
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproved antiparasitic activity by incorporation of organosilane entities into half-sandwich ruthenium(II) and rhodium(III) thiosemicarbazone complexes†
Muneebah
Adams
a,
Carmen
de Kock
b,
Peter J.
Smith
b,
Kirkwood M.
Land
c,
Nicole
Liu
c,
Melissa
Hopper
c,
Allyson
Hsiao
c,
Andrew R.
Burgoyne
a,
Tameryn
Stringer
a,
Mervin
Meyer
d,
Lubbe
Wiesner
b,
Kelly
Chibale
aef and
Gregory S.
Smith
*a
aDepartment of Chemistry, University of Cape Town, Rondebosch 7701, Cape Town, South Africa. E-mail: Gregory.Smith@uct.ac.za; Fax: +27-21-6505195
bDivision of Clinical Pharmacology, Department of Medicine, University of Cape Town, Observatory 7925, South Africa
cDepartment of Biological Sciences, University of the Pacific, Stockton, CA 95211, USA
dDepartment of Biotechnology, University of the Western Cape, Bellville 7535, Cape Town, South Africa
eInstitute of Infectious Disease and Molecular Medicine, University of Cape Town, Rondebosch 7701, South Africa
fSouth African Medical Research Council Drug Discovery & Development Research Unit, University of Cape Town, Rondebosch 7701, South Africa
First published on 5th January 2015
Abstract
A series of ferrocenyl- and aryl-functionalised organosilane thiosemicarbazone compounds was obtained via a nucleophilic substitution reaction with an amine-terminated organosilane. The thiosemicarbazone (TSC) ligands were further reacted with either a ruthenium dimer [(η6-iPrC6H4Me)Ru(μ-Cl)Cl]2 or a rhodium dimer [(Cp*)Rh(μ-Cl)Cl]2 to yield a series of cationic mono- and binuclear complexes. The thiosemicarbazone ligands, as well as their metal complexes, were characterised using NMR and IR spectroscopy, and mass spectrometry. The molecular structure of the binuclear ruthenium(II) complex was determined by single-crystal X-ray diffraction analysis. The thiosemicarbazones and their complexes were evaluated for their in vitro antiplasmodial activities against the chloroquine-sensitive (NF54) and chloroquine-resistant (Dd2) Plasmodium falciparum strains, displaying activities in the low micromolar range. Selected compounds were screened for potential β-haematin inhibition activity, and it was found that two Rh(III) complexes exhibited moderate to good inhibition. Furthermore, the compounds were screened for their antitrichomonal activities against the G3 Trichomonas vaginalis strain, revealing a higher percentage of growth inhibition for the ruthenium and rhodium complexes over their corresponding ligand.
Introduction
Parasitic diseases such as malaria and trichomoniasis are rife due to the emergence of strains that are resistant to widely used, cost-effective drugs. Malaria, a highly infectious parasitic disease caused by the plasmodium protozoan, is responsible for over 2 million cases worldwide.1 Chemotherapeutic drugs used to treat the disease consist mainly of quinoline-based drugs (e.g. Chloroquine) and artemisinin combination therapies (ACTs). ACTs are drug regimens in which one compound, an artemisinin derivative, is a fast but short-acting drug while the other drug has a longer half-life, enabling it to clear any remaining parasites.2Trichomoniasis is the most common sexually transmitted parasitic disease caused by the protozoal parasite Trichomonas vaginalis. Trichomoniasis is generally easily treatable; however, in most cases the infected individual is asymptomatic. If left untreated, the disease could spread or make the infected individuals more susceptible to HIV, cervical and aggressive prostate cancer.3–7 Antitrichomonal drugs consist mainly of 5-nitroimidazole compounds whereas current treatments are the FDA approved drugs such as metronidazole or tinidazole.8,9
However, a common problem pertaining to the treatment of parasitic diseases is the tendency of a parasite to develop resistance. Therefore, alternative strategies have been employed to address this problem. One such strategy involves the incorporation of metals into drugs with known pharmacological properties. The clinical success of transition metal complexes such as cisplatin, as well as compounds currently in clinical trials such as the ruthenium complex NKP-1339 or ferroquine, has demonstrated that the use of transition metals is a promising strategy.10–12
In an effort to improve lipophilicity and concomitant drug accumulation, silicon has previously been incorporated into the scaffold of a known drug to either enhance the potency of the drug or remodel the drug for the treatment of another disease. The silicon-containing compounds generally exhibit enhanced pharmacological activity and reduced toxicity when compared to their corresponding non-silicon analogues.13–15 In the case when a compound enters the cell via membrane crossing, the more lipophilic nature of silicon-containing compounds has been associated with enhanced cell and tissue penetration.13 Smith et al. prepared a series of quinoline and ferroquine organosilane derivatives which were evaluated for their antiplasmodial activity against chloroquine-sensitive (NF54) and chloroquine-resistant (Dd2) strains.16,17 The quinoline derivatives exhibited activity in the low nanomolar range, whilst the ferroquine derivatives exhibited nanomolar range activities comparable to or significantly greater than both chloroquine and ferroquine.16,17
Thiosemicarbazones are Schiff-base type compounds known for their pharmacological properties, particularly as antiparasitic18–22 and antitumoural agents.18,23–26 Generally, thiosemicarbazones act as chelating agents for various endogenous metal ions by bonding through the sulfur atom and the imine nitrogen atom. The precise mode of action of thiosemicarbazones on the parasite is currently unknown. However, different modes of action have been hypothesised for antimalarial agents: mechanisms such as binding to cysteine proteases, forming toxic complexes when bound to the cellular metals (generation of reactive oxygen species), preventing the uptake of cellular iron thus inhibiting various enzymes including ribonucleotide reductase (DNA synthesis) as well as interfering with electron transport and pentose phosphate shunt enzymes.27–30 The formation of metal complexes may aid either the transport into, or the accumulation of the compound within the active site, due to the lipophilicity of the compound changing upon coordination.31
Studies previously conducted by our group based on non-chloroquine-based derivatives identified thiosemicarbazone-containing compounds18,21,32,33 which exhibited moderate antiplasmodial and antitrichomonal activity (Fig. 1). It was observed that the compounds containing chlorides on the aromatic ring generally displayed better activity against P. falciparum and T. vaginalis than the compounds with unsubstituted aromatic rings.21
 | ||
| Fig. 1 Ruthenium(II) and palladium(II) complexes, which exhibit antiparasitic activity.21,32 | ||
Herein, we now report on the synthesis and biological activities of a series of half-sandwich organometallic thiosemicarbazone complexes after incorporating an amine-terminated organosilane on the terminal nitrogen. This study aims to show their increased biological activity through increased lipophilicity when endowed with an organosilane entity.
Results and discussion
Schiff-base dithiocarbamates (1a–b; 2b) were prepared following published methods,34–36 and a similar method was followed to synthesise the new compound 2a (Scheme 1). The Schiff-base dithiocarbamates were reacted with the amines, (aminomethyl)trimethylsilane and N,N′-dimethylpropanamine, to afford the silicon-containing thiosemicarbazones 3a–b, 4a–b and compound 5 (Scheme 1). Ruthenium(II) and rhodium(III) complexes, containing either the p-cymene (iPrC6H4Me) or pentamethylcyclopentadienyl (Cp*) ancillary ligand respectively, were prepared and isolated as the chloride salts. Two equivalents of the thiosemicarbazone, which acts as an N,S-chelate, were reacted with the ruthenium dimer [(η6-iPrC6H4Me)Ru(μ-Cl)Cl]2 to yield complexes 6a–b, 7a–b and 8. In a similar manner, the rhodium complexes (9a–b; 10a–b; 11) were prepared by reacting the thiosemicarbazone (2 equiv.) with the rhodium dimer [(Cp*)Rh(μ-Cl)Cl]2.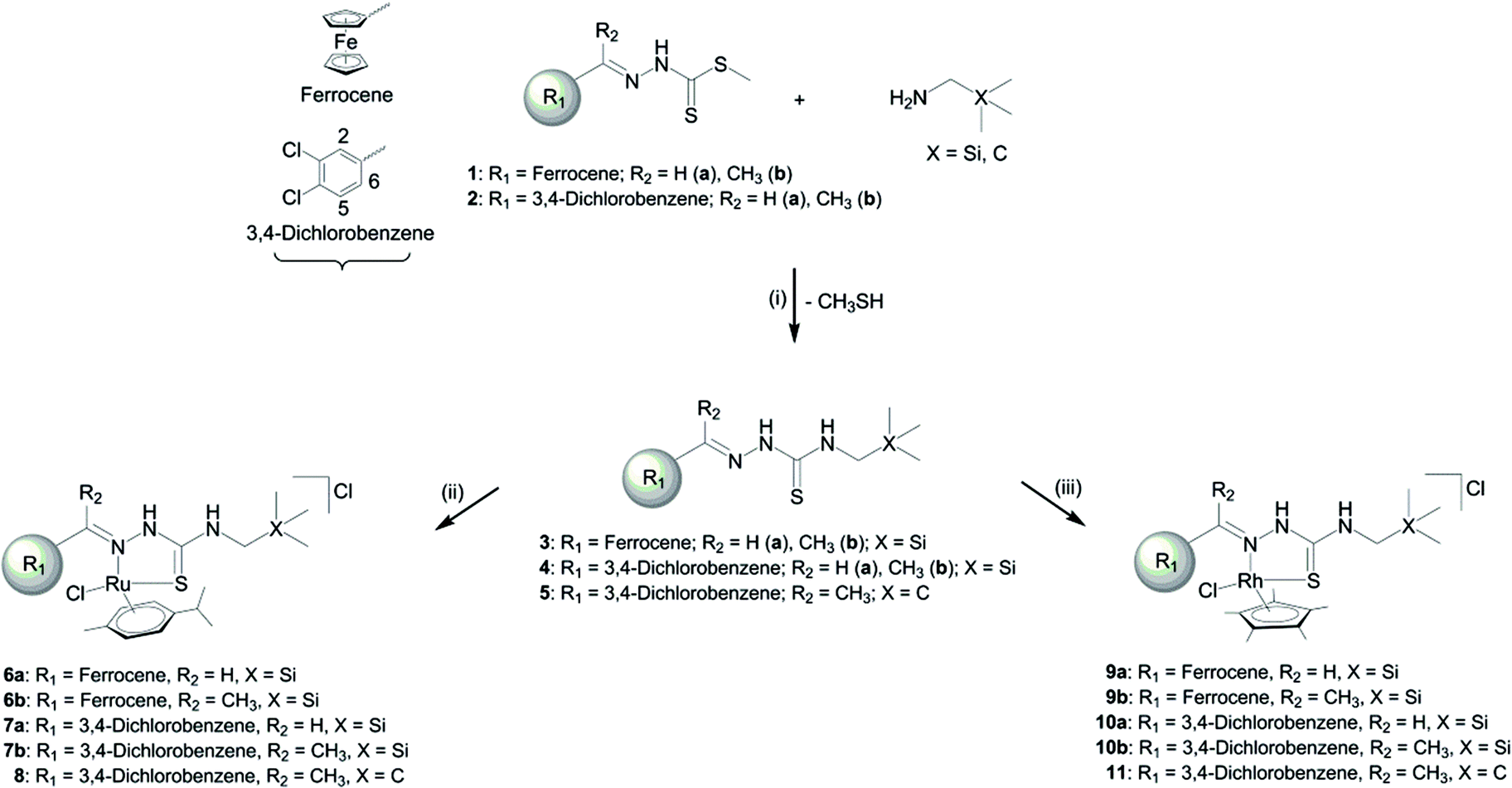 | ||
| Scheme 1 Synthesis of organosilane thiosemicarbazones and their metal complexes. (i) EtOH, reflux, N2; (ii) [(η6-iPrC6H4Me)Ru(μ-Cl)Cl]2, DCM, RT, 4 h; (iii) [(Cp*)Rh(μ-Cl)Cl]2, DCM, RT, 4 h. | ||
The compounds were characterised using 1H and 13C NMR spectroscopy. The presence of a singlet in the range of 0.10–0.18 ppm (3a–b; 4a–b) and a singlet at 1.04 ppm (5) confirms the displacement of the methanethiolate group (1a–b; 2a–b) with the Si(CH3)3 and C(CH3)3 containing amine groups. It has also been observed that the CH2 protons couple to the proton of the adjacent secondary amine (CH2NH), resonating as a doublet, while the amine proton resonates as a triplet. Additionally, in the 13C NMR spectra the displacement of the methanethiol group with the amine-terminated organosilane results in a more shielded thiocarbonyl carbon which resonates at ca. 178 ppm (e.g. 199.07 ppm for 2a). The 1H NMR spectra of these chiral ruthenium and rhodium complexes display peaks which are characteristics of these types of chiral N,S-chelate systems.21,37,38 The characteristic splitting pattern of the p-cymene arene ring is observed: methyl groups (isopropyl) resonate as two doublets; four doublets for the aromatic protons.21,37–39 A singlet is observed for the pentamethylcyclopentadienyl ring in the 1H NMR spectra of the rhodium complexes.
The thiosemicarbazone compounds (3a–b; 4a–b; 5) were analysed using EI+-MS, and the molecular ion peak is observed at m/z 373, 387, 333, 347 and 331 respectively. In the ESI+ mass spectra of the ruthenium complexes 6a–b, 7a–b and 8 the peak is observed at m/z 304, 311, 284, 291 and 283 respectively for the fragment [M − Cl]2+ (M refers to the cationic portion excluding the Cl counter-ion). The same fragment is observed at m/z 305, 312, 285, 292 and 284 for rhodium complexes 9a–b, 10a–b and 11 respectively.
Single crystals of compound 6b were grown by the slow diffusion of pentane into a solution of 6b in chloroform, and analysed using single crystal X-ray diffraction analysis. The compound crystallised as red blocks with a molecule of chloroform in a monoclinic crystal system with space group P21/c. Crystallographic data are presented in Table 1 and selected bond lengths and angles are listed in Table 2.
| 6b·CHCl3 | |
|---|---|
| Chemical formula | C27H39Cl2FeN3RuSSi.CHCl3 |
| Formula weight | 812.96 |
| Crystal system | Monoclinic |
| Space group | P21/c |
| Crystal size (mm) | 0.04 × 0.16 × 0.17 |
| a/Å | 13.8986(3) |
| b/Å | 22.6091(4) |
| c/Å | 11.4831(3) |
| α/° | 90 |
| β/° | 103.4820(10) |
| γ/° | 90 |
| V/Å3 | 3508.95(13) |
| Z | 4 |
| T/K | 173(2) |
| D c/g cm−3 | 1.539 |
| μ/mm−1 | 1.341 |
| Scan range/° | 3.5 < θ < 27.5 |
| Unique reflections | 8016 |
| Reflections used [I > 2σ(I)] | 6426 |
| R int | 0.061 |
| R indices (all data) | 0.0519, wR2 0.1587, S 1.06 |
| Goodness-of-fit | 1.012 |
| Max, Min Δρ/e Å | −1.96, 2.26 |
| Bond lengths (Å) | |
|---|---|
| Ru–N(3) | 2.158(4) |
| Ru–S | 2.3497(11) |
| Ru–Cl(1) | 2.4134(12) |
| N(1)–C(5) | 1.333(6) |
| N(2)–C(5) | 1.346(6) |
| N(3)–C(6) | 1.303(6) |
| C(5)–S | 1.701(4) |
| Bond angles (°) | |
|---|---|
| N(3)–Ru–S | 82.11(10) |
| N(3)–Ru–Cl(1) | 88.04(11) |
| Cl(1)–Ru–S | 86.07(4) |
| Ru–S–C(5) | 99.19(16) |
As seen in Fig. 2 and looking at the bond angles around the ruthenium metal centre (Table 2), the compound has a pseudo-tetrahedral geometry around the ruthenium(II) metal. The selected bond lengths and angles listed in Table 2 are similar to those observed for analogous compounds.23,37,40,41 The bond length of C(5)–S [1.701(4) Å] was compared to similar complexes, as well as similar ligands, and was found to be comparable, which suggests that the ligand chelates in the thione form (Table 2).23,37,42 This is also confirmed by comparison of the N–C bond lengths, where the N(2)–C(5) [1.346 Å] bond is longer than the imine bond N(3)–C(6) [1.303 Å], which suggests that N(2)–C(5) is not a double bond and thus the thiosemicarbazone chelates in the thione form.23,37
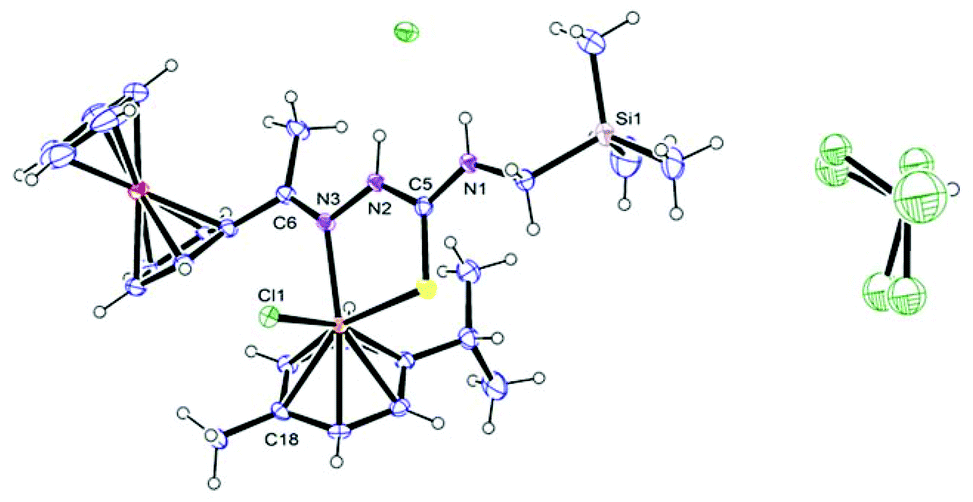 | ||
| Fig. 2 ORTEP diagram. The molecular structure of compound 6b. | ||
Predicting the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P values of dithiocarbamates and thiosemicarbazones
P values of dithiocarbamates and thiosemicarbazones
The incorporation of silicon into the framework of these compounds was carried out on the basis that an enhancement of the compound's lipophilicity may lead to the enhancement of biological activity as well as a reduced cytotoxicity to non-infected cells. Table 3 lists the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values either calculated using Marvinsketch or estimated using the method described in the Experimental section.
P values for the dithiocarbamates and thiosemicarbazones
P values either calculated using Marvinsketch or estimated using the method described in the Experimental section.
P values for the dithiocarbamates and thiosemicarbazones
| Compound number | logP |
Compound number | logP |
Compound number | logP |
|---|---|---|---|---|---|
|
a LogP values calculated using MarvinSketch V14.9.29.0.
b LogP of ferrocenyl compounds estimated using methods described in the Experimental section.
|
|||||
| 1a | 3.73b | 3a | 4.91b | FerrocenylTSC21 | 1.66b |
| 1b | 3.54b | 3b | 4.46b | ||
| 2a | 4.58a | 4a | 5.43a | ||
| 2b | 4.43a | 4b | 5.07a | 3,4-DichloroacetophenoneTSC32 | 2.72a |
| 5 | 4.56a | ||||
Comparison of the dithiocarbamate with the corresponding TSCs showed that incorporation of the side chain led to an increase in the logP value (Table 3). A more significant increase is observed for the organosilane TSCs than the non-silicon TSC (5). Furthermore, the logP values of two non-silicon thiosemicarbazones which are analogous to compounds 3a and 4b, and contain a terminal primary amine, were calculated. Their logP values were significantly lower than the thiosemicarbazones evaluated in this study and in certain cases, lower than chloroquine.43
In vitro antiplasmodial and cytotoxicity studies
The in vitro antiplasmodial activities of the ferrocenyl- (1a–b) and aryl-derived (2a–b) Schiff-base dithiocarbamates and their corresponding organosilanes (3a–b; 4a–b) and the non-silicon (5) thiosemicarbazone were evaluated against two P. falciparum strains, NF54 (chloroquine-sensitive) and Dd2 (chloroquine-resistant). The ruthenium (6b; 7a–b; 8) and rhodium (9a; 10a–b; 11) complexes were also evaluated against the two strains. Chloroquine diphosphate (CQDP) and Artesunate were used as the control drugs in this study, and the antiplasmodial data are listed in Table 4. Only compounds displaying IC50 values in the low micromolar range (against the NF54 strain) were further tested against the Dd2 strain, and the cytotoxicities of these compounds were evaluated against the Chinese hamster ovarian (CHO) cell line with Emetine as the control drug.| Compound number | IC50 values (μM) | RIa | SI1b |
SI2c |
||
|---|---|---|---|---|---|---|
| NF54 | Dd2 | CHO | ||||
| a RI = IC50(Dd2)/IC50(NF54). b SI1 = IC50(CHO)/IC50(NF54). c SI2 = IC50(CHO)/IC50(Dd2). d — = not determined. | ||||||
| 1a | 52.17 ± 11.69 | —d | — | — | — | — |
| 1b | 3.76 ± 1.35 | 13.70 ± 1.32 | 0.49 ± 0.21 | 3.64 | 0.13 | 0.036 |
| 2a | 27.33 ± 7.85 | — | — | — | — | — |
| 2b | 2.59 ± 0.89 | 10.95 ± 1.06 | 1.20 ± 0.41 | 4.23 | 0.46 | 0.11 |
| 3a | 1.65 ± 0.37 | 2.62 ± 0.29 | > 267.81 | 1.59 | — | — |
| 3b | 7.92 ± 1.99 | — | — | — | — | — |
| 4a | 1.26 ± 0.69 | 6.91 ± 0.48 | 25.10 ± 1.23 | 5.48 | 19.92 | 3.63 |
| 4b | 2.24 ± 0.26 | 2.40 ± 0.57 | 29.28 ± 0.89 | 1.07 | 13.07 | 12.20 |
| 5 | 175.74 ± 43.03 | — | — | — | — | — |
| 6b | 7.81 ± 0.56 | — | — | — | — | — |
| 7a | 2.92 ± 0.33 | 4.28 ± 0.33 | 71.82 ± 18.11 | 1.47 | 24.60 | 16.78 |
| 7b | 4.19 ± 0.12 | 6.66 ± 2.58 | 21.54 ± 3.80 | 1.59 | 5.14 | 3.23 |
| 8 | 2.57 ± 0.99 | 2.29 ± 0.25 | 3.65 ± 0.63 | 0.89 | 1.42 | 1.59 |
| 9a | 1.80 ± 0.04 | 2.27 ± 0.10 | 53.49 ± 1.45 | 1.26 | 29.72 | 23.56 |
| 10a | 3.73 ± 0.96 | 3.11 ± 0.58 | 14.89 ± 0.37 | 0.83 | 3.99 | 4.79 |
| 10b | 1.31 ± 0.29 | 1.18 ± 0.09 | 10.85 ± 1.00 | 0.90 | 8.28 | 9.19 |
| 11 | 3.41 ± 0.41 | 1.01 ± 0.16 | 4.10 ± 0.19 | 0.30 | 1.20 | 4.06 |
| CQDP | 0.0097 ± 0.0039 | 0.19 ± 0.06 | — | 19.59 | — | — |
| Artesunate | 0.0104 ± 0.0026 | 0.05 ± 0.02 | — | 4.81 | — | — |
| Emetine | — | — | 0.13 ± 0.0062 | — | — | — |
Comparing the data for the Schiff-base dithiocarbamates (1a–b; 2a–b) it is observed that 1b and 2b (R2 = CH3) are the most active with IC50 values 3.76 and 2.59 μM respectively. However, a loss of activity was observed towards the Dd2 strain, and 1b and 2b were found to be more toxic towards non-infected cells (CHO cell line). Incorporation of the organosilane moiety generally led to the enhancement of activity for thiosemicarbazones 3a, 4a and 4b compared to the dithiocarbamates. Previously evaluated thiosemicarbazones with unsubstituted terminal amines were less potent than the organosilane thiosemicarbazones mentioned herein.32,44 Even though a slight loss of activity was observed against the Dd2 strain, these organosilane thiosemicarbazones were found to be more selective towards the parasite strains than the non-infected cell-line. Compound 5, a non-silicon derivative of 4b, was tested to determine whether any observed activity can be attributed to the incorporation of the silicon atom or the entire group on the terminal nitrogen. Compound 5 (175.74 μM) was found to be significantly less potent compared to its silicon counterpart 4b (2.24 μM), which suggests that the incorporation of the silicon atom may play a role in improving the potency.
The logP values for the Schiff-base dithiocarbamates and the thiosemicarbazones were predicted (Table 3). The TSCs which displayed the higher logP values generally displayed the lowest IC50 values. Additionally, the ferrocenyl [3.43 μM (NF54); 6.40 μM (Dd2)] and 3,4-dichloroacetophenone [14.1 μM (NF54); 9.82 μM (Dd2)] analogues were found to have lower logP values (Table 3) and higher IC50 values than the corresponding TSCs evaluated in this study.21,32 This suggests that a difference in lipophilicity may assist in the compounds’ mode of action.
The ruthenium complexes were observed to be slightly less active than the corresponding thiosemicarbazones. A further loss of activity is observed when evaluated against the Dd2 strain. On the other hand, the potency of the non-silicon thiosemicarbazone is significantly enhanced upon complexation with ruthenium (8; 2.57 μM). However, compound 8 does not display selectivity towards the parasitic strains versus the mammalian cell line, whereas the silicon-containing ruthenium complexes are more selective towards the parasitic strains. The rhodium complexes exhibit activities in the low micromolar range, and the corresponding silicon-containing complexes are once again less cytotoxic towards the CHO cell line when compared to the non-silicon containing rhodium complex (11).
The resistance index [RI = IC50(Dd2)/IC50(NF54)] was calculated and the tested compounds had lower RI values than chloroquine (RI = 19.59). The rhodium complexes generally had RI values below 1. This also suggests that they may be less susceptible to cross resistance.
At this stage, no conclusive statement can be made regarding the use of ruthenium versus rhodium.
β-Haematin inhibition studies
Haemozoin is an important target of many antimalarials, including chloroquine. When chloroquine reaches the digestive vacuole of the malaria parasite, it binds to a toxic product of haemoglobin degradation known as haematin (ferriprotoporphyrin IX) and prevents conversion into less toxic haemozoin. Large amounts of haematin in the digestive vacuole will result in parasite damage. The NP-40 detergent mediated assay was used to establish whether selected thiosemicarbazone compounds prepared in this study inhibit β-haematin (synthetic haemozoin) formation.45 The neutral detergent, NP-40, is used to mimic lipids and mediate β-haematin formation in the assay. The amount of β-haematin was quantified using the colorimetric pyridine ferrochrome method developed by Egan et al.46 Three of the most active compounds (4b, 9a and 10b) were screened for their ability to inhibit β-haematin formation. Table 5 gives the IC50 values obtained for this study.Compounds 4b, 9a and 10b were screened with chloroquine for β-haematin inhibition activity. Ligand 4b did not exhibit activity at the tested concentration. Complex 10b was found to be the most active compound, exhibiting an IC50 value of approximately 38 μM, and was also more active than chloroquine. Complex 9a exhibited moderate β-haematin inhibition activity (IC50 = 150 μM). From the data it is observed that the complex possessing the chlorido moieties (10b) exhibits good β-haematin inhibition activity. The electron withdrawing chloride positioned on the quinoline ring of chloroquine and ferroquine is essential for β-haematin inhibition by providing the correct charge distribution. Therefore the presence of the increased number of chlorido moieties on 10b may enhance the β-haematin inhibition activity. The complex also displays enhanced activity compared to its free ligand (4b). This may be a consequence of the higher lipophilicity of the complex compared to the ligand. There also appears to be some relationship between the antiplasmodial activity and the ability of the tested compounds to inhibit haemozoin formation. Compound 4b was the least effective inhibitor of β-haematin formation and also displayed lower antiplasmodial activity than 9a and 10b. Complex 10b inhibited β-haematin formation to a greater extent and was also more active than 4b and 9a in the NF54 and Dd2 strains of P. falciparum. Since compound 4b exhibited good antiplasmodial activity but not very good β-haematin inhibition activity, these compounds may act by a different mechanism of action. This suggests that haemozoin may not be the only target, but may be one of the reasons for the activity of these compounds.
In vitro antitrichomonal studies
In vitro antitrichomonal screening was carried out for the synthesised compounds (50 μM samples) to determine general growth inhibition against the strain G3 of Trichomonas vaginalis. DMSO was employed as a control for the screening, and the antitrichomonal activity data are listed in Table 6.| Compound number | Percent inhibition ± standard error at 50 μM | Strain G3 IC50 (μM) |
|---|---|---|
| a — = Not determined. | ||
| 1a | 38.50 ± 4.90 | —a |
| 1b | 39.13 ± 3.24 | — |
| 2a | 34.27 ± 10.20 | — |
| 2b | 33.23 ± 3.62 | — |
| 3a | 41.17 ± 10.89 | — |
| 3b | 40.93 ± 7.63 | — |
| 4a | 51.10 ± 3.45 | — |
| 4b | 35.07 ± 1.57 | — |
| 5 | 15.56 ± 5.09 | — |
| 6b | 93.00 ± 5.66 | 35.21 ± 0.22 |
| 7a | 97.13 ± 1.62 | 17.53 ± 0.88 |
| 7b | 90.53 ± 5.00 | 12.55 ± 0.61 |
| 9a | 100.00 ± 0.00 | 7.51 ± 0.81 |
| 10a | 91.57 ± 1.89 | 7.28 |
| 10b | 72.97 ± 9.75 | — |
| 11 | 50.60 ± 16.61 | — |
| Metronidazole | 100 | 0.72 |
The Schiff-base dithiocarbamates (1a–b, 2a–b) and thiosemicarbazones (3a–b, 4a–b, 5) generally displayed poor inhibitory effects (<50%, Table 6). However, complexation of the thiosemicarbazones with ruthenium and rhodium led to a significant enhancement of the inhibitory effects (>90%). As proposed, the non-silicon containing thiosemicarbazone ligand (5) and rhodium complex (11) displayed the lowest inhibitory effects when compared to the silicon containing thiosemicarbazones and the rhodium complexes respectively (Table 6).
Only compounds with percent inhibition above 90% were further analysed by determining their IC50 values against the G3 strain. Of the IC50 values listed in Table 5, the ferrocenyl-ruthenium complex 6b (IC50 = 35.21 μM) showed significantly less activity compared to its aryl-derived ruthenium counterparts 7a (IC50 = 17.53 μM) and 7b (IC50 = 12.55 μM). Rhodium complexes 9a and 10a exhibit similar activities with IC50 values of 7.51 μM and 7.28 μM, respectively.
In vitro anti-tumour studies
Ferrocenyl compounds such as ferrocifen and its analogues have displayed significant antiproliferative effects on human cancer cell-lines.47,48 Therefore, preliminary anti-tumour studies were carried out on selected ferrocenyl compounds to determine whether they display anti-tumour activities. The ferrocenyl Schiff-base dithiocarbamates (1a–b), the organosilane thiosemicarbazones (3a–b), and a ruthenium complex (6a) were screened against cisplatin-sensitive (A2780) and cisplatin-resistant (A2780cisR) human ovarian carcinoma cell lines, as well as non-tumourigenic human fibroblast cells (KMST-6). The anti-tumour activities are listed in Table 7.| Compound number | IC50 (μM) | SI1a |
SI1b |
||
|---|---|---|---|---|---|
| A2780 | A2780cisR | KMST-6 | |||
| a SI1 = IC50(KMST-6)/IC50(A2780). b SI2 = IC50(KMST-6)/IC50(A2780cisR). | |||||
| 1a | 42.67 ± 5.03 | 76.52 ± 4.15 | 73.86 ± 4.15 | 1.73 | 0.96 |
| 1b | 120.77 ± 2.96 | 150.23 ± 3.85 | 85.83 ± 3.85 | 0.71 | 0.57 |
| 3a | 111.13 ± 5.87 | 110.25 ± 10.58 | 104.65 ± 10.58 | 0.94 | 0.95 |
| 3b | 82.14 ± 12.12 | 113.37 ± 4.02 | 132.27 ± 4.02 | 1.61 | 1.17 |
| 6b | 12.45 ± 2.50 | 18.91 ± 3.67 | 17.23 ± 3.67 | 1.38 | 0.91 |
| Cisplatin | 1.97 ± 3.41 | 18.90 ± 0.81 | 44.04 ± 0.81 | 22.35 | 2.33 |
The Schiff-base dithiocarbamates 1a (R2 = H) appear to be slightly more active than 1b (R2 = CH3) against both A2780 and A2780cisR cell lines (Table 7). Upon the formation of the organosilane thiosemicarbazone a loss of activity is observed for 3a, whilst 3b [R2 = CH3; 82.13 μM (A2780); 113.4 μM (A2780cisR)] displays a slight enhancement of activity. A significant enhancement of activity is observed for the ruthenium complex [6b; R2 = CH3; 12.45 μM (A2780); 18.91 μM (A2780cisR)] when compared to the TSC 3b. The activity against the resistant strain is comparable to the activity of cisplatin (18.90 μM). In general, all the compounds exhibited similar cytotoxicities against the tumourigenic cell-lines and the non-tumourigenic cells, which leads to unfavourably low selectivity index values.
Conclusions
We have introduced a series of organosilane-containing thiosemicarbazone ruthenium(II) and rhodium(III) complexes as antiparasitic agents. The compounds generally exhibited activities in the low micromolar range when screened against chloroquine-sensitive (NF54) and chloroquine-resistant (Dd2) P. falciparum strains. Two rhodium(III) complexes inhibited β-haematin formation, suggesting that this may be a mode of action for their antiplasmodial activity. The organosilane compounds were also found to be less cytotoxic towards the Chinese hamster ovarian cell line than the non-silicon containing complexes.Antitrichomonal data suggested that ruthenium(II) and rhodium(III) complexes generally display greater inhibitory effects against the G3 strain of Trichomonas vaginalis when compared to the uncoordinated compounds. Furthermore, rhodium(III) complexes demonstrated higher inhibitory action compared to ruthenium(II) complexes, and the silicon-containing compounds were more effective than the non-silicon compounds.
The ruthenium-arene and rhodium-cyclopentadienyl fragments are believed to enhance the lipophilicity of the compounds to which they coordinate by imparting hydrophobic character to the molecule, which may assist in passive diffusion through cell membranes, thus enhancing cellular accumulation. The presence of the ring may also facilitate interactions of the complexes with DNA or proteins.19,49 Perhaps these factors play a role in the inhibition observed for the ruthenium and rhodium complexes evaluated in this study.
Overall, the silicon-containing compounds displayed greater activity towards the P. falciparum parasitic strains compared to the non-infected cell line, and the proposed idea that silicon-containing compounds are more effective and less cytotoxic has been demonstrated.
Experimental
General details
All reagents and solvents were purchased from commercial suppliers. All reagents were purchased from Sigma-Aldrich or ABCR Chemicals and were used without further purification. Metal salts were kindly donated by AngloAmerican Platinum Limited. Methyl hydrazinecarbodithioate,50 Schiff-base dithiocarbamates [ferrocenyl (1a),34 acetylferrocenyl (1b)35 and 3,4-dichloroacetophenone (2b)36], [(η6-iPrC6H4Me)Ru(μ-Cl)Cl]251 and [(Cp*)Rh(μ-Cl)Cl]252 were synthesised by means of published methods. NMR spectra were recorded on either a Varian Mercury XR300 MHz (1H: 300.08 MHz), XR400 MHz (1H at 399.95 MHz) spectrometer or a Bruker Biospin GmbH (1H at 400.22 MHz, 13C at 100.65 MHz, 31P at 162.00 MHz) spectrometer at ambient temperature. 1H and 13C{1H} NMR chemical shifts are referenced to the deuterated solvent. Infrared (IR) spectra were determined using a Perkin-Elmer Spectrum 100 FT-IR spectrometer and were recorded using KBr pellets or ATR. Elemental analyses (C, H and N) were performed on a Thermo Flash 1112 Series CHNS-O analyser. Mass spectrometry was either carried out on a JEOL GCmateII and data were recorded using the Electron Impact (EI) mode or on a Waters API Quattro Micro triple quadrupole mass spectrometer (samples were injected into a stream of 50% acetonitrile and 0.1% formic acid) and data were recorded using electrospray ionisation (ESI) mass spectrometry in the positive mode. Melting points were determined using the Büchi Melting Point apparatus B-540.
Schiff-base dithiocarbamates
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); 811 (CS). δH (399.95 MHz, DMSO-d6): 13.4 (1H, s, NH); 8.21 (1H, s, HCN); 7.94 (1H, s, C(2)H); 7.71 (2H, m, C(5)H & C(6)H); 2.53 (3H, s, SCH3). δC (100.64 MHz, DMSO-d6): 199.0 (CS); 143.4 (CN); 134.2, 132.8, 131.8, 131.1, 128.7, 127.0 (CAr); 16.7 (SCH3). MS (EI+, m/z): 278 ([M]+, 100%).
N); 811 (CS). δH (399.95 MHz, DMSO-d6): 13.4 (1H, s, NH); 8.21 (1H, s, HCN); 7.94 (1H, s, C(2)H); 7.71 (2H, m, C(5)H & C(6)H); 2.53 (3H, s, SCH3). δC (100.64 MHz, DMSO-d6): 199.0 (CS); 143.4 (CN); 134.2, 132.8, 131.8, 131.1, 128.7, 127.0 (CAr); 16.7 (SCH3). MS (EI+, m/z): 278 ([M]+, 100%).
Functionalised thiosemicarbazones
N); 856 (CS). δH (300.07 MHz, CDCl3): 9.22 (1H, s, NNH); 7.68 (1H, s, HCN); 7.32 (1H, br s, NH); 4.57 (2H, t, JHH 2.0, C5H4); 4.42 (2H, t, JHH 2.0, C5H4); 4.21 (5H, s, C5H5); 3.26 (2H, d, JHH 5.6, CH2); 0.18 (9H, s, Si(CH3)3). δC (100.64 MHz, CDCl3): 166.4 (CS); 143.1 (CN); 78.0, 70.5, 69.3, 67.6 (Fc); 34.9 (CH2); −2.48 (Si(CH3)3). MS (EI+, m/z): 373 ([M]+, 100%).
N); 851 (CS). δH (300.07 MHz, DMSO-d6): 9.88 (1H, s, NNH); 7.95 (1H, t, 3JHH 5.7, NH); 4.74 (2H, t, 3JHH 1.8, C5H4); 4.38 (2H, t, 3JHH 1.8, C5H4); 4.17 (5H, s, C5H5); 3.18 (2H, d, 3JHH 6.0, CH2); 2.19 (3H, s, CH3); 0.10 (9H, s, Si(CH3)3). δC (100.64 MHz, CDCl3): 178.1 (CS); 148.0 (CN); 82.5, 70.3, 69.3, 66.8 (Fc); 34.7 (CH2); 14.2 (CH3); −2.50 (Si(CH3)3). MS (EI+, m/z): 387 ([M]+, 100%).
General procedure for the synthesis of compounds 4a–b
An excess of (aminomethyl)trimethylsilane was added to the flask under N2. The dithiocarbamates and dry ethanol (15.0 mL) were added to the silane. The reaction mixture was refluxed under Ar for 24 h, after which water (10.0 mL) was added to the flask, thus precipitating a solid. The ethanol was removed, and the white solid was collected by suction filtration and washed with water, followed by minimal ethanol.N); 854 (CS). δH (300.07 MHz, CDCl3): 9.84 (1H, s, NNH); 7.78 (1H, s, HCN); 7.69 (1H, d, 4JHH 1.8, C(2)H); 7.48 (1H, d, 3JHH 8.4, C(5)H); 7.41 (1H, dd, 4JHH 1.8, 3JHH 8.4, C(6)H); 7.38 (1H, br s, NH); 3.28 (2H, d, 3JHH 6.0, CH2); 0.18 (9H, s, Si(CH3)3). δC (100.64 MHz, CDCl3): 177.7 (CS); 139.3 (CN); 134.1, 133.6, 133.3, 130.9, 128.5, 126.0 (CAr); 35.0 (CH2); −2.44 (Si(CH3)3). MS (EI+, m/z): 333 ([M]+, 76%).
N); 847 (CS). δH (300.07 MHz, DMSO-d6): 10.2 (1H, s, NNH); 8.41 (1H, t, 3JHH 5.7, NH); 8.13 (1H, d, 3JHH 2.4, C(2)H); 7.85 (1H, dd, 4JHH 2.4, 3JHH 8.4, C(6)H; 7.65 (1H, d, 3JHH 8.4, C(5)H); 3.24 (2H, d, 3JHH 6.3, CH2); 2.28 (3H, s, CH3); 0.10 (9H, s, Si(CH3)3). δC (100.64 MHz, DMSO-d6): 177.3 (CS); 144.3 (CN); 138.4, 131.4, 131.2, 130.2, 127.9, 126.4 (CAr); 34.5 (CH2); 13.7 (CH3); −1.84 (Si(CH3)3). MS (EI+, m/z): 347 ([M]+, 31%).
N); 858 (CS). δH (399.95 MHz, CDCl3): 8.63 (1H, s, NNH); 7.77 (1H, d, 3JHH 1.6, C(2)H); 7.74 (1H, br s, NH); 7.50 (2H, m, C(5)H & C(6)H); 3.60 (2H, d, 3JHH 5.6, CH2); 2.26 (3H, s, CH3); 1.04 (9H, s, C(CH3)3). δC (100.64 MHz, CDCl3): 178.4 (CS); 143.7 (CN); 137.4, 133.7, 133.0, 130.6, 128.0, 125.1 (CAr); 55.8 (CH2); 32.2 (C(CH3)3); 27.4 (C(CH3)3); 13.3 (CH3). MS (EI+, m/z): 331 ([M]+, 64%).
Organosilane thiosemicarbazones ruthenium(II) complexes
General method
The ruthenium dimer [(η6-iPrC6H4Me)Ru(μ-Cl)Cl]2 was dissolved in DCM (5.00 mL) followed by the addition of the organosilane thiosemicarbazone. The reaction mixture was stirred at room temperature for 4 h. The solvent was reduced (∼1.00 mL) and added with stirring to diethyl ether to precipitate a solid. The solid was collected by suction filtration and washed with diethyl ether and pentane.N); 6.06 (1H, s, C5H4); 5.52 (1H, d, 3JHH 6.0, p-cym); 5.20 (2H, br s, p-cym); 5.13 (1H, d, 3JHH 6.0, p-cym); 4.65 (3H, br s, C5H4); 4.34 (5H, s, C5H5); 2.99 (2H, t, 3JHH 6.3, CH2); 2.66 (1H, m, CH(CH3)2); 2.13 (3H, s, CH3(p-cym)); 1.19 (3H, d, 3JHH 6.9, CH(CH3)2); 1.14 (3H, d, 3JHH 6.9, CH(CH3)2); 0.21 (9H, s, Si(CH3)3). δC (100.64 MHz, CDCl3): 158.6 (CN); 101.6 (p-cymquatern.); 88.5, 87.3, 83.8, 81.6 (p-cym); 70.0–72.6 (Fc); 36.1 (CH2); 30.5 (CH(CH3)2); 22.7 (CH(CH3)2); 21.7 (CH(CH3)2); 18.5 (CH3(p-cym)); −2.56 (Si(CH3)3). MS (ESI+, m/z): 608 ([M − H − Cl]+, 40%); 304 ([M − Cl]2+, 100%).
S); 168.2 (CN); 102.2, 102.1 (p-cymquatern.); 89.5, 87.3 (p-cym); 86.5 (Fcquatern.); 84.1, 81.3 (p-cym); 72.9, 71.9, 71.4, 70.5, 69.8 (Fc); 36.6 (CH2); 30.5 (CH(CH3)2); 27.7 (CH3); 22.8 (CH(CH3)2); 21.4 (CH(CH3)2); 18.4 (CH3(p-cym)); −2.26 (Si(CH3)3). MS (ESI+, m/z): 622 ([M − H − Cl]+, 100%); 311 ([M − Cl]2+, 98%).
N); 8.67 (1H, s, C(2)H); 7.88 (1H, d, 3JHH 8.4, C(5)H); 7.64 (1H, d, 3JHH 8.4, C(6)H); 5.49 (1H, d, 3JHH 5.7, p-cym); 4.98 (1H, d, 3JHH 6.0, p-cym); 4.90 (1H, d, 3JHH 6.0, p-cym); 4.86 (1H, d, 3JHH 5.7, p-cym); 3.03 (2H, d, 3JHH 6.0, CH2); 2.64 (1H, m, CH(CH3)2); 2.12 (3H, s, CH3(p-cym)); 1.18 (3H, d, 3JHH 6.9, CH(CH3)2); 1.12 (3H, d, 3JHH 6.9, CH(CH3)2); 0.18 (9H, s, Si(CH3)3). δC (100.64 MHz, CDCl3): δ (ppm) = 177.7 (CS); 156.4 (CN); 136.2, 133.4, 132.7, 132.6, 130.9, 129.8 (CAr); 104.2, 103.7 (p-cymquatern.); 88.5, 88.2, 82.7, 81.2 (p-cym); 36.5 (CH2); 30.8 (CH(CH3)2); 22.9 (CH(CH3)2); 21.5 (CH(CH3)2); 18.7 (CH3(p-cym)); −2.49 (Si(CH3)3). MS (ESI+, m/z): 568 ([M − H − Cl]+, 100%); 284 ([M − Cl]2+, 57%).
S); 165.9 (CN); 141.2, 134.7, 133.2, 131.5, 130.6, 128.1 (CAr); 105.9, 102.5 (p-cymquatern.); 88.9, 88.1, 84.0, 82.5 (p-cym); 36.8 (CH2); 30.6 (CH(CH3)2); 26.8 (CH3); 23.5 (CH(CH3)2); 21.1 (CH(CH3)2); 18.7 (CH3(p-cym)); −2.30 (Si(CH3)3). MS (ESI+, m/z): 582 ([M − H − Cl]+, 100%); 291 ([M − Cl]2+, 42%).
S); 166.85 (CN); 141.24, 134.83, 133.29, 131.50, 130.64, 128.18 (CAr); 106.14, 102.73 (p-cymquaternary); 89.00, 88.02, 84.07, 82.47 (p-cym); 57.81 (CH2); 32.50 (C(CH3)3); 30.63 (CH(CH3)3); 27.13 (CH3); 23.54 (CH(CH3)2); 22.26 (C(CH3)3); 21.15 (CH(CH3)2); 18.73 (CH3(p-cym)). MS (ESI+, m/z): 566 ([M − H − Cl]+, 100%); 283 ([M − Cl]2+, 68%).
Organosilane thiosemicarbazones rhodium(III) complexes
General method
The rhodium dimer [(Cp*)Rh(μ-Cl)Cl]2 was dissolved in DCM (5.00 mL), followed by the addition of the ligand. The solution was stirred at room temperature for 4 h. The solvent was reduced (∼1.00 mL) and added with stirring to pentane to precipitate a solid.N); 6.25 (1H, s, C5H4); 4.64 (1H, s, C5H4); 4.60 (2H, s, C5H4); 4.34 (5H, s, C5H5); 2.99 (2H, m, CH2); 1.51 (15H, s, Cp*); 0.240 (9H, s, Si(CH3)3). δC (100.64 MHz, CDCl3): 176.2 (CS); 158.5 (CN); 97.3 (d, 1JRhC 7.45, Cp*quatern.); 75.6, 74.1, 73.2, 72.3, 70.4, 69.1 (Fc); 36.3 (CH2); 9.48 (CH3(Cp*)); −2.42 (Si(CH3)3). MS (ESI+, m/z): 610 ([M − H − Cl]+, 88%); 305 ([M − Cl]2+, 100%).
S); 153.18 (CN); 94.58 (d, 1JRhC 7.75, Cp*quatern.); 82.59, 70.18, 69.41, 67.35 (Fc); 34.51 (CH2); 17.06 (CH3); 8.16 (CH3(Cp*)); −2.81 (Si(CH3)3). MS (ESI+, m/z): 624 ([M − H − Cl]+, 25%); 312 ([M − Cl]2+, 100%).
N); 7.67 (1H, d, 3JHH 2.0, C(2)H); 7.44 (1H, d, 3JHH 8.8, C(5)H); 7.36 (1H, dd, 4JHH 1.6, 3JHH 8.0, C(6)H); 7.10 (1H, br s, NH); 3.20 (2H, d, 3JHH 5.6, CH2); 1.70 (15H, s, Cp*); 0.20 (9H, s, Si(CH3)3). δC (100.64 MHz, CDCl3): 173.95 (CS); 143.57 (CN); 134.30; 133.61; 133.20; 130.81; 128.53; 126.66; 95.43 (d, 1JRhC 8.05, Cp*quatern.); 34.80 (CH2); 8.89 (CH3(Cp*)); −2.41 (Si(CH3)3). MS (ESI+, m/z): 570 ([M − H − Cl]+, 100%); 285 ([M − Cl]2+, 30%).
S); 149.4 (CN); 137.9; 133.5; 132.7; 130.3; 128.2; 125.5; 95.2 (d, 1JRhC 8.09, Cp*quatern.); 34.5 (CH2); 16.7 (CH3); 8.78 (CH3(Cp*)); −2.48 (Si(CH3)3). MS (ESI+, m/z): 584 ([M − H − Cl]+, 100%); 292 ([M − Cl]2+, 98%).
S); 149.8 (CN); 137.9; 133.6; 132.8; 130.4; 128.3; 125.5; 95.4 (d, 1JRhC 8.05, Cp*quatern.); 55.5 (CH2); 31.9 (C(CH3)3); 27.5 (C(CH3)3); 16.9 (CH3).8.79 (CH3(Cp*)). MS (ESI+, m/z): 568 ([M − H − Cl]+, 100%); 284 ([M − Cl]2+, 62%).
Single crystal X-ray diffraction
X-ray single crystal intensity data were collected on a Nonius Kappa-CCD diffractometer using graphite monochromated MoKα radiation (λ = 0.71073 Å). The temperature was controlled using an Oxford Cryostream cooling system (Oxford Cryostat). The strategy for the data collection was evaluated using the Bruker Nonius “Collect” program. Data were scaled and reduced using DENZO-SMN software.53 Absorption correction was performed using SADABS.54 The structure was solved by direct methods and refined by full-matrix least-squares using the program SHELXL-9755 refining on F2. The diagram was produced using the program PovRay and the graphic interface X-seed.56 All non-hydrogen atoms, except those of the solvent molecule, were refined anisotropically. The solvent molecules exhibit high thermal motions and were disordered. Therefore only the carbon atom C28 was refined anisotropically. The chlorine atoms were modelled over two positions each, with site occupancy factors of 0.55 and 0.45 respectively and were refined with isotropic temperature factors. All hydrogen atoms, except H1 and H2, were placed in idealised positions and refined in riding models with Uiso assigned the values of 1.2 or 1.5 times the Ueq of the atoms to which they are attached and the constraint distances of C–H ranging from 0.95 Å to 1.00 Å. The hydrogens H1 and H2 were located in the difference Fourier maps and refined with the bond length constraint d(N–H) = 0.97 Å. The structure was refined to an R factor of 0.0519. The parameters for crystal data collection and structure refinements, bond lengths, angles, and torsion angles are contained in the file MA218.SUP.Calculation of logP values
The modified method put forward by Stringer et al. combines the fragmental approach proposed by Rekker et al.57 and Lanez et al.58–60 with logP data from Marvinsketch V14.9.29.0 to predict the logP of the Fc compounds.61 Stringer et al. used Marvinsketch (weighted method) to predict the logP of a benzene derivative. The data were then used in the following equation:| logP(Fc derivative) = logP(benzene derivative) − f(benzene fragment) + f(fc-H) |
P(benzene derivative) is calculated using Marvinsketch, f(benzene fragment) is obtained from Marvinsketch, f(fc-H) is obtained from the reference by Lanez et al.58 to predict the logP values of ferrocenyl compounds with published experimental logP data.
The calculated logP versus experimental logP data was plotted, and the graph equation was determined.
The logP values were corrected using the following straight line equation:
| y = 0.8238x + 0.9425 R2 = 0.9676 |

Corrected: (4.017–0.9425)/0.8238 = 3.732.

Antiplasmodial assay
Continuous in vitro cultures of asexual erythrocyte stages of P. falciparum were maintained using a modified method of Trager and Jensen.62 A quantitative assessment of antiplasmodial activity in vitro was determined via the parasite lactate dehydrogenase assay using a modified method described by Makler.63 The antiplasmodial assay was conducted according to previously published methods.63 A full dose–response was performed for all compounds to determine the concentration inhibiting 50% of parasite growth (IC50-value). Test samples were tested at a starting concentration of 100 μg ml−1, which was then serially diluted 2-fold in complete medium to give 10 concentrations, with the lowest concentration being 0.2 μg ml−1. Reference drugs were tested at a starting concentration of 1000 ng ml−1. Active compounds were retested at a starting concentration of 10 μg ml−1 or 1000 ng ml−1. The highest concentration of the solvent to which the parasites were exposed had no measurable effect on the parasite viability (data not shown). The IC50 values were obtained using a non-linear dose–response curve fitting analysis via Graph Pad Prism v.4.0 software.Cytotoxicity assay
The MTT-assay is used as a colorimetric assay for cellular growth and survival, and compares well with other available assays.64,65 The test samples were tested in triplicate on one occasion. The same stock solutions prepared for the antiplasmodial activity testing were used for the cytotoxicity tests. Dilutions were prepared on the day of the experiment in complete medium. Emetine was used as the reference drug. The initial concentration of emetine was 100 μg ml−1, which was serially diluted in complete medium with 10-fold dilutions to give 6 concentrations, the lowest being 0.001 μg ml−1. The same dilution technique was applied to all the test samples. The highest concentration of the solvent to which the cells were exposed had no measurable effect on the cell viability (data not shown). The IC50 values were obtained from full dose–response curves, using a non-linear dose–response curve fitting analysis via GraphPad Prism v.4 software.β-Haematin inhibition assay
The β-haematin inhibition assay was adapted from the method described by Wright and co-workers.45 Compounds were prepared as a 10 mM stock solution in DMSO. The samples were tested at various concentrations between 5 and 500 μM. The stock solution was serially diluted to give 12 concentrations in a 96 well flat-bottom assay plate. NP-40 detergent was added to mediate the formation of β-haematin (305.5 μM). A 25 mM stock solution of haematin was prepared by dissolving haemin (16.3 mg) in dimethyl sulfoxide (1 mL). A 177.76 μL aliquot of the haematin stock was suspended in 20 ml of a 2 M acetate buffer at pH 4.7. The suspension was then added to the plate to give a final haematin concentration of 100 μM. The plate was then incubated for 16 hours at 37 °C. The assay was analysed using the pyridine-ferrochrome method developed by Ncokazi and Egan.46 32 μL of a solution of 50% pyridine, 20% acetone, 20% water and 10% 2 M HEPES buffer (pH 7.4) was added to each well. To this, 60 μL acetone was added to each well and mixed. The absorbance of the resulting complex was measured at 405 nm on a SpectraMax 340PC plate reader. The IC50 values were obtained using a non-linear dose–response curve fitting analysis via GraphPad Prism v.5.00 software.Antitrichomonal assay
Cultures of the G3 strain of T. vaginalis were grown in 5 ml complete TYM Diamond's media in a 37 °C incubator for 24 hours. 50 mM stocks of the compounds were prepared by dissolving in DMSO and were screened against the G3 strain of T. vaginalis. Untreated cells and those inoculated with 5 μl DMSO (0.1%) were used as controls. 5 μL of 50 mM stocks of the compound library were inoculated at a final concentration of 50 μM. The results were calculated based on cell counts utilising a haemocytometer after 24 hours. IC50 values were determined using serial dilution of the compounds. The calculated IC50 values were then confirmed using the same assay described above.Antitumour assay
Human A2780 and A2780cisR ovarian cancer cells and KMST-6 cells were obtained from the European Collection of Cell Cultures (Salisbury, UK). A2780 and A2780cisR cells were grown routinely in RPMI–1640 medium and KMST-6 cells in DMEM medium. Both media were supplemented with 10% fetal calf serum (FCS) and antibiotics (penicillin and streptomycin) at 37 °C and 5% CO2. Cytotoxicity was determined using the WST-1 assay (WST-1 = (4-[3-(4-iodophenyl)-2-(4-nitrophenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate). The cells were seeded in 96-well plates as monolayers with 100 μL of cell solution (approximately 5000 cells) per well and pre-incubated for 24 h in a medium supplemented with 10% FCS. The compounds were prepared as DMSO solutions, dissolved in the culture medium and serially diluted to the appropriate concentration to give a final DMSO concentration of 0.5%. 100 μL of drug solution was added to each well and the plates were incubated for another 24 h. Subsequently, WST-1 (10 μL solution) was added to the cells and the plates were incubated for a further 3 h. The WST-1 tetrazolium salt is cleaved to a soluble formazan by a cellular mechanism, the succinate–tetrazolium reductase system (EC 1.3.99.1), which occurs primarily at the cell surface. This bioreduction depends largely on the cellular production of NAD(P)H within metabolically intact and viable cells. The optical density, directly proportional to the number of surviving cells, was quantified at 450 nm, background correction was performed at 600 nm using a multiwell plate reader, and the fraction of surviving cells was calculated from the absorbance of untreated control cells. Evaluation is based on the means from three microcultures per concentration level and is analysed via GraphPad Prism v.5.00 software.Acknowledgements
Financial support was received from the University of Cape Town, the National Research Foundation (NRF) of South Africa, and the South African Medical Research Council (MRC). The South African Research Chairs initiative of the Department of Science and Technology administered by the NRF is gratefully acknowledged for support (KC). Even though the work was supported by the MRC, the views and opinions expressed are not those of the MRC but of the authors of the material produced or published. Professor Timothy J. Egan (University of Cape Town) is gratefully acknowledged for his assistance with β-haematin inhibition assays and the University of Western Cape is gratefully acknowledged for assistance with anti-tumour studies.Notes and references
- The World Health Organization, Malaria http://www.who.int/malaria/media/world_malaria_report_2013/ (accessed 16 October 2014).
- J. N. Burrows, E. Burlot, B. Campo, S. Cherbuin, S. Jeanneret, D. Leroy, T. Spangenberg, D. Waterson, T. N. Wells and P. Willis, Parasitology, 2014, 141, 128–139 CrossRef PubMed.
- D. Petrin, K. Delgaty, R. Bhatt and G. Garber, Clin. Microbiol. Rev., 1998, 11, 300–317 CAS.
- C. M. Ryan, N. De Miguel and P. J. Johnson, Essays Biochem., 2011, 51, 161–175 CAS.
- P. Kissinger and A. Adamski, Sex. Transm. Infect., 2013, 89, 426–433 CrossRef PubMed.
- Z. F. Zhang, S. Graham, S. Z. Yu, J. Marshall, M. Zielezny, Y. X. Chen, M. Sun, S. L. Tang, C. S. Liao, J. L. Xu and X. Z. Yang, Ann. Epidemiol., 1995, 5, 325–332 CrossRef CAS.
- S. Sutcliffe, E. Giovannucci, J. F. Alderete, T.-H. Chang, C. A. Gaydos, J. M. Zenilman, A. M. De Marzo, W. C. Willett and E. A. Platz, Cancer Epidemiol. Biomarkers Prev., 2006, 15, 939–945 CAS.
- J. R. Schwebke and D. Burgess, Clin. Microbiol. Rev., 2004, 17, 794–803 CrossRef PubMed.
- K. Madhuri, M. S. Kumar and L. Kalyani, Int. J. Pharm., Chem. Biol. Sci., 2011, 1, 38–42 CAS.
- B. Lippert, Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, VCHA & Wiley-VCH, Zurich, 1999 Search PubMed.
- R. Trondl, P. Heffeter, C. R. Kowol, M. A. Jakupec, W. Berger and B. K. Keppler, Chem. Sci., 2014, 5, 2925–2932 RSC.
- C. Biot, G. Glorian, L. A. Maciejewski and J. S. Brocard, J. Med. Chem., 1997, 2623, 3715–3718 CrossRef PubMed.
- A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388–405 CrossRef CAS PubMed.
- M. Blunder, N. Hurkes, S. Spirk, M. List and R. Pietschnig, Bioorg. Med. Chem. Lett., 2011, 21, 363–365 CrossRef CAS PubMed.
- I. Segal, A. Zablotskaya and E. Lukevics, Chem. Heterocycl. Compd., 2005, 41, 713–725 CrossRef PubMed.
- Y. Li, C. de Kock, P. J. Smith, H. Guzgay, D. T. Hendricks, K. Naran, V. Mizrahi, D. F. Warner, K. Chibale and G. S. Smith, Organometallics, 2013, 32, 141–150 CrossRef CAS.
- Y. Li, C. de Kock, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2014, 33, 4345–4348 CrossRef CAS.
- P. Chellan, K. M. Land, A. Shokar, A. Au, S. H. An, C. M. Clavel, P. J. Dyson, C. De Kock, P. J. Smith, K. Chibale and G. S. Smith, Organometallics, 2012, 31, 5791–5799 CrossRef CAS.
- B. Demoro, M. Rossi, F. Caruso, D. Liebowitz, C. Olea-Azar, U. Kemmerling, J. D. Maya, H. Guiset, V. Moreno, C. Pizzo, G. Mahler, L. Otero and D. Gambino, Biol. Trace Elem. Res., 2013, 153, 371–381 CrossRef CAS PubMed.
- A. Moreno-Rodríguez, P. M. Salazar-Schettino, J. L. Bautista, F. Hernández-Luis, H. Torrens, Y. Guevara-Gómez, S. Pina-Canseco, M. B. Torres, M. Cabrera-Bravo, C. M. Martinez and E. Pérez-Campos, Eur. J. Med. Chem., 2014, 87, 23–29 CrossRef PubMed.
- M. Adams, Y. Li, H. Khot, C. De Kock, P. J. Smith, K. Land, K. Chibale and G. S. Smith, Dalton Trans., 2013, 42, 4677–4685 RSC.
- A. Walcourt, J. Kurantsin-Mills, J. Kwagyan, B. B. Adenuga, D. S. Kalinowski, D. B. Lovejoy, D. J. R. Lane and D. R. Richardson, J. Inorg. Biochem., 2013, 129, 43–51 CrossRef CAS PubMed.
- W. Su, Q. Qian, P. Li, X. Lei, Q. Xiao, S. Huang, C. Huang and J. Cui, Inorg. Chem., 2013, 52, 12440–12449 CrossRef CAS PubMed.
- K. Sampath, S. Sathiyaraj and C. Jayabalakrishnan, Med. Chem. Res., 2013, 23, 958–968 CrossRef.
- A. Q. Ali, S. G. Teoh, N. E. Eltayeb, M. B. K. Ahamed and A. A. Majid, J. Coord. Chem., 2014, 67, 3380–3400 CrossRef CAS.
- D. Palanimuthu, S. V. Shinde, K. Somasundaram and A. G. Samuelson, J. Med. Chem., 2013, 56, 722–734 CrossRef CAS PubMed.
- G. H. G. Trossini, R. V. C. Guido, G. Oliva, E. I. Ferreira and A. D. Andricopulo, J. Mol. Graphics Modell., 2009, 28, 3–11 CrossRef CAS PubMed.
- D. C. Greenbaum, Z. Mackey, E. Hansell, P. Doyle, J. Gut, C. R. Caffrey, J. Lehrman, P. J. Rosenthal, J. H. McKerrow and K. Chibale, J. Med. Chem., 2004, 47, 3212–3219 CrossRef CAS PubMed.
- J. Shao, B. Zhou, A. J. Di Bilio, L. Zhu, T. Wang, C. Qi, J. Shih and Y. Yen, Mol. Cancer Ther., 2006, 5, 586–592 CrossRef CAS PubMed.
- R. Bailey-Wood and L. Blayney, Br. J. Exp. Pathol., 1975, 56, 193–198 CAS.
- N. Farrell, Coord. Chem. Rev., 2006, 232, 2–5 Search PubMed.
- M. Adams, C. De Kock, P. J. Smith, K. Chibale and G. S. Smith, J. Organomet. Chem., 2013, 736, 19–26 CrossRef CAS PubMed.
- P. Chellan, T. Stringer, A. Shokar, P. J. Dornbush, G. Vazquez-Anaya, K. M. Land, K. Chibale and G. S. Smith, J. Inorg. Biochem., 2011, 105, 1562–1568 CrossRef CAS PubMed.
- G. Zhao and C. Yuan, Transition Met. Chem., 1994, 19, 218–220 CrossRef.
- K. Z. Ismail, Transition Met. Chem., 1997, 22, 565–569 CrossRef CAS.
- C. Biot, B. Pradines, M.-H. Sergeant, J. Gut, P. J. Rosenthal and K. Chibale, Bioorg. Med. Chem. Lett., 2007, 17, 6434–6438 CrossRef CAS PubMed.
- T. Stringer, B. Therrien, D. T. Hendricks, H. Guzgay and G. S. Smith, Inorg. Chem. Commun., 2011, 14, 956–960 CrossRef CAS PubMed.
- L. Glans, A. Ehnbom, C. de Kock, A. Martínez, J. Estrada, P. J. Smith, M. Haukka, R. A. Sánchez-Delgado and E. Nordlander, Dalton Trans., 2012, 41, 2764–2773 RSC.
- W. Nkoana, D. Nyoni, P. Chellan, T. Stringer, D. Taylor, P. J. Smith, A. T. Hutton and G. S. Smith, J. Organomet. Chem., 2014, 752, 67–75 CrossRef CAS PubMed.
- F. Beckford, D. Dourth, M. Shaloski, J. Didion, J. Thessing, J. Woods, V. Crowell, N. Gerasimchuk, A. Gonzalez-Sarrías and N. P. Seeram, J. Inorg. Biochem., 2011, 105, 1019–1029 CrossRef CAS PubMed.
- F. Beckford, G. Leblanc, J. Thessing, M. Shaloski, B. J. Frost, L. Li and N. P. Seeram, Inorg. Chem. Commun., 2009, 12, 1094–1098 CrossRef CAS PubMed.
- P. Bourosh and M. Revenko, J. Struct. Chem., 2009, 50, 532–535 CrossRef.
- H. Lullmann and M. Wehling, Biochem. Pharmacol., 1979, 28, 3409–3415 CrossRef CAS.
- M. Adams, C. De Kock, P. J. Smith, P. Malatji, A. T. Hutton, K. Chibale and G. S. Smith, J. Organomet. Chem., 2013, 739, 15–20 CrossRef CAS PubMed.
- R. D. Sandlin, M. D. Carter, P. J. Lee, J. M. Auschwitz, S. E. Leed, J. D. Johnson and D. W. Wright, Antimicrob. Agents Chemother., 2011, 55, 3363–3369 CrossRef CAS PubMed.
- K. K. Ncokazi and T. J. Egan, Anal. Biochem., 2005, 338, 306–319 CrossRef CAS PubMed.
- I. Zanellato, J.-M. Heldt, A. Vessières, G. Jaouen and D. Osella, Inorg. Chim. Acta, 2009, 362, 4037–4042 CrossRef CAS PubMed.
- D. Plazuk, A. Vessières, E. A. Hillard, O. Buriez, E. Labbé, P. Pigeon, M.-A. Plamont, C. Amatore, J. Zakrzewski and G. Jaouen, J. Med. Chem., 2009, 52, 4964–4967 CrossRef CAS PubMed.
- A. Casini, C. Gabbiani, F. Sorrentino, M. P. Rigobello, A. Bindoli, T. J. Geldbach, A. Marrone, N. Re, C. G. Hartinger, P. J. Dyson and L. Messori, J. Med. Chem., 2008, 51, 6773–6781 CrossRef CAS PubMed.
- D. Klayman and J. Scovill, J. Med. Chem., 1979, 22, 1367–1373 CrossRef CAS.
- M. Bennett and A. Smith, J. Chem. Soc., Dalton Trans., 1974, 233–241 RSC.
- C. White, A. Yates and P. M. Mailtlis, Inorg. Synth., 1992, 29, 228–234 CrossRef CAS.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, ed. C. W. Carter and R. M. Sweet, Academic Press, 1997 Search PubMed.
- G. M. Sheldrick, SADABS, University of Göttinger, Göttinger, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189–191 CrossRef CAS.
- R. Mannhold and R. Rekker, Perspect. Drug Discovery Des., 2000, 18, 1–18 CrossRef CAS.
- R. Ahmedi and T. Lanez, Asian J. Chem., 2010, 22, 299–306 CAS.
- R. Ahmedi and T. Lanez, Int. J. Pharm. Pharm. Sci., 2009, 1, 182–189 CAS.
- R. Ahmedi and T. Lanez, Rev. Sci. Fond. App., 2011, 3, 57–67 Search PubMed.
- T. Stringer, D. Taylor, C. de Kock, H. Guzgay, A. Au, S. H. An, B. Sanchez, R. O'Connor, N. Patel, K. M. Land, P. J. Smith, D. T. Hendricks, T. J. Egan and G. S. Smith, Eur. J. Med. Chem., 2013, 69, 90–98 CrossRef CAS PubMed.
- W. Trager and J. B. Jensen, Science, 1976, 193, 673–675 CAS.
- M. T. Makler, J. M. Ries, J. A. Williams, J. E. Bancroft, R. C. Piper, B. L. Gibbins and D. J. Hinrichs, Am. J. Trop. Med. Hyg., 1993, 48, 739–741 CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- L. V. Rubinstein, R. H. Shoemaker, K. D. Paull, R. M. Simon, S. Tosini, P. Skehan, D. A. Scudiero, A. Monks and M. R. Boyd, J. Natl. Cancer Inst., 1990, 82, 1113–1118 CrossRef CAS PubMed.
Footnote |
| † CCDC 1013088 for complex 6b. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03234a |
| This journal is © The Royal Society of Chemistry 2015 |