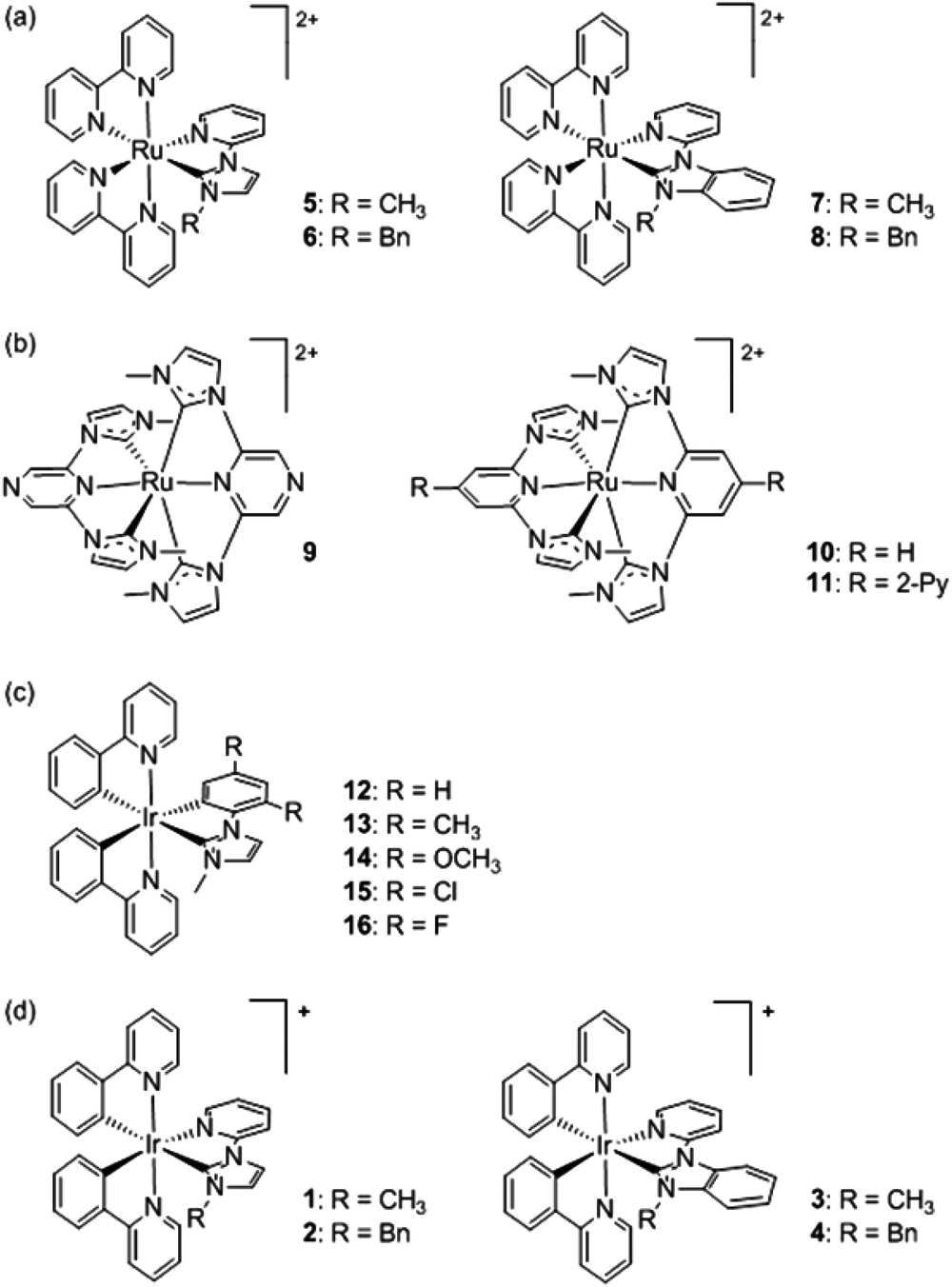 | ||
| Scheme 1 (a–c) Representative Ru(II) and Ir(III) complexes incorporating NHC ligands that have been previously examined for their ECL activity.26,32,33 (d) The Ir(III) complexes examined in this study. | ||
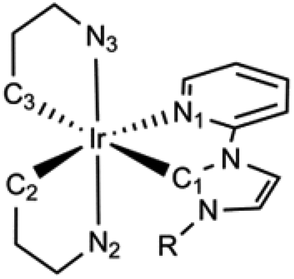
We also recently reported the ECL properties of five Ir(III) complexes that each contained two cyclometalated 2-(phenyl)pyridine (ppy) ligands and a 2,4-disubstituted phenyl-imidazoylidene NHC ligand (12–16, Scheme 1c).33 The addition of electron withdrawing halogen substituents on the phenyl ring of the NHC ligand alone shifted the emission maxima from 532 nm to 524 nm (Cl) or 490 nm (F), which was predominantly attributed to their stabilising inductive effects on the substantially metal-based HOMO, compared to the ppy-based LUMO. In contrast, resonance electron-donation of the methoxy substituents was not significant due to its meta position. Reasonably intense annihilation ECL was observed from each of the Ir(III) complexes, but only the difluoro derivative produced significant ECL via the co-reactant pathway.33 These findings, in conjunction with other ECL investigations,47 led us to propose a plot of oxidation potential versus emission colour as a convenient guide to the energy sufficiency of novel metal complexes for co-reactant ECL. Herein, we report the first examination of ECL with Ir(III) complexes containing a pyridyl-imidazoylidene- or pyridyl-benzimidazolylidene-NHC ligand (1–4, Scheme 1d). This not only provides new insight into the potential application of NHC ligands in ECL systems, but also aptly illustrates the additional factors apart from energetics that must be considered, when using an oxidation potential versus emission wavelength plot, as a predictor of co-reactant ECL ability.
Results and discussion
Synthesis
The desired azolium salts I·I, II·Br, III·I and IV·Br (Scheme 2), were prepared by heating either 1-(2-pyridyl)imidazole or 1-(2-pyridyl)benzimidazole with the appropriate alkyl halide (methyl iodide or benzyl bromide) in CH3CN as has been described previously.32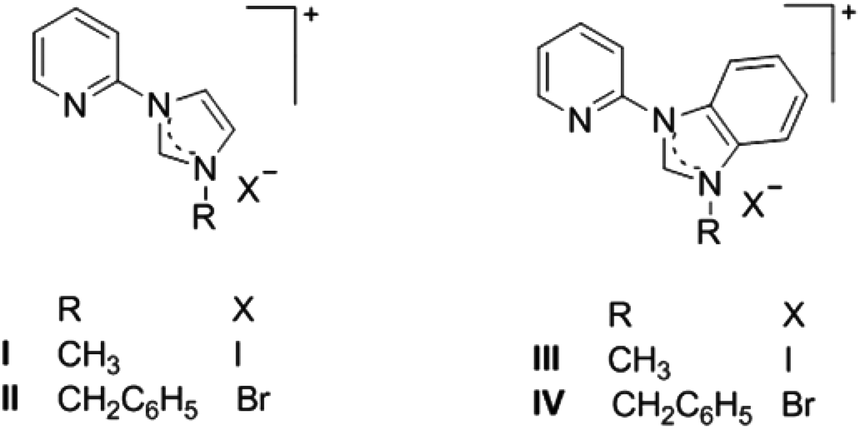 | ||
| Scheme 2 Structures of the azolium salts I·I, II·Br, III·I and IV·Br. | ||
Formation of the [Ir(ppy)2(C^N)]+ complexes (where C^N is a bidentate NHC ligand derived from the azolium salts I·I, II·Br, III·I and IV·Br) was carried out using Ag(I) transmetallation utilising Ag2O and the dinuclear Ir(III) precursor compound, [Ir(ppy)2Cl]2, in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of CH2Cl2 and CH3OH (Scheme 3).18 Initial attempts to synthesise the Ir(III) complexes utilising the hexafluorophosphate salts of the azolium cations, resulted is very low yields (3–8%). In subsequent attempts the halide salts of the azolium cations (I·I, II·Br, III·I and IX·Br) were used and increased yield of the recrystallised Ir(III) complexes were obtained (20–54%).
:1 mixture of CH2Cl2 and CH3OH (Scheme 3).18 Initial attempts to synthesise the Ir(III) complexes utilising the hexafluorophosphate salts of the azolium cations, resulted is very low yields (3–8%). In subsequent attempts the halide salts of the azolium cations (I·I, II·Br, III·I and IX·Br) were used and increased yield of the recrystallised Ir(III) complexes were obtained (20–54%).
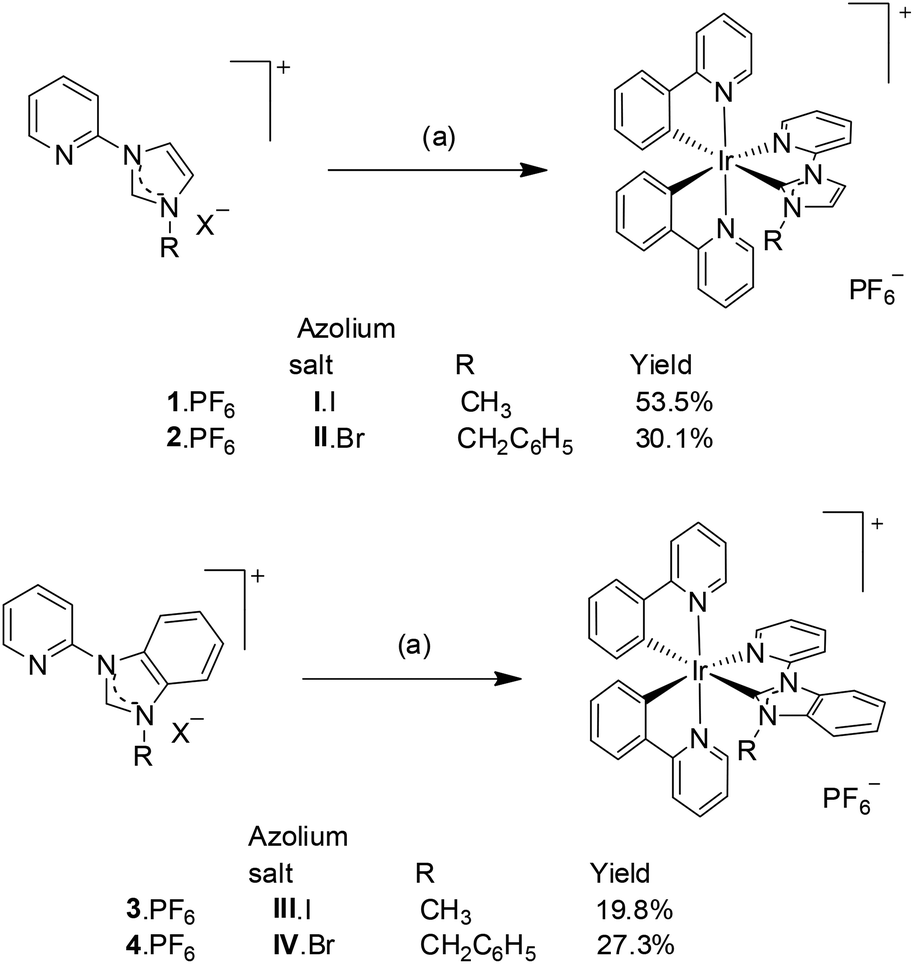 | ||
| Scheme 3 Synthesis of iridium(III) complexes 1·(PF6)–4·(PF6). Reagents and conditions: (a) 1. azolium salt, Ag2O in a mixture of CH2Cl2 and CH3OH (1:1), 80 °C, 4 h; 2. [Ir(ppy)2Cl]2, 17 h; 3. aq. KPF6. | ||
Characterisation
The structure of the azolium salts and the [Ir(ppy)2(C^N)]+ complexes were confirmed by 1H and 13C NMR spectroscopy and in the case of the 1·PF6, 3·PF6 and 4·PF6, by X-ray crystallography. The predicted number of signals were obtained in the 1H and 13C spectra for the synthesised [Ir(ppy)2(C^N)]+ complexes, consistent with the low symmetry structures (C1 point group). Fig. S1 and S2† show representative 1H and 13C NMR spectra for compounds 2·PF6 and 3·PF6 (ESI†). As expected, upon coordination of the NHC unit, the signal for the azolium salt procarbenic proton was lost from the 1H NMR spectra and a characteristic down-field chemical shift was observed for the carbenic (NHC) carbon atom, occurring at 177.5, 178.7, 187.4 and 188.8 ppm for the 13C spectra of complexes 1·PF6–4·PF6, respectively. For complexes 3·PF6 and 4·PF6, where the NHC is substituted with a benzyl group, the protons of the methylene linker are non-equivalent and a characteristic AX pattern was observed. These methylene protons are enantiotopic and non-equivalent as a result of the chirality associated with the octahedral tris(bidentate) Ir(III) complexes (Δ and Λ).Single crystals of the Ir(III) complexes 1·PF6, 3·PF6 and 4·PF6 were grown by slow evaporation of methanol solutions of each compound. The structure of 1·PF6 has been reported previously18 and is included here for the purpose of comparison. The X-ray crystal structures of the Ir(III) cations 1+, 3+ and 4+ are shown in Fig. 1, with selected bond distances collated in Table 1 and crystal refinement data summarised in Table 2. The molecular structures all display slightly distorted octahedral coordination geometries (C1 point group) around the Ir(III) centres with two cyclometalating ppy ligands and the bidentate NHC-pyridine unit. In each case the pyridine groups of the cyclometallated ppy ligands are trans to each other, while the ppy phenyl rings are cis to each other.
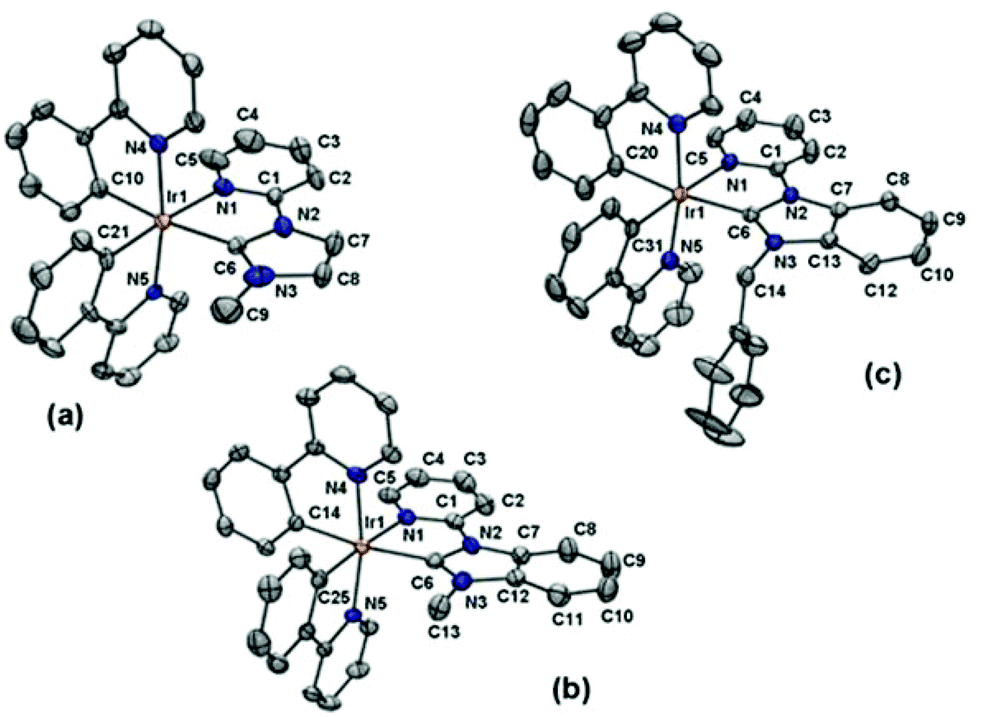 | ||
| Fig. 1 ORTEP51 representations of the X-ray crystal structures of the Ir(III) cations: (a) 1+, (b) 3+ and (c) 4+. The PF6− anions have been omitted for clarity. Thermal ellipsoids are shown at 40% probability. Labelled according to cif files. Selected bond distances given in Table 1. | ||
| Bond distance | 1 + | 3 + | 4 + |
|---|---|---|---|
| a For the purpose of comparison, a common labelling is used here that differs slightly from Fig. 1 (cif file labelling). | |||
| Ir–N1 | 2.140(5) | 2.170(4) | 2.147(2) |
| Ir–N2 | 2.050(4) | 2.058(4) | 2.050(2) |
| Ir–N3 | 2.044(5) | 2.049(3) | 2.035(2) |
| Ir–C1 | 2.075(5) | 2.061(4) | 2.046(2) |
| Ir–C2 | 2.007(5) | 2.015(4) | 2.009(2) |
| Ir–C3 | 2.051(5) | 2.046(4) | 2.053(2) |
| Group | Complex |
E
oxa (V) |
E
reda,b (V) |
ΔE (V) | ECL λmax (nm) | Rel. ECL annihil. | Rel. ECL coreact. | PL λmax (nm) | PL Quantum yield (ϕPL) | Excited state lifetime (μs) |
|---|---|---|---|---|---|---|---|---|---|---|
| a E° or E½ values for reversible or irreversible peaks, as stated in original papers. In the case of 1·PF6–4·PF6, the values are E½ taken at a scan rate of 0.1 V s−1 (see Fig. 3a). b Only first reduction potential listed. | ||||||||||
| [Ru(bpy)3](PF6)2 | 0.89 | −1.75 | 2.64 | 618 | 100 | 100 | 0.087 | 0.850 | ||
| [Ir(ppy)2(N^C:)]+ | 1·PF6 | 0.76 | −2.43 | 3.19 | 499 | 7.4 | 1.5 | 501 | <0.005 | 0.034 |
| 2·PF6 | 0.80 | −2.39 | 3.19 | 499 | 2.0 | 1.0 | 502 | <0.005 | 0.033 | |
| 3·PF6 | 0.92 | −2.30 | 3.22 | 498 | 7.0 | 3.2 | 499 | <0.005 | 0.030 | |
| 4·PF6 | 0.89 | −2.30 | 3.19 | 499 | 3.4 | 0.4 | 499 | <0.005 | 0.026 | |
| [Ru(bpy)2(N^C:)]2+ | 5·(PF6)2 | 0.85 | −1.78 | 2.62 | 628 | 68 | 622 | 0.01 | ||
| 6·(PF6)2 | 0.84 | −1.78 | 2.63 | 633 | 95 | 629 | 0.021 | |||
| 7·(PF6)2 | 0.90 | −1.75 | 2.66 | 613 | 7 | 611 | 0.007 | |||
| 8·(PF6)2 | 0.92 | −1.76 | 2.68 | 608 | 52 | 619 | 0.004 | |||
| [Ru(:C^N^C:)2]2+ | 9·(PF6)2 | 0.91 | −1.84 | 2.75 | 613 | 563 | 0.013 | |||
| 10·(PF6)2 | 0.65 | −2.41 | 2.79 | 536 | 532 | 0.008 | ||||
| 11·(PF6)2 | 0.68 | −1.96 | 2.64 | 631 | 584 | 0.0008 | ||||
| [Ir(ppy)2(C^C:)] | 12 | 0.24 | −2.72 | 2.96 | 532 | 31 | 0 | 529 | 0.498 | 1.98 |
| 13 | 0.21 | −2.73 | 2.95 | 537 | 10 | 0 | 533 | 0.420 | 1.59 | |
| 14 | 0.25 | −2.72 | 2.98 | 531 | 48 | 0 | 525 | 0.621 | 1.97 | |
| 15 | 0.36 | −2.61 | 2.97 | 524 | 17 | 0.1 | 507 | 0.683 | 2.03 | |
| 16 | 0.66 | −2.57 | 3.23 | 490 | 103 | 20 | 487 | 0.531 | 2.06 | |
For the benzimidazolylidene-containing complexes the Ir–CNHC bond distances are 2.061(4) and 2.046(2) Å for 3+ and 4+, respectively, which are relatively consistent and also similar to that determined for the imidazolylidene containing complex 1+. Previously reported benzimidazolylidene-containing structures include a series of complexes closely related to 2+ with a variety of cyclometalating 2-(phenyl)pyridine ligands, with Ir–CNHC bond distances in the range of 2.060(5) to 2.076(4) Å.19,20 For a homoleptic Ir(III) complex of a cyclometalated benzimidazolylidene-based NHC ligand (fac-Ir(pmb)3), the average Ir–CNHC bond distance was 2.026(7) Å.52
A marked trans influence from the carbene is apparent, with a significant elongation of the Ir–CPPY bond trans to the CNHC donor (2.051(5), 2.046(4) and 2.053(2) Å for 1+, 3+ and 4+, respectively) when compared to the other Ir–CPPY bond distances of 2.007(5), 2.015(4) and 2.009(2) Å for 1+, 3+ and 4+, respectively. The trans effect is observed in similar NHC-containing iridium(III) complexes.18–20,48,53
Absorption spectroscopy
The absorption spectra of the complexes 1·PF6–4·PF6 were recorded in acetonitrile solutions at room temperature (Fig. 2a and Table S7†). The absorption bands at high energy (<340 nm) can be assigned to π→π* transitions involving the co-ordinated heterocyclic ligands, whereas the weaker bands (>340 nm) can be assigned to spin-allowed metal-to-ligand charge-transfer (1MLCT) transitions in addition to spin-forbidden 3MLCT transitions promoted by the strong spin–orbit coupling of the heavy-atom metal centre.54,55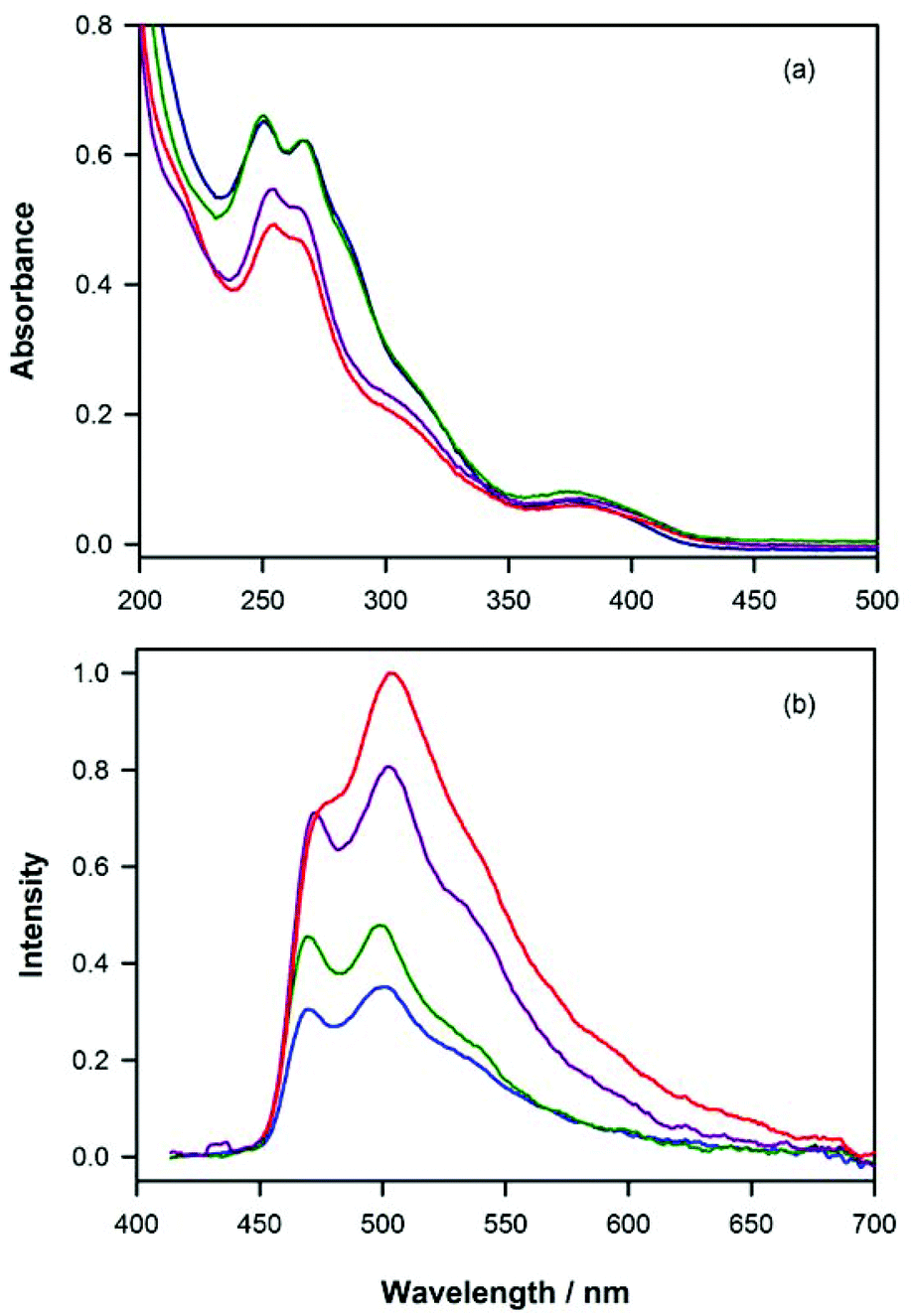 | ||
| Fig. 2 (a) Absorption spectra and (b) photoluminescence emission spectra (λex = 400 nm) of complexes 1·PF6 (purple line), 2·PF6 (red), 3·PF6 (green) and 4·PF6 (blue) at 10 μM and 1 μM, respectively, in acetonitrile at room temperature. | ||
Photoluminescence spectroscopy
Complexes 1·PF6–4·PF6 each showed blue-green phosphorescent emission at room temperature in acetonitrile under deaerated conditions. Despite the differences in the structure of the NHC ligand, the photoluminescence spectra of 1·PF6–4·PF6 (Fig. 2b) were quite similar, exhibiting maximum intensity at 499–502 nm, with a prominent shoulder or peak at ∼470 nm and a broad shoulder at ∼540 nm. These emissions have much more structure than those of the [Ru(bpy)2(N^C:)]2+ complexes containing pyridyl-imidazoylidene (Fig. 1a: 5·(PF6)2 and 6·(PF6)2) or pyridyl-benzimidazolylidene-NHCs (7·(PF6)2 and 8·(PF6)2),32 or the [Ir(ppy)2(C^C:)] complexes containing phenyl-imidazoylidene-NHCs (Fig. 1c: 12–16),33 which is indicative of weak MLCT character and a pronounced LC π→π* contribution.18,55 The photoluminescence quantum yields of the [Ir(ppy)2(N^C:)]+ complexes 1·PF6–4·PF6 in acetonitrile were all less than 1%, which is poor compared to the [Ir(ppy)2(C^C:)] complexes 12–16 (42–68%),33 but close to those of the [Ru(bpy)2(N^C:)]2+ complexes 5·(PF6)2–8·(PF6)2 (0.4–2%).32 Similarly, Zhang et al.18 reported a low quantum yield of 0.5% for complex 1·PF6 in degassed acetonitrile at room temperature, which was attributed to significant non-radiative energy-transfer to solvent. Baranoff and co-workers20 reported a quantum yield of 0.6% for a complex that differed from 3·PF6 by a methyl group on the pyridyl ring of the pyridylbenzimidazolylidene NHC ligand. The difference in quantum yields between the C^C coordinated and N^C coordinated iridium NHC complexes is striking. For example, complexes 12 and 1 differ by only one atom, yet their efficiencies of photoluminescence are 0.498 and 0.006 respectively. This may be explained by differing abilities of the two types of NHC ligand to de-stabilise thermally accessible metal centred (3MC) non-emissive states. The short lifetimes of complexes 1–4 compared with those of 12–16 tend to support this conclusion. A similar theory was suggested by Thompson and co-workers56 for tris-cyclometalated (N^C) iridium complexes and by Baranoff and co-workers20 on the basis of temperature-dependent excited-state lifetime measurements on complexes with similar NHC ligands to ours.Electrochemistry
The redox properties of 1·PF6–4·PF6 were studied using cyclic voltammetry and the results are compared with selected data from previous papers26,32,33 in Table 2. All complexes displayed reduction peaks between −1.30 and −1.43 V vs. Fc, which were chemically irreversible (i.e. no return peak) at all scan rates tested, (Fig. 3a). Similarly, at 0.1 V s−1, irreversible peaks due to oxidation of the complexes were seen between 0.76 and 0.92 V vs. Fc. However, these oxidation processes became more chemically reversible at slightly elevated scan rates. For example, when complexes 1·PF6 and 2·PF6 were scanned at rates ≥0.1 V s−1 and complexes 3·PF6 and 4·PF6 at rates ≥0.5 V s−1, they exhibited responses that were semi-reversible chemically (ip,ox > ip,red) and became fully reversible (ip,ox/ip,red = 1) at scan rates of greater than or equal to 1, 5, 3 and 5 V s−1 for complexes 1·PF6, 2·PF6, 3·PF6 and 4·PF6, respectively (Fig. 3b).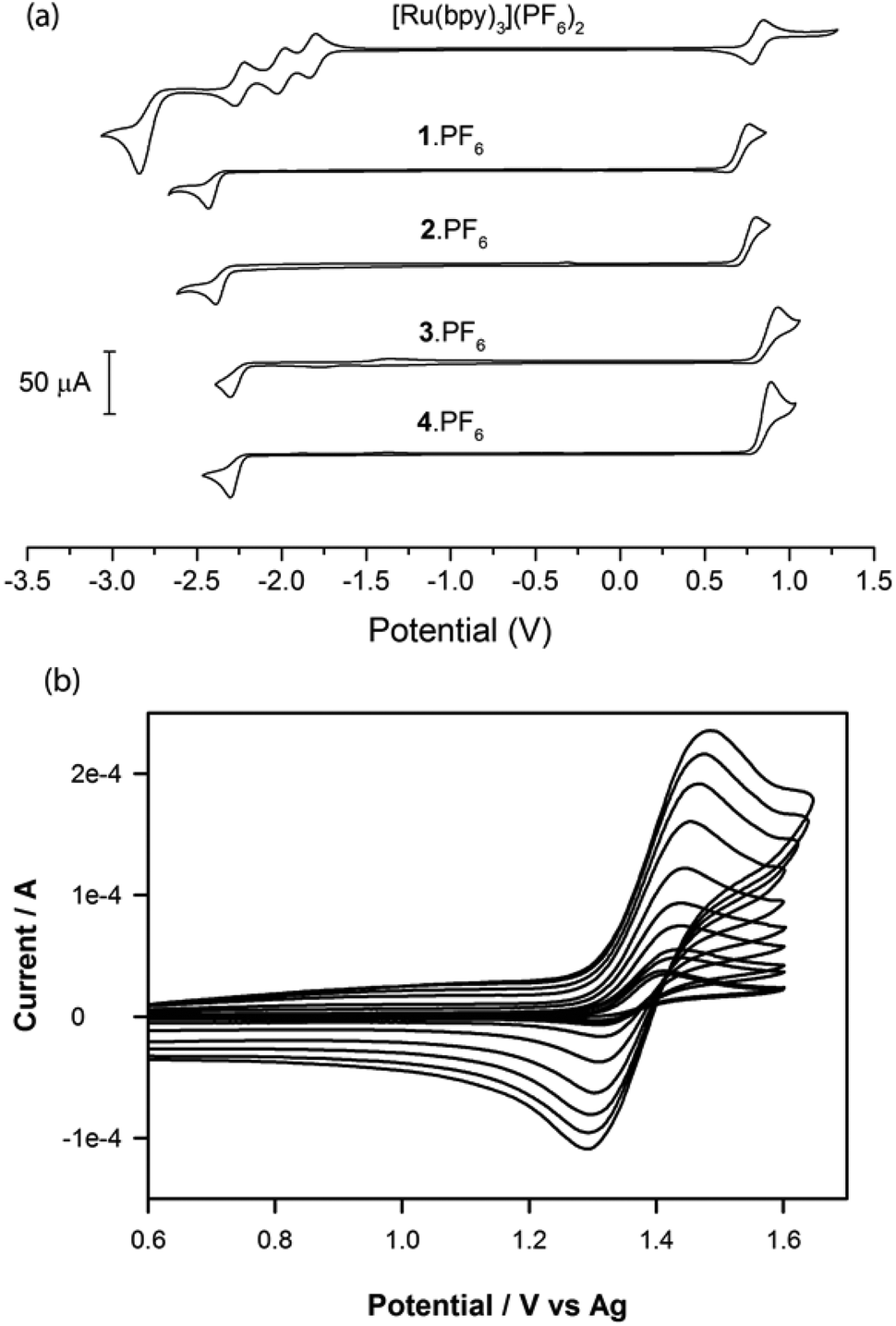 | ||
| Fig. 3 (a) Cyclic voltammetric responses (scan rate = 0.1 V s−1) for complexes 1·PF6–4·PF6 and [Ru(bpy)3](PF6)2 (1 mM) dissolved in acetonitrile containing 0.1 M [Bu4N]PF6. (b) Change in the oxidative cyclic voltammetric response from irreversible to reversible for complex 4·PF6 as the scan rates were increased from 0.08 to 25.00 V s−1. | ||
The oxidation potentials, which in this case can be formally assigned to the Ir3+/4+ redox couple (although the HOMO is delocalised over the phenyl ring of the ppy ligands), were similar in magnitude to those of the Ru2+/3+ couple in [Ru(bpy)3]2+ and the previously reported [Ru(bpy)2(N^C:)]2+ complexes 5·(PF6)2–8·(PF6)2, but much more positive than the Ir3+/4+ couple of the [Ir(ppy)2(C^C:)] complexes 12–16. This is due to the much greater electron donation of the orthometalating ring of the phenyl-imidazoylidene or phenyl-benzimidazolylidene NHC ligands than the nitrogen lone pair of their pyridyl analogues.
As observed for the [Ru(bpy)2(N^C:)]2+ complexes,32 the benzimidazolylidene-NHC ligand of complexes 3·PF6 and 4·PF6 resulted in more positive oxidation potentials and larger HOMO–LUMO gaps than the analogous imidazolylidene-NHC ligand of 1·PF6 and 2·PF6, which can be attributed to stabilisation of the HOMO by benzimidazolylidene π-backbonding. In contrast to the oxidation potentials, the (primarily ligand-based) first reduction potentials of complexes 1·PF6–4·PF6 were more akin to those of the [Ir(ppy)2(C^C:)] series than the [Ru(bpy)3]2+ and [Ru(bpy)2(N^C:)]2+ complexes, because the bipyridine ligands are much more easily reduced than the phenylpyridine or NHC ligands.
Electrogenerated chemiluminescence
Under oxidative potential with TPA as a co-reactant, the relative ECL intensities of the [Ir(ppy)2(N^C:)]+ complexes 1·PF6–4·PF6 were 0.4–3.2% that of [Ru(bpy)32+]. These are higher than that of [Ir(ppy)2(C^C:)] complexes 12–15 (≤0.1%; Table 2), but much lower than that of the difluoro derivative 16 (20%).33 In contrast, under annihilation conditions, the ECL intensities of complexes 1·PF6–4·PF6 (≤7.4%) were poorer than those of either the [Ir(ppy)2(C^C:)] (10–103%) or [Ru(bpy)2(N^C:)]2+ (7–95%) series. These findings can be effectively rationalised by considering the dominant factors that determine the relative intensities in each mode of ECL. In the case of co-reactant ECL using relatively high concentrations of TPA, the major reaction pathway can be summarised as follows,57 where M is the metal-complex electrochemiluminophore, and P is other products.| M − e− → M+ | (1) |
| TPA − e− → TPA˙+ | (2) |
| TPA˙+ → TPA˙ + H+ | (3) |
| M+ + TPA˙ → M* + P | (4) |
| M* → M + hν | (5) |
Intense co-reactant ECL with TPA could be expected from complexes that: (i) have an appropriately high oxidation potential, so that there is sufficient excess energy in reaction (4) to generate the product in an electronically excited state; (ii) are sufficiently stable in their oxidised form; and (iii) have a high luminescence quantum yield. With respect to factor (i), we recently introduced a graph of oxidation potential versus emission wavelength indicating the ‘wall of energy sufficiency’ for co-reactant ECL with TPA,33 which can be derived from the following relationship for the free energy of reaction (4):
| ΔG = Ered − Eox + EES | (6) |
In the case of co-reactant ECL with TPA, the Ered is E(TPA˙), which has been previously reported as −2.1 V (vs. Fc).57 The energy of the excited state, EES, is best taken from the maximum emission wavelength at low temperature, but may be estimated using room temperature data to a first approximation. As illustrated by Fig. 4, there is clearly sufficient free energy for the generation of the excited states in the [Ir(ppy)2(N^C:)]+ (1·PF6–4·PF6), [Ru(bpy)2(N^C:)]2+ (5·(PF6)2–8·(PF6)2), and [Ru(:C^N^C:)2]2+ (9·(PF6)2–11·(PF6)2) complexes, but members of the previously investigated [Ir(ppy)2(C^C:)] series fall on both sides of the ‘wall’. Complexes 12–14, which showed no co-reactant ECL with TPA (Table 2) were found to be energy insufficient. Complex 15, which exhibited weak co-reactant ECL intensity, was adjacent to the wall, whereas complex 16, which gave the most intense co-reactant ECL, was positioned well within the energy sufficient zone. Complexes 1·PF6–4·PF6 were positioned even further away from the wall (near the [Ru(bpy)3]2+ reference complex), but their corresponding co-reactant ECL intensities were much poorer than those of either complex 16 or [Ru(bpy)3]2+. Although complexes 1·PF6–4·PF6 have oxidation potentials that would certainly provide adequate free energy upon reaction with TPA˙ to generate the excited state, their co-reactant ECL is limited by factors (ii) and (iii). Due to the apparent instability of their dications, the voltammetric responses for the oxidation of complexes 1·PF6–4·PF6 were only reversible at relatively fast scan rates (Fig. 3b), whereas the oxidation of [Ru(bpy)3]2+ (Fig. 3a) and complex 16 are fully chemically reversible at relatively low scan rates.33 Moreover, the photoluminescence quantum yields of 1·PF6–4·PF6 (<1%) were poor compared to [Ru(bpy)3]2+ (9%) and the [Ir(ppy)2(C^C:)] complexes 12–16 (42–68%).
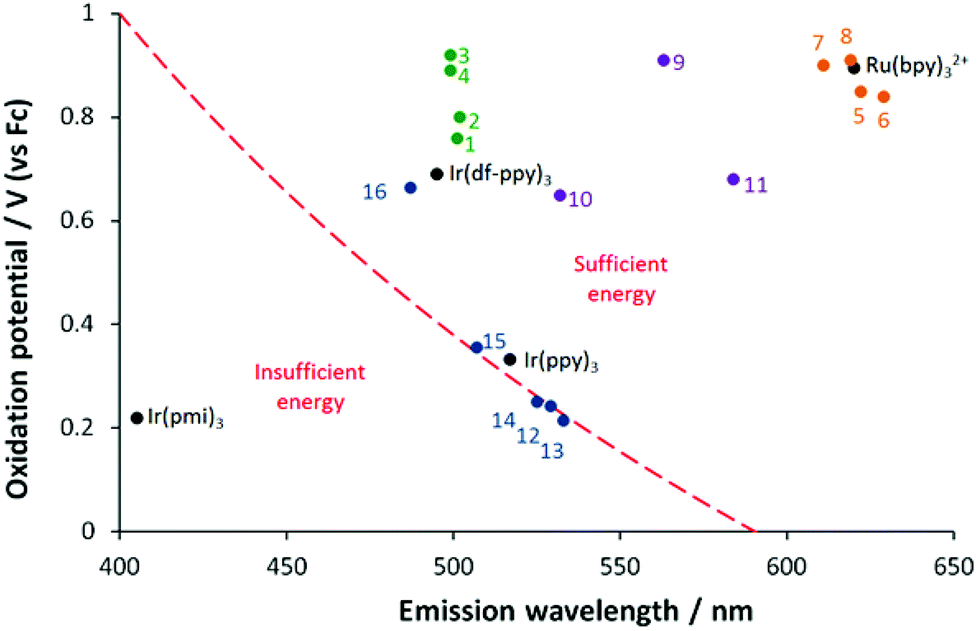 | ||
| Fig. 4 ‘Wall of energy sufficiency’ for co-reactant ECL with TPA. The dashed line represents the critical oxidation potential required for ECL for a luminophore with a given emission colour. Solid circles show the position of [Ir(ppy)2(N^C:)]+ complexes 1·PF6–4·PF6 (green circles), [Ru(bpy)2(N^C:)]2+ complexes325·(PF6)2–8·(PF6)2 (orange), [Ru(:C^N^C:)2]2+ complexes269·(PF6)2–11·(PF6)2 (purple), and [Ir(ppy)2(C^C:)] complexes3312–16 (blue). | ||
In the case of annihilation ECL, the reductant utilised to generate the electronically excited state is derived from electrolysis of the metal complex:
| M + e− → M− | (7) |
| M+ + M− → M* + M | (8) |
Entering the Eox, Ered and EES data from Table 2 into eqn (6) shows that the annihilation reaction (8) of each complex has sufficient free energy to generate the product in an electronically excited state. Here, however, the reversibility of both the oxidation and reduction potentials must be considered, in addition to the luminescence quantum yield. Complex 16 for example, exhibited an annihilation ECL intensity that was 103% that of [Ru(bpy)3]2+, which can be attributed at least in part to its greater quantum yield, as both complexes have highly reversible oxidation and reduction potentials.33 Although complexes 12–15 also have high quantum yields, their oxidation potentials were less reversible, which can account for their moderate annihilation ECL intensities (10–48%). The [Ru(bpy)2(N^C:)]2+ complexes 5·(PF6)2–8·(PF6)2 exhibit reversible Ru2+/3+-oxidation and bipyridine-ligand-reduction potentials, but they have poorer luminescence quantum yields than [Ru(bpy)3]2+, somewhat accounting for their reduced but still significant annihilation ECL intensities (7–95%). In contrast, weak annihilation ECL was observed from complexes 1·PF6–4·PF6, due to a combination of their low quantum yields and their instability in both the oxidised and reduced form as revealed by the limited reversibility of their reduction and oxidation processes.
Theoretical studies
The mPW1PW91/SDD,6-31+G(d) gas-phase optimized geometries (Table 2) of the iridium complexes 1–4 (omitting PF6− counter-ions) are in good agreement with the X-ray structures (Table 1), with differences between calculated and experimental Ir–N and Ir–C bond distances for 1+, 3+, and 4+ all less than 0.07 Å (mean absolute deviation of 0.02 Å).The mPW1PW91 calculated bond distances and Wiberg bond indices (WBI) for Ir–N and Ir–C reflect the difference between the imidazolylidene (1+ and 2+) and benzimidazolylidene (3+ and 4+) containing complexes (see Table 3). In 1+ and 2+, the Ir–CNHC bond distance is slightly longer than those for 3+ and 4+, and this small difference is reflected in the WBI values (indicative of bond order). For the other coordination bonds (Ir–N and Ir–CNHC), there is no appreciable difference in bond distances and WBI values between the imidazolylidene (1+ and 2+) and benzimidazolylidene (3+ and 4+) containing complexes. The well-known trans influence58–61 from the NHC group is replicated in the theoretical results, with calculated Ir–CPPY(trans) bond distances (2.048–2.051 Å) significantly longer than the other Ir1–CPPY(cis) bond distances (2.004–2.008 Å) (Table 3).
| Ir–CNHC | Ir–NNHC | Ir–CPPYtrans | Ir–CPPY | |||||
|---|---|---|---|---|---|---|---|---|
| Å | WBI | Å | WBI | Å | WBI | Å | WBI | |
| 1 2+ | 2.075 | 0.47 | 2.201 | 0.24 | 2.048 | 0.55 | 2.004 | 0.71 |
| 2 2+ | 2.075 | 0.47 | 2.200 | 0.24 | 2.048 | 0.55 | 2.004 | 0.71 |
| 3 2+ | 2.064 | 0.49 | 2.188 | 0.24 | 2.051 | 0.55 | 2.007 | 0.71 |
| 4 2+ | 2.069 | 0.49 | 2.189 | 0.24 | 2.051 | 0.55 | 2.008 | 0.70 |
Analysis of the frontier MOs (Fig. 5, S4–7†) indicates that for all compounds, the singlet HOMO is comprised of contributions from the iridium d and phenyl π orbitals, distributed across both ppy ligands. The singlet LUMO is comprised of a majority contribution from the auxiliary NHC ligand, with a smaller contribution from the ppy ligands. In the imidazolylidene complexes (1+–2+) the contribution is ∼30%, whereas in the benzimidazolylidene complexes (3+–4+) it is ∼15%. Similar subtle differences are noted in the LUMO+1 between 1+–2+ (∼60% NHC) and 3+–4+ (∼80% NHC), and also in the HOMO−1 for 1+–2+ (∼15% Ir) and 3+–4+ (7% Ir). For other relevant frontier orbitals in 1+–4+, there is no discernible difference between contributions from the different fragments.
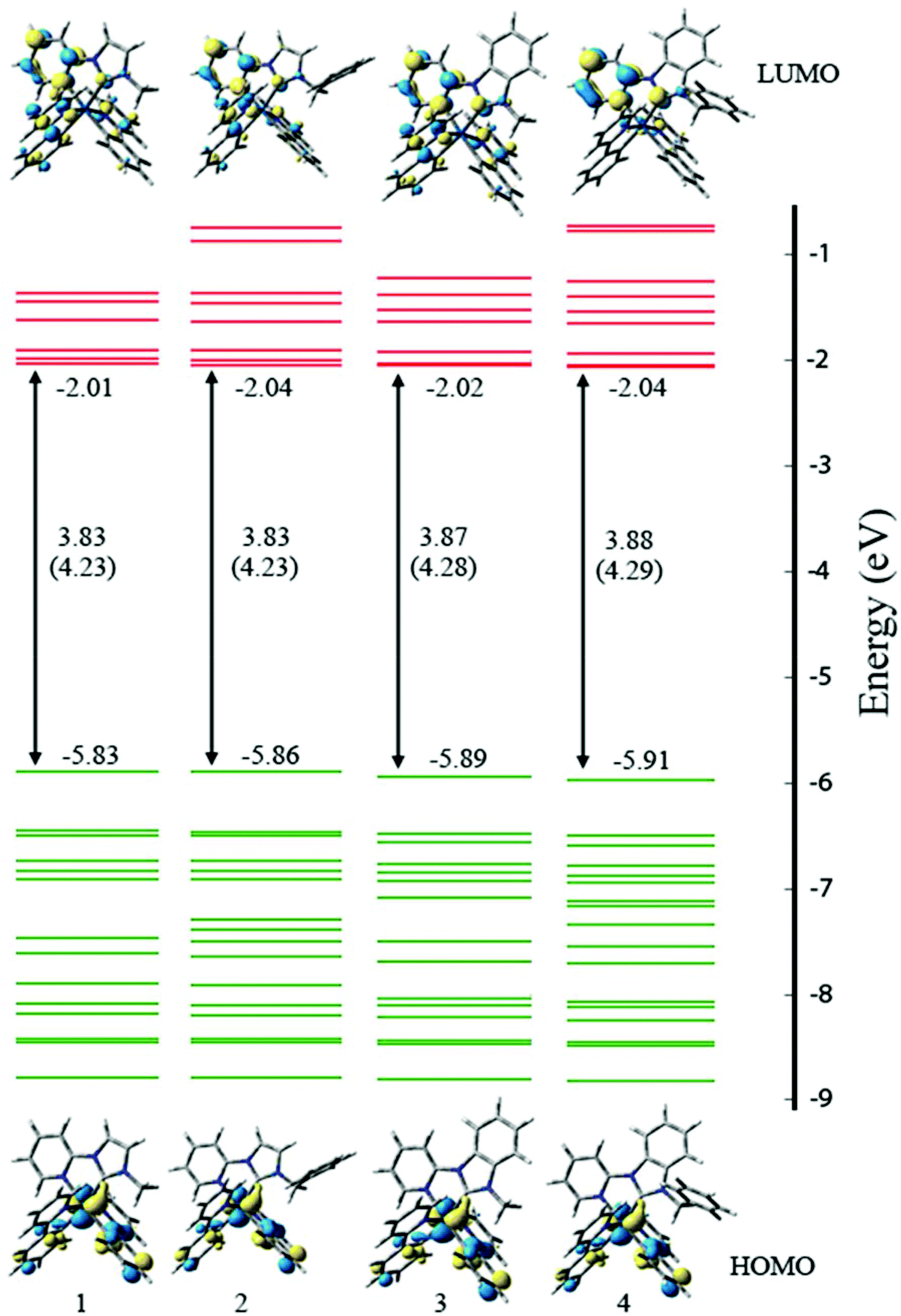 | ||
| Fig. 5 B3LYP/SDD,tzvp//mPW1PW91/SDD,6-31+G(d) calculated molecular orbital energy diagram including HOMO–LUMO gaps (mPW1PW91/SDD,tzvp result in parentheses), and surface plots of the HOMO and LUMO for 1–4. Acetonitrile solvent correction included with single-pint SCRF. Units of eV. | ||
For all of 1+–4+, the singlet-state HOMO and LUMO do overlap (i.e. they are not entirely orthogonal), which suggests that the HOMO and LUMO energies cannot be easily ‘tuned’ by substitution of the ligands. Moreover, the triplet state density (Fig. S8†) does not share the same spatial extent as the singlet HOMO and LUMO.
It is instructive to compare the characteristics of the MOs of the Ir compounds considered here and the analogous Ru compounds (bpy in place of ppy) considered previously.32 With Ru, the HOMO, HOMO−1 and HOMO−2 are all largely metal based, whereas in the Ir compounds the HOMO−1 is mostly associated with the ppy ligand (the HOMO and HOMO−2 are metal based). For the unoccupied MOs, the density plots are qualitatively similar however there is a different energy ordering. With Ru, the LUMO and LUMO+1 are associated with the bpy ligands and the LUMO+2 resides on the auxiliary NHC ligand, whereas in the Ir compounds the LUMO and LUMO+2 are associated with the ppy ligand and the LUMO+1 resides on the NHC ligand. That is, with Ir the NHC π* MO is stabilised relative to the Ru compounds and hence may be expected to have a greater impact on photophysical and electrochemical properties.
Calculated energies of valence orbitals of complexes 1+–4+ are included in Table 4. The energy of the HOMOs of 3+ and 4+ are slightly lower than those for 1+ and 2+. It is suggested that the benzimidazolylidene exerts a greater stabilising influence on the metal-based HOMO than does the imidazolylidene. The blue-shifted MLCT bands from the absorption spectra of 3+ and 4+ are consistent with the benzimidazolylidene exerting a stabilising influence on the metal-based HOMO. There is no discernible LUMO energy difference. The HOMO–LUMO energy gap of 1+–2+ is less than that of 3+–4+, which is consistently reproduced by mPW1PW91, B3LYP and M06 density functionals. The larger HOMO–LUMO gap for 3+ and 4+ (consistent with the analysis of luminescence spectra) arises from the lower HOMO energies of 3+ and 4+.
| mPW1PW91 | B3LYP | |||||
|---|---|---|---|---|---|---|
| HOMO | LUMO | ΔE | HOMO | LUMO | ΔE | |
| a SDD,TZVP basis set and effective core potential with acetonitrile PCM SCRF. | ||||||
| 1 2+ | −6.12 | −1.89 | 4.23 | −5.83 | −2.01 | 3.82 |
| 2 2+ | −6.13 | −1.90 | 4.23 | −5.86 | −2.04 | 3.82 |
| 3 2+ | −6.18 | −1.89 | 4.28 | −5.89 | −2.02 | 3.87 |
| 4 2+ | −6.20 | −1.91 | 4.29 | −5.91 | −2.04 | 3.87 |
Calculation of electronic excitations with TD-DFT (mPW1PW91) supports the above discussion of the spectroscopic and electrochemical results (Fig. S9, Table S5†), with transitions in the visible region best described as MLCT, and transitions in the UV region of the spectrum dominated by intra-ligand charge transfer (ILCT) and ligand-centred (LC) transitions.
Conclusions
A family of four heteroleptic iridium(III) complexes of the form [Ir(ppy)2(C^N)]PF6 (where C^N is a pyridyl-imidazolylidene or pyridyl-benzimidazolylidene NHC ligand) were synthesized in a relatively straight forward manner using a Ag(I) transmetallation protocol. A combined theoretical and experimental investigation into the spectroscopic and electrochemical properties of the series was performed with a view to gaining a better understanding into reasons behind the varying efficiency of the photoluminescence and electrochemically generated luminescence observed for such complexes. To this end, the properties of the four complexes were compared with those of similar NHC complexes of iridium and ruthenium recently reported by ourselves and others in the literature.The photoluminescence quantum yields for 1·PF6–4·PF6 were all found to be below 1%, contrasting with the strongly luminescent complexes 12–16. The lesser abilities of the N^C coordinated ligands to de-stabilise thermally accessible non-emissive states, compared to the C^C variety, is the most likely reason for this.20 Varying the structure and substituents on the NHC ligand in compounds 1·PF6-4·PF6 had a negligible effect on the colour of the emission with λmax being virtually identical in each case. This is explained by the fact that neither of the frontier orbitals is located on or in close proximity to the NHC ring of the auxiliary ligand, as determined from DFT calculations, therefore electron donating or withdrawing effects on these orbitals tend to be weak and to be directed with approximately equal influence toward the HOMO and LUMO. In the case of the ruthenium analogues 5–8, where moderate differences in λmax were observed, the HOMO was substantially metal based, allowing this orbital to be directly influenced by structural differences in the NHC moiety. The electrochemical results bear out this analysis; for example, although the oxidation potentials of 1·PF6-4·PF6 differ by up to 160 mV, the gap between oxidation and reduction processes (the electrochemical HOMO–LUMO gap) is almost invariant at about 3.20 ± 0.01 V. The electrochemical responses for 1·PF6-4·PF6 are characterised by chemical irreversibility of both the oxidative and reductive processes, pointing to instability in both the oxidised and reduced forms of the complexes, though the oxidation couple become reversible at faster scan rates. The electrochemiluminescence intensities (both annihilation and co-reactant modes) observed for compounds 1·PF6–4·PF6 were quite weak. This is due a combination of low photoluminescence quantum yield and instability in the oxidised and reduced forms of the complex. A plot of oxidation potential versus emission wavelength was constructed to rationalise the influence of energetics on ECL intensity for these and the previously investigated NHC complexes (5–16). This plot demonstrates that favourable energetics is necessary, but is not the only condition required for intense ECL. For example, 1·PF6-4·PF6 have favourable energetics for ECL but their efficiency is confounded by unfavourable kinetic and photophysical factors. On the other hand, compounds 12–15 exhibit high photoluminescence quantum yields and reversible electrochemistry, but unfavourable energetics. In the cases of 16 and [Ru(bpy)3]2+, however, all constraints are satisfied and intense ECL is observed.
Experimental section
General
All reagents were purchased from Sigma-Aldrich and were of analytical grade or higher and were used without further purification unless otherwise stated. Dry CH3CN and THF were distilled from CaH2 and sodium benzophenone ketyl under nitrogen, respectively. Ligands I–IV were prepared using previously published procedures.32 NMR spectra were recorded using Bruker ARX-300 (300.14 MHz for 1H, 75.48 MHz for 13C) and referenced to solvent resonances. Microanalyses were performed by the Microanalytical Laboratory at the ANU Research School of Chemistry, Canberra, Australia. All compounds were prepared in air unless otherwise specified.Absorption and photoluminescence spectroscopy
UV-visible absorption spectra were collected using a Cary 300 Bio UV-Vis spectrophotometer (Varian Australia, Mulgrave, Vic., Australia) with 1 cm path length quartz cells. Photoluminescence spectra were collected with a Cary Eclipse Spectrofluorimeter (Varian Australia), using a 1 cm quartz cuvette (5 nm bandpass, 1 nm data interval, PMT voltage: 800 V). Emission spectra correction factors were established using an Optronic Laboratories spectral irradiance standard (model OL 245M) with constant current source (model OL 65A). UV-visible and photoluminescence spectra were recorded at 10 μM and 1 μM concentrations respectively in acetonitrile. Lifetimes were measured with a Nanolog (HORIBA Jobin Yvon IBH) spectrometer using the time correlated single photon counting (TCSPC) option and correlated by a time-to-amplitude converter (TAC) in forward TAC mode. 10 μM solutions were prepared in an air-tight four-sided quartz cuvette in a oxygen-free glovebox using acetonitrile which had been purged using the freeze–pump–thaw method. The complexes were excited using a Nanoled 340 (344 nm) laser pulsed at 100 kHz repetition rate. The emitted photons were detected by a thermoelectrically cooled TBX picosecond single-photon detector with the emission band width set to 10 nm. Signals were collected using a FluoroHub counter and the decay curves were fitted to a single exponential using DAS6 software (HORIBA Jobin Yvon IBH). Weighted residuals averaging around zero and a χ2 < 1.2 were used to judge a good fit.Electrochemistry and electrogenerated chemiluminescence
Experiments were performed using Autolab PGSTAT 101 (for cyclic voltammograms) and Autolab PGSTA12 (for chronoamperometry and cyclic voltammograms with ECL detection) potentiostats (Metrohm Autolab B.V., Netherlands). The electrochemical cell consisted of a cylindrical glass cell with a quartz window base and Teflon cover with spill tray. The cell was encased in a custom-built light-tight faraday cage. A conventional three electrode configuration, consisting of a glassy carbon 3 mm diameter working electrode, shrouded in Teflon (CH Instruments, Austin, TX, USA), reference electrode and gold wire counter electrode. Potentials were referenced to the ferrocene/ferrocenium couple measured in situ (1 mm) in each case. ECL spectra were obtained using an Ocean Optics CCD, model QE65pro, interfaced with our electrochemical cell using an optic fibre (1.0 m, 1.0 mm core diameter), collimating lens, and custom cell holder, and the acquisition was triggered using a HR 4000 Break-Out box in conjunction with the potentiostat. The complexes were prepared in freshly distilled acetonitrile at a concentration of 1 mm for voltammetric and ECL measurements, with 0.1 m tetrabutylammonium hexafluorophosphate (TBAPF6) as the supporting electrolyte, with a co-reactant (TPA) concentration of 10 mm for ECL experiments. Prior to each experiment, the working electrode was polished using 0.3 μm and then 0.05 μm alumina with water on a felt pad, sonicated in MilliQ water (1 min), rinsed in freshly distilled acetonitrile and dried with a stream of N2. The working electrode was then positioned at an appropriate distance (∼2 mm) from the bottom of the cell for detection of the ECL signal, and the solution was purged with grade 5 argon for 15 min prior to measurement. ECL spectra were recorded using a 45 s integration time, and single 40 s chronoamperometry pulse at 1.4 V. Spectra were integrated to determine the relative ECL intensities and compared with the ECL spectrum of the standard ([Ru(bpy)3]2+) measured under identical conditions.X-ray crystallography
Single crystals of the iridium complexes 1·(PF6), 3·(PF6) and 4·(PF6) suitable for X-ray diffraction studies were grown by slow evaporation of methanol solutions of each compound. Crystallographic data for all structures determined are given in Table 2. Crystals were mounted on a glass fibre in paratone oil and cooled rapidly to 173 K in a stream of cold N2 using an Oxford low-temperature device. Diffraction data were measured using an Oxford Gemini dual-wavelength X-ray diffractometer mounted with molybdenum (Mo) and copper (Cu) X-ray sources (graphite-monochromated Mo-Kα and Cu-Kα radiation, λ = 0.71073 Å and λ = 1.54184 Å, respectively). Data were reduced and corrected for absorption using the CrysAlis Pro program.61 The SHELXL2013-2 program62 was used to solve the structures with Direct Methods, with refinement by the Full-Matrix Least-Squares refinement techniques on F2. The non-hydrogen atoms were refined anisotropically and hydrogen atoms were placed geometrically and refined using the riding model. Coordinates and anisotropic thermal parameters of all non-hydrogen atoms were refined. All calculations were carried out using the program Olex2.63 Images were generated by using ORTEP-3.51 Further XRD details are provided in the ESI.† CCDC 1031786–1031788 contains the supplementary crystallographic data for this paper.Theoretical calculations
Density functional theory (DFT) calculations were carried out within the Gaussian 09 suite of programs.64 Ground state geometries were optimised in the absence of solvent with B3LYP65–67 and mPW1PW9168,69 functionals in conjunction with the 6-31+G(d) basis set70–72 for non-metal atoms and the SDD basis set and MWB core potential for iridium.73,74 Only mPW1PW91 results are presented since it has been shown previously that this functional yields reliable results.32,75,76 Single-point energy calculations were carried out at the optimised geometries using the SDD basis and core potential (MWB)73,74 for Ir and the TZVP basis set77 for all other atoms. The polarisable continuum model (PCM)78 self-consistent reaction field (SCRF) was used to model solvent effects at the gas-phase optimised geometries with a solvent of acetonitrile, consistent with the experimental system. Frontier MO energies were calculated using DFT MOs. Excitation energies to singlet and triplet excited states were investigated with TD-DFT79 with 40 states calculated. An SCF convergence criteria of 10−8 a.u. was employed throughout. Molecular orbital analysis was carried out with the AOMix program80 and NBO 5.9.81Synthesis
1·PF6: A mixture of I·I (0.2 g, 0.66 mmol) and Ag2O (0.23 g, 0.94 mmol) in 1:1 CH2Cl2–CH3OH (50 mL) was heated in the dark at 80 °C for 4 h under N2. [Ir(ppy)2Cl]2 (0.37 g, 0.35 mmol) was added and this temperature was maintained for 17 h. The hot reaction mixture was filtered through celite and water (30 mL) and KPF6 (0.6 g, 3.2 mmol) were added to the filtrate. After 1 h the bright green precipitate was collected and recrystallised from methanol giving the product as a bright green solid (Yield: 0.3 g, 53.5%). 1H NMR (d6-DMSO): δ(ppm) = 8.49 (d, J = 2.1 Hz, 1H), 8.32 (d, J = 8.1 Hz, 1H), 8.17–8.24 (m, 3H), 7.96 (d, J = 5.4 Hz, 1H), 7.84–7.92 (m, 4H), 7.57 (t, J = 5.4 Hz, 2H), 7.50 (d, J = 2.1 Hz, 1H), 7.40 (t, J = 6.3, 6.6 Hz, 1H), 7.12 (t, J = 6.9, 8.4 Hz, 2H), 7.00 (t, J = 7.2, 6.9 Hz, 1H), 6.91 (dd, J = 6.9, 5.4 Hz, 2H), 6.77 (t, J = 7.2, 6.9 Hz, 1H), 6.24 (d, J = 7.2 Hz, 1H), 6.09 (d, J = 6.9 Hz, 1H), 3.07 (s, 3H, CH3). 13C NMR(d6-DMSO): δ(ppm) = 177.5, 167.9, 166.6, 164.0, 153.6, 153.2, 149.2, 149.1, 149.0, 144.6, 142.8, 141.8, 138.5, 137.7, 130.5, 130.3, 129.7, 125.5, 125.0, 124.6, 124.3, 123.6, 122.8, 121.1, 120.2, 119.9, 117.9, 112.9, 36.3. Found: C, 46.43; H, 3.16; N, 8.74%. C31H25N5PF6Ir requires C, 46.27; H, 3.13; N, 8.70%.
2·PF6: This compound was prepared as described for 1·(PF6) from II·Br (0.2 g, 0.63 mmol), Ag2O (0.22 g, 0.95 mmol) and [Ir(ppy)2Cl]2 (0.34 g, 0.32 mmol) in 1:1 CH2Cl2–CH3OH (50 mL). The product was obtained as a bright yellow solid after recrystallisation from a mixture of methanol and water. (Yield: 0.17 g, 30.1%). 1H NMR (d6-DMSO): δ(ppm) = 8.60 (s, 1H), 8.35 (d, J = 8.4 Hz, 1H), 8.16–8.24 (m, 2H), 7.99 (d, J = 5.7 Hz, 1H), 7.81–7.93 (m, 4H), 7.62 (d, J = 5.7 Hz, 1H), 7.49–7.57 (m, 3H), 7.40 (t, J = 6.6, 6.3 Hz, 1H), 7.11 (t, J = 7.2, 6.3 Hz, 3H), 6.97 (t, J = 7.5, 7.2 Hz, 3H), 6.89 (t, J = 6.6, 7.2 Hz, 1H), 6.70–6.76 (m, 2H), 6.31 (d, J = 7.5 Hz, 2H), 6.19 (d, J = 6.9 Hz, 1H), 6.05 (d, J = 7.2 Hz, 1H), 4.80 (dd, J = 15.3 Hz, 2H, –CH2). 13C NMR (d6-DMSO): δ(ppm) = 178.7, 167.9, 166.5, 163.7, 153.5, 153.2, 149.1, 148.6, 144.5, 143.0, 141.8, 138.6, 137.5, 136.4, 130.5, 130.4, 130.2, 129.6, 128.2, 127.4, 125.8, 125.0, 124.9, 124.4, 123.4, 122.8, 120.9, 120.3, 119.9, 119.0, 113.1, 52.1. Found: C, 48.39; H, 3.27; N, 8.09%. C35H27N5PF6Ir·CH3OH requires C, 48.76; H, 3.52; N, 7.90%.
3·PF6: This compound was prepared as described for 1·(PF6) from III·I (0.2 g, 0.59 mmol), Ag2O (0.2 g, 0.89 mmol) and [Ir(ppy)2Cl]2 (0.32 g, 0.29 mmol) in 1:1 CH2Cl2–CH3OH (50 mL). The product was obtained as a bright green solid after recrystallisation from hot methanol (Yield: 0.09 g, 19.8%). 1H NMR (d6-DMSO): δ(ppm) = 8.71 (d, J = 8.7 Hz, 1H), 8.48–8.52 (m, 1H), 8.18–8.25 (m, 4H), 7.87–7.90 (m, 4H), 7.67–7.70 (m, 2H), 7.63 (d, J = 5.1 Hz, 1H), 7.51–7.56 (m, 2H), 7.43 (t, J = 6 Hz, 1H), 6.99–7.10 (m, 3H), 6.92 (dd, J = 5.7, 4.2 Hz, 2H), 6.84 (t, J = 6 Hz, 1H), 6.28 (d, J = 6.6 Hz, 1H), 6.06 (d, J = 6.3 Hz, 1H), 3.31 (s, 3 H, CH3). 13C NMR (d6-DMSO): δ(ppm) = 187.4, 167.5, 166.5, 164.5, 154.3, 153.8, 149.6, 149.2, 144.4, 142.9, 142.0, 138.7, 137.9, 136.4, 131.2, 130.6, 130.5, 130.3, 129.9, 125.2(2), 125.1, 124.9, 124.7, 123.9, 123.6, 123.0, 121.4, 120.3(2), 114.0, 112.9, 112.5, 33.4. Found: C, 50.45; H, 3.32; N, 7.95%. C37H29N5PF6Ir requires C, 50.54; H, 3.52; N, 8.09%.
4·PF6: This compound was prepared as described for 1·(PF6) from IV·Br (0.2 g, 0.55 mmol), Ag2O (0.19 g, 0.82 mmol) and [Ir(ppy)2Cl]2 (0.3 g, 0.27 mmol) in 1:1 CH2Cl2–CH3OH (50 mL). The product was obtained as a bright yellow solid after recrystallisation from acetonitrile (Yield: 0.14 g, 27.3%). 1H NMR (d6-DMSO): δ(ppm) = 8.76 (d, J = 8.4 Hz, 1H), 8.57 (d, J = 8.4 Hz, 1H), 8.17–8.25 (m, 3H), 7.82–7.90 (m, 4H), 7.72 (t, J = 6, 6.3 Hz, 2H), 7.53 (t, J = 6.4 Hz, 1H), 7.35–7.47 (m, 4H), 7.00–7.11 (m, 4H), 6.85–6.93 (m, 3H), 6.71–6.74 (m, 2H), 6.15–6.21 (m, 3H), 6.02 (d, J = 6.6 Hz, 1H), 5.19 (dd, J = 16.5 Hz, 2H, –CH2). 13C NMR (d6-DMSO): δ(ppm) = 188.8, 167.5, 166.5, 164.4, 154.2, 153.9, 149.5, 149.4, 148.7, 144.4, 143.0, 141.9, 138.8, 137.7, 135.5, 135.3, 131.9, 130.5, 130.3, 130.2, 129.7, 128.7, 128.1, 127.1, 125.3, 125.1, 124.9, 124.6, 124.0, 123.4, 123.1, 121.1, 120.3, 120.1, 117.5, 114.3, 113.4, 112.8, 49.9. Found: C, 53.49; H, 3.67; N, 7.78%. C41H31N5PF6Ir·0.5CH3CN requires C, 53.02; H, 3.44; N, 8.10%.
Acknowledgements
This work has been supported by the Australian Research Council (FT100100646, LE120100213, DP150102741). The Victorian Partnership for Advanced Computing (VPAC), the National Computational Infrastructure National Facility (NCI-NF) and La Trobe University are acknowledged for substantial computing resources.Notes and references
- L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC.
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed.
- R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC.
- S. Bellemin-Laponnaz and S. Dagorne, Chem. Rev., 2014, 114, 8747–8774 CrossRef CAS PubMed.
- K. Matsumoto, N. Matsumoto, A. Ishii, T. Tsukuda, M. Hasegawa and T. Tsubomura, Dalton Trans., 2009, 6795–6801 RSC.
- V. J. Catalano, L. B. Munro, C. E. Strasser and A. F. Samin, Inorg. Chem., 2011, 50, 8465–8476 CrossRef CAS PubMed.
- X. Zhang, S. Gu, Q. Xia and W. Chen, J. Organomet. Chem., 2009, 694, 2359–2367 CrossRef CAS PubMed.
- A. Poethig and T. Strassner, Organometallics, 2011, 30, 6674–6684 CrossRef CAS.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price, B. W. Skelton and A. H. White, Dalton Trans., 2004, 1038–1047 RSC.
- V. J. Catalano and A. O. Etogo, J. Organomet. Chem., 2005, 690, 6041–6050 CrossRef CAS PubMed.
- P. J. Barnard, L. E. Wedlock, M. V. Baker, S. J. Berners-Price, D. A. Joyce, B. W. Skelton and J. H. Steer, Angew. Chem., Int. Ed., 2006, 45, 5966–5970 CrossRef CAS PubMed.
- F.-F. Hung, W.-P. To, J.-J. Zhang, C. Ma, W.-Y. Wong and C.-M. Che, Chem. – Eur. J., 2014, 20, 8604–8614 CrossRef CAS PubMed.
- Q.-X. Liu, F.-B. Xu, Q.-S. Li, H.-B. Song and Z.-Z. Zhang, Organometallics, 2004, 23, 610–614 CrossRef CAS.
- Q.-X. Liu, H.-B. Song, F.-B. Xu, Q.-S. Li, X.-S. Zeng, X.-B. Leng and Z.-Z. Zhang, Polyhedron, 2003, 22, 1515–1521 CrossRef CAS.
- Y. Unger, D. Meyer and T. Strassner, Dalton Trans., 2010, 39, 4295–4301 RSC.
- Y. Unger, D. Meyer, O. Molt, C. Schildknecht, I. Münster, G. Wagenblast and T. Strassner, Angew. Chem., Int. Ed., 2010, 49, 10214–10216 CrossRef CAS PubMed.
- R. Wai-Yin Sun, A. Lok-Fung Chow, X.-H. Li, J. J. Yan, S. Sin-Yin Chui and C.-M. Che, Chem. Sci., 2011, 2, 728–736 RSC.
- F. Zhang, L. Duan, J. Qiao, G. Dong, L. Wang and Y. Qiu, Org. Electron., 2012, 13, 1277–1288 CrossRef CAS PubMed.
- F. Kessler, R. D. Costa, D. Di Censo, R. Scopelliti, E. Orti, H. J. Bolink, S. Meier, W. Sarfert, M. Gratzel, M. K. Nazeeruddin and E. Baranoff, Dalton Trans., 2012, 41, 180–191 RSC.
- F. Monti, F. Kessler, M. Delgado, J. Frey, F. Bazzanini, G. Accorsi, N. Armaroli, H. J. Bolink, E. Ortí, R. Scopelliti, M. K. Nazeeruddin and E. Baranoff, Inorg. Chem., 2013, 52, 10292–10305 CrossRef CAS PubMed.
- Y. Zhou, J. Jia, W. Li, H. Fei and M. Zhou, Chem. Commun., 2013, 49, 3230–3232 RSC.
- S. U. Son, K. H. Park, Y.-S. Lee, B. Y. Kim, C. H. Choi, M. S. Lah, Y. H. Jang, D.-J. Jang and Y. K. Chung, Inorg. Chem., 2004, 43, 6896–6898 CrossRef CAS PubMed.
- G. Duan and V. W.-W. Yam, Chem. – Eur. J., 2010, 16, 12642–12649 CrossRef CAS PubMed.
- J. Dinda, S. Liatard, J. Chauvin, D. Jouvenot and F. Loiseau, Dalton Trans., 2011, 40, 3683–3688 RSC.
- D. G. Brown, N. Sanguantrakun, B. Schulze, U. S. Schubert and C. P. Berlinguette, J. Am. Chem. Soc., 2012, 134, 12354–12357 CrossRef CAS PubMed.
- H.-J. Park, S. Yoo, I.-S. Shin, Y. K. Chung and J. Kim, Electroanalysis, 2013, 25, 1111–1115 CrossRef CAS PubMed.
- V. Leigh, W. Ghattas, R. Lalrempuia, H. Muller-Bunz, M. T. Pryce and M. Albrecht, Inorg. Chem., 2013, 52, 5395–5402 CrossRef CAS PubMed.
- L.-H. Chung, S.-C. Chan, W.-C. Lee and C.-Y. Wong, Inorg. Chem., 2012, 51, 8693–8703 CrossRef CAS PubMed.
- L. A. Casson, S. Muzzioli, P. Raiteri, B. W. Skelton, S. Stagni, M. Massi and D. H. Brown, Dalton Trans., 2011, 40, 11960–11967 RSC.
- X.-W. Li, H.-Y. Li, G.-F. Wang, F. Chen, Y.-Z. Li, X.-T. Chen, Y.-X. Zheng and Z.-L. Xue, Organometallics, 2012, 31, 3829–3835 CrossRef CAS.
- C. Romain, S. Choua, J.-P. Collin, M. Heinrich, C. Bailly, L. Karmazin-Brelot, S. Bellemin-Laponnaz and S. Dagorne, Inorg. Chem., 2014, 53, 7371–7376 CrossRef CAS PubMed.
- G. J. Barbante, P. S. Francis, C. F. Hogan, P. R. Kheradmand, D. J. D. Wilson and P. J. Barnard, Inorg. Chem., 2013, 52, 7448–7459 CrossRef CAS PubMed.
- B. D. Stringer, L. M. Quan, P. J. Barnard, D. J. D. Wilson and C. F. Hogan, Organometallics, 2014, 33, 4860–4872 CrossRef CAS.
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS PubMed.
- B. A. Gorman, P. S. Francis and N. W. Barnett, Analyst, 2006, 131, 616–639 RSC.
- W. Miao, Chem. Rev., 2008, 108, 2506–2553 CrossRef CAS PubMed.
- X. Zhou, D. Zhu, Y. Liao, W. Liu, H. Liu, Z. Ma and D. Xing, Nat. Protoc., 2014, 9, 1146–1159 CrossRef CAS PubMed.
- G. J. Barbante, C. F. Hogan and A. B. Hughes, J. Solid State Electrochem., 2009, 13, 599–608 CrossRef CAS.
- T. Joshi, G. J. Barbante, P. S. Francis, C. F. Hogan, A. M. Bond, G. Gasser and L. Spiccia, Inorg. Chem., 2012, 51, 3302–3315 CrossRef CAS PubMed.
- T. Joshi, G. J. Barbante, P. S. Francis, C. F. Hogan, A. M. Bond and L. Spiccia, Inorg. Chem., 2011, 50, 12172–12183 CrossRef CAS PubMed.
- E. F. Reid, V. C. Cook, D. J. D. Wilson and C. F. Hogan, Chem. – Eur. J., 2013, 19, 15907–15917 CrossRef CAS PubMed.
- R. V. Kiran, C. F. Hogan, B. D. James and D. J. D. Wilson, Eur. J. Inorg. Chem., 2011, 4816–4825 CrossRef CAS PubMed.
- E. H. Doeven, E. M. Zammit, G. J. Barbante, C. F. Hogan, N. W. Barnett and P. S. Francis, Angew. Chem., Int. Ed., 2012, 51, 4354–4357 CrossRef CAS PubMed.
- E. H. Doeven, E. M. Zammit, G. J. Barbante, P. S. Francis, N. W. Barnett and C. F. Hogan, Chem. Sci., 2013, 4, 977–982 RSC.
- G. J. Barbante, N. Kebede, C. M. Hindson, E. H. Doeven, E. M. Zammit, G. R. Hanson, C. F. Hogan and P. S. Francis, Chem. – Eur. J., 2014, 20, 14026–14031 CrossRef CAS PubMed.
- E. Kerr, E. H. Doeven, G. J. Barbante, C. F. Hogan, D. Bower, P. S. Donnelly, T. Connell and P. S. Francis, Chem. Sci., 2015, 6, 472–479 RSC.
- A. Kapturkiewicz and G. Angulo, Dalton Trans., 2003, 3907–3913 RSC.
- F. Zhang, L. Duan, J. Qiao, G. Dong, L. Wang and Y. Qiu, Org. Electron., 2012, 13, 2442–2449 CrossRef CAS PubMed.
- C.-H. Chien, S. Fujita, S. Yamoto, T. Hara, T. Yamagata, M. Watanabe and K. Mashima, Dalton Trans., 2008, 916–923 RSC.
- A. Sinha, S. M. Wahidur Rahaman, M. Sarkar, B. Saha, P. Daw and J. K. Bera, Inorg. Chem., 2009, 48, 11114–11122 CrossRef CAS PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau, M. E. Thompson, R. J. Holmes and S. R. Forrest, Inorg. Chem., 2005, 44, 7992–8003 CrossRef CAS PubMed.
- R. D. Costa, R. Casillas and J. Cano, J. Phys. Chem. C, 2013, 117, 8545–8555 CAS.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377–7387 CrossRef CAS PubMed.
- M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. De Cola, Adv. Funct. Mater., 2010, 20, 1812–1820 CrossRef CAS PubMed.
- T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed.
- W. Miao, J.-P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef CAS PubMed.
- Y. Cheng, H.-J. Xu, J.-F. Sun, Y.-Z. Li, X.-T. Chen and Z.-L. Xue, Dalton Trans., 2009, 7132–7140 RSC.
- W. Ghattas, H. Müller-Bunz and M. Albrecht, Organometallics, 2010, 29, 6782–6789 CrossRef CAS.
- J. J. Concepcion, J. W. Jurss, M. R. Norris, Z. Chen, J. L. Templeton and T. J. Meyer, Inorg. Chem., 2010, 49, 1277–1279 CrossRef CAS PubMed.
- H.-S. Chen, W.-C. Chang, C. Su, T.-Y. Li, N.-M. Hsu, Y. S. Tingare, C.-Y. Li, J.-H. Shie and W.-R. Li, Dalton Trans., 2011, 40, 6765–6770 RSC.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 rev. D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- A. D. Becke, Phys. Rev. A: Gen. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS PubMed.
- J. P. Perdew, in Electronic Structure of Solids ‘91, ed. P. Ziesche and H. Eschrig, Akademie Verlag, Berlin, 1991, pp. 11–20 Search PubMed.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. v. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS PubMed.
- T. H. Dunning Jr. and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer, III Plenum, New York, 1976, vol. 3, pp. 1–28 Search PubMed.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- J. Lin, K. Wu and M. Zhang, J. Comput. Chem., 2009, 30, 2056–2063 CrossRef CAS PubMed.
- G. J. Barbante, C. F. Hogan, D. J. D. Wilson, N. A. Lewcenko, F. M. Pfeffer, N. W. Barnett and P. S. Francis, Analyst, 2011, 136, 1329–1338 RSC.
- A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2517 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS PubMed.
- S. I. Gorelsky, AOMix: Program for Molecular Orbital Analysis, Version 6.46, University of Ottawa, http://www.sg-chem.net/, 2010 Search PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales and F. Weinhold, NBO 5.9, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2001 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallography data, molecular orbital analysis and plots, TD-DFT calculated absorption spectra. CCDC 1031786–1031788. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03378g |
| This journal is © The Royal Society of Chemistry 2015 |