
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural and luminescence studies of the new nitridomagnesoaluminate CaMg2AlN3
M.
Kubus
*a,
K.
Levin
b,
S.
Kroeker
*b,
D.
Enseling
c,
T.
Jüstel
c and
H.-J.
Meyer
a
aSection for Solid State and Theoretical Inorganic Chemistry, Institute of Inorganic Chemistry, Eberhard-Karls-Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: mariusz.kubus@anorg.uni-tuebingen.de; Fax: +49-(0)7071-29-5702
bDepartment of Chemistry, University of Manitoba, Winnipeg, Manitoba, R3T 2N2, Canada. E-mail: scott.kroeker@umanitoba.ca
cLabor für Angewandte Materialwissenschaften, Fachhochschule Münster, Stegerwaldstrasse 39, 48565 Steinfurt, Germany
First published on 17th December 2014
Abstract
CaMg2AlN3 was synthesized in a closed system by solid state reaction from binary nitrides. Structure refinements based on powder X-ray diffraction data suggested ambiguity about the occupancy of magnesium and aluminum tetrahedral sites. Solid-state 27Al and 25Mg NMR studies were used to adjudicate amongst possible space groups. With reference to projector augmented wave calculations of the quadrupolar coupling constants, the measured values of CQ and the numbers of crystallographically inequivalent Al and Mg sites indicate that CaMg2AlN3 crystallizes in the space group P63/mmc with partial occupancy of the distorted tetrahedral Al site and possibly also mixing of Mg2+ and Al3+ ions on opposite sites. The compound obtained by synthesis with a flux shows orange defect-related luminescence at room temperature.
Introduction
Many new ternary and quaternary nitrides have been obtained in recent years, however their synthesis is very challenging because of required synthesis conditions and reaction partners. The reactants and products are often easily hydrolyzed or oxidized in air.1 Preparations of nitrides with a highly condensed network structure are mainly performed by high-temperature solid state syntheses.2 However, it is also possible to synthesize such nitrides at low temperature via solid state metathesis reactions.3 A steady increase in the importance of nitride-based materials is devoted to the employment of these materials as host structures of phosphors. The temperature and pressure conditions for the production of the nitride-based phosphors are crucial because the starting materials behave chemically inert in reactions.4 A flux agent can be used to enhance the yield of nitrides synthesis, e.g. molten sodium,5 or liquid lithium.6 In many cases pure nitrides without oxygen impurities are difficult to make and special attention should be paid to the choice of substrates.7Ternary nitrides can be divided into two groups, intermetallic with dominant metal–metal interactions and ionic/covalent where metal–nitrogen bonding is dominant. Intermetallic nitrides with ionic or covalent bonds may have many different metal–anion coordination environments with structures related to oxides or sulfides.8
Nitridosilicates, nitridoaluminosilicates, oxonitridosilicates, and oxonitridoaluminosilicates are usually constructed of [M(N,O)4] tetrahedra (M = Al, Si) which are connected with each other to form isolated polyhedra, or one-, two- and three-dimensional structures.2,9 For example edge-bridged dimers of tetrahedra were reported as [Al2N6]12− in the structure of Ca6Al2N6.10 One-dimensional chains of edge sharing [AlN4] units contain M3Al2N4 structures with M = Sr and Ba.10 Two-dimensional sheets built from corner- and edge-sharing tetrahedra are found in MMg2N2 with Ca and Sr.11 Layered structures also appear for β-Ca3Al2N4 and LiCaAlN2 containing edge- and vertex-sharing [AlN4]-tetrahedra.11b,12 An important nitride is CaAlSiN3 with a three-dimensional network structure built from vertex-sharing [(Si,Al)N4]-tetrahedra forming [(Si,Al)6N6] rings.2 CaAlSiN3 doped with Eu2+ ions is one of the most successfully commercialized nitride which can be used in LEDs.13 In view of the technological significance of this material, we have targeted the closely related compound, CaMg2AlN3, by complete substitution of silicon by magnesium, resulting in a unique luminescent nitride which does not require doping by activator elements.
Solid-state NMR is becoming increasingly common as a complementary tool in the determination of crystal structures, particularly for materials possessing some degree of disorder or lacking sufficiently large crystals for single-crystal X-ray crystallography.14 Along with high-level computations of magnetic shielding and electric field gradients, precisely measured NMR parameters can provide key constraints on atomic positions, local symmetry elements and the number of crystallographically inequivalent sites. Improvements in hardware and methodology continue to expand the range of nuclei for which structurally informative NMR spectra may be obtained.
Experimental section
Synthesis
Preparation of the reaction mixture for the synthesis of CaMg2AlN3 was performed under argon atmosphere inside a glove box. The reaction carried out in a fused niobium reaction container, sealed into a silica ampoule. 128 mg Mg3N2 (Alfa Aesar, 99.6%) with 94 mg Ca3N2 (Aldrich, 99%), and 78 mg AlN (ABCR, grade C) were mixed in an agate mortar. For luminescent applications the material was synthesized additionally with 10 wt% of a flux. The flux was a mixture of LiCl (Sigma, 99.0%) with KCN (Merck, 96%) in molar ratio one to one.The mixtures were heated in a furnace to 1100 °C with a heating rate of 2 °C min−1, held at this temperature for 48 h, and cooled to room temperature with rate 2 °C min−1. The containers were opened under argon atmosphere, and the products were quickly flushed with dry ethanol in order to remove any possible impurities. The reaction product was analyzed by powder X-ray diffraction measurements.
X-Ray diffraction
Powder X-ray diffraction measurements were carried out with a powder diffractometer (Stoe, STADI-P, Ge-monochromator) using CuKα1 (λ = 154.06 pm) radiation.N/O analysis
All nitrogen and oxygen contents were determined using a TC-400C from Leco Corporation, equipped with TCWin Analysis software, version 4.03, and Helium 4.6 (Westfalen) as purge gas. For every analysis between 10–20 mg of the material were weighed, transferred into tin capsules and sealed. For a more homogeneous combustion three scraps of highly pure nickel wire were added. The subsequent analysis was conducted at 1000 A in high purity carbon crucibles and repeated three times.Nuclear magnetic resonance spectroscopy
27Al MAS NMR was performed on a Varian Inova 600 (B0 = 14.1 T) spectrometer (ν0 = 156.3 MHz) with a 1.6 mm TR Varian-Chemagnetics MAS probe at a 30 kHz rotational frequency. 200k transients were co-added using the Bloch-decay sequence with an optimized recycle delay of 1 s and excitation pulse of 0.5 μs (ca. 40° tip angle). The frequency axis is referenced to 1 M Al(NO3)3 (aq.) at 0 ppm.25Mg NMR was done on a Bruker Avance II 900 MHz (B0 = 21.1 T) spectrometer. The rotor-synchronized spin-echo MAS NMR spectrum was collected with a 4 mm low-γ probe at a rotor spinning frequency of 18 kHz, a recycle delay of 1 second and 60k scans. 25Mg quadrupole-echo NMR spectra of a non-spinning sample were collected with a 5 mm low-γ probe using an optimized recycle delay of 1 second and 54k scans. Chemical shifts are referenced to 1 M MgCl2 (aq.) at 0 ppm.
Spectral fits were achieved using WSOLIDS and dmfit.15 All samples were handled in a glove box under a dry nitrogen atmosphere.
Quantum chemical calculations
Density functional theory (DFT) calculations were performed using CASTEP,16 which incorporates GIPAW optimized for the calculation of electronic properties in periodic solids.17 Calculations implemented Perdew–Burke–Ernzerhof (PBE) functionals. Fine accuracy plane-wave basis sets with a 400 eV cutoff were used. The CASTEP code was run in the Materials Studio 4.4 environment on an HP xw4400 workstation with a single Intel Dual-Core 2.67 GHz processor and 8 GB DDR RAM.The input data for the GIPAW calculations used the experimental lattice parameters and atomic coordinates. Geometry optimizations allowed atomic positions to change within fixed lattice parameters and space-group symmetry. The construction of supercells maintained the atomic positions found by X-ray refinement but removed some atoms to mimic partial occupancy.
Luminescence spectroscopy
Excitation and emission spectra were recorded with an Edinburgh Instruments FSL920 spectrometer equipped with a 450 W Xe arc lamp, mirror optics for powder samples and a cooled (−20 °C) single-photon counting photomultiplier from Hamamatsu (R2658P). The correction file for the emission spectra was obtained from a tungsten incandescent lamp certified by the NPL (National Physics Laboratory, UK).Results and discussion
In the past, relatively little attention has been paid to magnesium nitride compounds and many of them may not be structurally characterized. As expected magnesium nitrides should form a similar structural diversity as the Ca, Sr or Ba compounds and only recently new materials such as SrMg3GeN4, SrMg2Ga2N4, MMg2Al2N4 (M = Ca, Sr, Ba, Eu), BaMg2Ga2N4, Mg3GaN3, Li2−xTa2+xN4, Mg2.6−xTa1.3+xN4, BaMg3.33M0.67N4 (M = Nb, Ta), Li0.51Mg2.49N1.83 or Li1.12Mg0.88N0.96 were obtained and described.1,9a,18The new nitridomagnesoaluminate compound CaMg2AlN3 was prepared at 1100 °C in a straightforward manner from Mg3N2, Ca3N2, and AlN (1).
| 2Mg3N2 + Ca3N2 + 3AlN → 3CaMg2AlN3 | (1) |
The obtained product exhibited sensitivity to moist air and had to be kept under inert air conditions. An N/O-analysis did not indicate a significant substitution of nitride versus oxide in the structure of CaMg2AlN3. The overall oxygen content according to the measurement was about 0.6%.
The crystal structure of CaMg2AlN3 is closely related to the structures of ScAl3C3 (a = 3.355(1) Å, c = 16.776(3) Å)19 and UAl3C3 (a = 3.389(1) Å, c = 17.394(3) Å).19 The crystal structure determination of these compounds was not straightforward. Reminiscent of previous work on carbides, the refinements of the ScAl3C3 and UAl3C3 were performed in the high-symmetry space group P63/mmc with a large displacement parameter for one aluminum position or, alternatively, with a split aluminum position.20 Afterwards, the crystal structures were redetermined with full occupancy of the aluminum sites in the space group P63mc.19 The main difference between the structural results is a trigonal bipyramidal, or split tetrahedral environment of aluminum in the space group P63/mmc, and a tetrahedral environment when the structures are refined in the space group P63mc.19
A similar picture is evident in the case of aluminum ions in the structure of CaMg2AlN3. An analysis of the extinction conditions in the X-ray diffraction data set clearly leads to the space groups P63/mmc and P63mc, including related subgroups and supergroups. Structure refinements for CaMg2AlN3 were carried out in both space groups by the Rietveld method using the FullProf-WinPLOTR software, with the results displayed in Table 1 and Fig. 1.21
 | ||
| Fig. 1 Projection of the crystal structure of CaMg2AlN3, space group P63/mmc (left) and P63mc (right), with highlighted [MgN4] tetrahedra (grey), [AlN4] tetrahedra (dark grey), and [CaN6] octahedra (transparent). Al and Mg occupy two crystallographic (4f) sites in P63/mmc, of which the Al position is split (occupancy 1/2) above and below the (mz) mirror plane (left). In P63mc Al1, Mg1, and Mg2 occupy three crystallographic (2b) sites (right). Atom types: Mg (red and brown), Al (grey with white front ellipsis), Ca (blue) and N (black). Unit cells are outlined with solid grey lines. | ||
| Crystal data | |||||
|---|---|---|---|---|---|
| a Split position: 1/3, 2/3, z; 1/3, 2/3, −z + 1/2, with 50% occupancy. | |||||
| Space group | P63/mmc (194), Z = 2 | ||||
| Unit cell dimensions | a = 3.4087(1) Å, c = 17.3760(5) Å, γ = 120° | ||||
| Cell volume | 174.84(1) Å3 | ||||
| Data range | 5° ≤ 2θ ≤ 100° | ||||
| Temperature | 293 K | ||||
| Atomic coordinates | |||||
| Atom | Wyck. | x | y | z | B iso (Å2) |
| Ca | 2a | 0 | 0 | 0 | 2.22(7) |
| Mg | 4f | 1/3 | 2/3 | 0.1330(2) | 2.16(8) |
| Al | 4fa | 1/3 | 2/3 | 0.7347(5) | 2.5(2) |
| N1 | 2b | 1/3 | 2/3 | 0.5950(3) | 1.9(1) |
| N2 | 4f | 1/3 | 2/3 | 1/4 | 1.9(1) |
| Rietveld parameters | |||||
| RBragg/RF | 0.0498/0.0928 | ||||
| Crystal data | |||||
| Space group | P63mc (186), Z = 2 | ||||
| Unit cell dimensions | a = 3.4087(1) Å, c = 17.3758(4) Å, γ = 120° | ||||
| Cell volume | 174.84(1) Å3 | ||||
| Data range | 5° ≤ 2θ ≤ 100° | ||||
| Temperature | 293 K | ||||
| Atomic coordinates | |||||
| Atom | Wyck. | x | y | z | B iso (Å2) |
| Ca | 2a | 0 | 0 | 0 | 1.95(6) |
| Mg1 | 2b | 1/3 | 2/3 | 0.2575(6) | 2.65(9) |
| Mg2 | 2b | 1/3 | 2/3 | 0.6317(5) | 2.65(9) |
| Al | 2b | 2/3 | 1/3 | 0.3652(4) | 2.1(1) |
| N1 | 2b | 1/3 | 2/3 | 0.0972(7) | 1.6(1) |
| N2 | 2b | 1/3 | 2/3 | 0.3960(7) | 1.6(1) |
| N3 | 2b | 1/3 | 2/3 | 0.7437(7) | 1.6(1) |
| Rietveld parameters | |||||
| RBragg/RF | 0.0807/0.0748 | ||||
The structure refinement in P63/mmc resulted in a 50% split position of the Al3+ ions on a 4f site, introducing alternating occupations of two closely adjacent Al3+ positions (0.44(2) Å apart from each other) as a result of presence of a mirror plane perpendicular to the crystallographic c-axis at z = 1/4, 3/4. Mg2+ ions occupy another 4f site in a regular manner (Fig. 1, at left). The coordination environment of the aluminum ion in the structure of CaMg2AlN3 is trigonal pyramidal with one long distance to the N1 atom parallel to the c-axis direction (Table 2). The combination of both closely adjacent (split) Al3+ sites into one would introduce a trigonal bipyramidal environment. A trigonal coordination environment of Al ions in nitrides appears unknown to us, but has been reported for [GaN3] units in nitrides, e.g. Sr3GaN3 or Sr6GaN5.22 A similar trigonal bipyramidal coordination was found for magnesium ions in Mg3BN3 (P63/mmc) whose structure appears somehow related to that of CaMg2AlN3.23
| Ca–N1 (6×) | 2.5767(4) |
| Mg–N1(3×) | 2.0746(5) |
| Mg–N2 | 2.0243(1) |
| Al–N1 | 2.4208(1) |
| Al–N3(3×) | 1.9851(6) |
| N1–Mg–N2 | 108.447(5) |
| N1–Al–N2 | 97.524(5) |
According to the structure refinement of CaMg2AlN3 with the non-centrosymmetric space group P63mc, two Mg2+ ions occupy two 2b sites, and one Al3+ ion occupies another 2b site, resulting in essentially tetrahedral coordination environments for all cations.
In order to validate the probability of one or the other space group by energy measures, the Madelung part of the lattice energy (MAPLE) was calculated for both structural models.24 Calculated MAPLE values for space group P63/mmc and P63mc are 25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 903 and 25926 kJ mol−1, respectively. Due to the similarity of both structural models, these values are quite close to each other and may indicate the possibility of a coexistence of both arrangements. Moreover, these MAPLE values represent the sum of the constituting binary nitrides with a deviation of less than 2%.
903 and 25926 kJ mol−1, respectively. Due to the similarity of both structural models, these values are quite close to each other and may indicate the possibility of a coexistence of both arrangements. Moreover, these MAPLE values represent the sum of the constituting binary nitrides with a deviation of less than 2%.
The NMR analysis described in the following part of this article indicates that the correct space group is P63/mmc. Therefore, we focus our structural view on the assignment of the space group P63/mmc (no. 194) with lattice parameters a = 3.4087(1) Å and c = 17.3760(5) Å for two formula units CaMg2AlN3. The recorded diffraction pattern was fitted with two phases: CaMg2AlN3 and AlN as shown in Fig. 2. The relative weight proportions of CaMg2AlN3 and AlN in the sample determined by Rietveld refinement were 97 and 3%, respectively. The parameters of the refinement, obtained crystal data, and atomic coordinates are presented in Table 2. A pseudo-Voigt profile function was used to fit the reflection peaks.
 | ||
| Fig. 2 Experimental XRD pattern and the corresponding reflexes for CaMg2AlN3 (P63/mmc) and AlN. Observed powder XRD pattern (grey circles), calculated pattern (black line), Bragg positions (grey ticks), and difference curve (black bottom line) after Rietveld refinement. | ||
The crystal structure of CaMg2AlN3 is based on a hexagonal closest packing of nitride ions (N1 and N2). One third of octahedral sites are occupied by Ca2+ and one half of tetrahedral sites are occupied by Mg2+ and Al3+ ions. The distributions of these cations result in a layered arrangement with two alternating layers, [CaN]+ and [Mg2AlN2]−, the first of which corresponds to the NaCl type, and the second corresponds to an Al4C3-type structure. These layers are stacked in an alternating fashion along the crystallographic c-axis direction. Projections of related CaMg2AlN3, ScAl3C3, Al4C3 and Mg3BN3 structures are shown in Fig. 3.
 | ||
Fig. 3 Crystal structure of (a) CaMg2AlN3P63/mmc, (b) ScAl3C3P63mc,24 (c) Al4C3R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m25 and (d) Mg3BN3P63/mmc30 projected on the (110) plane. Atoms connected by solid lines represent NaCl-type layers, atom connected by dashed lines can be related to Al4C3-type layers. Unit cells are outlined with solid grey lines. m25 and (d) Mg3BN3P63/mmc30 projected on the (110) plane. Atoms connected by solid lines represent NaCl-type layers, atom connected by dashed lines can be related to Al4C3-type layers. Unit cells are outlined with solid grey lines. | ||
The bond lengths are summarized in Table 2. The distances between Ca2+ and N1 are in line with corresponding Ca–N distances in CaMg2N3 (2.596 Å).11a The Mg–N distances compare well with Mg–N contacts in [MgN4] tetrahedra of Mg3N2 (2.085–2.179 Å), or CaMg2N3 (2.128–2.296 Å). Aluminum ions are known to have tetrahedral coordination environments with N3− ions in nitrides. The Al–N2 distance for the trigonal environment of the [AlN4] trigonal pyramid of CaMg2AlN3 is close to the Al–N distance in AlN (1.889–1.902 Å) and in Ca3Al2N4 (1.98 Å). However, the Al–N1 distance with the pyramidal N1 is significantly longer. A mixed metal occupation is known for some nitride compounds, for example Mg3GaN3,with Mg2+ and Ga3+ ions occupying the same crystallographic site.9a A similar situation appears to exist in CaMg2AlN3, where partial occupation of the Al3+ site by Mg2+ ions (and vice versa) could result in an apparent lengthening of the Al–N1 contact.
Second harmonic generation measurement
A measurement of the second harmonic generation (SHG) effect was performed to get information about the presence or absence of inversion symmetry in the structure. A SHG signal of the powder sample was not observed, in comparison to other noncentrosymmetric powders which clearly did show such an effect. This “observation” is consistent with the other evidence suggesting a centrosymmetric space group (P63/mmc). However, it must be noted that only the opposite case, i.e., the appearance of a SHG signal (beyond surface SHG), can provide definitive proof of a noncentrosymmetric structure.27Al and 25Mg nuclear magnetic resonance
In order to resolve the ambiguity between space groups, solid-state NMR and theoretical calculations were used to probe the coordination environments and measure the number of crystallographically inequivalent Al3+ and Mg2+ sites in CaMg2AlN3. Whereas these two elements have similar X-ray scattering cross-sections, their NMR spectroscopic responses are completely different and can be probed independently. 27Al NMR is well established and readily available on most solid-state NMR spectrometers, but 25Mg NMR is less common and remains experimentally challenging even on state-of-the-art ultrahigh-field NMR spectrometers.26 Besides its 10% natural abundance and quadrupolar nature, the low magnetogyric ratio of 25Mg makes it inherently insensitive relative to many other typical NMR nuclei. To mitigate this limitation, all 25Mg NMR spectra were obtained using a 21.1 T magnet at the Canadian Ultrahigh-Field NMR Facility for Solids.From an NMR perspective (i.e., short-range order), the structural differences between the proposed space groups are straightforward. P63/mmc (no. 194) is characterized by a single [AlN4] site with highly distorted tetrahedral geometry and a single, slightly distorted [MgN4] tetrahedron. P63mc (no. 186) possesses a single [AlN4] site in a trigonal pyramidal environment and two distinct Mg sites, one in standard tetrahedral environment (Mg2) and the other in trigonal pyramidal environment (Mg1). NMR is capable of quantifying the number of crystallographically inequivalent sites for each nucleus and measuring the quadrupole coupling constants (CQ) at 27Al and 25Mg as a good indication of the degree of deviation from ideal tetrahedral symmetry. Pure cubic point symmetry produces a constant electric field at the cubic site, whereas geometric distortions generate progressively larger electric field gradients which couple with the nuclear electric quadrupole moment and thus produce the quadrupolar interaction. To assist in predicting the CQs associated with a given geometric arrangement, theoretical calculations of the proposed structures were employed.
27Al MAS NMR reveals a single peak with a chemical shift associated with the [AlN4] environment (Fig. 4).27 The peak shape indicates the presence of structural disorder, appearing more like a typical glassy 27Al NMR signal than a crystalline line shape. This spectrum was fit using a Czjzek model to account for the presence of local geometric distributions,15b,28 yielding an average quadrupole coupling constant in the range of 5.4 to 5.8 MHz. Long acquisitions times and a wide spectral window provided no evidence of a second aluminum site with a larger quadrupolar interaction.
 | ||
| Fig. 4 27Al MAS NMR spectrum of CaMg2AlN3 acquired at 14.1 T. The top trace is calculated using a Czjzek distribution with δiso = 120 ± 2 ppm and CQ = 5.6 ± 0.2 MHz. | ||
Likewise, the 25Mg MAS NMR spectrum consists of a single peak with a line shape indicative of disorder (Fig. 5). The chemical shift is consistent with a previous measurement of tetrahedral [MgN4] in Mg3N229 and the line shape may be modelled as above using a single Mg site with CQ = 6.5 ± 0.5 MHz. Considering the low intrinsic sensitivity of 25Mg NMR and the possibility of broad signals exceeding the spinning rate, a quadrupole-echo experiment on a non-spinning sample was acquired to seek additional intensity signifying a high-CQ site (Fig. 6). The spectrum obtained can be successfully modelled using the same parameters as the 25Mg MAS NMR spectrum and provides no evidence of additional sites. While the presence of structural disorder – and hence, a distribution of NMR parameters – implies that this fit may not be unique, it places an upper limit on the average value of CQ.
 | ||
| Fig. 5 25Mg MAS NMR spectrum of CaMg2AlN3 acquired at 21.1 T. The top trace is calculated using a Czjzek distribution with δiso = 100 ± 5 ppm and CQ = 6.5 ± 0.5 MHz. | ||
 | ||
| Fig. 6 25Mg quadrupole echo NMR spectrum of non-spinning CaMg2AlN3 acquired at 21.1 T. The top trace is calculated using a Czjzek distribution of the central transition only with δiso = 100 ± 5 ppm and CQ = 6.5 ± 0.5 MHz. | ||
Even without calculations, these NMR results point toward P63/mmc (194) as the correct space group, since there is no experimental evidence of the second Mg2+ site required by P63mc (186). Gauge-including projector augmented wave (GIPAW) calculations17 provide a firmer basis for this conclusion by estimating the quadrupolar interactions expected for each of the putative space groups. GIPAW calculations of P63mc CaMg2AlN3 predicts a CQ(27Al) of 6.4 MHz and CQ(25Mg) values of 2.9 and 17 MHz for the four- and three-coordinate Mg, respectively. Unusually large residual forces in the c-dimension resulting from direct usage of the X-ray coordinates suggested that geometry optimization may provide a more physically realistic structure. After energy minimization, the quadrupolar coupling constants changed to CQ(27Al) = 9.2 MHz and CQ(25Mg) = 8 and 19 MHz. While these values are substantially different than the unoptimized CQs, the main conclusion remains the same: two very different Mg sites are expected to be observed for this space group.
Due to the partial Al occupancy, GIPAW calculations of P63/mmc (194) necessitated the use of a 2 × 2 × 1 supercell. Various vacancy configurations gave a range of CQ(25Mg) values between 1.3 and 6.2 MHz, while CQ(27Al) of the highly distorted [AlN4] tetrahedron ranged from 9.5 to 10.3 MHz. While none of these calculations is expected to yield precise CQ values – especially those for the disordered model – they serve as useful guidelines for the relative magnitudes associated with particular local geometries. As such, they provide a perspective on the experimental results, indicating that for this space group a single disordered Mg2+ and a single disordered Al3+ site is predicted, each with moderate quadrupolar coupling constants. The unusual geometry adopted by Al in the crystallographic refinement leads to calculated CQ values which are likely overestimated since local distortions are expected to maximize tetrahedral regularity, consequently leading to reduced CQs. It is worth noting that the 27Al chemical shift is in excellent agreement with that of pure wurtzite AlN,27 suggesting that the local Al symmetry is approaching tetrahedral. A full energy minimization of the supercell would be required to test this hypothesis, but is beyond our current computational capacity. The experimental data confirm that a single type of each cation is observed with moderate CQ values, consistent with the P63/mmc space group. Perhaps most importantly, the spectral observation of a distribution of local Mg and Al geometric environments is compatible only with the space group implicating partial Al site occupancy, as subtle geometric distortions would undoubtedly accompany such vacancies and produce the observed line shapes. An alternate or additional explanation for this observation is that some degree of Mg/Al antisite disorder is present in the structure, which is only expected in the P63/mmc space group.
Luminescence spectroscopy
The excitation, emission and reflection spectra measured at room temperature of CaMg2AlN3 are presented in Fig. 7. | ||
| Fig. 7 Excitation, emission, and reflection spectra of CaMg2AlN3 synthesized with a flux, λEm = 630 nm and λEx = 330 nm. | ||
The reflection spectrum is composed of an absorption edge around 350 nm (3.54 eV), with an onset at 370 nm. The main feature of the excitation spectrum is a broad band with a maximum at 330 nm. Since excitation above 350 nm is not efficiently possible, the material is for the moment not of interest for solid state lighting, since (In,Ga)N chips emit between 370 nm and 500 nm. The Gaussian-type emission band is likely caused by defects introduced by the flux during the solid state reaction. The possible origins of the luminescence are structural and point defects as well as impurities incorporated during the synthesis process which affect the electrical and optical properties of the nitride host.30 The full-width-half-maximum (FWHM) obtained for CaMg2AlN3 is about 152 nm, much larger than for typical emission bands of Eu2+ ions in nitrides, for example 75 to 90 nm from CaAlSiN3:Eu2+ prepared by different methods.31 It is possible to draw a comparison between CaMg2AlN3 and the related compounds MMg2Al2N4:Eu2+ (M = Ca,Sr,Ba). BaMg2Al2N4:Eu2+ has similar luminescent properties even at room temperature with the large spectral width of emission (FWHM ∼ 115 nm) and large Stokes shift.18c These properties were ascribed to the emission from the trapped exciton states, for the calcium and strontium compound at higher temperatures a normal emission from Eu2+ ions was dominant.
Decay of the defect transition was investigated (Fig. 8), and the kinetics were determined by a multi-exponential decay curve fitted with three time constants τ1 = 52 ± 3 (7%), τ2 = 308 ± 21 (34%) and τ3 = 817 ± 35 μs (59%). The values are significantly higher than for the [Xe]4f65d1–[Xe]4f7 transitions of Eu2+, usually about 1 μs.32 Defects in the crystal lattice lead usually to many pathways for the excitation energy and for this reason the luminescence lifetime of emission is long for such structure. A detailed explanation of the character of the luminescent centers with the measured lifetimes needs more study on structurally related and/or doped compounds.
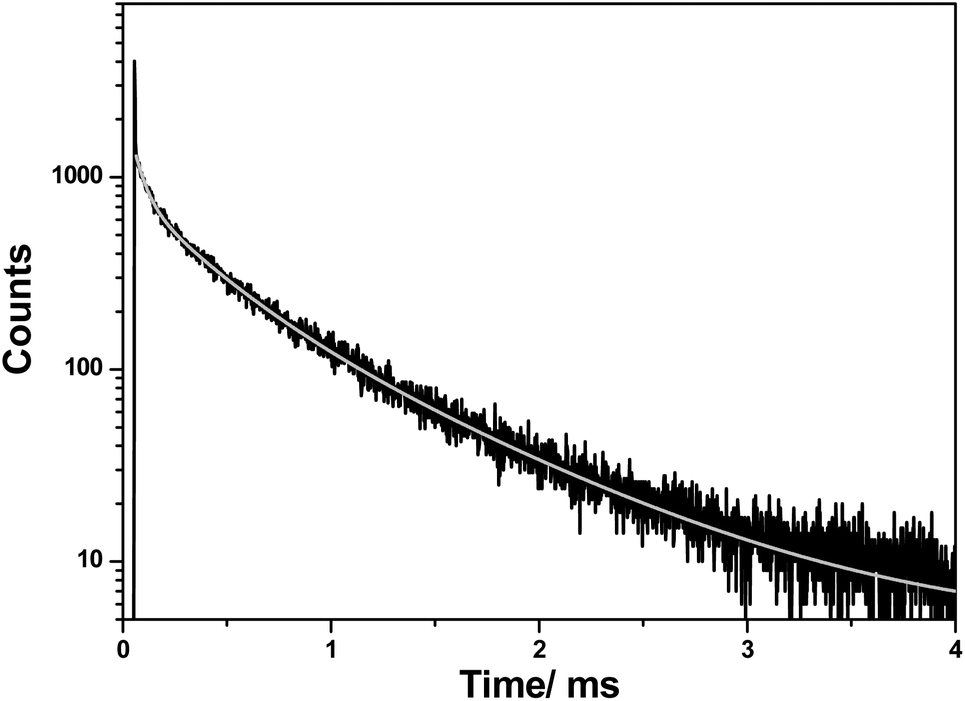 | ||
| Fig. 8 Decay curve of CaMg2AlN3 monitored at 630 nm upon 330 nm excitation. | ||
Conclusions
CaMg2AlN3 was successfully synthesized in a closed system by solid state reaction, leading to a layered crystal structure. X-ray powder diffraction indicates two possible space groups: P63mc and P63/mmc. The NMR results clearly indicate that the compound crystallizes in the space group P63/mmc (194) with a distorted tetrahedral environment around the aluminum atoms, including the possibility of a disordering of Mg2+ and Al3+ ions over opposite lattice sites in the structure. CaMg2AlN3 can undergo hydrolysis and oxidation, the same as most of the nitrides having low condensed network structures. The material prepared with a flux shows orange luminescence at room temperature. Its luminescence properties of this new material without a luminescent activator may open up a range of possible applications of undoped materials with advantages including broad, eye-friendly emission and cost efficiency. However, this requires a primary radiation source which emits below 350 nm.Funding sources
SK acknowledges the Natural Science and Engineering Research Council (NSERC) of Canada for operating support, and the Canada Foundation for Innovation (CFI) and the Manitoba Research Innovation Foundation (MRIF) for infrastructure funds. Access to the 900 MHz NMR spectrometer was provided by the National Ultrahigh-Field NMR Facility for Solids (Ottawa, Canada), a Canadian research facility funded by CFI, the Ontario Innovation Trust, Recherche Québec, the National Research Council Canada, and BrukerBioSpin and managed by the University of Ottawa (http://www.nmr900.ca). NSERC is acknowledged for a Major Resources Support grant.Acknowledgements
We like to express our gratitude to Dr Markus Ströbele and Dr Jochen Glaser for their help with the Rietveld refinement of the powder XRD data, and Dr Xiao Wang and Prof. Alfred Meixner from Eberhard-Karls-Universität Tübingen for SHG measurements. We are grateful to Drs Victor Terskikh and John Wren for technical assistance and helpful discussions.Notes and references
- D. G. Park, Y. Dong and F. J. DiSalvo, J. Solid State Chem., 2008, 10, 1846 CAS.
- M. Zeuner, S. Pagano and W. Schnick, Angew. Chem., Int. Ed., 2011, 50, 7754 CrossRef CAS PubMed.
- M. Kubus and H.-J. Meyer, Z. Anorg. Allg. Chem., 2013, 639, 669 CrossRef CAS.
- B. Lei, K. Machida, T. Horikawa and H. Hanzawa, Chem. Lett., 2010, 39, 104 CrossRef CAS.
- Z. A. Gal, P. M. Mallinson, H. J. Orchard and S. J. Clarke, Inorg. Chem., 2004, 43, 3998 CrossRef CAS PubMed.
- S. Pagano, S. Lupart, M. Zeuner and W. Schnick, Angew. Chem., Int. Ed., 2009, 121, 6453 CrossRef.
- R. J. Bruls, H. T. Hintzen and R. Metselaar, J. Mater. Sci., 1999, 4, 4519 CrossRef.
- S. T. Oyama, The Chemistry of Transition Metal Carbides and Nitrides, Blackie Academic & Professiona, 1996 Search PubMed.
- (a) F. Hintze, N. W. Johnson, M. Seibald, D. Muir, A. Moewes and W. Schnick, Chem. Mater., 2013, 25, 4044 CrossRef CAS; (b) W. Schnick and H. Huppertz, Chem. – Eur. J., 1997, 3, 679 CrossRef CAS.
- (a) M. Ludwig, R. Niewa and R. Kniep, Z. Naturforsch., B: Chem. Sci., 1999, 54, 461 CAS; (b) W. Blase, G. Cordier, M. Ludwig and R. Kniep, Z. Naturforsch., B: Chem. Sci., 1994, 49, 501 CAS.
- (a) O. Reckeweg and F. J. Disalvo, Z. Anorg. Allg. Chem., 2001, 6, 371 CrossRef; (b) P. Pust, S. Pagano and W. Schnick, Eur. J. Inorg. Chem., 2013, 1157 CrossRef CAS.
- M. Ludwig, J. Jäger, R. Niewa and R. Kniep, Inorg. Chem., 2000, 39, 5909 CrossRef CAS.
- C. C. Lin and R. Liu, J. Phys. Chem. Lett., 2011, 2, 1268 CrossRef CAS.
- R. K. Harris, R. E. Wasylishen and M. J. Duer, NMR Crystallography, John Wiley and Sons, Chichester, UK, 2009 Search PubMed.
- (a) K. Eichele and R. E. Wasylishen, WSolids1 – Solid State NMR Simulations, Dalhousie University, Halifax, Canada, 2001 Search PubMed; (b) D. Massiot, F. Fayon, M. Capron, I. King, S. Le Calve, B. Alonso, J. O. Durand, B. Bujoli, Z. H. Gan and G. Hoatson, Magn. Reson. Chem., 2002, 40, 70 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. J. Probert, K. Refson and M. C. Payne, Z. Kristallogr., 2005, 220, 567 CrossRef CAS.
- C. J. Pickard and F. Mauri, Phys. Rev. B: Condens. Matter, 2001, 63, 245101 CrossRef.
- (a) O. Reckeweg, J. C. Molstad and F. J. DiSalvo, J. Alloys Compd., 2001, 315, 134 CrossRef CAS; (b) A. N. Zhukov, K. P. Burdina and K. N. Semenenko, Russ. J. Gen. Chem., 1996, 66, 1046 Search PubMed; (c) P. Pust, F. Hintze, C. Hecht, V. Weiler, A. Locher, D. Zitnanska, S. Harm, D. Wiechert, P. J. Schmidt and W. Schnick, Chem. Mater., 2014, 26, 6113 CrossRef CAS; (d) T. Brokamp and H. Jacobs, J. Alloys Compd., 1992, 183, 325 CrossRef CAS; (e) H. Yamane, T. H. Okabe, O. Ishiyama, Y. Waseda and M. Shimada, J. Alloys Compd., 2001, 319, 124 CrossRef CAS.
- T. M. Gesing and W. Jeitschko, J. Solid State Chem., 1998, 140, 396 CrossRef CAS.
- (a) A. O. Tsokol’, O. I. Bodak, E. P. Marusin and M. G. Baivel'man, Sov. Phys. Crystallogr. (Engl. Transl.), 1986, 31, 467 Search PubMed; (b) T.-M. Gesing, R. Pöttgen, W. Jeitschko and U. Wortmann, J. Alloys Compd., 1992, 186, 321 CrossRef CAS.
- (a) T. Roisnel and J. Rodriguez-Carvajal, WinPLOTR: A Windows Tool for Powder Diffraction Pattern Analysis, Paper presented at the Proceedings of the Seventh European Powder Diffraction Conference (EPDIC 7), 2000 Search PubMed; (b) J. Rodriguez-Carvajal, Phys. B, 1993, 192, 55 CrossRef CAS.
- D. G. Park, Z. A. Gal and F. J. DiSalvo, Inorg. Chem., 2003, 42, 1779 CrossRef CAS PubMed.
- H. Hiraguchi, H. Hashizume, O. Fukunaga, A. Tekenaka and M. Sakato, J. Appl. Crystallogr., 1991, 24, 286 CrossRef CAS.
- R. Hübbenthal, MAPLE, Programm zur Berechnung des Madelunganteils der Gitterenergie, Version 4, Universität Gießen, Germany, 1993 Search PubMed.
- T. M. Gesing and W. Jeitschko, Z. Naturforsch., B: Chem. Sci., 1995, 50, 196 CAS.
- I. L. Moudrakovski, Annu. Rep. NMR Spectrosc., 2013, 79, 129 CrossRef CAS.
- R. Dupree, M. H. Lewis and M. E. Smith, J. Appl. Crystallogr., 1988, 21, 109 CrossRef CAS.
- D. R. Neuville, L. Cormier and D. Massiot, Geochim. Cosmochim. Acta, 2004, 68, 5071 CrossRef CAS PubMed.
- P. J. Pallister, I. L. Moudrakovskii and J. A. Ripmeester, Phys. Chem. Chem. Phys., 2009, 11, 11487 RSC.
- M. A. Reshchikov and H. Morkoc, J. Appl. Phys., 2005, 97, 061301 CrossRef PubMed.
- (a) M. Mikami, H. Watanabe, K. Uheda, S. Shimooka, Y. Shimomura, T. Kurushima and N. Kijima, IOP Conf. Ser.: Mater. Sci. Eng., 2009, 1, 1–10 Search PubMed; (b) X. Piao, K. Machida, T. Horikawa and H. Hanzawa, Chem. Mater., 2007, 19, 4592 CrossRef CAS.
- S. H. M. Poort, A. Meyerink and G. Blasse, J. Phys. Chem. Solids, 1997, 58, 1451 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |