
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of phosphinoferrocene amides and thioamides from carbamoyl chlorides and the structural chemistry of Group 11 metal complexes with these mixed-donor ligands†
Tiago A.
Fernandes
a,
Hana
Solařová
a,
Ivana
Císařová
a,
Filip
Uhlík
b,
Martin
Štícha
c and
Petr
Štěpnička
*a
aDepartment of Inorganic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 12840 Prague 2, Czech Republic. E-mail: stepnic@natur.cuni.cz
bDepartment of Physical and Macromolecular Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 12840 Prague 2, Czech Republic
cDepartment of Organic Chemistry, Faculty of Science, Charles University in Prague, Hlavova 2030, 12840 Prague 2, Czech Republic
First published on 24th December 2014
Abstract
The reaction of in situ generated 1′-(diphenylphosphino)-1-lithioferrocene with carbamoyl chlorides, ClC(E)NMe2, affords the corresponding (thio)amides, Ph2PfcC(E)NMe2 (E = O (2), S (3); fc = ferrocene-1,1′-diyl). These compounds as well as their analogues, Ph2PfcC(O)NHMe (4) and Ph2PfcC(O)NH2 (5), prepared from 1′-(diphenylphosphino)ferrocene-1-carboxylic acid (Hdpf) were studied as ligands for the Group 11 metal ions. In the reactions with [Cu(MeCN)4][BF4], the amides give rise to bis-chelate complexes of the type [Cu(L-κ2O,P)2][BF4]. Similar products, [Ag(L-κ2O,P)2]ClO4, are obtained from silver(I) perchlorate and 2, 4 or 5. In contrast, the reaction of AgClO4 with 3 produces a unique molecular dimer [Ag(3)(ClO4-κO)]2, where the metal centres are bridged by the sulfur atoms of the P,S-chelating thioamides. The reactions of 2–5 with [AuCl(tht)] (tht = tetrahydrothiophene) afford the expected gold(I)–phosphine complexes, [AuCl(L-κP)], containing uncoordinated (thio)amide moieties. Hemilabile coordination of the phosphinoamide ligands in complexes with the soft Group 11 metal ions is established by the crystal structure of a solvento complex, [Cu(5-κ2O,P)(5-κP)(CHCl3-κCl)][BF4], which was isolated serendipitously during an attempted crystallisation of [Cu(5-κ2O,P)2][BF4]. All of the compounds are characterised by spectroscopic methods, and the structures of several representatives of both the free phosphinoamides and their complexes are determined by X-ray diffraction analysis and further studied by DFT calculations and cyclic voltammetry.
Introduction
Phosphine donors modified with carboxamide substituents have evolved into a specific class of functional phosphine ligands with applications in coordination and supramolecular chemistry, catalysis, biomedical research, etc.1,2 The attractiveness of these compounds lies mainly in their structural modularity and facile synthesis, especially via amide coupling reactions.1During our studies3 on phosphinoferrocene carboxamides, we have also typically relied on the amide coupling reactions,4 employing 1′-(diphenylphosphino)ferrocene-1-carboxylic acid (Hdpf),5 suitable functional amines and conventional peptide coupling agents (route A in Scheme 1). Although this synthetic strategy proved very efficient, we felt that the search for alternative synthetic routes that are more straightforward and avoid the use of expensive stoichiometric reagents was still desirable. Thus far, we have demonstrated that phosphinoferrocene carboxamides can be synthesised equally well by lithiation of 1′-(diphenylphosphino)-1-bromoferrocene (1)6 and subsequent reaction of the lithiated intermediate with isocyanates (route B in Scheme 1).7
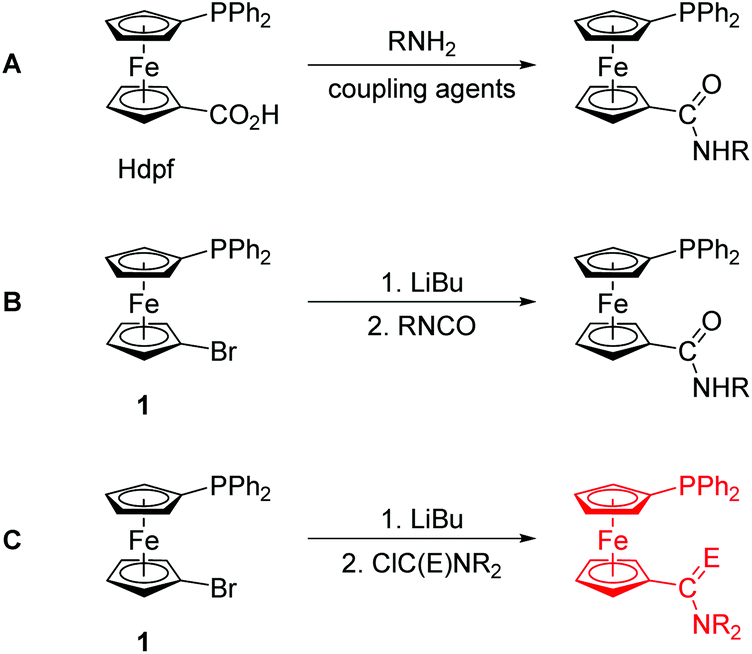 | ||
| Scheme 1 Synthetic routes to phosphinoferrocene carboxamides: conventional amidation (A) and lithiation/electrophilic quenching (B and C; E = O and S). | ||
In an attempt to extend this preparative strategy, we decided to replace isocyanates with carbamoyl chlorides (route C in Scheme 1).8 Although the choice of substituents is inherently limited in carbamoyl halides because of their high reactivity, we reasoned that a reaction of lithiated intermediates with these reagents9 could possibly offer an alternative direct route to phosphinoamides and their corresponding thioamides that do not require protection of the already present phosphine moiety from an undesired oxidation.10 In this regard, a practical synthesis of phosphine-thioamides is particularly attractive as it may provide access to this type of mixed-donor ligands and thus initiate investigations into their coordination properties, which still remain largely unexplored.11 To the best of our knowledge, there is only one report on the synthesis of a phosphine-thioamide donor via the rather unconventional Diels–Alder [4 + 2]-cycloaddition of N,N-dimethylthioacrylamide across 3,4-dimethyl-1-phenyl-1H-phosphole bonded to a Pd(II) centre.12 This solitary example markedly contrasts with the numerous studies that focus on the chemistry of phosphine-carbothioamides, Ph2PC(S)NR2.13
In this contribution, we describe the synthesis of a phosphinoferrocene carboxamide and thioamide via lithiation and electrophilic functionalisation of 1, and their model compounds obtained by the conventional amidation of Hdpf. The resulting hybrid ligands14 are structurally characterised through a combination of physicochemical and computational methods and further employed as donors for the soft Group 11 metal ions in order to investigate their coordination properties.
Results and discussion
Preparation and characterisation of phosphino-amide donors
Phosphinoferrocene carboxamide 2 and thioamide 3 were prepared from 1via a one-pot, two-step procedure consisting of lithiation and subsequent quenching of the in situ generated lithio intermediate with N,N-dimethylcarbamoyl chloride and N,N-dimethylthiocarbamoyl chloride, respectively (Scheme 2). The targeted amides were purified by column chromatography and isolated in moderate to good yields (2: 46%; 3: 81%).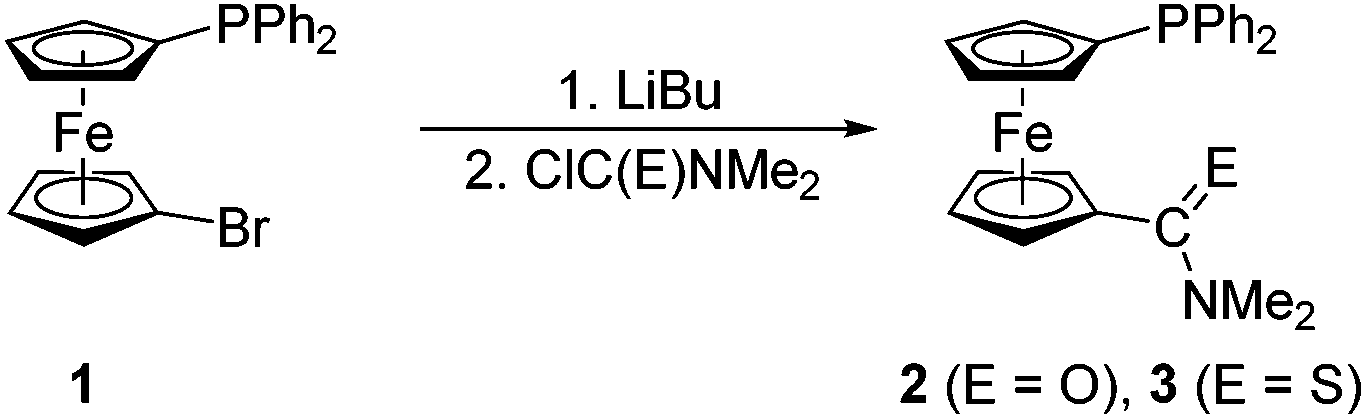 | ||
| Scheme 2 Preparation of phosphinoferrocene amide 2 and thioamide 3 from 1 and the corresponding carbamoyl halides. | ||
In view of the intended coordination study, the series of phosphinoamide donors was extended by the homologous secondary amide 4 (Scheme 3) and the known primary amide 1′-(diphenylphosphino)-1-(aminocarbonyl)ferrocene (5)7 for the purpose of comparison. The former compound was synthesised by the conventional amidation of Hdpf with methylamine in the presence of 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide (EDC) and 1-hydroxybenzotriazole (HOBt), resulting in an 87% yield after isolation by column chromatography. Amide 4 was further converted to the corresponding phosphine oxide (4O) and sulfide (4S) via standard oxidations with hydrogen peroxide and elemental sulfur, respectively.
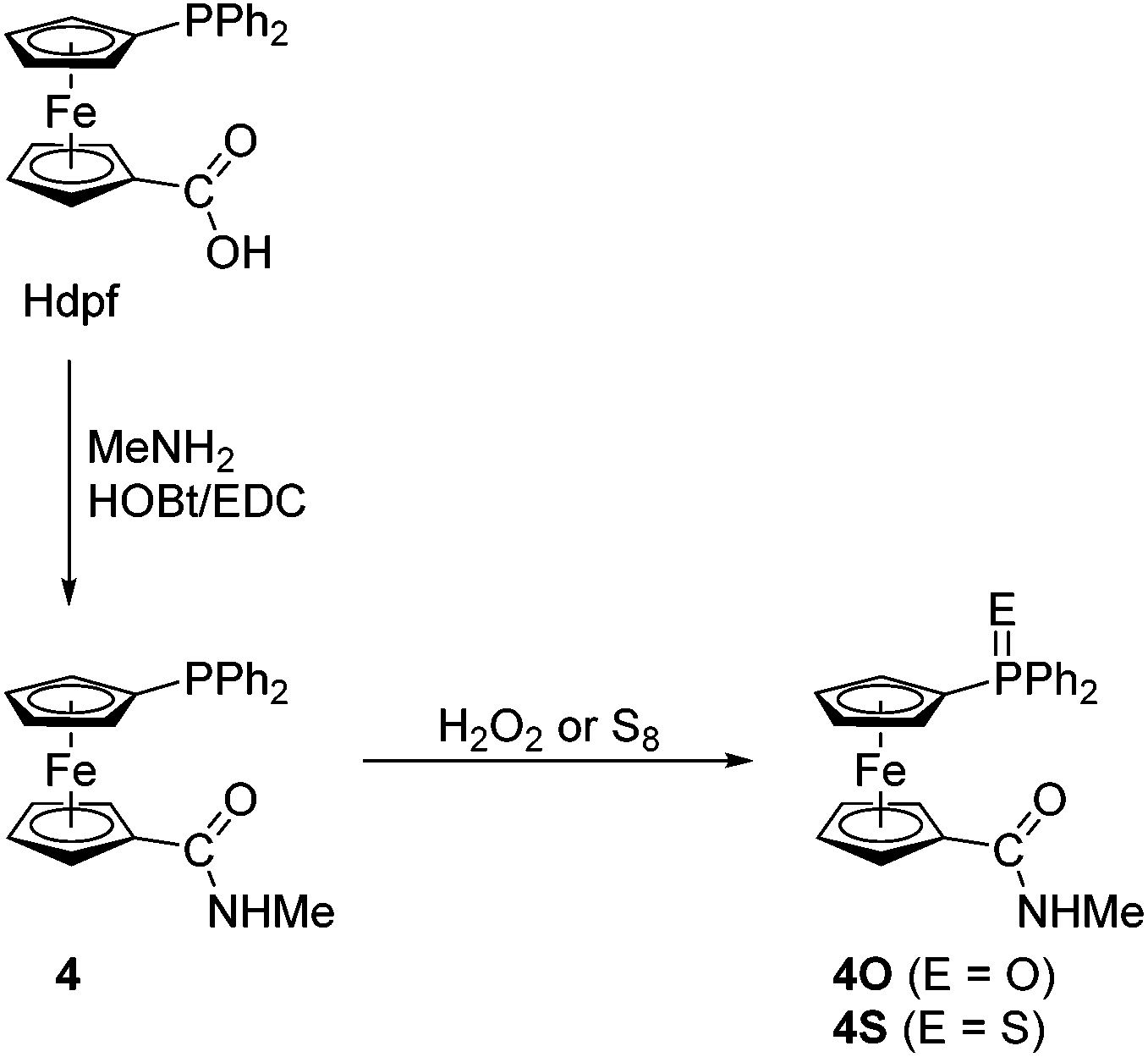 | ||
| Scheme 3 Preparation of amide 4 and its corresponding P-oxide and P-sulfide. Legend: EDC = 1-ethyl-3-[3-(dimethylamino)propyl]carbodiimide, HOBt = 1-hydroxybenzotriazole. | ||
Amides 2–4 have been characterised by multinuclear NMR and IR spectroscopy, electrospray ionisation (ESI) mass spectrometry and elemental analysis. Phosphines 2–4 display singlets in their 31P{1H} NMR spectra at δPca. 17, close to that of Hdpf itself,5 while the signals of the P-oxidised derivatives appear shifted to lower fields (δPca. 32 and 43 for 4O and 4S, respectively).5,15 The 1H and 13C{1H} NMR spectra reveal signals typical for the 1′-(diphenylphosphino)ferrocenyl moieties. The amide resonances are observed at δCca. 170 for the amides and at δC 199 for thioamide 3. As a result of the limited molecular mobility typical for conjugated amides, the signals of the methyl substituents in the spectra of the tertiary amides are observed either as a broadened singlet (2) or a pair of non-equivalent signals (3) at room temperature. On the contrary, the 1H NMR signals of the methyl groups in the spectra of secondary amides 4, 4O and 4S are seen as NH-coupled doublets associated with quartets attributed to the NH proton at a lower field.
The type of amide pendant is manifested in the IR spectra, showing bands resulting from the C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E16 stretching vibrations (2: 1502 and 1650 cm−1, 3: 1508 cm−1). The spectra of the secondary amides further display bands due to NH stretching modes above 3000 cm−1. Additional characterisation of 2 and 3 by DFT computations, UV-vis spectroscopy and by cyclic voltammetry are described below.
E16 stretching vibrations (2: 1502 and 1650 cm−1, 3: 1508 cm−1). The spectra of the secondary amides further display bands due to NH stretching modes above 3000 cm−1. Additional characterisation of 2 and 3 by DFT computations, UV-vis spectroscopy and by cyclic voltammetry are described below.
Molecular structures of uncoordinated amides
The molecular structures of all tertiary amides (2, 3 and 3O) and secondary amides (4, 4O·CHCl3 and 4S) have been determined by single-crystal X-ray diffraction analysis. The crystals of 3O were isolated during an attempted complexation experiment with 3, whereas those of all other compounds were grown by crystallisation of authentic samples.The molecular structures of 2, 3 and 3O are depicted in Fig. 1. Selected geometric parameters are summarised in Table 1. The ferrocene units in the structures of these tertiary amides exert the typical regular geometries with similar Fe–C distances and tilt angles below ca. 6° (maximum: 5.8(1)° for 3). The ferrocene cyclopentadienyl rings in 2 and 3 assume similar anticlinal eclipsed conformations that divert the substituents into mutually distant positions. The amide plane in 2 is rotated by as much as 47.4(2)° from an arrangement coplanar with its parent cyclopentadienyl ring (Cp1) with the NMe2 group pointing away from the ferrocene unit and the PPh2 substituent. The thioamide moiety in 3 is twisted considerably less (26.7(2)°) and adopts the opposite orientation with respect to the PPh2 moiety (i.e., with the CS bond more distant). In contrast, the ferrocene unit in 3O has a synclinal eclipsed conformation, which results in a rather compact structure in which both substituents are located on the same side of the ferrocene scaffold. Consequently, the rotation of the thioamide plane is increased to 41.8(2)° and the orientation of the C(S)NMe2 pendant unit is changed so that the more bulky NMe2 unit is directed away from the ferrocene unit (though on the same side as the phosphine substituent). Parameters pertaining to the amide/thioamide and (diphenylphosphino)-ferrocenyl moieties appear unexceptional in view of the data reported earlier for FcCSNH2,17 fc[CSNMe2]2,9a Hdpf and its P-oxide,5 Ph2P(O)fcCONHCy,7 and bromide 118 (Fc = ferrocenyl, fc = ferrocene-1,1′-diyl).
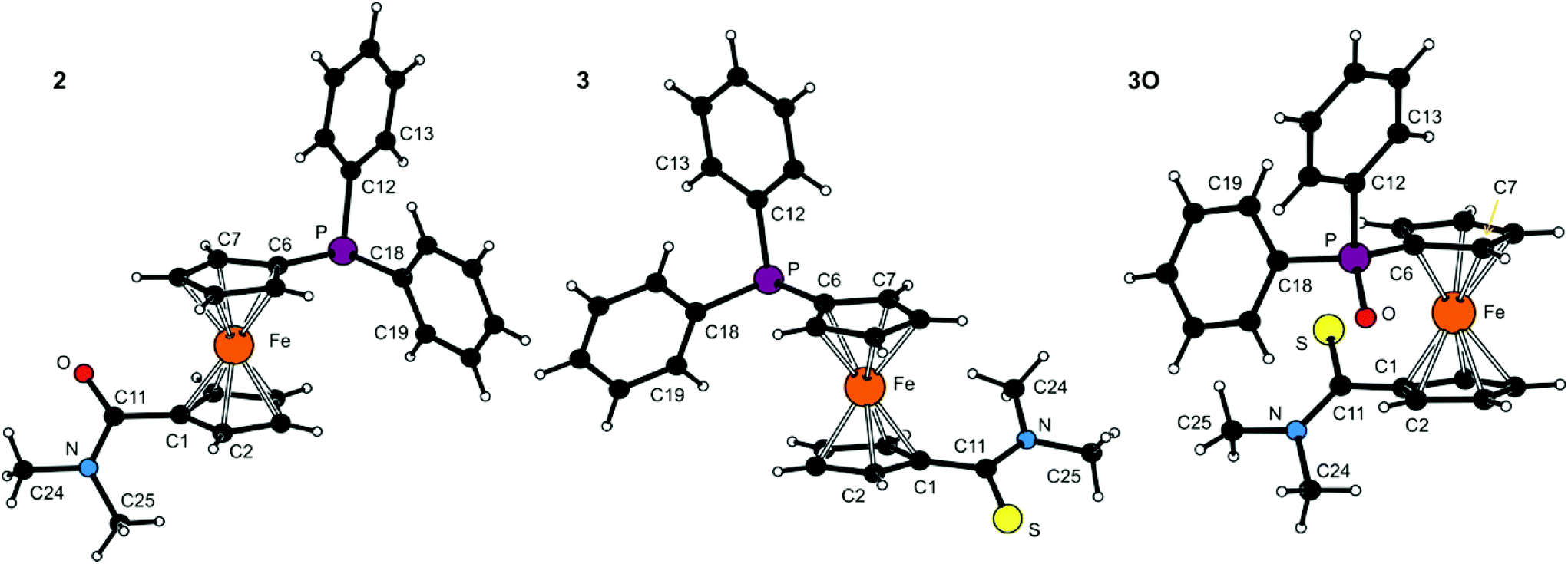 | ||
| Fig. 1 PLATON plots of the molecular structures of 2, 3 and 3O (for displacement ellipsoid plots, see ESI†). | ||
| Parameter | 2 (E = O) | 3 (E = S) | 3O (E = S)b |
|---|---|---|---|
|
a Definitions: Cp1 and Cp2 are the cyclopentadienyl rings C(1–5) and C(6–10), respectively. Cg1/2 denote their centroids. τ is the torsion angle C1–Cg1–Cg2–C6 and φ is the dihedral angle subtended by the amide unit (C11, E, N) and the plane of its parent ring Cp1.
b Further data: PO = 1.536(1) Å.
|
|||
| Fe–Cg1 | 1.6462(8) | 1.6544(8) | 1.6413(7) |
| Fe–Cg2 | 1.6464(8) | 1.6486(8) | 1.6402(7) |
| ∠Cp1, Cp2 | 2.8(1) | 5.8(1) | 4.12(9) |
| τ | −151.1(1) | 143.5(1) | 71.8(1) |
| C11E |
1.225(2) | 1.689(2) | 1.672(2) |
| C11–N | 1.356(2) | 1.333(2) | 1.337(2) |
| EC11–N |
121.7(2) | 121.4(1) | 122.3(1) |
| φ | 47.4(2) | 26.7(2) | 41.8(2) |
| N–C24 | 1.460(2) | 1.465(2) | 1.468(2) |
| N–C25 | 1.460(2) | 1.464(2) | 1.461(2) |
| C24–N–C25 | 116.3(1) | 113.7(2) | 113.0(2) |
| P–C6 | 1.820(2) | 1.811(2) | 1.788(2) |
| P–C12 | 1.832(2) | 1.835(2) | 1.811(2) |
| P–C18 | 1.841(2) | 1.839(2) | 1.808(2) |
Molecular structures of the secondary amides 4, 4O·CHCl3 and 4S are shown in Fig. 2. Principal geometric data are given in Table 2. The molecular parameters compare well with those reported for other structurally characterised secondary amides derived from Hdpf.19 Similar to the tertiary amides, the ferrocene units in the structures of the secondary amides are typical with tilt angles not exceeding ca. 5°. A most notable difference observed across the series is again associated with the mutual orientation of the ferrocene substituents, which are near synclinal eclipsed in 4 and 4O, staggered anticlinal in molecule 2 of 4S, or assume an intermediate conformation between anticlinal staggered and eclipsed anticlinal eclipsed (molecule 1 of 4S).
 | ||
| Fig. 2 PLATON plots of the molecular structures of 4, 4O·CHCl3 and 4S (molecule 1). Labelling of the second independent molecule in the structure of 4S is strictly analogous. Displacement ellipsoid plots are available in the ESI.† The N–H⋯O and C–H⋯Cl hydrogen bonds in the structure of 4O·CHCl3 are shown as dashed lines. | ||
| Parameter | 4 (E = void) | 4O (E = O1P)b | 4S (E = S1/S2)c |
|---|---|---|---|
| a The parameters are defined as for the tertiary amides (see Table 1). n.a. = not applicable. b The compound crystallised in the form of stoichiometric solvate 4O·CHCl3. c Data for the two structurally independent molecules (molecule 1/molecule 2). | |||
| Fe–Cg1 | 1.6462(8) | 1.650(1) | 1.640(4)/1.662(4) |
| Fe–Cg2 | 1.6420(7) | 1.641(1) | 1.643(4)/1.639(4) |
| ∠Cp1, Cp2 | 3.2(1) | 1.2(1) | 3.2(5)/4.7(5) |
| τ | −82.6(1) | 71.3(2) | −162.3(5)/145.4(5) |
| C11O |
1.242(2) | 1.233(3) | 1.222(6)/1.233(7) |
| C11–N | 1.330(2) | 1.343(3) | 1.326(8)/1.309(7) |
| O–C11–N | 122.1(2) | 123.3(2) | 123.6(5)/121.9(6) |
| φ | 6.1(2) | 25.3(3) | 11.3(8)/13.6(7) |
| N–C24 | 1.453(2) | 1.456(3) | 1.457(9)/1.45(1) |
| PE |
n.a. | 1.495(2) | 1.951(3)/1.960(3) |
| P–C6 | 1.822(2) | 1.785(2) | 1.785(8)/1.792(8) |
| P–C12 | 1.835(2) | 1.809(2) | 1.826(9)/1.813(9) |
| P–C18 | 1.839(2) | 1.809(2) | 1.819(7)/1.818(7) |
The rotation of the amide unit with respect to the parent cyclopentadienyl ring varies from ca. 6° in amide 4 to ca. 25° in 4O. However, these structural changes seem to be (at least partly) induced by different intermolecular interactions because, unlike the tertiary amides whose solid-state structures are essentially molecular,20 the molecules of the secondary amides associate in the solid state by means of hydrogen bonds formed by the NH hydrogens. Compounds 4 and 4S assemble into infinite chains consisting of molecules located around the crystallographic glide planes via N–H⋯OC hydrogen bonds (N⋯O = 2.835(2) Å for 4 and 2.797(6)/2.834(6) for molecules 1/2 of 4S). Conversely, the solvated phosphine oxide 4O forms an intramolecular hydrogen N–H⋯OP bond towards the highly polarised21 phosphoryl oxygen (N⋯O = 2.838(2) Å) rather than the amide CO moiety, while the CO group is employed in binding the solvent molecule via the soft C–H⋯O interaction (Cl3C–H⋯OC, C⋯O = 3.013(3) Å).
DFT and electrochemical study of amides 2 and 3
Geometries of 2 and 3 computed by DFT methods for isolated molecules in vacuum reproduce very well those determined by X-ray crystallography in the solid state (for an overlap of the computed and experimentally determined molecular structures, see ESI†). Whereas the calculated interatomic distances differ from the experimental ones by less than ca. 0.04 Å (the mean difference being only 0.01 Å), the interatomic angles show more pronounced variation (maximum 14°, average difference: 1–3°) due to changes in conformation.22 The dihedral angle of the cyclopentadienyl planes and τ parameter calculated for amide 2 are 2° and −174°, respectively. The amide moiety in the DFT optimised structure has similar orientation to the solid state structure with twist angle φ of 39°. In the case of thioamide 3, the τ and φ angles of 149° and 26°, respectively, correspond also well with the crystal structure data (cf. data in Table 1).The LUMO, HOMO and two next molecular orbitals below HOMO are depicted in Fig. 3. In the case of 2, the HOMO and HOMO−1 show dominant contributions from d orbitals on Fe, while for 3, the HOMO consists mainly of non-bonding orbital located on sulfur (lone pair). The next two lower molecular orbitals of 3 are of similar nature as HOMO and HOMO−1 of 2 (similar to diagonal relationship), but with certain contribution from atomic orbitals located on the sulfur atom and with somewhat lower energies. This corresponds well with the lower ionisation potential and higher softness of sulfur with respect to oxygen. This principal difference in the structure of the highest occupied molecular orbitals is most likely responsible for the different electrochemical behaviour of 2 and 3 (vide infra).
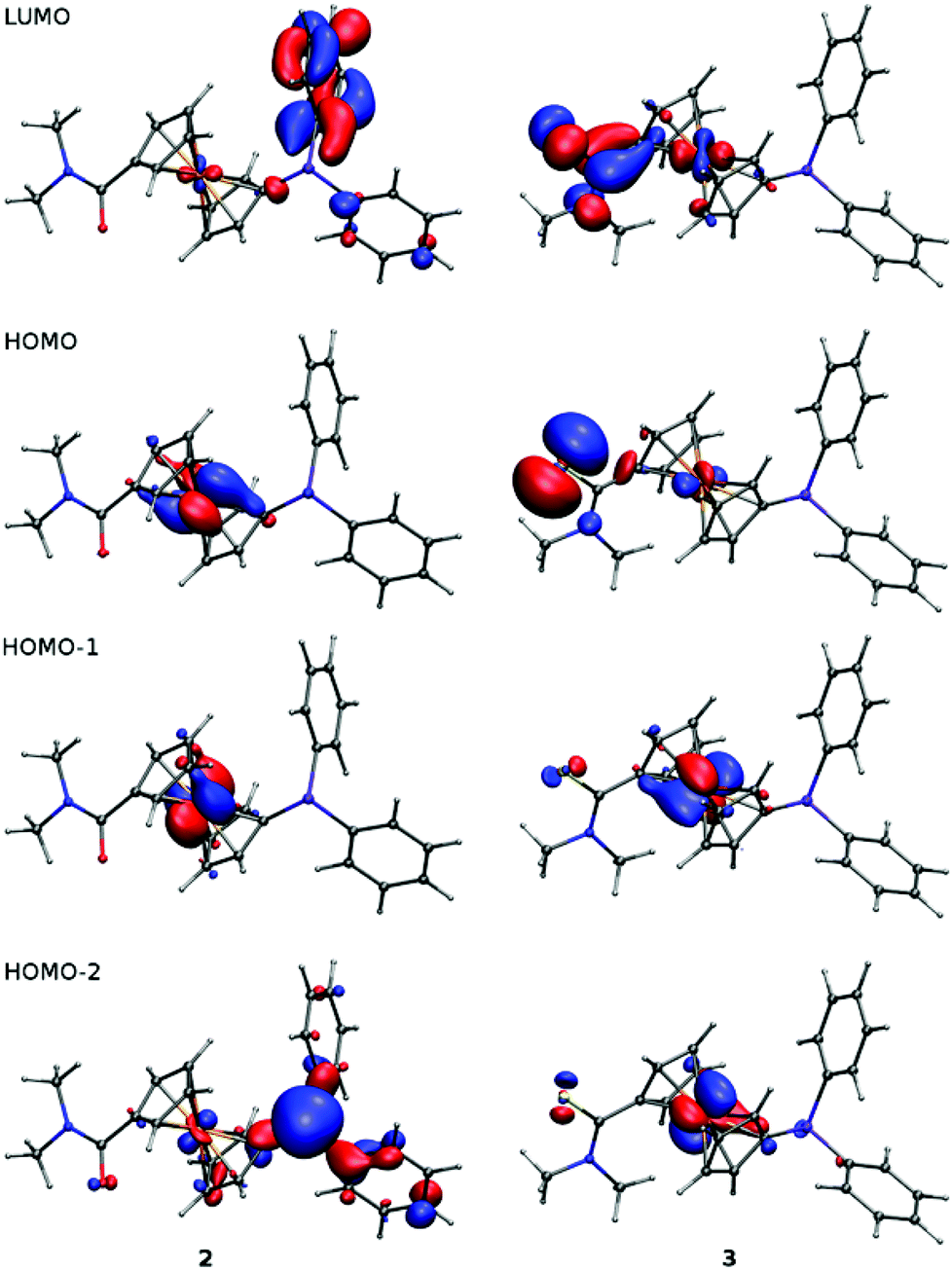 | ||
| Fig. 3 Plots of LUMO, HOMO and two lower molecular orbitals of 2 and 3 showing contours at the ±0.05 a.u. level. | ||
Changes in electronic structure associated with the (formal) replacement of the amide oxygen with sulfur prompted us to investigate the representative amides 2 and 3 by UV-vis spectroscopy and by electrochemical methods. The UV-vis spectra (Fig. 4) comprise single bands (with a shoulder at lower energies) located at the foot of a more intense bands extending from the UV region. This band in the spectrum of 2 is observed at 445 nm, shifted slightly to lower energies as compared with ferrocene itself (440 nm; forbidden d–d transition).23 In the spectrum of 3, the absorption band is significantly red-shifted (458 nm) and also more intense, presumably owing to a more extensive conjugation.24
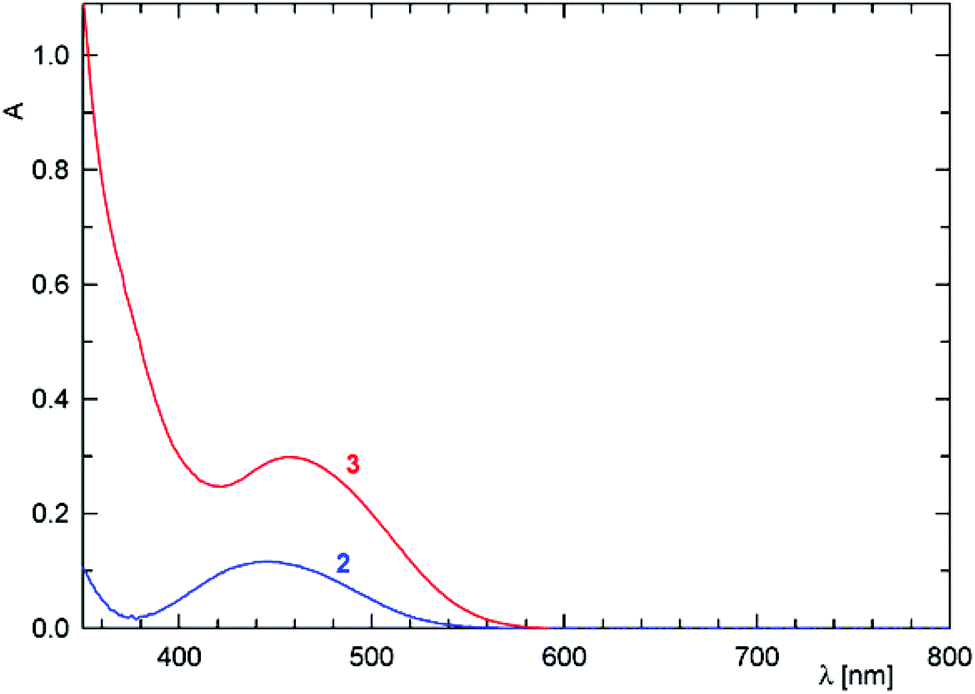 | ||
| Fig. 4 UV-vis spectra of 2 (blue line) and 3 (red line) recorded in 1,2-dichloroethane solutions (c = 5 × 10−4 M, optical path 10 mm). | ||
Cyclic voltammogram of amide 2 (Fig. 5) displays a one-electron oxidation at E°′ ca. 0.17 V vs. ferrocene/ferrocenium. The oxidation, which can be attributed to the FeII/FeIII couple (electron removal from HOMO located predominantly at the ferrocene unit, see above), is associated with some follow-up processes that render it quasi-reversible and also give to rise to additional redox waves at higher potentials. Nonetheless, chemical reactions of the electrochemically generated species are relatively slow because the ratio of the cathodic and anodic peak currents (ipc/ipa) significantly increases with increasing scan rate (see inset in Fig. 5), limiting to unity. Such behaviour resembles that of the parent acid Hdpf.5 The fact that the oxidation of the ferrocene unit is shifted to less positive potential than that of Hdpf (E°′ ca. 0.31 V in MeCN) is in accordance with the lower electron-withdrawing ability of the amide moiety as compared with the carboxyl group (cf. the Hammett σp constants: 0.36 for CONH2, and 0.45 for CO2H).25
 | ||
| Fig. 5 Cyclic voltammogram of amide 2 as recorded at Pt disc electrode in 1,2-dichloroethane at 0.1 V s−1 scan rate (the arrow indicates the scan direction). The inset shows partial cyclic voltammograms recorded at different scan rates (black: 20 mV s−1, blue: 50 mV s−1, green: 0.1 mV s−1, red: 0.2 V s−1, and cyan: 0.5 V s−1). | ||
The redox behaviour of thioamide 3 is much less clear-cut (Fig. 6). The compound undergoes an irreversible oxidation at ca. 0.02 V (anodic peak potential, Epa, is given), which is followed by several ill-defined irreversible oxidations that replace the original composite oxidative wave during the second and following scans (even at 1 V s−1). Such a response may well correspond with the properties of the HOMO orbital, which encompasses both ferrocene unit and the thioamide moiety.
 | ||
| Fig. 6 Full (red line) and partial (black line) cyclic voltammograms of 3 as recorded at Pt disc electrode in 1,2-dichloroethane at 0.1 V s−1 scan rate. The arrow indicates the scan direction and the second scan is distinguished by a dashed line. | ||
DFT study of amide group conformation
The relatively high and varying twisting of the amide pendant observed in the solid-state structures of free phosphinoamides led us further to investigate the influence of the dihedral angle subtended by the amide plane {C, N, E} and its parent cyclopentadienyl ring on the overall energy of the isolated model molecules of FcCONR2 and FcCSNR2 (R = H and Me) by DFT calculations. Attention was paid to this parameter mainly because it could significantly affect the coordination properties of the phosphinoamides, being responsible for an efficient approach of the amide moieties to a metal centre.For the sake of a simpler definition, the dihedral angle φ was replaced with the torsion angle C2–C1–C11–E (ψ). The energy profiles calculated as a function of this angle for FcCONH2 and FcCSNH2 (Fig. 7) show two equivalent minima corresponding to enantiomers. The maxima belong to conformations with amide groups perpendicular to the parent cyclopentadienyl rings, with the higher in energy corresponding to the conformation in which the NH2 unit is directed closer to the Fe atom. The practically coincident minima for both amides exhibit twists of the amide group of 18°. In the case of the more bulky N,N-dimethyl derivatives, FcC(E)NMe2, the twisting increases to 35° and 37° for E = O and S, respectively, presumably for steric reasons. The former value corresponds with that determined in the solid state (FcCONMe2: 36/37° for two independent molecules).26,27
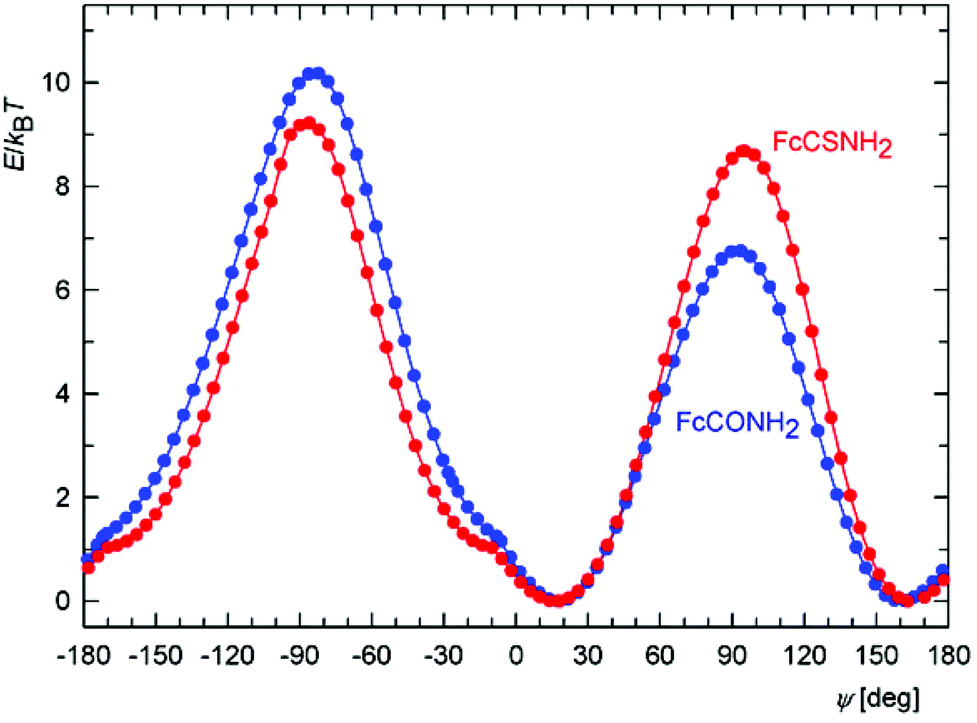 | ||
| Fig. 7 Calculated energy dependence on the torsion angle ψ for FcCONH2 (blue) and FcCSNH2 (red) at T = 300 K. | ||
Importantly, the curvature of the energy landscape near the minima for both FcCONH2 and FcCSNH2 allows for an essentially free change of ψ by approximately 20° in both directions at room temperature, i.e., within the energy change of 1 kBT (where kB is the Boltzmann constant). In this interval, the twist angle can thus be controlled via an interplay between energy changes reflecting the extent of conjugation, steric effects, coordination and intermolecular interactions (the latter in the solid state).
Another notable feature in the energy profiles concerns small changes in the slope for conformers whose amide groups are nearly coplanar with their bonding cyclopentadienyl rings (ψ ≈ 10°). These changes result from the potential energy surface (PES) crossing other surface corresponding to an inversion of the pyramidal NH2 moiety because the two surfaces coincide for the planar arrangement of the NH2 groups (for clarity, only the PES with the lower energy is shown in Fig. 7).
Synthesis of copper(I) and silver(I) complexes
In order to fully exploit the donor moieties available in 2–5, coordination study with the soft Cu(I) and Ag(I) ions was undertaken using metal precursors devoid of any firmly bound ligands (e.g., halides) and coordinating anions that could possibly compete with the donor groups offered by the amidophosphine ligands. Hence, the complexation reactions were performed at the ligand-to-metal ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 using [Cu(MeCN)4][BF4] and AgClO4 as the metal sources (Scheme 4).
:1 using [Cu(MeCN)4][BF4] and AgClO4 as the metal sources (Scheme 4).
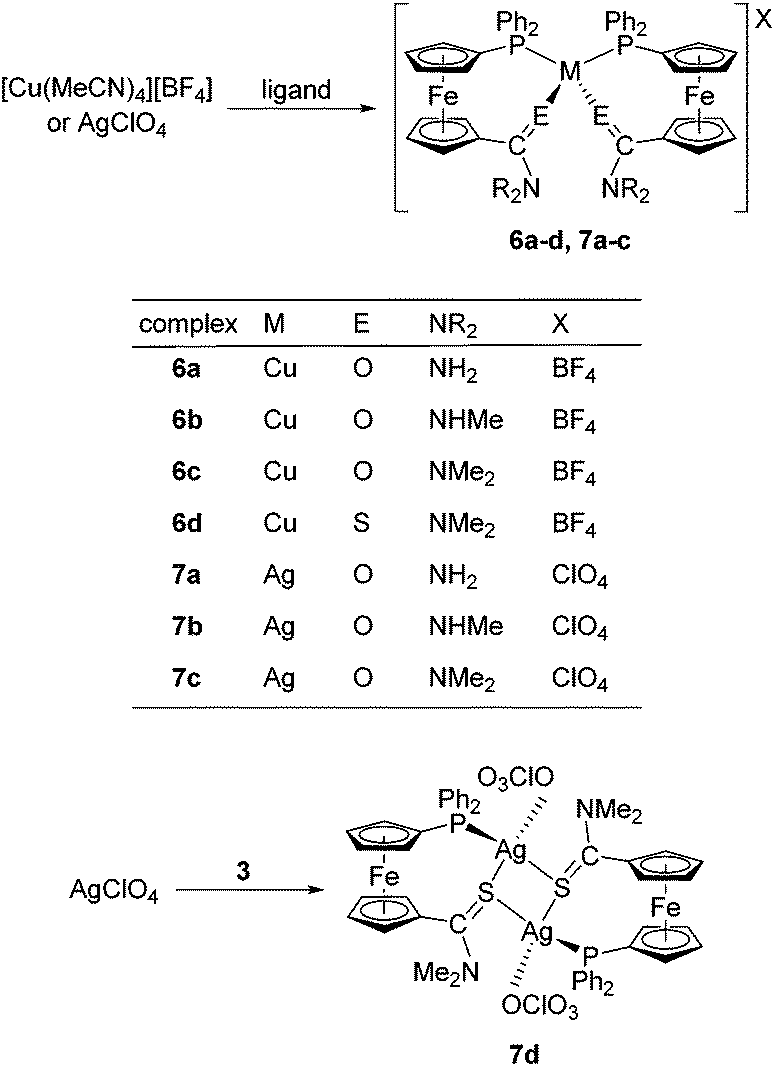 | ||
| Scheme 4 Synthesis of Cu(I) and Ag(I) complexes. | ||
These precursors reacted identically with all of the amides to afford bis-chelate complexes of the type [M(L-κ2O,P)2]X. The reaction of thioamide 3 with [Cu(MeCN)4][BF4] afforded an analogous compound 6d, the structure of which, together with other representatives (6b, 7b and 6d; vide infra), was unambiguously confirmed by single-crystal X-ray diffraction analysis. In contrast, the reaction of 3 with silver(I) perchlorate produced an evidently different product (7d) exhibiting properties very different from those of the rest of the series: the compound readily precipitated from the reaction mixture and was practically insoluble in common organic solvents (including DMSO-d6), which in turn precluded its detailed characterisation by solution techniques (e.g., NMR spectroscopy). This finding alerted us to investigate this material in more detail. Fortunately, X-ray quality crystals were obtained when the educts were allowed to mix slowly by liquid-phase diffusion. The isolated crystals proved to be identical with 7d obtained by direct mixing of the starting materials, as evidenced by the IR spectra.
The structure determination revealed 7d to be a “dimer”, wherein the thioamide coordinates in a P,S-bidentate fashion to one silver(I) centre and simultaneously acts as a bridge towards the other Ag(I) ion through its sulfur atom. The coordination sphere is completed by O-bonded perchlorate, resulting in tetrahedral coordination around the chemically equivalent metal centres. As the consequence, the complex has an overall 1:1 ligand-to-metal stoichiometry, which clearly differentiates 7d from the rest of the Cu(I) and Ag(I) complexes.
Complexes 6 and 7 were characterised by elemental analysis, IR and NMR spectroscopy, and ESI MS spectrometry. The coordination of the amidophosphine ligands in these compounds is clearly manifested in the 31P NMR spectra via a shift of the 31P NMR signal to lower fields. The 31P NMR signals are observed as singlets at approximately −11 ppm for the Cu(I) complexes 6a–d, and as 107/109Ag-coupled doublets at ca. 3–4 ppm for 7a–c. Compound 7d gives rise to a broad doublet at δP −0.3 with 1J(Ag,P) ≈ 510 Hz. It is also noteworthy that the 31P and 1H NMR signals are typically broadened, indicating that some dynamic processes are taking place in the solution. In their ESI mass spectra, the “mononuclear” complexes 6a–d and 7a–c exhibit signals of the cations [ML2]+ and their fragments [ML]+. The spectrum of the disilver(I) species 7d is dominated by the ions due to [Ag(3)]+ at m/z 563/566 and further shows additional signals attributable to [Ag(3)(CH3OH)]+ (m/z 596/599). All these results (shifts of the 1H and 31P NMR signals and species observed in the ESI MS spectra) suggest the solid state structures to be retained even in solution, though perhaps with some structural dynamics.
Finally, the presence of the counter anions is reflected in the IR spectra, showing composite intense bands resulting from the ν3 vibrations28 of the tetrahedral ions BF4− and ClO4− at ca. 1030–1095 cm−1 and 1050–1130 cm−1, respectively.
The anticipated dynamic and possibly hemilabile coordination29 of the phosphinoamide donors was proven by a serendipitous isolation of several crystals of a solvento complex, [Cu(5-κ2O,P)(5-κP)(CHCl3-κCl)][BF4] (6a′), which was isolated during an attempted crystallisation of 6a and structurally characterised. It is noteworthy that this solvento complex ensued from a copper(I) complex whose amide substituents (NH2) provide the lowest steric protection and donating ability among the amides studied and comprise oxygen as a hard donor atom (N.B. Cu(I) is softer than Ag(I) according to the absolute hardness scale30).
Molecular structures of the Cu(I) and Ag(I) complexes
Crystallisation of the “bulk” samples provided single crystals of 6b·1/4CHCl3, 6d·2CHCl3, and 7b·CHCl3, which were used for structure determination. The crystals of 7d had to be grown by reactive diffusion because of poor solubility, making any recrystallisation impossible (see Experimental), whereas those of 6a′·CHCl3 were obtained unintentionally upon attempted crystallisation of 6a.As indicated by the formulae given above, the compounds tend to retain crystallisation solvents in their structure, which often become disordered in structural voids defined by the bulky complex molecules. A similar effect affects the counter anions, which are considerably smaller than the cations they are associated with, and even some peripheral molecular parts (e.g., phenyl rings).
The crystal structure of 6d·2CHCl3 is shown in Fig. 8 (structural drawings of the analogous complexes 6b·1/4CHCl3 and 7b·CHCl3 are presented in the ESI†). Relevant geometric parameters for all three complexes are provided in Table 3. The structures support the formulation, revealing tetrahedral coordination environments around the metal centres constituted by two P,E-chelating (E = O or S) amidophosphine ligands. An inspection of the interligand angles reveals pronounced angular distortion of the coordination sphere resulting from the different steric demands of the donor moieties. In the pair of complexes derived from ligand 4 (i.e., 6b·1/4CHCl3 and 7b·CHCl3), the interligand angles increase from O–M–O via O–M–P to P–M–P. Such a feature, as well as the individual ligand–donor bond lengths, correspond with those reported for [Cu(Ph2PfcCONHCH2CO2Me-κ2O,P)2](CF3SO3).31
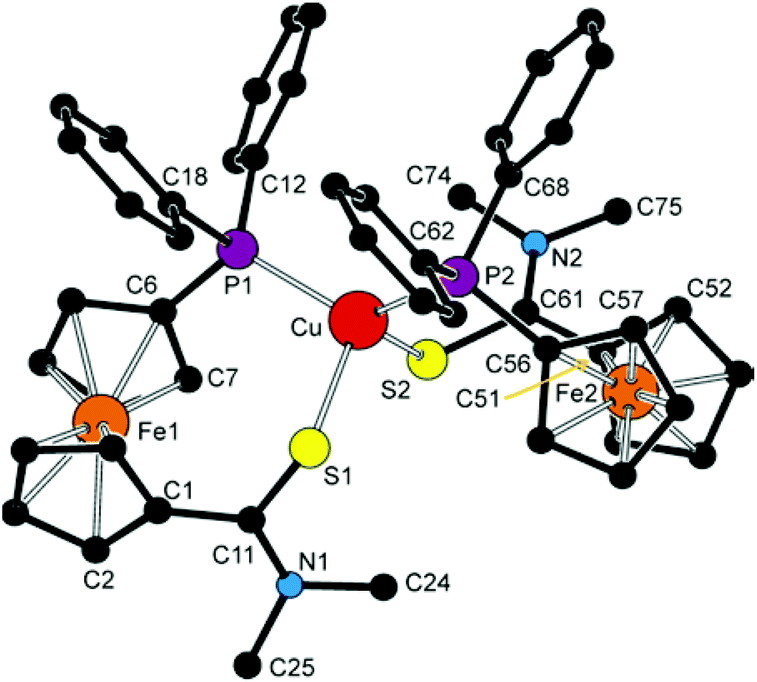 | ||
| Fig. 8 View of the cation in the structure of 6d·2CHCl3. The hydrogen atoms are omitted for clarity. | ||
| Compound | 6b·1/4CHCl3 (M/E = Cu/O1, O2) | 6d·2CHCl3 (M/E = Cu/S1, S2) | 7b·CHCl3 (M/E = Ag/O) | 6a′·CHCl3 (M/E = Cu/O1, O2)c | |||
|---|---|---|---|---|---|---|---|
| Parameter | Ligand 1 | Ligand 2 | Ligand 1 | Ligand 2 | Ligand 1b | Ligand 1d | Ligand 2d |
|
a Definitions: Cp1 and Cp2 are the amide- and phosphine-substituted cyclopentadienyl rings, respectively. Cg1/2 are their centroids. τ is the torsion angle C1–Cg1–Cg2–C6 and φ is the dihedral angle subtended by the amide unit (EC1–N) and the plane of its parent ring Cp1. n.a. = not applicable.
b Only one set of distances and angles available because of the imposed symmetry.
c Further data: Cu–Cl1 = 3.138(2); Cl1–Cu–P1 = 107.90(4), Cl1–Cu–P2 = 86.96(4), Cl1–Cu–O2 = 91.8(1).
d Ligand 1 = P-monodentate 5, ligand 2 = O,P-chelating 5.
|
|||||||
| M–P | 2.2469(7) | 2.2256(8) | 2.2842(5) | 2.2636(5) | 2.4256(6) | 2.251(1) | 2.233(1) |
| M–E | 2.174(2) | 2.102(2) | 2.3866(6) | 2.3951(7) | 2.468(2) | n.a. | 2.006(3) |
| P–M–P [E–M–E] | 125.79(3) | [93.06(9)] | 127.32(2) | [108.24(2)] | 142.99(3) [80.20(6)] | 128.17(4) | n.a. |
| P–M–E1 | 104.09(6) | 109.40(6) | 105.59(2) | 100.17(2) | 115.50(5) | n.a. | n.a. |
| P–M–E2 | 101.29(7) | 117.74(6) | 104.98(2) | 109.33(2) | 93.16(5) | 110.95(9) | 118.04(9) |
| Fe–Cg1 | 1.645(2) | 1.648(1) | 1.647(1) | 1.643(1) | 1.651(1) | 1.652(2) | 1.652(2) |
| Fe–Cg2 | 1.639(1) | 1.646(1) | 1.644(1) | 1.647(1) | 1.640(1) | 1.649(2) | 1.646(2) |
| ∠Cp1, Cp2 | 1.0(2) | 4.0(2) | 3.5(2) | 7.5(2) | 1.0(2) | 2.7(2) | 2.2(2) |
| τ | –49.4(2) | –68.2(2) | –59.5(2) | –15.2(2) | –69.5(2) | 101.0(3) | –59.4(3) |
| CE (amide) |
1.240(4) | 1.228(3) | 1.711(2) | 1.714(2) | 1.231(3) | 1.250(4) | 1.271(3) |
| C–N (amide) | 1.324(4) | 1.331(3) | 1.325(3) | 1.323(3) | 1.337(4) | 1.320(5) | 1.312(4) |
| EC–N (amide) |
121.1(3) | 121.4(2) | 120.0(2) | 121.1(2) | 122.0(2) | 121.9(3) | 118.6(3) |
| φ | 19.3(4) | 27.9(3) | 11.1(2) | 39.1(2) | 15.5(3) | 9.0(4) | 19.8(3) |
Upon going from 6b·1/4CHCl3 to 7b·CHCl3, a lengthening of the M–P (by ca. 0.2 Å) and, particularly, the M–O bonds (from ca. 2.10/2.17 Å for Cu(I) to 2.47 Å for Ag(I)) is observed owing to the presence of a larger central atom in the Ag(I) complex. The fact that the Cu–O distances in the structure of the former compound differ significantly (0.07 Å) can be associated with a relatively weaker coordination of the hard donor group, which may in turn allow for structural distortions without any dramatic destabilisation (increase in the overall energy; cf. the DFT calculations above). On the other hand, the bond lengths within the amide pendant change only marginally upon coordination (see the data for 4 above) but the ligand undergoes conformational reorganisation. The ferrocene-bound donor moieties are rotated closer to each other and the amide planes are twisted so as their oxygen atoms can reach the metal centre.
The Cu(I) complex bearing the thioamide ligands, 6d·2CHCl3, also possesses a tetrahedral structure, but because of longer Cu–S bonds, it appears to be more sterically relaxed. This is manifested in the interligand angles among which the S–Cu–S is no longer the most acute. Notably, the two structurally independent P,S-chelating ligands in the structure of 6d differ by conformation as evidenced by the τ and φ angles (Table 3; N.B. similar though less pronounced differences can be observed in the structure of 6b).
Opening of one of the chelate rings, such in the structure of 6a′·CHCl3 (Fig. 9 and Table 3), does not result in an equalisation of the interligand angles (ca. 87–128°), presumably because the amide oxygen is replaced with a relatively bulky chloroform molecule. The Cu–Cl1 distance of 3.138(2) Å approaches the threshold of the van der Waals contacts (3.15 Å32). According to a search in the Cambridge Structural Database (CSD),33 analogous Cu⋯Cl–CHCl2 interactions are rare and can be detected in the crystal structures of chloroform solvates of molecular triangles (Cu3)34 and squares (Cu4)35 built up from bis(acetylacetonate) ligands and Cu(II) ions, in which the chloroform occupies an apical position in a square pyramid around the Cu(II) ions (Cu⋯Cl ≈ 3.11–3.25 Å).
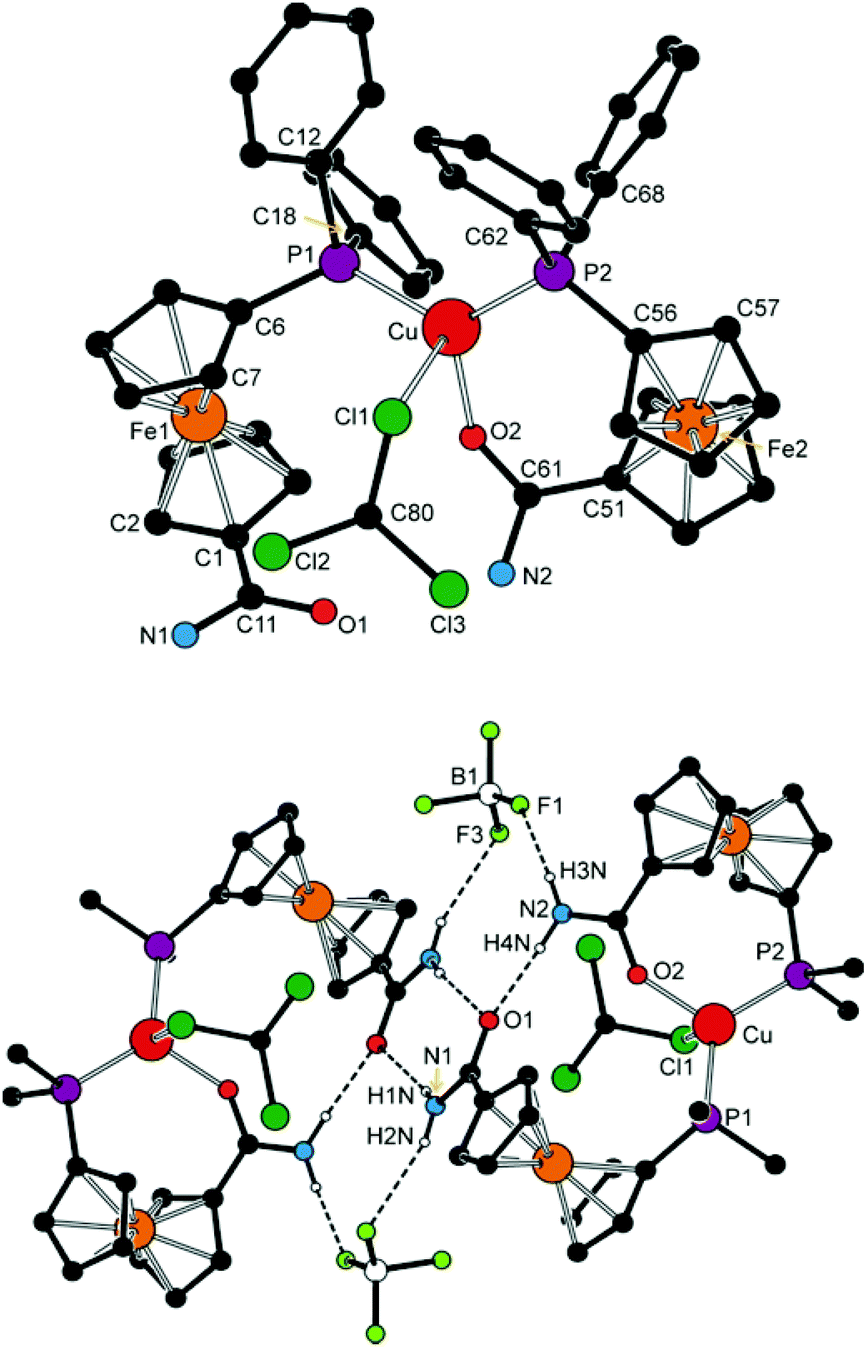 | ||
| Fig. 9 Top: view of the complex molecule in the structure of 6a′·CHCl3. All CH hydrogen atoms are omitted for clarity. Bottom: packing diagram for 6a′·CHCl3. Only the pivotal carbon atoms from the phenyl rings and NH hydrogens are shown for clarity. Hydrogen bond parameters are as follows (in Å and °): N1–H1N⋯O1, N1⋯O1 = 2.914(5), angle at H1N = 158; N1–H2N⋯F3, N1⋯F3 = 3.15(1), angle at H2N = 160; N2–H4N⋯O1, N2⋯O1 = 2.914(4), angle at H4N = 152; N2–H3N⋯F1, N2⋯F1 = 2.872(8), angle at H3N = 152. | ||
The different roles that the two amidophosphine ligands play in the complex cation of 6a′ are reflected in their conformations. Thus, the donor substituents in the chelating ligand adopt a conformation between synclinal staggered and synclinal eclipsed (τ ≈ −59°), and the amide planes are rotated by ca. 20° with respect to the parent cyclopentadienyl ring. On the other hand, the P-bound ligand assumes a more opened anticlinal conformation (τ ≈ 101°) and the amide plane is twisted by only ca. 9°. The CO distances are affected only marginally but in the expected manner as the coordinated CO bond is ca. 0.02 Å longer than the uncoordinated one.
NH protons in the structures of complexes with coordinated 4 (i.e., 6b and 7b) are involved in hydrogen bonding to the respective counter anion in the crystal state. Those in molecules of 6a′ interconnect the complex units into dimers positioned around the inversion centres (Fig. 9). In the latter case, each P-bound ligand is linked to its inversion-related counterpart via a pair of N–H⋯OC hydrogen bonds from the NH hydrogen closer to the amide oxygen, whereas the other hydrogen forms an N–H⋯F hydrogen bridge to one of the BF4− anions. Amide hydrogens in the P,O-chelating ligand participate in similar interactions towards the oxygen in the P-monodentate ligand (O1) bonded to the same metal centre and towards another BF4 fluorine, respectively.
As it was stated above, complex 7d (Fig. 10 and Table 4) is a dimer, in which the phosphinoamide ligands coordinate in a chelating manner and are further involved in bridging of the “other” Ag(I) ion through the sulfur atom. The donor set around each Ag(I) is supplemented with an O-bonded perchlorate anion into a distorted tetrahedron. The Ag–O distance of 2.814(6) Å falls well below the sum of the van der Waals radii (3.24 Å), suggesting a relatively weaker yet significant interaction between the anion and Ag(I).
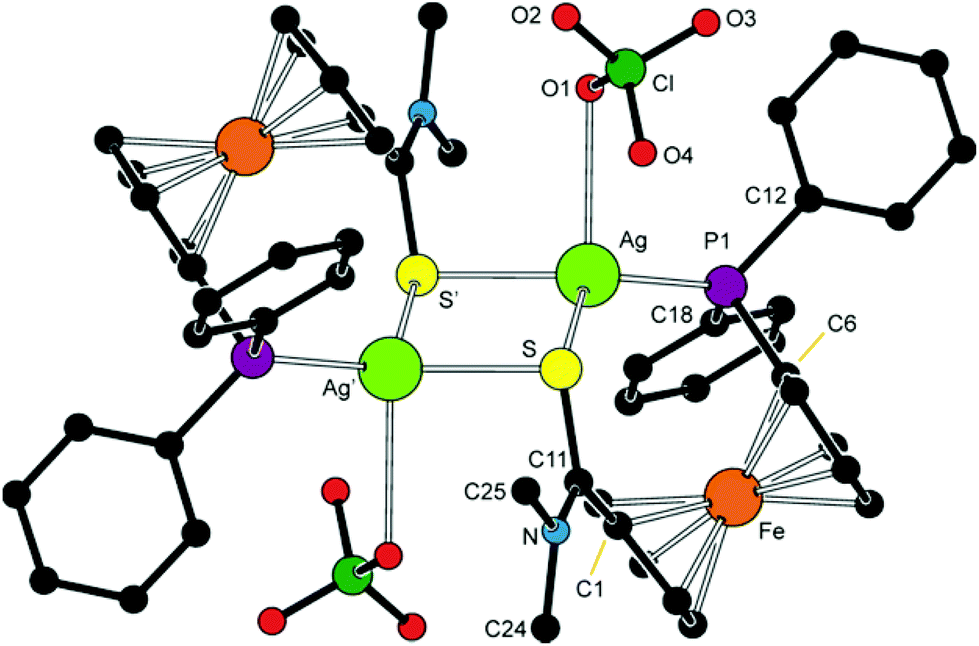 | ||
| Fig. 10 View of the molecular structure of 7d. The prime-labelled atoms are generated by the crystallographic inversion. | ||
| a All parameters are defined as for the free ligand (see Table 1). The prime-labelled atoms are generated by the (1 − x, 2 − y, 2 − z) symmetry operation. | |||
|---|---|---|---|
| Ag–P | 2.426(1) | P–Ag–S | 121.68(4) |
| Ag–S | 2.663(1) | P–Ag–S′ | 143.55(5) |
| Ag–S′ | 2.572(1) | P–Ag–O1 | 95.9(1) |
| Ag–O1 | 2.814(6) | S–Ag–S′ | 85.44(4) |
| Ag⋯Ag′ | 3.8469(7) | S–Ag–O1 | 117.6(1) |
| S⋯S′ | 3.552(2) | S′–Ag–O1 | 90.9(1) |
| Fe–Cg1 | 1.647(2) | ∠Cp1, Cp2 | 5.1(3) |
| Fe–Cg2 | 1.651(2) | τ | –80.9(3) |
| C11S |
1.719(4) | S–C11–N | 120.3(3) |
| C11–N | 1.306(6) | φ | 37.8(5) |
The Ag–S distance pertaining to the sulfur atom from the chelating ligand is ca. 0.1 Å longer than the Ag–S distance to the bridging sulfur, and the central Ag4S4 ring has a twisted rhomboidal shape (S–Ag–S′ = 85.44(4)°, Ag–S–Ag′ = 94.56(4)°). The ferrocene ligands adopt a conformation near to synclinal eclipsed (ideal value: τ = 72°) and their amide pendants are twisted by ca. 38°.
Synthesis of chloridogold(I) complexes
For the sake of completeness, we have synthesised a series of chloridogold(I) complexes of the type [AuCl(L-κP)] (8; L = 2–5, Scheme 5), in which the amidophosphine ligands employ only their phosphorus donor moieties for coordination. These compounds were readily prepared via displacement of the tetrahydrothiophene (tht) ligand in [AuCl(tht)] by the stoichiometric amount of the respective amidophosphine, resulting in good to excellent yields depending on the isolation procedure.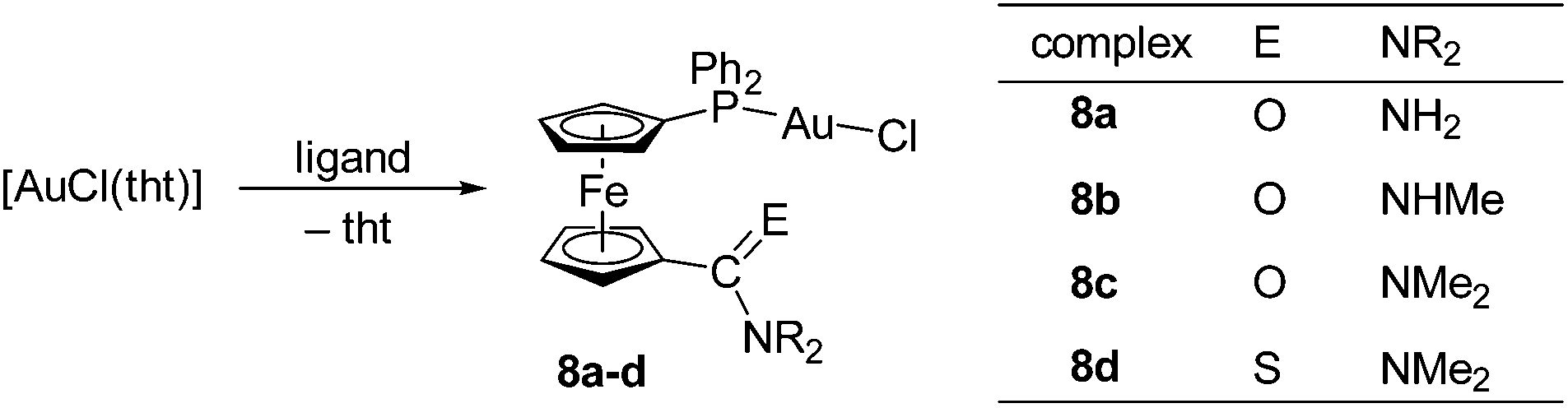 | ||
| Scheme 5 Synthesis of Au(I) complexes 8a–d (tht = tetrahydrothiophene). | ||
Compounds 8a–d were characterised similarly to the Cu(I) and Ag(I) complexes discussed above. In addition, the molecular structure of 8d was determined by single-crystal X-ray diffraction analysis. The 1H and 31P{1H} NMR spectra of 8a–d show signals of the phosphinoferrocene ligands and sharp singlets at ca. δP +29, respectively. The ESI MS spectra of these complexes display signals attributable to cationic fragments resulting from the loss of chloride ion ([M − Cl]+) and, for 8a–c, also the signals due to the pseudomolecular ions ([M + Na]+ and [M + K]+).
Molecular structure of 8d
Complex 8d (Fig. 11) crystallises with two independent but otherwise similar36 molecules per asymmetric unit (for an overlap, see ESI†). The molecules comprise the typical, practically linear Cl–Au–P moieties with the Au–P and Au–Cl bond lengths being close to those previously determined for [AuCl(FcPPh2)]37 and the related AuCl complexes with 1′-functionalised phosphinoferrocene ligands.38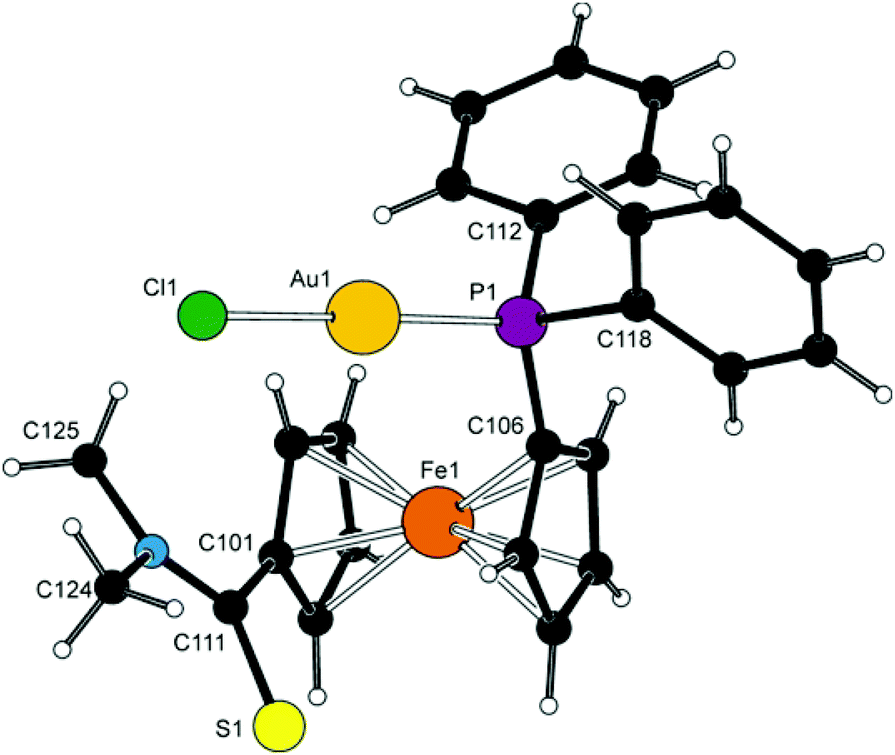 | ||
| Fig. 11 View of molecule 1 in the crystal structure of 8d. The labelling of molecule 2 is analogous with the respective labels having 2 as the first numeral. Selected distances and angles (in Å and °) for molecule 1 [molecule 2]: Au–Cl 2.294(2) [2.278(2)], Au–P 2.230(2) [2.226(2)], Cl–Au–P 178.3(1) [174.91(7)], Fe–Cg1 1.653(4) [1.654(4)], Fe–Cg2 1.640(3) [1.628(4)], τ −78.9(5) [–83.0(5)], CS 1.663(9) [1.669(8)], C–N 1.34(1) [1.34(1)], S–C–N 123.2(6) [122.1(6)], φ 33.2(8) [39.9(8)]. Note: the parameters are defined as for the free ligand. | ||
The amidophosphine ligands in the two molecules exert negligible tilting on the ferrocene unit (1.9(5)° and 3.4(5)°) and adopt conformations close to synclinal eclipsed (cf. τ with the ideal value of 72°). The amide substituents are rotated by ca. 33° and 40° (for molecules 1/2) from the planes of their parent cyclopentadienyl rings so that the bulky NMe2 unit are directed away from the ferrocene unit. The structure of 8d is essentially molecular; no Au⋯Au contacts indicative of possible aurophilic interactions39 were detected.
Electrochemical study of representative complexes
In addition to the characterization discussed above, complexes 6c, 6d, 7c and 8c as the representatives were studied by voltammetric methods similarly to the free ligands. Attention was paid mostly to the behaviour in the anodic region.Thus, in cyclic voltammetry, compound 6c undergoes an oxidation which can be tentatively attributed to the oxidation of its ferrocene ligands (Fig. 12). However, the observed redox wave is composite, presumably owing to a convolution of two narrow-spaced oxidations (peak potentials: anodic 0.49 V, cathodic 0.34 V). This is clearly manifested in differential pulse voltammograms (see Figure in the ESI†). The associated redox process appears to be reversible, though only when the wave is scanned separate over a narrow range. If the scan is extended beyond this first redox event, an irreversible oxidative and two reduction waves appear, replacing the original redox response during the second and following scans (Fig. 12). The response of the analogous Ag(I) complex 7c in cyclic voltammetry is very similar (peak potentials for the composite wave: approx. 0.50 and 0.37 V).
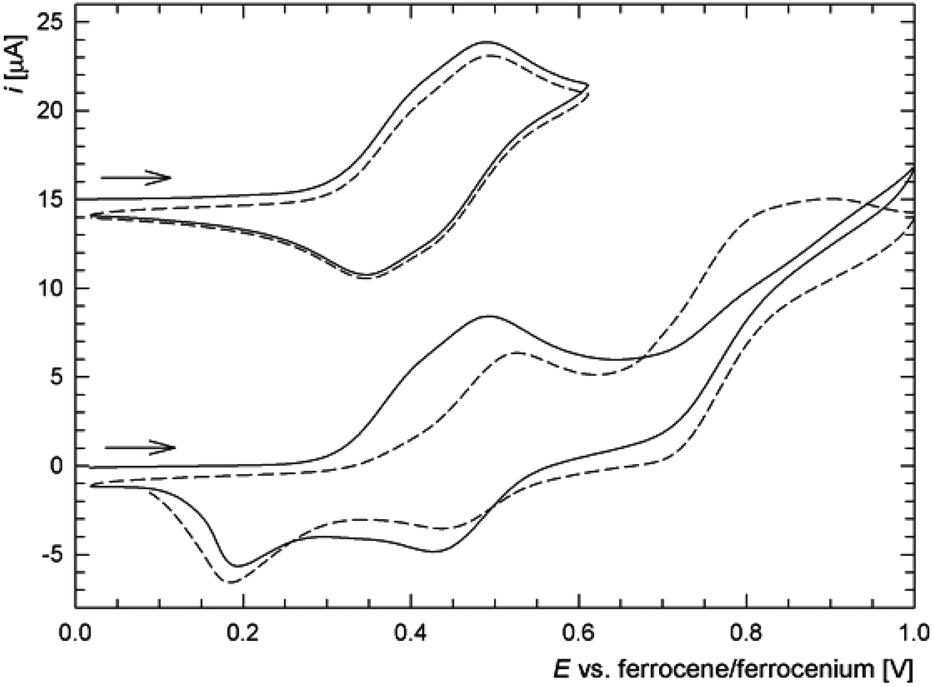 | ||
| Fig. 12 Full (bottom) and partial (top) cyclic voltammograms of 6c as recorded at a glassy carbon electrode and with 0.1 V s−1 scan rate (second scans are shown as dashed lines). For clarity, the partial cyclic voltammogram is shifted by +15 μA (to avoid overlaps) and the scan direction is indicated with an arrow. | ||
On the other hand, the redox behaviour of the Cu(I)–thioamide complex 6d differs from that of 6c (Fig. 13) in that compound 6d undergoes an irreversible oxidation at ca. 0.41 V (peak potential). When the scan window is enlarged, a pair of redox waves appears at ca. 0.66 and 0.32 V that practically supersede the original oxidative wave. Finally, the gold(I) monophosphine complex 8c undergoes a standard one-electron oxidation at E°′ = 0.375 V. It is noteworthy that the wave, which can be ascribed to the ferrocene/ferrocenium couple, is electrochemically reversible, very likely because the lone pair at phosphorus as a reactive site is no longer available.5 The shift of the redox wave towards more positive potential with respect to free 2 corresponds with the expected electron density lowering at the ferrocene unit associated with coordination.
 | ||
| Fig. 13 Representative cyclic voltammograms of 6d as recorded at a glassy carbon electrode and 0.1 V s−1 scan rate (the second scan is shown as a dashed line). | ||
Conclusions
The results presented in this paper demonstrate that the reaction of 1′-(diphenylphosphino)-1-lithioferrocene with carbamoyl and thiocarbamoyl chlorides is a viable synthetic route to new phosphinoamide ligands, offering an alternative to other commonly employed preparative methods such as the amidation of carboxylic acids. The reaction appears to be particularly attractive for the synthesis of phosphine-thioamides because it eliminates the additional protection/deprotection steps required during the conventional thionations.As evidenced by the structures of the free donors and their complexes, the molecules of 1′-(diphenylphosphino)ferrocene amides and thioamides are flexible, allowing for a pre-organisation of the donor moieties into positions suitable for coordination via rotation of the ferrocene cyclopentadienyls and twisting of the amide unit around the pivotal C–C bond at no substantial energy cost. The hybrid nature of these donors, particularly the amides combining hard and soft donor groups, results in hemilabile coordination in complexes with the soft Group 11 metal ions that in turn affects the stability and structural dynamics of these coordination compounds. The coordination variability is in no way reduced upon the replacement of amide oxygen with the soft sulfur atom. The thioamides may thus not only simply parallel the behaviour of their amide analogues but can also behave differently, taking advantage of the specific qualities of the thioamide moiety (longer CS bond, softer and less electronegative donor atom, etc.) and thus give rise to new and unique structural motifs (cf. the structure of 7d). All of these factors render the coordination chemistry of phosphino-thioamides an attractive research target that has not yet been explored in great detail.
Experimental
Materials and methods
All syntheses were performed under an argon atmosphere in the absence of direct daylight. Compounds 140 and [AuCl(tht)]41 were synthesised according to the literature. Dichloromethane and tetrahydrofuran were dried with a Pure Solv MD-5 Solvent Purification System (Innovative Technology, USA). Benzene and toluene were dried over sodium metal and distilled under argon. Other chemicals and solvents utilised for crystallisations and during column chromatography were used as received (Sigma-Aldrich; solvents from Lachner, Czech Republic).IR spectra were recorded with an FTIR Nicolet 760 instrument in the range 400–4000 cm−1. NMR spectra were obtained on a Varian UNITY Inova 400 spectrometer at 25 °C unless noted otherwise. Chemical shifts (δ/ppm) are referenced to internal tetramethylsilane (for 1H and 13C NMR spectra) and external 85% aqueous H3PO4 (31P NMR spectra). Alongside the standard notation of signal multiplicity, vq and vt are used to distinguish virtual quartets and triplets arising from the magnetically non-equivalent protons at the phosphine- and carbamoyl-substituted cyclopentadienyl rings, respectively.
Electrospray ionisation mass spectra (ESI MS) were recorded with a Bruker Esquire 3000 spectrometer using samples dissolved in HPLC-grade methanol. High-resolution (HR) ESI mass spectra were measured on a LTQ Orbitrap XL spectrometer. UV-vis spectra were recorded with a Unicam UV300 spectrometer in 1,2-dichloroethane. Elemental analyses were determined with a PE 2400 Series II CHNS/O Elemental Analyser (Perkin Elmer). The amount of clathrated solvent (if any) was verified by NMR analysis.
Safety note. Caution! Although we have not encountered any problems, it must be noted that perchlorate salts of complexes with organic ligands are potentially explosive and should be handled with care and only in small quantities.
Synthesis of the amidophosphine ligands
:1) and then ethyl acetate–hexane (3:1) as the eluents. Subsequent evaporation under vacuum afforded amide 2 as an orange solid. Yield: 0.41 g (46%).
1H NMR (399.95 MHz, CDCl3): δ 3.04 (br s, 6 H, NMe2), 4.14 (vq, J′ ≈ 1.7 Hz, 2 H, fc), 4.19 (vt, J′ = 1.9 Hz, 2 H, fc), 4.47 (vt, J′ ≈ 1.9 Hz, 2 H, fc), 4.52 (vt, J′ ≈ 1.9 Hz, 2 H, fc), 7.28–7.39 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, CDCl3): δ −16.8 (s). 13C{1H} NMR (100.58 MHz, CDCl3): δ 37.5 (very br, NMe2), 70.73 (CH of fc), 71.44 (CH of fc), 73.46 (d, JPC = 4 Hz, CH of fc), 74.18 (d, JPC = 15 Hz, CH of fc), 79.20 (C-CONMe2 of fc), 128.17 (d, 3JPC = 7 Hz, PPh2 CHmeta), 128.58 (PPh2 CHpara), 133.43 (d, 2JPC = 20 Hz, PPh2 CHortho), 138.60 (d, 1JPC = 9 Hz, PPh2 Cipso), 170.07 (CO). The signal due to C-PPh2 was not observed. IR (Nujol): νmax 1615 vs (amide I), 1502 s (amide II) cm−1. ESI+ MS: m/z 442 ([M + H]+). HR MS (ESI) calcd for C25H25FeNOP ([M + H]+) 442.1018, found 442.1019. Anal. Calcd for C25H24FeNOP (441.3): C 68.04, N 3.17, H 5.48%. Found: C 67.74, N 3.04, H 5.32%.
:1) and (in the second run) pure dichloromethane as the eluents. Following evaporation under vacuum, thioamide 3 was isolated as an a dark orange-red solid. Yield: 0.74 g, 81%.
1H NMR (399.95 MHz, CDCl3): δ 3.31 (br s, 3 H, NMe), 3.43 (br s, 3 H, NMe), 4.12 (vq, J′ ≈ 1.8 Hz, 2 H, fc), 4.24 (vt, J′ ≈ 1.9 Hz, 2 H, fc), 4.50 (vt, J′ ≈ 1.8 Hz, 2 H, fc), 4.59 (vt, J′ ≈ 1.9 Hz, 2 H, fc), 7.29–7.39 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, CDCl3): δ −16.8 (s). 13C{1H} NMR (100.58 MHz, CDCl3): δ 44.19 (NMe), 45.08 (NMe), 70.62 (CH of fc), 72.99 (CH of fc), 75.39 (d, JPC = 14 Hz, CH of fc), 75.81 (d, JPC = 4 Hz, CH of fc), 88.02 (C-CSNMe2 of fc), 128.17 (d, 3JPC = 7 Hz, PPh2 CHmeta), 128.57 (PPh2 CHpara), 133.46 (d, 2JPC = 20 Hz, PPh2 CHortho), 138.73 (d, 1JPC = 10 Hz, PPh2 Cipso), 199.24 (CS). The signal due to C-PPh2 was not observed. IR (Nujol): νmax 1508 (νC–N) cm−1. ESI+ MS: m/z 480 ([M + Na]+). HR MS (ESI) calcd for C25H25NSPFe ([M + H])+ 458.0789, found 458.0789.
:1) as the eluent. The first minor band was discarded and the second one was collected and evaporated to afford amide 4 as an orange yellow solid. Yield: 0.75 g, 87%.
1H NMR (399.95 MHz, CDCl3): δ 2.83 (d, 3JHH = 4.9 Hz, 3 H, NMe), 4.07 (vq, J′ ≈ 1.9 Hz, 2 H, fc), 4.22 (vt, J′ ≈ 1.9 Hz, 2 H, fc), 4.44 (vt, J′ ≈ 1.8 Hz, 2 H, fc), 4.55 (vt, J′ ≈ 1.9 Hz, 2 H, fc), 5.60 (br q, 3JHH ≈ 5 Hz, 1 H, NH), 7.31–7.41 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, CDCl3): −16.7 (s). 13C{1H} NMR (100.58 MHz, CDCl3): δ 26.50 (NMe), 69.38 (CH of fc), 71.31 (CH of fc), 72.71 (d, JPC = 4 Hz, CH of fc), 74.31 (d, JPC = 14 Hz, CH of fc), 128.30 (d, 3JPC = 7 Hz, PPh2 CHmeta), 128.80 (PPh2 CHpara), 133.48 (d, 2JPC = 20 Hz, PPh2 CHortho), 138.34 (d, 1JPC = 9 Hz, PPh2 Cipso), 170.30 (CO). Signals due to ferrocene Cipso were not observed. IR (Nujol): νmax 3308 m (νN–H), 1628 s (amide I), 1545 s (amide II) cm−1. ESI+ MS: m/z 428 ([M + H]+). HR MS (ESI) calcd for C24H22FeNNaOP ([M + Na]+) 450.0681, found 450.0680. Anal. Calcd for C24H22FeNO (427.3): C 67.46, N 3.28, H 5.19%. Found: C 67.07, N 3.24, H 5.03%.
:1). Following evaporation, phosphine oxide 4O was isolated as a dark yellow solid. Yield: 40 mg, 82%.
1H NMR (399.95 MHz, CDCl3): δ 2.93 (d, 3JHH = 4.7 Hz, 3 H, NMe), 4.07 (vt, J′ ≈ 1.9 Hz, 2 H, fc), 4.14 (vq, J′ ≈ 1.9 Hz, 2 H, fc), 4.58 (vq, J′ ≈ 1.8 Hz, 2 H, fc), 4.99 (vt, J′ ≈ 1.9 Hz, 2 H, fc), 7.44–7.58 (m, 6 H, PPh2), 7.65–7.73 (m, 4 H, PPh2), 8.76 (br q, 3JHHca. 4.5 Hz, 1 H, NH). 31P{1H} NMR (161.90 MHz, CDCl3): δ 31.6 (s). 13C{1H} NMR (100.58 MHz, CDCl3): δ 26.46 (NMe), 70.26 (CH of fc), 70.74 (CH of fc), 72.56 (d, JPC = 11 Hz, CH of fc), 73.07 (d, 1JPC = 114 Hz, C-PPh2 of fc), 74.91 (d, JPC = 13 Hz, CH of fc), 79.63 (C-CONH of fc), 128.45 (d, JPC = 12 Hz, PPh2 CH), 131.43 (d, JPC = 10 Hz, PPh2 CH), 131.94 (d, 4JPC = 2 Hz, PPh2 CHpara), 133.10 (d, 1JPC = 108 Hz, PPh2 Cipso), 170.35 (CO). IR (Nujol): νmax 3246 m, 1657 s (amide I), 1556 s (amide II) cm−1. ESI+ MS: m/z 466 ([M + Na]+). HR MS (ESI) calcd for C24H23FeNO2P ([M + H]+) 444.0810, found 444.0809. Anal. Calcd for C24H22FeNO2·1/4CH2Cl2 (464.5): C 62.71, N 3.02, H 4.88%. Found: C 62.70, N 2.81, H 4.73%.
:1). Subsequent evaporation afforded the product as a yellow glassy solid. Yield of 4S: 40 mg, 62%.
1H NMR (399.95 MHz, CDCl3): δ 2.92 (d, 3JHH = 4.8 Hz, 3 H, NMe), 3.96 (vt, J′ ≈ 2.0 Hz, 2 H, fc), 4.23 (vq, J′ ≈ 2.0 Hz, 2 H, fc), 4.62 (vq, J′ ≈ 2.0 Hz, 2 H, fc), 4.90 (vt, J′ ≈ 2.0 Hz, 2 H, fc), 7.41 (br q, 3JHH ≈ 5 Hz, 1 H, NH), 7.43–7.56 (m, 6 H, PPh2), 7.68–7.76 (m, 4 H, PPh2). 31P{1H} NMR (161.90 MHz, CDCl3): δ 42.9 (s). 13C{1H} NMR (100.58 MHz, CDCl3): δ 26.13 (NMe), 70.86 (CH of fc), 71.00 (CH of fc), 73.05 (d, JPC = 10 Hz, CH of fc), 74.81 (d, JPC = 12 Hz, CH of fc), 75.98 (d, 1JPC = 97 Hz, C-PPh2 of fc), 79.01 (C-CONH of fc), 128.40 (d, JPC = 13 Hz, PPh2 CH), 131.60 (d, JPC = 10 Hz, PPh2 CH), 131.62 (d, 4JPC = 4 Hz, PPh2 CHpara), 133.51 (d, 1JPC = 88 Hz, PPh2 Cipso), 169.88 (CO). IR (Nujol): νmax 3298 m, 3229 m, 1628 s (amide I), 1558 s (amide II) cm−1. ESI+ MS: m/z 482 ([M + Na]+). HR MS (ESI) calcd for C24H23FeNOPS ([M + H]+) 460.0582, found 460.0583. Anal. Calcd for C24H22FeNOPS·1/2CH2Cl2 (501.2): C 58.64, N 2.79, H 4.62%. Found C 58.57, N 2.71, H 4.53%.
Synthesis of Cu(I) complexes
1H NMR (399.95 MHz, dmso-d6): δ 4.15 (br s, 2 H, fc), 4.19 (vt, 2 H, J′ = 1.7 Hz, fc), 4.51 (vt, 2 H, J′ = 1.7 Hz, fc), 4.82 (br s, 2 H, fc), 7.40 (s, 1 H, NH2), 7.42–7.52 (m, 10 H, PPh2), 7.71 (s, 1 H, NH2). 31P{1H} (161.90 MHz, dmso-d6): δ −10.1 (br s). IR (Nujol): νmax 3450 (m), 3362 (m), 1650 (s) 1583 (s), 1481 (m), 1437 (m), 1405 (m), 1167 (m), 1095 (s), 1069 (s), 1029 (s), 837 (m), 748 (m), 697 (m), 517 (m), 502 (m) cm−1. ESI+ MS: m/z 889 ([Cu(5)2]+), 476 ([Cu(5)]+). Anal. calcd for C46H40BCuF4Fe2N2O2P2·1/2CH2Cl2 (1019.3): C 54.79; N 2.75, H 4.05%. Found C 54.51; N 3.00, H 4.27%.
1H NMR (399.95 MHz, dmso-d6): δ 2.73 (d, 3JHH = 4.5 Hz, 3 H, NMe), 4.15 (vt, J′ = 1.8 Hz, 2 H, fc), 4.18 (vt, J′ = 1.8 Hz, 2 H, fc), 4.52 (vt, J′ = 1.8 Hz, 2 H, fc), 4.75 (vt, J′ = 1.8 Hz, 2 H, fc), 7.38–7.50 (m, 10 H, PPh2), 8.24 (br d, 3JHH ≈ 4.5 Hz, 1 H, NH). 31P{1H} NMR (161.90 MHz, dmso-d6): δ −11.5 (br s). IR (Nujol): ν 3397 (m), 1618 (s), 1601 (w), 1558 (s), 1435 (m), 1411 (m), 1310 (m), 1165 (w), 1061 (s), 1029 (s), 830 (m), 816 (m), 747 (s), 699 (s), 519 (s), 510 (m), 488 (s), 462 (m) cm−1. ESI+ MS: m/z 917 ([Cu(4)2]+), 490 ([Cu(4)]+). Anal. Calcd for C48H44BCuF4Fe2N2O2P2·CH2Cl2 (1089.8): C 54.00, N 2.57, H 4.25%. Found C 54.28, N 2.47, H 4.20%.
1H NMR (399.95 MHz, dmso-d6): δ 2.93 (br s, 3 H, NMe), 3.02 (br s, 3 H, NMe), 4.19 (vt, J′ = 1.8 Hz, 2 H, fc), 4.21 (vt, J′ = 1.8 Hz, 2 H, fc), 4.53 (br s, 2 H, fc), 4.60 (vt, J′ = 1.8 Hz, 2 H, fc), 7.42–7.53 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, dmso-d6): δ −10.5 (br s). IR (Nujol): νmax 1582 (s), 1575 (s), 1505, (m), 1435 (w), 1401 (m), 1377 (m), 1112 (m), 1096 (m), 1050 (s) 1032 (s), 745 (m), 696 (s), 511 (s), 495 (s) cm−1. ESI+ MS: m/z 945 ([Cu(2)2]+), 504 ([Cu(2)]+). Anal. calcd for C50H48BCuF4Fe2N2O2P2 (1032.9): C 58.14, N 2.71; H 4.68%. Found: C 58.63, N 2.52, H 4.94%.
1H NMR (399.95 MHz, dmso-d6, 25 °C): δ 3.28 (br s, 3 H, NMe), 3.53 (s, 3 H, NMe), 4.15 (br s, 2 H, fc), 4.35 (br s, 2 H, fc), 4.60 (br s, 2 H, fc), 4.64 (br s, 2 H, fc), 7.30–7.52 (m, 10 H, PPh2). 1H NMR (399.95 MHz, dmso-d6, 50 °C): δ 3.19 (s, 3 H, NMe), 3.53 (s, 3 H, NMe), 4.16 (br s, 2 H, fc), 4.35 (vt, J′ = 1.9 Hz, 2 H, fc), 4.59 (vt, J′ = 1.9 Hz, 2 H, fc), 4.74 (br s, 2 H, fc), 7.35–7.50 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, dmso-d6, 25 °C): δ −11.0 (br s). IR (Nujol): νmax 1712 (w), 1526 (m), 1436 (w), 1277 (m), 1059 (s) 1135 (m), 825 (m) 745 (m), 697 (m), 511 (m), 491 (w) cm−1. ESI+ MS: m/z 977 ([Cu(3)2]+), 520 ([Cu(3)]+). Anal. Calcd for C50H48BCuF4Fe2N2P2S2·1/4CH2Cl2 (1086.3): C 55.56, N 2.58, H 4.50%. Found: C 55.53, N 2.57, H 4.77%.
Synthesis of Ag(I) complexes
1H NMR (399.95 MHz, CDCl3): δ 4.11 (br vt, J′ = 1.9 Hz, 2 H, fc), 4.46 (br s, 2 H, fc), 4.72 (vt, J′ = 1.9 Hz, 2 H, fc), 4.84 (br vt, J′ = 1.8 Hz, 2 H, fc), 6.24 (br s, 1 H, NH), 6.75 (br s, 1 H, NH), 7.39–7.56 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, CDCl3): δ 3.1 (broad d, 1J(107/109Ag, 31P) ≈ 510 Hz). IR (Nujol): νmax 3444 (m), 3340 (m), 1645 (s), 1591 (s), 1555 (m), 1479 (w), 1436 (s), 1396 (m), 1168 (m), 1098 (br s), 1027 (s), 910 (m), 837 (m), 826 (m), 747 (s), 735 (s), 697 (s), 624 (m), 532 (w), 506 (s), 489 (m), 465 (m) cm−1. ESI+ MS: m/z 933 ([Ag(5)2]+), 520 ([Ag(5)]+). Anal. Calc. for C46H40AgClFe2N2O6P2·CH2Cl2 (1118.7): C 50.46, H 3.76, N 2.50%. Found: C 49.95, H 3.58, N 2.62%.
1H NMR (399.95 MHz, dmso-d6): δ 2.67 (d, 3JHH = 4.6 Hz, 3 H, NMe), 4.12 (vt, J′ = 1.9 Hz, 2 H, fc), 4.29 (vt, J′ = 1.9 Hz, 2 H, fc), 4.60 (vt, J′ = 1.9 Hz, 2 H, fc), 4.85 (vt, J′ = 1.9 Hz, 2 H, fc), 7.44–7.60 (m, 10 H, PPh2), 8.09 (q, 3JHH = 4.6 Hz, 1 H, NH). 31P{1H} NMR (161.90 MHz, dmso-d6): δ 2.6 (pair of d, 1J(107Ag, 31P) = 543 Hz, 1J(109Ag, 31P) = 471 Hz). IR (Nujol): νmax 3389 (m), 3079 (w), 1629 (s), 1615 (s), 1544 (s), 1480 (w), 1435 (m), 1412 (m), 1301 (m), 1194 (m), 1174 (m), 1095 (s), 1067 (br s), 1050 (w), 1031 (m), 914 (m), 816 (m), 748 (s), 697 (s), 623 (m), 529 (m), 509 (s), 492 (m), 467 (m) cm−1. ESI+ MS: m/z 961 ([Ag(4)2]+), 534 ([Ag(4)]+). Anal. Calc. for C48H44AgClFe2N2O6P2·CH2Cl2 (1146.7): C 51.32, H 4.04, N 2.44%. Found: C 50.90, H 3.75, N 2.13%.
1H NMR (399.95 MHz, dmso-d6): δ 2.91 (br s, 3 H, NMe), 2.99 (br s, 3 H, NMe), 4.22 (br s, 2 H, fc), 4.32 (br s, 2 H, fc), 4.61 (vt, J′ = 1.9 Hz, 2 H, fc), 4.65 (vt, J′ = 1.9 Hz, 2 H, fc), 7.49–7.56 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, dmso-d6): δ 4.2 (pair of concentric d, 1J(107Ag, 31P) = 770 Hz, 1J (109Ag, 31P) = 685 Hz). IR (Nujol): νmax 1557 (s), 1492 (s), 1436 (m), 1127 (m), 1109 (s), 1097 (m), 1071 (w), 1050 (s), 824 (w), 758 (m), 747 (m), 695 (m), 679 (s), 623 (s), 514 (w), 503 (w), 490 (m), 467 (w) cm−1. ESI+ MS: m/z 989 ([Ag(2)2]+), 548 ([Ag(2)]+). Anal. Calc. for C50H48AgClFe2N2O6P2·1/2CH2Cl2 (1132.3): C 53.56, H 4.36, N 2.47%. Found: C 53.30, H 4.15, N 2.35%.
1H NMR (399.95 MHz, dmso-d6): δ 3.39 (s, 3 H, NMe), 3.56 (s, 3 H, NMe), 4.12 (dt, J = 3.0, 1.8 Hz, 2 H, fc), 4.30 (vt, J′ = 1.9 Hz, 2 H, fc), 4.73 (vt, J′ = 1.8 Hz, 2 H, fc), 5.06 (vt, J′ = 1.9 Hz, 2 H, fc), 7.50–7.57 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, dmso-d6): δ −0.3 (broad d, 1J(107/109Ag, 31P) ≈ 510 Hz). IR (Nujol): νmax 1551 (s), 1436 (m), 1280 (m), 1144 (m), 1108 (s), 1096 (s), 1061 (m), 1030 (m), 979 (m), 851 (m), 818 (w), 749 (m), 695 (m), 623 (m), 507 (m), 465 (m) cm−1. ESI+ MS: m/z 564 ([Ag(3)]+), 597 ([Ag(3)(CH3OH)]+). Anal. Calc. for C50H48Ag2Cl2Fe2N2O8P2S2 (1329.3): C 45.17, H 3.64, N 2.11%. Found: C 44.83, H 3.81, N 1.90%.
Synthesis of chloridogold(I) complexes
1H NMR (399.95 MHz, dmso-d6): δ 4.28 (vt, J′ = 1.9 Hz, 2 H, fc), 4.48 (dt, J = 3.0, 1.9 Hz, 2 H, fc), 4.59–4.61 (m, 2 H, fc), 4.78 (vt, J′ = 1.9 Hz, 2 H, fc), 7.02 (br s, 1 H, NH), 7.37 (br s, 1 H, NH), 7.54–7.64 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, dmso-d6): δ 28.7 (s). IR (Nujol): νmax 3421 (m), 1667 (s), 1612 (m), 1432 (m), 1349 (m), 1176 (m), 1099 (m), 1027 (m), 843 (m), 822 (m), 745 (m), 688 (s), 526 (m), 511 (m), 496 (w), 477 (s) cm−1. ESI+ MS: m/z 610 ([8a − Cl]+), 668 ([8a + Na]+), 684 ([8a + K]+). Anal. Calcd for C23H20AuClFeNOP (645.6): C 41.75, N 2.12, H 3.05%. Found: C 41.56, N 1.93, H 2.96%.
1H NMR (399.95 MHz, CDCl3): δ 2.93 (d, 3JHH = 4.8 Hz, 3 H, NMe), 4.19 (vt, J′ = 1.8 Hz, 2 H, fc), 4.23 (dt, J = 2.9, 1.9 Hz, 2 H, fc), 4.68–4.70 (m, 2 H, fc), 4.87 (vt, J′ = 1.9 Hz, 2 H, fc), 6.07 (q, 3JHH = 4.8 Hz, 1 H, NH), 7.44–7.61 (m, 10H, PPh2). 31P{1H} NMR (161.90 MHz, CDCl3): δ 28.8 (s). IR (Nujol): νmax 3369 (m), 1628 (s), 1544 (m), 1440 (m), 1298 (m), 1173 (m), 1103 (w), 1029 (m), 1000 (w), 842 (m), 743 (m), 691 (m), 628 (w), 555 (w), 529 (w), 515 (w), 494 (w), 480 (w) cm−1. ESI+ MS: m/z 624 ([8b − Cl]+), 680 ([8b + Na]+), 698 ([8b + K]+). Anal. Calcd for C24H22AuClFeNOP·1/2CHCl3 (719.3): C 40.90, N 1.95, H 3.15%. Found C 41.04, N 1.79, H 3.09% (analytical sample crystallised from chloroform/pentane).
1H NMR (399.95 MHz, dmso-d6): δ 2.85 (s, 3 H, NMe), 3.00 (s, 3 H, NMe), 4.30 (vt, J′ = 1.9 Hz, 2 H, fc), 4.43 (dt, J = 3.0, 1.8 Hz, 2 H, fc), 4.61 (vt, J′ = 1.8 Hz, 2 H, fc), 4.73–4.76 (m, 2 H, fc), 7.54–7.65 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, dmso-d6): δ 28.9 (s). IR (Nujol): νmax 3103 (m), 1622 (s), 1585 (m), 1499 (m), 1439 (s), 1393 (m), 1308 (m), 1265 (m), 1225 (m), 1180 (m), 1173 (m), 1107 (s), 1039 (m), 1032 (m), 840 (m), 821 (m), 758 (s), 752 (s), 704 (s), 558 (m), 535 (m), 526 (s), 504 (s), 490 (s), 474 (m) cm−1. ESI+ MS: m/z 638 ([8c − Cl]+), 696 ([8c + Na]+), 712 ([8c + K]+). Anal. Calcd for C25H24AuClFeNOP (673.7): C 44.57, N 2.08, H 3.59%. Found: C 44.22, N 2.01, H 3.50%.
1H NMR (399.95 MHz, dmso-d6): δ 3.27 (s, 3 H, NMe), 3.29 (s, 3 H, NMe), 4.34 (vt, J′ = 1.9 Hz, 2 H, fc), 4.35 (dt, J = 3.0, 1.8 Hz, 2 H, fc), 4.73 (vt, J′ = 1.9 Hz, 2 H, fc), 4.76–4.79 (m, 2 H, fc), 7.54–7.65 (m, 10 H, PPh2). 31P{1H} NMR (161.90 MHz, dmso-d6): δ 28.7 (s). IR (Nujol): νmax 1712 (w), 1512 (s), 1435 (m), 1309 (m), 1274 (m) 1174 (m), 1138 (m), 1101 (s), 1062 (w), 1034 (w), 1025 (w), 989 (w), 973 (w), 838 (m), 824 (m), 814 (w), 764 (w), 749 (m), 695 (s), 556 (m), 530 (m), 509 (s), 498 (w), 477 (m) cm−1. ESI+ MS: m/z 654 ([8d − Cl]+. Anal. calcd for C25H24AuClFeNPS (689.8): C 43.53, N 2.03, H 3.51%. Found: C 43.88, N 1.87, H 3.45%.
X-ray crystallography
Crystallisation conditions are described in the ESI.† Full set diffraction data (± h ± k ± l, θmax = 26.0–27.5°, completeness ≥99.3%) were collected with a Nonius Kappa CCD diffractometer equipped with an Apex II image plate detector and Cryostream Cooler (Oxford Cryosystems) using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). The data were analysed and corrected for absorption by methods included in the diffractometer software. Details on the data collection, structure solution and refinement are available in the ESI (Table S1†), which also contains conventional displacement ellipsoid plots.All structures were solved by direct methods (SHELXS9742) and refined by full-matrix least-squares based on F2 (SHELXL9742). The non-hydrogen atoms were refined with anisotropic displacement parameters. The amide hydrogen atoms (NH; if present) were located on the difference electron density maps and refined as riding atoms with Uiso(H) set to a 1.2Ueq(N). Hydrogens residing on the carbon atoms were included in their calculated positions and refined similarly with Uiso(H) = 1.5Ueq(C) for the methyl groups and 1.2Ueq(C) for all other CHn moieties. Particular details of the structure refinement are as follows.
Compound 4S crystallises in the chiral Pc space group with two molecules per asymmetric unit. However, all atoms except for the amide moiety in these two independent molecules match each other because of an inversion centre from the space group P21/c (the maximum distance of the overlapping atoms in the phosphinoferrocenyl units is 0.53 Å). Such a virtually higher symmetry results in large correlations during the refinement and, therefore, restrictions to anisotropic displacement parameters had to be applied to several atoms.
Parts of some structures (7b·CHCl3: the solvent and the perchlorate anion; 6b·1/4CHCl3 and 6a′·CHCl3: one of the phosphorus-bound phenyl rings and the BF4− anion) are disordered and were refined over two positions (with isotropic displacement parameters, if necessary). Moreover, the solvating molecules in the structures of 6b·1/4CHCl3 and 6a′·CHCl3 are severely disordered in structural voids and were modelled by PLATON/SQUEEZE.43
All geometric calculations were performed and the diagrams were obtained with the PLATON program.44 All numerical values were rounded with respect to their estimated deviations (ESDs) given to one decimal place. Parameters relating to atoms in constrained positions (hydrogens) are given without ESDs.
DFT computations
Calculations were performed using the density-functional theory (DFT) with Becke's three-parameter functional45 employing the non-local Lee–Yang–Parr correlation functional (B3LYP)46 and the 6-31G* basis set for FcC(E)NH2 and 6-311G** for 2 and 3 with an analytically constructed energy gradient as implemented in the Gaussian 09 program package.47 Geometry optimisations were started from the experimentally determined solid-state structures of 2, 3 and FcCONH2.27b The stationary points of the potential energy surface (PES) were located and harmonic vibrational analysis was performed using the analytically calculated force-constant matrix. For the case of FcC(E)NH2 (E = O, S), relaxed PES scan using ψ as a single variable was performed, starting from the respective stationary points.Electrochemistry
Electrochemical measurements were carried out with a μAUTOLAB III instrument (Eco Chemie) at room temperature (23 °C) using a standard three-electrode cell (Metrohm) equipped with a glassy carbon disc working electrode (2 mm diameter), platinum sheet auxiliary electrode, and a double-junction Ag/AgCl (3 M KCl) reference electrode. The samples were dissolved in anhydrous 1,2-dichloroethane (Sigma-Aldrich; absolute) to give a solution containing 1 × 10−3 M of the analysed compound and 0.1 M Bu4N[PF6] (Fluka, p. a. for electrochemistry). The solutions were deaerated by bubbling with argon and then kept under an argon blanket during the measurement. The redox potentials are given relative to the ferrocene/ferrocenium reference.Acknowledgements
This research was supported through the grants from the Czech Science Foundation (project no. 13-08890S) and the Grant Agency of Charles University in Prague (project no. 643012).Notes and references
- P. Štěpnička, Chem. Soc. Rev., 2012, 41, 4273 RSC.
- For recent examples, see: (a) N. Nasser, P. D. Boyle and R. J. Puddephatt, Organometallics, 2013, 32, 5504 CrossRef CAS; (b) N. Nasser and R. J. Puddephatt, Inorg. Chim. Acta, 2014, 409, 238 CrossRef CAS PubMed; (c) E. C. Constable, N. Hostettler, C. E. Housecroft, N. S. Murray, J. Schönle, U. Soydaner, R. M. Walliser and J. A. Zampese, Dalton Trans., 2013, 42, 4970 RSC; (d) N. Ye and W.-M. Dai, Eur. J. Org. Chem., 2013, 831 CrossRef CAS; (e) I. Philipova, G. Stavrakov and V. Dimitrov, Tetrahedron: Asymmetry, 2012, 23, 927 CrossRef CAS PubMed.
- For recent examples, see: (a) J. Schulz, I. Císařová and P. Štěpnička, Organometallics, 2012, 31, 729 CrossRef CAS; (b) J. Tauchman, B. Therrien, G. Süss-Fink and P. Štěpnička, Organometallics, 2012, 31, 3985 CrossRef CAS; (c) J. Schulz, I. Císařová and P. Štěpnička, Eur. J. Inorg. Chem., 2012, 5000 CrossRef CAS; (d) J. Tauchman, I. Císařová and P. Štěpnička, Dalton Trans., 2011, 40, 11748 RSC; (e) P. Štěpnička, B. Schneiderová, J. Schulz and I. Císařová, Organometallics, 2013, 32, 5754 CrossRef; (f) J. Schulz, J. Tauchman, I. Císařová, T. Riedel, P. J. Dyson and P. Štěpnička, J. Organomet. Chem., 2014, 751, 604 CrossRef CAS PubMed; (g) P. Štěpnička, M. Verníček, J. Schulz and I. Císařová, J. Organomet. Chem., 2014, 755, 41 CrossRef PubMed; (h) M. Semler, J. Čejka and P. Štěpnička, Catal. Today, 2014, 227, 207 CrossRef CAS PubMed; (i) H. Solařová, I. Císařová and P. Štěpnička, Organometallics, 2014, 33, 4131 CrossRef; (j) J. Tauchman, I. Císařová and P. Štěpnička, Dalton Trans., 2014, 43, 1599 RSC.
- (a) A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557 CrossRef CAS PubMed; (b) E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606 RSC.
- J. Podlaha, P. Štěpnička, J. Ludvík and I. Císařová, Organometallics, 1996, 15, 543 CrossRef CAS.
- This method represents a versatile route to phosphinoferrocenes functionalised in position 1′ of the ferrocene unit. For examples, see: (a) I. R. Butler and R. L. Davies, Synthesis, 1996, 1350 CrossRef CAS PubMed; (b) P. Štěpnička, in Ferrocenes: Ligands, Materials and Biomolecules, Wiley, Chichester, 2008, ch. 5, pp. 177–204 Search PubMed.
- P. Štěpnička, H. Solařová, M. Lamač and I. Císařová, J. Organomet. Chem., 2010, 695, 2423 CrossRef PubMed.
- For representative examples of (thio)amide synthesis from organolithium reagents and (thio)carbamoyl halides, see: (a) E. G. Doadt and V. Snieckus, Tetrahedron Lett., 1985, 26, 1149 CrossRef CAS; (b) M. P. Sibi, S. Chattopadhyay, J. W. Dankwardt and V. Snieckus, J. Am. Chem. Soc., 1985, 107, 6312 CrossRef CAS; (c) E. J. Bures and B. A. Keay, Tetrahedron Lett., 1988, 29, 1247 CrossRef CAS; (d) R. J. Mills, N. J. Taylor and V. Snieckus, J. Org. Chem., 1989, 54, 4372 CrossRef CAS; (e) D. L. Comins and H. Hong, J. Am. Chem. Soc., 1991, 113, 6672 CrossRef CAS; (f) P. Gros, Y. Fort, G. Queguiner and P. Caubere, Tetrahedron Lett., 1995, 36, 4791 CrossRef CAS; (g) J. Clayden, N. Westlund and F. X. Wilson, Tetrahedron Lett., 1999, 40, 7883 CrossRef CAS; (h) F. Wudl, S. D. Cox and D. E. Wellman, Mol. Cryst. Liq. Cryst., 1985, 125, 181 CrossRef CAS.
- For examples of the reactions of lithioferrocenes with thiocarbamoyl chlorides, see: (a) K. Hamamura, M. Kita, M. Nonoyama and J. Fujita, J. Organomet. Chem., 1993, 463, 169 CrossRef CAS; (b) S. Jautze, P. Seiler and R. Peters, Angew. Chem., Int. Ed., 2007, 46, 1260 CrossRef CAS PubMed; (c) S. Jautze, P. Seiler and R. Peters, Chem. – Eur. J., 2008, 14, 1430 CrossRef CAS PubMed; (d) T. Hellmuth, S. Rieckhoff, M. Weiss, K. Dorst, W. Frey and R. Peters, ACS Catal., 2014, 4, 1850 CrossRef CAS N.B. The amidation of ferrocene with carbamoyl chlorides has been typically achieved through Friedel–Crafts reactions: (e) W. F. Little and R. Eisenthal, J. Am. Chem. Soc., 1960, 82, 1577 CrossRef CAS; (f) J. Hu, L. J. Barbour and G. W. Gokel, New J. Chem., 2004, 28, 907 RSC.
- Thioamides are typically synthesised through thionation of amides with Lawesson's reagent or P2S5 which, however, convert phosphines to phosphine sulfides: (a) M. Jesberger, T. P. Davis and L. Barner, Synthesis, 2003, 1929 CrossRef CAS PubMed; (b) T. Ozturk, E. Ertas and O. Mert, Chem. Rev., 2010, 110, 3419 CrossRef CAS PubMed; (c) A. B. Charette and M. Grenon, J. Org. Chem., 2003, 68, 5792 CrossRef CAS PubMed and references cited therein.
- This contrasts with the attention dedicated to simple and multi-donor (non-phosphine) thioamide donors. For selected examples, see: E. S. Raper, Coord. Chem. Rev., 1994, 129, 91 CrossRef CAS; W. Zhang and M. Shi, Synlett, 2007, 19 Search PubMed; K. Belhamel, T. K. D. Nguyen, M. Benamor and R. Ludwig, Eur. J. Inorg. Chem., 2003, 4110 CrossRef; M. A. Hossain, S. Lucarini, D. Powell and K. Bowman-James, Inorg. Chem., 2004, 43, 7275 CrossRef PubMed; R. A. Begum, D. Powell and K. Bowman-James, Inorg. Chem., 2006, 45, 964 CrossRef PubMed; N. K. Singh, M. Singh, P. Tripathi, A. K. Srivastava and R. J. Butcher, Polyhedron, 2007, 27, 375 CrossRef PubMed; K. Ahlford, M. Livendahl and H. Adolfsson, Tetrahedron Lett., 2009, 50, 6321 CrossRef PubMed; J. Kulesza, M. Guzinski, V. Hubscher-Bruder, F. Arnaud-Neu and M. Bocheńska, Polyhedron, 2011, 30, 98 CrossRef PubMed; T. Teratani, T. Koizumi, T. Yamamoto, K. Tanaka and T. Kanbara, Dalton Trans., 2011, 40, 8879 RSC and references therein; T. Suzuki, Y. Kajita and H. Masuda, Dalton Trans., 2014, 43, 9732 RSC.
- P.-H. Leung, Y. Qin, G. He, K. F. Mok and J. J. Vittal, J. Chem. Soc., Dalton Trans., 2001, 309 RSC.
- For representative examples, see: (a) K. Issleib and G. Harzfeld, Chem. Ber., 1964, 97, 3430 CrossRef CAS; (b) K. Issleib and G. Harzfeld, Z. Anorg. Allg. Chem., 1967, 351, 18 CrossRef CAS; (c) E. W. Abel and I. H. Sabherwal, J. Chem. Soc. A, 1968, 1105 RSC; (d) A. W. Gal, J. W. Gosselink and F. A. Vollenbroek, J. Organomet. Chem., 1977, 142, 357 CrossRef CAS; (e) H. P. M. M. Ambrosius, F. A. Cotton, L. R. Falvello, H. T. J. M. Hintzen, T. J. Melton, W. Schwotzer, M. Tomas and J. G. M. Van der Linden, Inorg. Chem., 1984, 23, 1611 CrossRef CAS; (f) U. Kunze, H. Jawad, R. Burghardt, R. Tittmann and V. Kruppa, J. Organomet. Chem., 1986, 302, C30 CrossRef CAS and previous articles in this series; (g) A. Gutierrez-Alonso, L. Ballester-Reventos, V. Perez-Garcia and C. Ruiz-Valero, Polyhedron, 1990, 9, 2163 CrossRef CAS; (h) D. Clajus, R. Kramolowsky, G. Siasios and E. R. T. Tiekink, Inorg. Chim. Acta, 1998, 281, 64 CrossRef CAS; (i) O. Crespo, E. J. Fernandez, P. G. Jones, A. Laguna, J. M. Lopez de Luzuriaga, M. Monge, M. E. Olmos and J. Perez, Dalton Trans., 2003, 1076 RSC; (j) A. C. Behrle and J. A. R. Schmidt, Organometallics, 2013, 32, 1141 CrossRef CAS.
- (a) A. Bader and E. Lindner, Coord. Chem. Rev., 1991, 108, 27 CrossRef CAS; (b) C. S. Slone, D. A. Weinberger and C. A. Mirkin, Prog. Inorg. Chem., 1999, 48, 233 CrossRef CAS; (c) P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680 CrossRef CAS.
- P. Štěpnička, J. Schulz, I. Císařová and K. Fejfarová, Collect. Czech. Chem. Commun., 2007, 72, 453 CrossRef.
- The CS stretching band is probably obscured by Nujol. See, for instance, the data for PhC(S)NC5H10: K. A. Jensen and P. H. Nielsen, Acta Chem. Scand., 1966, 20, 597 CrossRef CAS PubMed.
- D. Plażuk, J. Zakrzewski, A. Rybarczyk-Pirek and S. Domagała, J. Organomet. Chem., 2005, 690, 4302 CrossRef PubMed.
- P. Štěpnička and I. Císařová, J. Organomet. Chem., 2006, 691, 2863 CrossRef PubMed.
- (a) J. Kühnert, M. Dušek, J. Demel, H. Lang and P. Štěpnička, Dalton Trans., 2007, 2802 RSC; (b) J. Kühnert, I. Císařová, M. Lamač and P. Štěpnička, Dalton Trans., 2008, 2454 RSC; (c) J. Schulz, I. Císařová and P. Štěpnička, J. Organomet. Chem., 2009, 694, 2519 CrossRef CAS PubMed; (d) J. Tauchman, I. Císařová and P. Štepnička, Organometallics, 2009, 28, 3288 CrossRef CAS and also ref. 3a, c and e.
- A C7–H7⋯SC interaction between molecules related by elemental translation along the crystallographic a-axis can be found in the structure of 3O: C7⋯S = 3.689(2) Å, angle at H7 = 151°.
- The difference in electronegativity is considerably higher in the P/O pair than in the C/O pair.
- Change in the mutual orientation of the cyclopentadienyl ring (i.e., conformation of the 1,1′-disubstited ferrocene scaffold) results in changes in the C(ring1)–Fe–C(ring2) angles, which is also the present case. This concerns mainly amide 2; the maximum difference in interatomic angles for 3 is only 5°.
- (a) D. R. Scott and R. S. Becker, J. Chem. Phys., 1961, 35, 516 CrossRef CAS PubMed (erratum: D. R. Scott and R. S. Becker, J. Chem. Phys., 1961, 35, 2246 CrossRef CAS PubMed; (b) U. Salzner, J. Chem. Theor. Comput., 2013, 9, 4064 CrossRef CAS.
- The position of the absorption band is of course reflected in the colours of the compounds. Whereas amide 2 is orange (orange brown as a bulk solid), thioamide 3 is intensely red-orange (red in the solid state).
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- J. A. S. Howell, P. C. Yates, N. Fey, P. McArdle, D. Cunningham, S. Parsons and D. W. H. Rankin, Organometallics, 2002, 21, 5272 CrossRef CAS (Note: the value was calculated from the data deposited in CSD).
- In the case of primary amides, FcC(E)NH2, the comparison is not possible because the individual molecules are involved in hydrogen-bonding interactions that influence molecular conformations. For instance, the ψ angles determined from the crystal structure data of FcCONH2 are −13.9°, −9.7(2)° and −8.4(2) ° for different polymorphs: (a) D. Salazar-Mendoza, S. A. Baudron, M. W. Hosseini, N. Kyritsakas and A. De Cian, Dalton Trans., 2007, 565 RSC (Note: the first value was calculated from the data deposited with CCDC); (b) P. Štěpnička, I. Císařová, D. Nižňanský and S. Bakardjieva, Polyhedron, 2010, 29, 134 CrossRef PubMed The corresponding thioamide FcCSNH2 shows a lower tilting in the solid state (ψ ≈ –3.7°; ref. 17, Note: the value was calculated from the data deposited with CCDC).
- K. Nakamoto, in Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 5th edn, 1997, Part A: Theory and Applications in Inorganic Chemistry, ch. II-6, pp. 189–201, and Part B: Applications in Coordination, Organometallic and Bioinorganic Chemistry, ch. III-12, pp. 82–83 Search PubMed.
- The M–P bonds between the soft phosphine donor group and the soft metal centre can be expected to be stronger than the M–O bonds involving the amide moiety.
- R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS.
- J. Tauchman, I. Císařová and P. Štěpnička, Eur. J. Org. Chem., 2010, 4276 CrossRef CAS.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- Cambridge Structural Database version 5.35 of November 2013 with updates from November 2013, February 2014 and May 2014.
- C. Pariya, F. R. Fronzek and A. W. Maverick, Inorg. Chem., 2011, 50, 2748 CrossRef CAS PubMed.
- C. Paryia, C. R. Sparrow, C.-K. Back, G. Sandí, F. R. Fronczek and A. W. Maverick, Angew. Chem., Int. Ed., 2007, 46, 6305 CrossRef PubMed.
- The molecules differ by the overall conformation, mainly the phenyl rings (see an overlap in the ESI†).
- K. Rössler, T. Rüffer, B. Walfort, R. Packheiser, R. Holze, M. Zharnikov and H. Lang, J. Organomet. Chem., 2007, 692, 1530 CrossRef PubMed.
- (a) J. E. Aguado, S. Canales, M. C. Gimeno, P. G. Jones, A. Laguna and M. D. Villacampa, Dalton Trans., 2005, 3005 RSC; (b) J. Kühnert, P. Ecorchard and H. Lang, Eur. J. Inorg. Chem., 2008, 5125 CrossRef; (c) A. Hildebrandt, N. Wetzold, P. Ecorchard, B. Walfort, T. Rüffer and H. Lang, Eur. J. Inorg. Chem., 2010, 3615 CrossRef CAS; (d) U. Siemeling, T. Klemann, C. Bruhn, J. Schulz and P. Štěpnička, Z. Anorg. Allg. Chem., 2011, 637, 1824 CrossRef CAS; (e) P. Štěpnička, M. Zábranský and I. Císařová, ChemistryOpen, 2012, 1, 71 CrossRef PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC and references cited therein.
- I. R. Butler and R. L. Davies, Synthesis, 1996, 1350 CrossRef CAS PubMed.
- R. Uson, A. Laguna and M. Laguna, Inorg. Synth., 1989, 26, 85 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc, Wallingford CT, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Summary of crystallographic parameters, an overlap of the two independent molecules in the structure of 8d, displacement ellipsoid plots for all structurally characterised compounds, comparison of the DFT optimised and experimental structures of 2 and 3, and DFT calculated coordinates for compounds 2, 3 and FcC(E)NR2 (E = O, S; R = H, Me) as XYZ files. CCDC 1029509–1029520. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03279a |
| This journal is © The Royal Society of Chemistry 2015 |