
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence1,2,4-Triazole-derived carbene complexes of gold: characterization, solid-state aggregation and ligand disproportionation†
Shuai
Guo
,
Jan Christopher
Bernhammer
and
Han Vinh
Huynh
*
Department of Chemistry, National University of Singapore, 3 Science Drive 3, 117543 Singapore, Singapore. E-mail: chmhhv@nus.edu.sg; Fax: +65 6779 1691; Tel: +65 6516 2670
First published on 20th January 2015
Abstract
Ligand redistribution reactions are well documented for silver(I) N-heterocyclic carbene (NHC) complexes of the type [AgX(NHC)] (X = halido ligand), but only two reports have been described in the literature for gold analogues of the general formula [AuX(NHC)]. In both cases, the NHCs in question were exceptionally strong donors. To probe the dependence of ligand redistribution processes on NHC donor strength, a model study was conducted using a weakly donating 1,2,4-triazolin-5-ylidene (tazy) ligand and different halido coligands. For [AuX(tazy)] (X = Cl, Br, OAc, tazy = 4-benzyl-1-methyl-1,2,4-triazolin-5-ylidene), no ligand redistribution was found, while a reversible disproportionation between [AuI(tazy)] in solution and [Au(tazy)2][AuI2] in the solid state was observed and studied by means of X-ray crystallography, NMR and UV-Vis spectroscopy, as well as DFT calculations.
Introduction
Coinage metal complexes incorporating N-heterocyclic carbenes (NHCs) have received a great deal of interest1 due to their intriguing coordination chemistry as well as their applications in catalysis,2 luminescence,3 and biological science.4 In this context, much attention has been paid to Ag(I) mono-NHC complexes of the type [AgX(NHC)] (X = halido ligand), particularly due to their ability to serve as carbene transfer agents.5 The structural diversity of silver NHC complexes in this category has proved to be fairly broad. A large number of neutral (e.g. [AgX(NHC)], [AgX(NHC)]2) and ionic structures (e.g. [Ag(NHC)2][AgX2]) have been reported. Additionally, fluxional behaviour has been observed for different Ag–NHC species in solution, indicating rapid ligand disproportionations.6In contrast, only scattered examples of ligand disproportionations of complexes with the general formula [AuX(NHC)] have been reported, although such complexes have been extensively employed in homogenous catalysis.2 A DFT study by Frenking et al. has revealed that the bond dissociation energy (BDE) of group 11 metal–NHC bonds follows the trend Au > Cu > Ag.7 The disproportionation process of complexes [AuX(NHC)], obviously involving the breakage and reconstruction of Au–Ccarbene bonds, is probably less favourable due to the higher BDE of the Au–NHC bond.
To the best of our knowledge, there are only two examples in this regard.8 In 2008, Bertrand and co-workers reported an ionic complex [Au(CAAC)2][AuCl2] (A′),8a which can be generated via the autoionization of the neutral complex [AuCl(CAAC)] (A) in solution (I, CAAC = 2-(2,6-diisopropylphenyl)-3,3-dimethyl-2-azaspiro[4.5]decan-1-ylidene, Scheme 1). In 2012, we published the 2nd example for ligand disproportionation, involving an indazole-derived carbene complex [AuI(Indy)] (B, Indy = 6,7,8,9-tetrahydropyridazino[1,2-a]indazolin-3-ylidene).8b The latter was found to be solvent-dependant and led to the formation of two ionic species B′ and B′′ (II, Scheme 1).
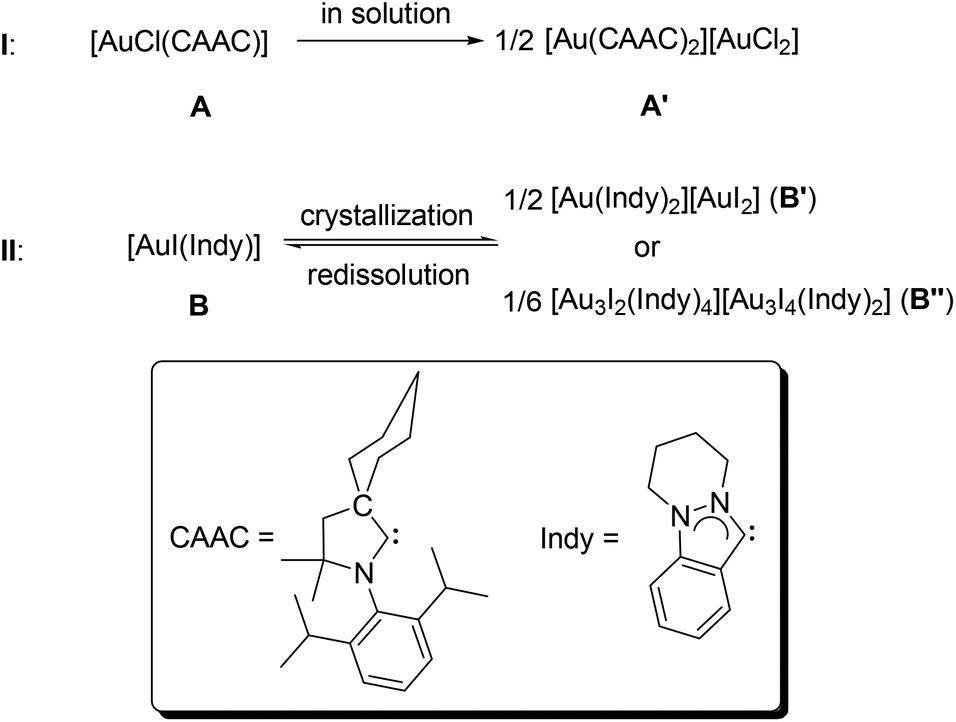 | ||
| Scheme 1 Previous reports on ligand disproportionations of complexes of the type [AuX(NHC)] (X = halido ligand). | ||
Notably, in both cases (I/II) non-classical carbenes (CAAC and Indy) were employed, which are known to be stronger σ-donors compared to classical NHCs.9 In order to gain better insights into the factors affecting such processes, complexes of a 1,2,4-triazolin-5-ylidene, as one of the most weakly donating classical NHCs,9b were prepared with the objective to study, if the NHC's donating ability plays a crucial role in ligand disproportionations.
Results and discussion
Syntheses of Au(I) triazolin-5-ylidene complexes
The carbene precursor salt tazy·HCl (1) (Scheme 2, tazy = 4-benzyl-1-methyl-1,2,4-triazolin-5-ylidene) was readily prepared by the alkylation of 1-methyl-1,2,4-triazole, with benzyl chloride acting both as alkylating agent and solvent. The subsequent auration via silver carbene transfer to [AuCl(SC4H8)] afforded the desired Au(I) chlorido mono-carbene complex [AuCl(tazy)] (2) in a good yield of 78%. Initial attempts to use the same metallation route with the bromide salt tazy·HBr gave a mixture of chlorido and bromido complexes due to halido scrambling, which was corroborated by X-ray diffraction analyses (vide infra).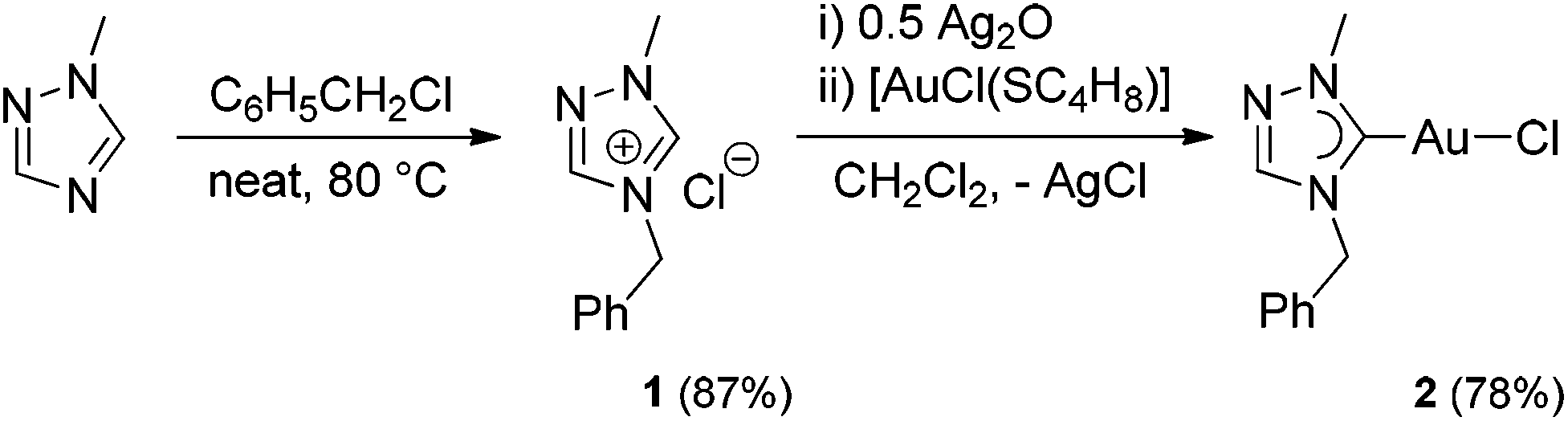 | ||
| Scheme 2 Synthesis of Au(I) chlorido mono-carbene complex 2. | ||
Complex 2 was isolated as white solid, which is readily soluble in common organic solvents, with exception of diethyl ether, n-hexane and toluene. In the 1H NMR spectrum of 2 in CDCl3, the benzylic protons and N-methyl group give singlets at 5.37 and 4.01 ppm, respectively, which show upfield shifts (Δδ = 0.40, 0.10 ppm) compared to those in precursor salt 1. The formation of 2 was further corroborated by the disappearance of the resonance at 11.61 ppm attributed to the acidic proton on C5 in 1, and the presence of a carbene signal at 174.8 ppm in the 13C NMR spectrum. This carbene resonance is found to be comparable to those of previously reported Au(I) mono-(1,2,4-triazolin-5-ylidene) analogues.10 The ESI mass spectrum of 2 displays a base peak at m/z 402 arising from [M − Cl + CH3OH]+.
The chlorido complex 2 can serve as the precursor for further ligand replacement reactions. Treating 2 with excess LiBr in acetone gave bromido complex [AuBr(tazy)] (3) and Finkelstein reaction with NaI led to the formation of iodido analogue [AuI(tazy)] (4) in good yields of 81% and 88%, respectively (Scheme 3).
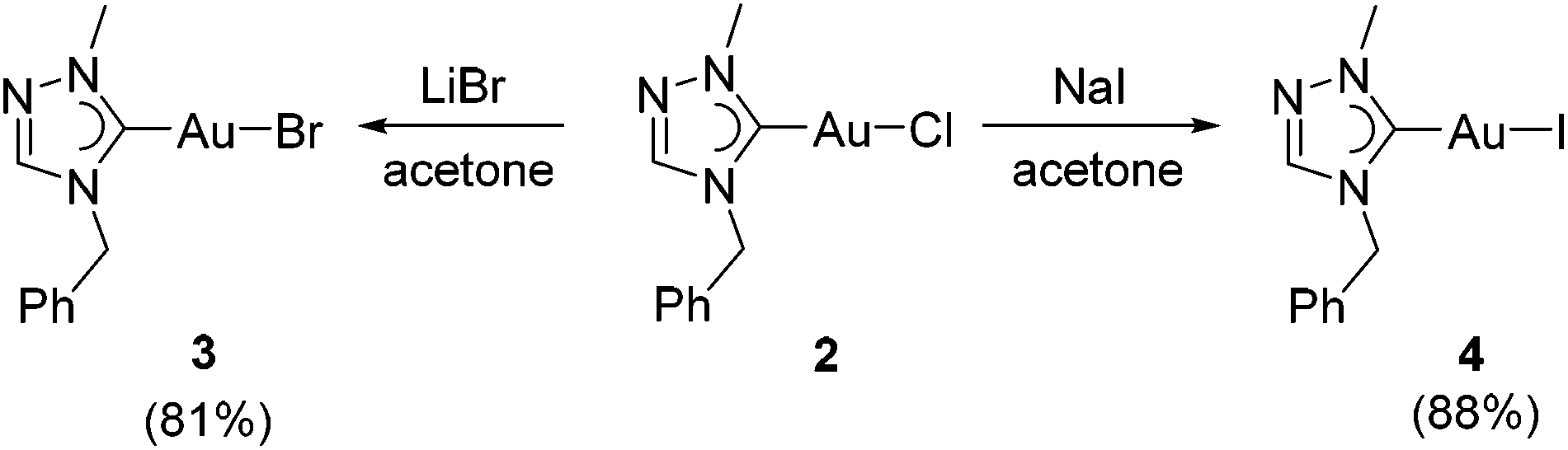 | ||
| Scheme 3 Syntheses of Au(I) bromido and iodido complexes 3 and 4. | ||
The solubilities of complexes 3 and 4 are similar to that of precursor 2. The 1H NMR spectra of 3 and 4 in CDCl3 resemble that of complex 2 and thus are non-indicative for these halido substitution reactions. However, in their 13C NMR spectra, the carbene signals of 3 and 4 are found to be highly sensitive to the trans-standing halido ligands. Complexes 2–4 show a gradual downfield shift of the 13Ccarbene signal in the order 174.8 ppm (Cl) < 178.3 ppm (Br) < 184.8 ppm (I), which correlates to the increasing net donating ability of the trans halido ligands. This is in line with the earlier studies from Baker et al.11 and our group,9b,c,12 which revealed that an increased Lewis acidity of the metal center will result in an upfield 13Ccarbene shift. The formation of complexes 2 and 3 was also supported by their ESI-MS spectra, where dominant peaks at m/z 543 corresponding to the monocationic species [Au(tazy)2]+ were observed.
The identity of complexes 2 and 3 was confirmed by X-ray diffraction analysis on single crystals obtained by slow evaporation of their concentrated solutions in CH2Cl2–n-hexane. The molecular structures are depicted in Fig. 1. The Au–Ccarbene distance of 3 is found to be longer than that of complex 2, which is due to the stronger trans influence of the bromido versus the chlorido ligand. As anticipated, the two benzyl substituents bend away from the coordination spheres to reduce steric repulsion. In both cases of 2 and 3, two mono-carbene molecules were found to dimerize in an antiparallel eclipsed manner through aurophilic interactions clearly indicated by the inter-gold distances of 3.389 Å (2) and 3.305 Å (3), respectively.13 A computational study by Pyykkö et al. on the dimerization of model complex [AuX(PH3)] (X = anionic ligand) concluded that a “softer” anionic ligand X leads to a stronger aurophilic bonding.14 In the present cases with NHCs as supporting ligands, a shorter inter-gold separation (i.e. stronger aurophilic interaction) was also found in complex 3, where the bromido is “softer” compared to the chlorido coligand in the corresponding complex 2. Notably, a deviation of the C1–Au1–Br1 vector from linearity was observed in the case of 3, while the Au(I) centre of complex 2 adopts an essentially linear geometry.
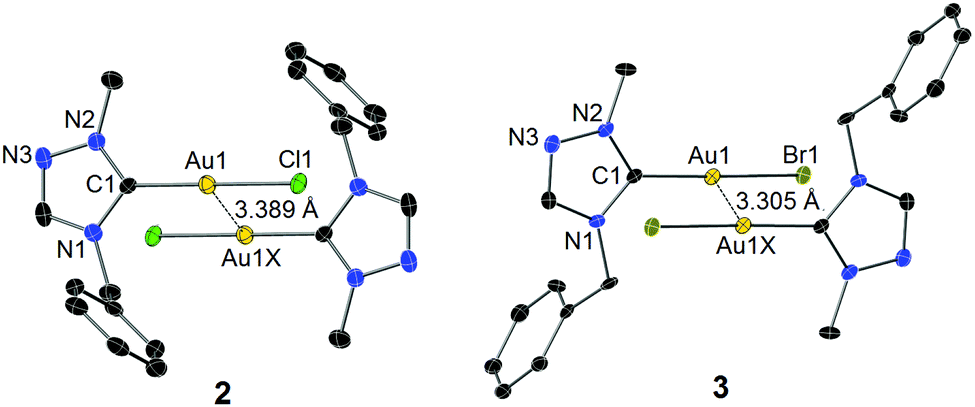 | ||
| Fig. 1 Molecular structures of 2 (left) and 3 (right) showing 50% probability ellipsoids. The other crystallographically independent molecule without aurophilic interactions observed in the solid state of 3 is not shown. Hydrogen atoms and a partial bromido ligand (19%) also found in the structure of 2 have been omitted for clarity. Selected bond lengths (Å) and bond angles (°) for complex 2: Au1–C1 1.970(3), Au1–Cl1 2.274(7), Au1–Au1X 3.389; C1–Au1–Cl1 179.9(3), N2–C1–N1 103.6(3), C1–Au1–Au1X 83.48, Cl1–Au1–Au1X 96.42. 3: Au1–C1 1.989(4), Au1–Br1 2.4057(5), Au1–Au1X 3.305; C1–Au1–Br1 176.15(10), N2–C1–N1 103.9(3), C1–Au1–Au1X 77.61, Br1–Au1–Au1X 106.23. | ||
Single crystals of iodido complex 4 suitable for X-ray diffraction study were obtained by slow evaporation of a concentrated solution of 4 in CH2Cl2. In contrast, the molecular structure was found to be the complex salt [Au(tazy)2][AuI2] (4′), indicating a ligand disproportionation of neutral complex 4. The ion pair depicted in Fig. 2 contains bis(carbene) [Au(tazy)2]+ as the cationic fragment and [AuI2]− as the counteranion. Notably, both Au(I) centers adopt a perfectly linear coordination geometry. The ions aggregate in a staggered orientation with a C1–Au1–Au2–I1 torsion angle of ∼81°, leading to the formation of a polymeric array of “crossed-sticks” with an inter-gold contact of 3.316 Å.
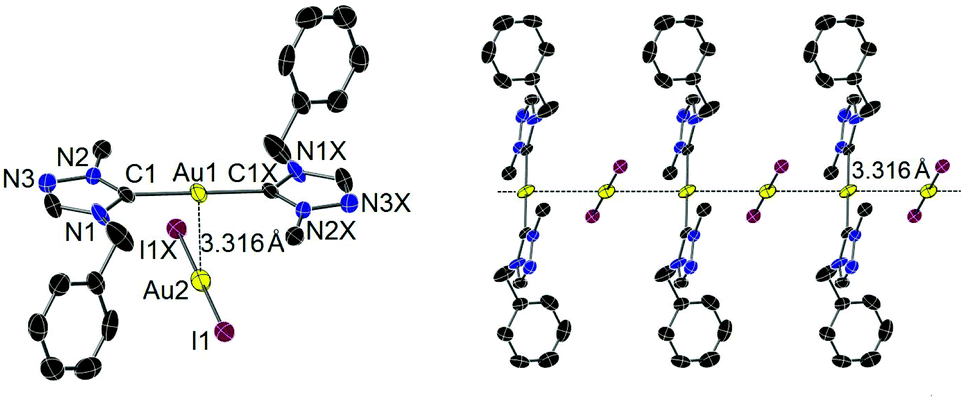 | ||
| Fig. 2 Molecular structure (left) and packing pattern (right) of 4′ showing 50% probability ellipsoids. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å) and bond angles (°): Au1–C1 2.024(5), Au2–I1 2.5586(4), Au1–Au2 3.3161(3); C1–Au1–C1X 180.0(3), I1–Au2–I1X 180.000(1), N2–C1–N1 104.3(4), C1–Au1–Au2 94.81(14), C1X–Au1–Au2 85.19(14), I1–Au2–Au1 83.545(9), I1X–Au2–Au1 96.455(9). | ||
X-ray powder diffraction was carried out to study the bulk of the crystalline material obtained from the CH2Cl2 solution of 4. The experimental diffraction pattern is in very good agreement with the simulated one based on the molecular structure of 4′ (Fig. 3), confirming that 4′ indeed represents the whole solid material. Nevertheless, it should also be noted that the neutral complex 4 may only undergo ligand disproportionation yielding the ionic isomer 4′ upon crystallization. This disproportionation behavior is similar to the one that has been investigated for an indazole-derived carbene complex [AuI(Indy)].8b
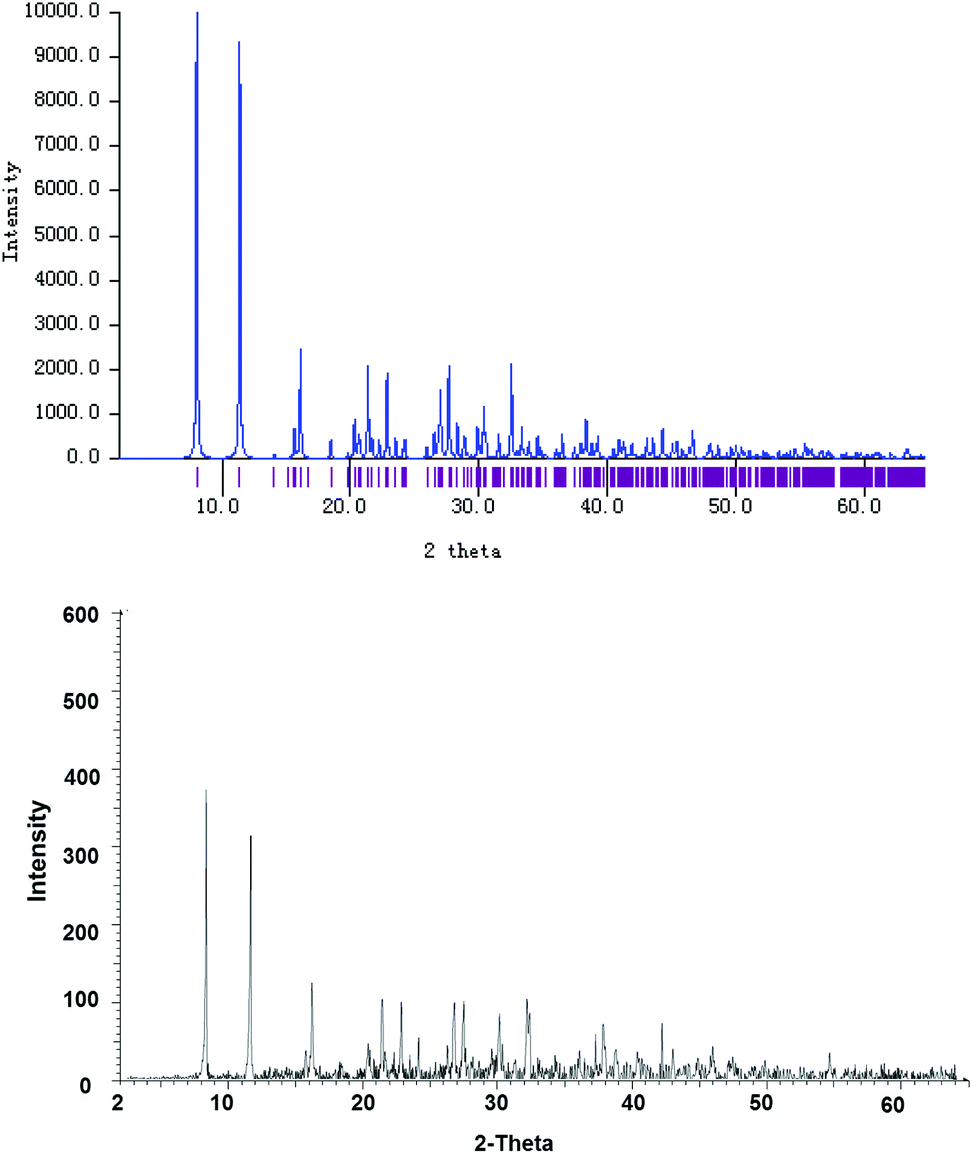 | ||
| Fig. 3 Simulated (top) and experimentally determined (bottom) X-ray powder diffraction patterns of bulk crystalline solids obtained from a saturated CH2Cl2 solution of complex 4. | ||
The Ccarbene signal of the tazy ligand is a diagnostic tool to distinguish tazy-containing complexes with different co-ligands (vide supra). Thus, to identify the prevalent species present in the solution, Au(I) bis(carbene) complex [Au(tazy)2]PF6 (6) was synthesized for comparison via a two-step protocol (Scheme 4). In the first step, the chlorido ligand of complex 2 was substituted by an acetato ligand with AgOAc. The obtained acetato complex [Au(CH3COO)(tazy)] (5) can serve as a basic precursor, which reacted with azolium salt tazy·HPF6 and gave the desired bis(NHC) complex 6 in a good yield of 88%.
 | ||
| Scheme 4 Syntheses of Au(I) acetato complex 5 and bis(NHC) complex 6. | ||
The solubilities of complexes 5 and 6 are very similar to that of the precursor 2. Their 1H NMR spectra also resemble that of 2 apart from the additional signals due to the acetato group in the case of 5. Again, the formation of 5 and 6 was better evidenced by their 13C NMR spectra, where the carbene resonances are found to shift markedly upfield (Δδ = 6.8 ppm, 5) and downfield (Δδ = 11.4 ppm, 6), respectively.
Single crystals of 5 and 6 for X-ray diffraction studies were obtained by slow evaporation of their concentrated CH2Cl2–n-hexane solutions. The molecular structures are shown in Fig. 4. In the structure of complex 5, the acetato moiety is coordinated to the Au(I) center in a monodentate fashion. In contrast to the chlorido and bromido counterparts 2 and 3 (vide supra), a weaker aurophilic interaction with a longer inter-gold separation of 3.471 Å was observed. This is again due to the increased “hardness” of acetato versus chlorido/bromido ligands (vide supra). In contrast to 5, no aurophilic interaction was found in complex 6, which likely results from the high steric demand of the two tazy ligands.
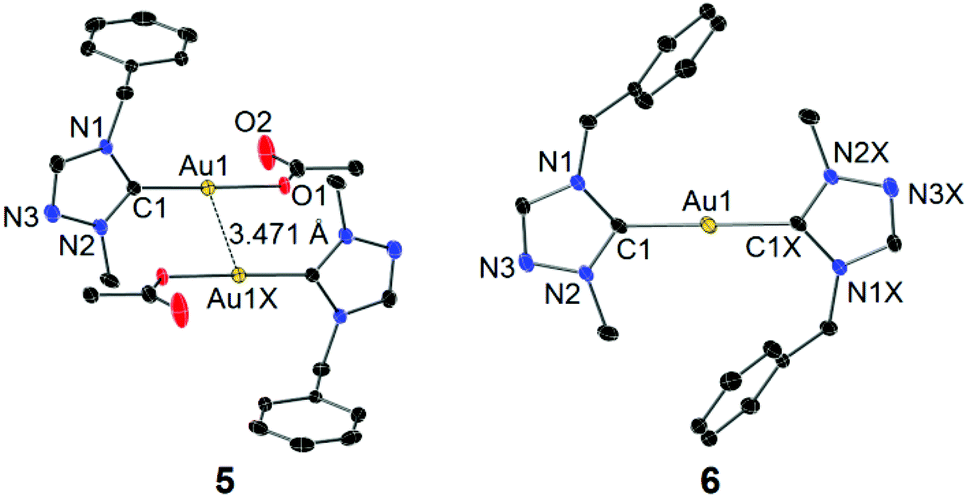 | ||
| Fig. 4 Molecular structures of 5 (left) and 6 (right) showing 50% probability ellipsoids. Hydrogen atoms and solvent molecules are omitted for clarity. Selected bond length (Å) and angles (°) for complex 5: Au1–C1 1.981(4), Au1–O1 2.039(3); C1–Au1–O1 178.12(13), N2–C1–N1 104.6(3). 6: Au1–C1 2.017(4), C1–Au1–C1X 180.0(2), N2–C1–N1 103.2(3). | ||
Comparison of diagnostic 13C NMR spectroscopic data
The carbene resonances of complexes 2–6 are illustrated in Fig. 5. It was found that the 13Ccarbene signal of the fixed tazy ligand correlates well with the net donating ability of the trans-standing ligand. The weakest donor, the acetato ligand, leads to the most upfield shift of the tazy carbene resonance, whereas the carbene signal of bis(carbene) complex 6 bearing an additional strong carbene ligand in the trans position is the most downfield. This observation is in line with previous reports.9b,c,11,12 | ||
| Fig. 5 13C carbene signals of tazy ligand in complexes 2–6. | ||
Notably, the 13Ccarbene (tazy) signal of bis(NHC) complex 6 at 186.2 ppm shows a clear downfield shift compared to resonance observed in complex 4 (184.8 ppm), which suggests that the mono-carbene complex is the prevalent species in solution. When 4′ was redissolved in CDCl3 and subjected to NMR spectroscopic analysis, the same spectroscopic features were observed as for the freshly prepared complex 4, which supports a full reversibility of the ligand disproportionation process.
UV-Vis absorption spectroscopy
To provide additional evidence that complex 4 is the prevalent species in solution instead of 4′, UV-Vis absorption spectroscopic analyses were performed. The absorption spectra of dihalido aurate species [AuX2]− (X = Cl, Br, I) have been thoroughly studied by Mason et al.15 It has been disclosed that the [AuI2]− anion features a characteristic LMCT band at 362 nm, which is a good indicator for the presence of [AuI2]−-containing species (such as 4′) in solution. As shown in Fig. 6, the absorption patterns of complexes 2–6 are quite similar and all absorption bands are within the high-energy region (λ < 300 nm). Notably, in the case of iodido complex 4, no LMCT absorption at ca. 362 nm characteristic for diiodidoaurate(I) is observed, which suggests the absence of the ionic species 4′ in solution.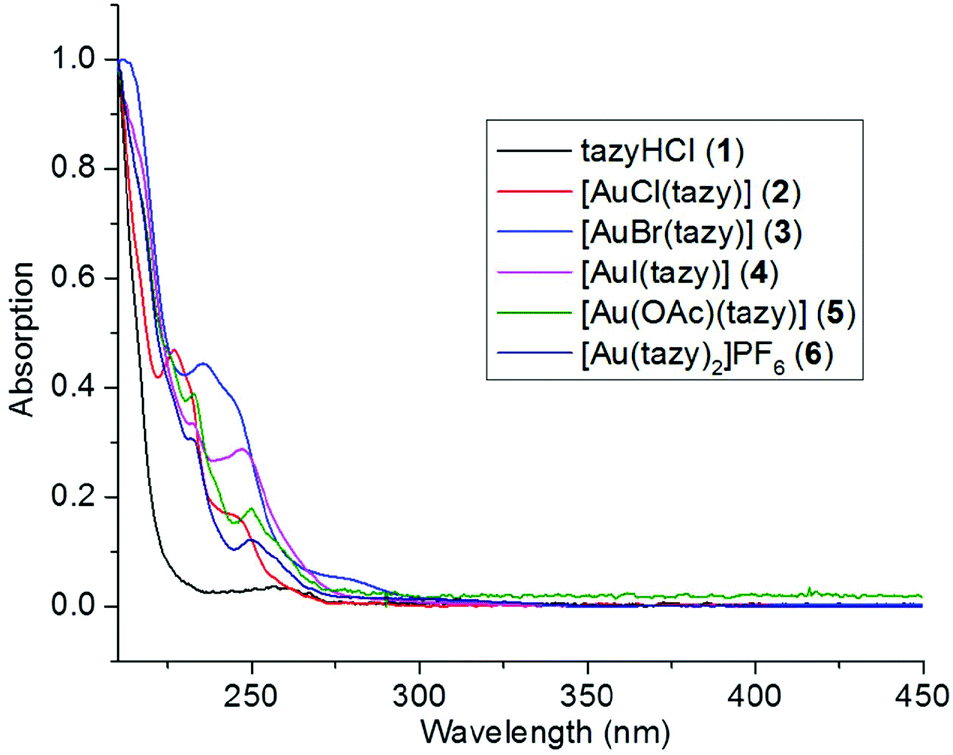 | ||
| Fig. 6 Normalized absorption spectra of precursor salt 1 and complexes 2–6. | ||
Oxidative addition
The identity of mono-carbene species 4 in solution can be also indirectly verified by its further transformation. The oxidative addition of I2 to complex 4 afforded the triiodido(monocarbene) complex [AuI3(tazy)] (7) in a very good yield of 93% (Scheme 5).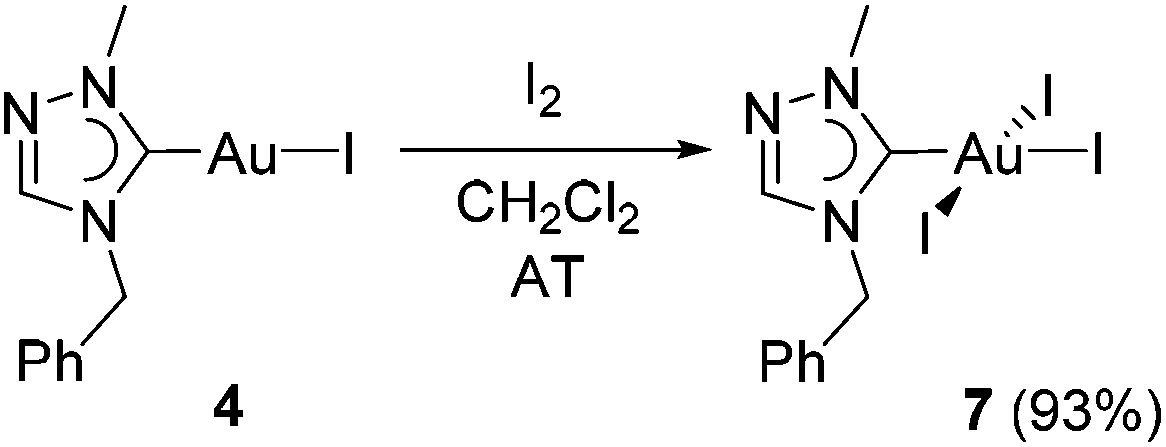 | ||
| Scheme 5 Synthesis of Au(III) triiodido complex 7. | ||
Complex 7 was isolated as dark-red solid. The color originates most likely from the ligand-to-metal-charge-transfer (LMCT) from a iodido ligand to the d8 Au(III) center.12a The formation of 7 was corroborated by its ESI mass spectrum, where a base peak at m/z 797 assignable to [M − I + tazy]+ was observed. The 1H NMR spectrum of 7 does not show pronounced differences despite the increased Lewis acidity of the metal center upon oxidation. Nevertheless, the Ccarbene signal is again a good indicator for the successful oxidation. This resonance has shifted significantly upfield (Δδ = 21.8 ppm) compared to that in Au(I) precursor 4.
X-ray diffraction study on single crystals obtained from a concentrated CH2Cl2–n-hexane solution of 7 confirmed the essentially square planner geometry adopted by the Au(III) center (Fig. 7). No adduct of complex 7 with I2 was found in the solid state molecular structure, which was previously observed for Au(I) phosphine analogues reported by Schmidbaur et al.16 The molecules of 7 were found to aggregate through an inter-iodine contact of 3.628 Å, which is within the sum of van der Waals radii for two iodine atoms (3.80 Å).17 The two cis-standing iodido ligands (I1, I3) bend to the more bulky tazy ligand with a I1–Au1–I3 bond angle of 173.26(3)°. Similar bending has been reported for a number of trihalido Au(III) carbene complexes,8b,12 which has been attributed to the electron donation of cis-halido ligand to the formally vacant pπ orbital at the Ccarbene atom, and the electrostatic repulsion between the trans- and cis-halido ligands. Another interesting feature in the structure of 7 is that the Au1–I2 bond distance is shorter compared to those of Au1–I1 and Au1–I3 bonds. In earlier studies on complexes of the type [AuX3(NHC)] (X = halido ligand), the more strongly donating carbene ligand always has a greater trans influence and lengthens the Au–Xtrans bond.12a,18 In the present case, the weakening of the Au–Xcis bond is likely to be due to the intermolecular I–I interaction (vide supra).
 | ||
| Fig. 7 Molecular structure and packing pattern of 7 showing 50% probability ellipsoids. Solvent molecules, disordered atoms are omitted for clarity. Selected bond length (Å) and angles (°): Au1–C1 2.038(11), Au1–I2 2.5888(10), Au1–I1 2.6183(8), Au1–I3 2.6259(7); C1–Au1–I2 177.1(3), I1–Au1–I3 173.26(3), C1–Au1–I1 85.8(3), C1–Au1–I3 88.9(3), I2–Au1–I1 92.27(3), I2–Au1–I3 93.11(3). | ||
Computational study
To gain further insight into the factors contribution or hampering ligand redistribution, the process was studied in greater detail by means of DFT calculations. The reaction between two molecules of [AuX(tazy)] can be formally decomposed into two bond-breaking and two bond-making steps, each of which contributes to the overall Gibbs free energy of the reaction (Scheme 6). While this decomposition most likely does not reflect the actual mechanism of the ligand redistribution process, it allows gauging the energetic contributions each broken and newly formed bond makes towards the overall Gibbs free energy.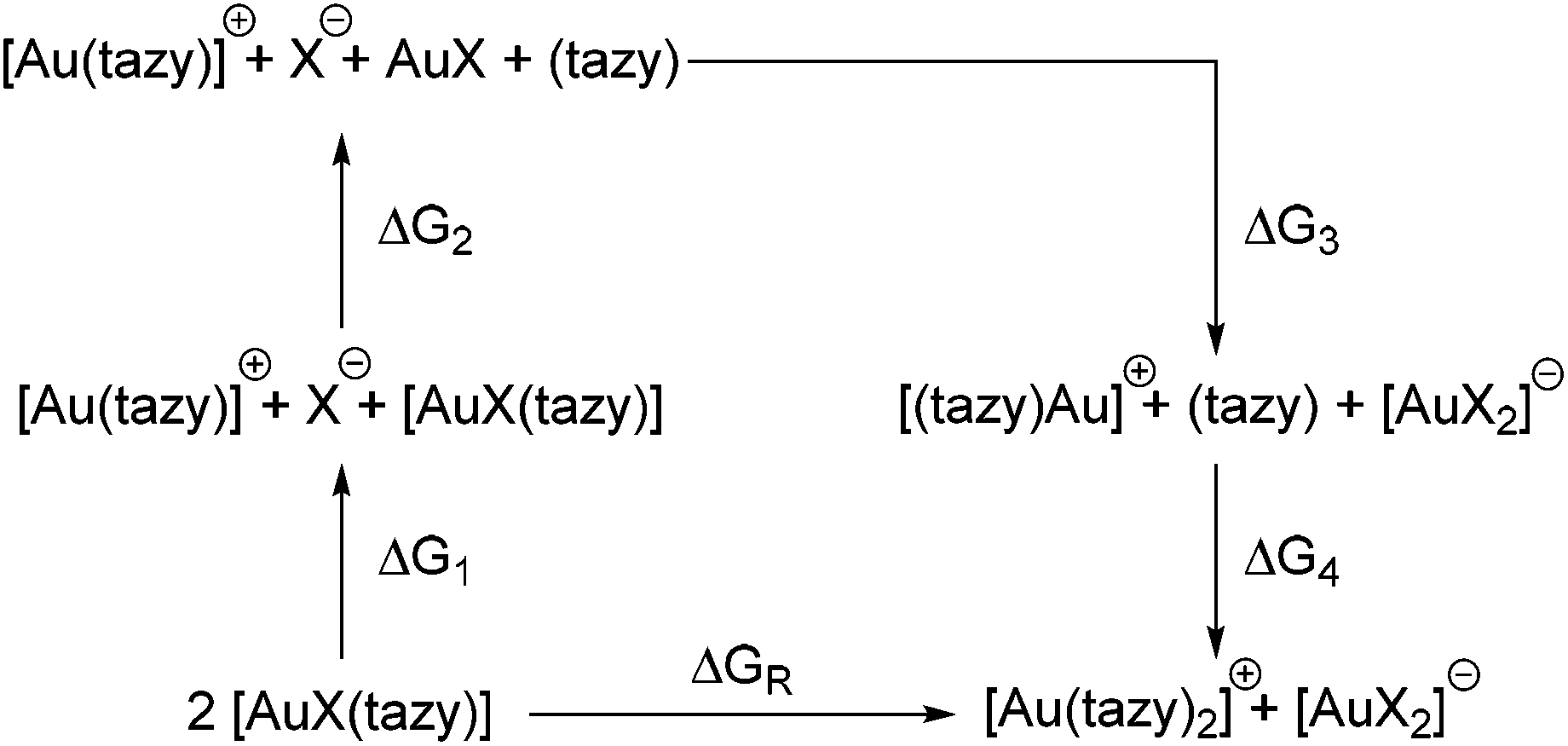 | ||
| Scheme 6 Ligand redistribution process and decomposition into individual steps. | ||
The geometries of all species involved in this hypothetical reaction outlined in Scheme 6 were optimised at the B3LYP/cc-pVDZ-(PP) level of theory. Initially, no solvent model was included in the optimization, but it became quickly apparent that this leads to a vast overestimation of the charge-separation step contributing to ΔG1. To mitigate this problem, a polarizable continuum model for dichloromethane, the solvent in which the ligand redistribution was observed, was included in the calculations.19
The optimized geometries are in good agreement with the molecular structures obtained by X-ray diffraction, insofar the latter are available. Bond angles and lengths are accurately reproduced, with Au–X and Au–C bonds being overestimated in the theoretical structures by less than 3%. The largest deviations were observed for species, which exhibit strong aurophilic interactions in the solid state. These are absent in the theoretical structures, explaining the slight changes in bond lengths and angles.
Regardless of the halido ligand, all reactions were found to be endothermic (Table 1). However, it should be noted that the energetic contributions from ion pairing and aurophilic interactions have not been accounted for. Aurophilic interactions can contribute up to 60 kJ mol−1 in additional stabilization, depending on the exact nature of the species involved,13,20 which is sufficient for the ligand redistribution to proceed upon crystallization.21
| X = Cl | X = Br | X = I | |
|---|---|---|---|
| ΔG1 | 215.5 | 213.5 | 157.9 |
| ΔG2 | 230.0 | 223.0 | 204.3 |
| ΔG3 | −183.7 | −171.7 | −112.3 |
| ΔG4 | −205.3 | −205.3 | −205.3 |
| ΔGR | 56.8 | 59.0 | 44.5 |
The calculated energy difference between 4 and 4′ is by ∼12 kJ mol−1 smaller for the iodido complexes when compared to the chlorido and bromido congeners, and aurophilic interactions are known to increase with softer coligands such as iodido. Hence it is very plausible that aurophilic interactions can offset the energy difference between 4 and 4′ in the solid state, while allowing the system to revert back to the mono-NHC form in solution. The larger energy difference and the weaker aurophilic interactions prevent a similar equilibrium for 2 and 3.
When examining the individual energetic contributions in detail, it becomes apparent that cleaving the Au–X bond in 2–4 requires considerably less energy for the iodido ligand than for the lighter homologues (ΔG1). The more polarizable iodide is a better leaving group than the harder bromide and chloride anions. However, this energetic difference is negated by a lower energy gain from aurate formation (ΔG3) due to the stronger trans-influence of the iodido ligands. This stronger trans-influence also makes the cleavage of the Au–C bond in 2–4 less endothermic for 4 (ΔG2), and indeed, it is energetically more favourable for the tazy ligand to bind to [Au(tazy)]+ than to AuI, while this is not the case for AuCl and AuBr.
Based on these results, it appears that the combination of a stronger aurophilic interaction in the solid state of 4′ and the stronger trans-influence of the iodido ligand in 4 renders the ligand redistribution energetically favorable upon crystallization, while the absence of aurophilic interaction in solution explains the reversible behavior observed upon dissolving the crystalline sample. Nevertheless, it must be noted that our calculation can only address (i) electronic effects of the ligands and (ii) solvent effects as two important factors contributing to the observed ligand disproportionation upon crystallization. The crystal lattice energy as the third major contributor, on the other hand, can currently not be accounted for within the scope of this study. Hence, this study provides a deeper insight into this phenomenon, but an exact prediction of the process is expectedly not possible.
Conclusions
Four mono-NHC complexes of the type [AuX(tazy)] (X = Cl, Br, I, OAc) have been synthesized and fully characterized. Only the iodido complex underwent a reversible ligand redistribution upon crystallization. By careful analysis of crystallographic data as well as NMR and UV-Vis spectroscopic data and comparison with spectra obtained from reference compound [Au(tazy)2]PF6 (6), it could be firmly established that the solid phase of the iodido complex exists exclusively as the disproportionated material 4′, while in solution the neutral form 4 is prevalent. Unlike previous reports of such redistributions, which occurred with unusually electron-rich NHC ligands, we used the weakly donating tazy ligand, thus ruling out a strong electron donor as requirement for such reactions. By contrast, a soft halido coligand with high trans-influence seems to be required for the reaction to proceed in this system, as evidenced by DFT calculations.Experimental section
General considerations
Unless otherwise noted all operations were performed without taking precautions to exclude air and moisture. All solvents and chemicals were used as received without any further treatment if not noted otherwise. [AuCl(SC4H8)] was synthesized according to the reported procedure.221H, 13C, 19F and 31P NMR spectra were recorded on a Bruker ACF 300 and Bruker AMX 500 spectrometer and the chemical shifts (δ) were internally referenced to the residual solvent signals relative to [Si(CH3)4] (1H, 13C) or externally to CF3CO2H (19F) and 85% H3PO4 (31P). ESI mass spectra were measured using a Finnigan MAT LCQ spectrometer. Elemental analyses were carried out on a Elementar Vario Micro Cube elemental analyzer at the Department of Chemistry, National University of Singapore. UV–Vis absorption spectroscopic analyses were carried out with a Shimadzu 2550 UV-Vis spectrometer. The measurement was performed using a 0.05 mM solution in CH3CN in a UV quartz cuvette. X-ray powder diffraction analyses were carried out with a Bruker D8 ADVANCE Powder X-ray diffractometer. 4-Benzyl-1-methyl-1,2,4-triazolium hexafluorophosphate was synthesized via a salt metathesis reaction of its chloride analogue and KPF6 in H2O.X-ray diffraction studies
X-ray data for 2, 3, 4, 5·H2O, 6 and 7 were collected with a Bruker AXS SMART APEX diffractometer, using Mo-Kα radiation at 100(2)K with the SMART suite of Programs.23 Data were processed and corrected for Lorentz and polarisation effects with SAINT,24 and for absorption effect with SADABS.25 Structural solution and refinement were carried out with the SHELXTL suite of programs.26 The structure was solved by direct methods to locate the heavy atoms, followed by difference maps for the light, non-hydrogen atoms. All non-hydrogen atoms were generally given anisotropic displacement parameters in the final model. All H-atoms were put at calculated positions.Computational methods
All calculations were carried out using hybrid density functional theory (DFT) employing the B3LYP functional27 and the Gaussian 09 software,28 using the IEFPCM approach to account for solvatation in dichloromethane.19 The nature of all stationary points was confirmed by frequency analysis, and all geometries were found to represent minima on the potential energy surface. For the optimisation of complex geometries and the calculation of their Gibbs free energies, gold and iodine were described with a cc-pVDZ-PP basis set in combination with the corresponding electronic core potential (ECP),29 while the lighter elements were treated with the cc-pVDZ basis set.30 The basis sets were all obtained from the EMSL Basis Set Library.31Acknowledgements
We thank the National University of Singapore and the Singapore Ministry of Education for financial support (WBS R-143-000-483-112). Technical assistance from staff at the CMMAC of our department is appreciated. In particular, we thank Ms Geok Kheng Tan, Ms Hong Yimian and Prof. Lip Lin Koh for determining the X-ray molecular structures.Notes and references
- For a review on coinage metal–NHCs, see: J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561 CrossRef CAS PubMed.
- For catalytic applications, see: (a) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776 RSC; (b) J. D. Egbert, C. S. J. Cazin and S. P. Nolan, Catal. Sci. Technol., 2013, 3, 912 RSC; (c) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (d) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180 CrossRef CAS PubMed.
- For luminescence applications, see: (a) L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903 RSC; (b) C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409 RSC.
- For biological applications, see: (a) W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755 RSC; (b) L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269 RSC; (c) A. Gautier and F. Cisnetti, Metallomics, 2012, 4, 23 RSC; (d) C. G. Hartinger, N. Metzler-Nolte and P. J. Dyson, Organometallics, 2012, 31, 5677 CrossRef CAS.
- For reviews on Ag–NHCs, see: (a) J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978 CrossRef CAS PubMed; (b) I. J. B. Lin and C. S. Vasam, Coord. Chem. Rev., 2007, 251, 642 CrossRef CAS PubMed.
- For selected examples, see: (a) H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972 CrossRef CAS; (b) O. Guerret, S. Solé, H. Gornitzka, M. Teichert, G. Trinquier and G. Bertrand, J. Am. Chem. Soc., 1997, 119, 6668 CrossRef CAS.
- C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801 CrossRef CAS.
- (a) G. D. Frey, R. D. Dewhurst, S. Kousar, B. Donnadieu and G. Bertrand, J. Organomet. Chem., 2008, 693, 1674 CrossRef CAS PubMed; (b) H. Sivaram, R. Jothibasu and H. V. Huynh, Organometallics, 2012, 31, 1195 CrossRef CAS.
- (a) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 CrossRef CAS PubMed; (b) H. V. Huynh, Y. Han, R. Jothibasu and J. A. Yang, Organometallics, 2009, 28, 5395 CrossRef CAS; (c) S. Guo, H. Sivaram, D. Yuan and H. V. Huynh, Organometallics, 2013, 32, 3685 CrossRef CAS.
- H. M. J. Wang, C. S. Vasam, T. Y. R. Tsai, S.-H. Chen, A. H. H. Chang and I. J. B. Lin, Organometallics, 2005, 24, 486 CrossRef CAS.
- M. V. Baker, P. J. Barnard, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, J. Chem. Soc., Dalton Trans., 2005, 37 RSC.
- (a) H. V. Huynh, S. Guo and W. Wu, Organometallics, 2013, 32, 4591 CrossRef CAS; (b) J. C. Bernhammer and H. V. Huynh, Dalton Trans., 2012, 41, 8600 RSC.
- For reviews on aurophilic interactions, see: (a) H. Schmidbaur, Chem. Soc. Rev., 1995, 24, 391 RSC; (b) H. Schmidbaur, Gold Bulletin, 2000, 33, 1 CrossRef; (c) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370 RSC.
- P. Pyykkö, N. Runeberg and F. Mendizabal, Chem. – Eur. J., 1997, 3, 1451 CrossRef.
- (a) M. M. Savas and W. R. Mason, Inorg. Chem., 1987, 26, 301 CrossRef CAS; (b) M. E. Koutek and W. R. Mason, Inorg. Chem., 1980, 19, 648 CrossRef CAS.
- D. Schneider, O. Schuster and H. Schmidbaur, Dalton Trans., 2005, 1940 RSC.
- P. H. Svensson and L. Kloo, Chem. Rev., 2003, 103, 1649 CrossRef CAS PubMed.
- (a) P. de Frémont, R. Singh, E. D. Stevens, J. L. Petersen and S. P. Nolan, Organometallics, 2007, 26, 1376 CrossRef; (b) S. Gaillard, A. M. Z. Slawin, A. T. Bonura, E. D. Stevens and S. P. Nolan, Organometallics, 2010, 29, 394 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- (a) P. Pyykkö, Chem. Rev., 1997, 97, 597 CrossRef PubMed; (b) H. Schmidbaur, S. Cronje, B. Djordjevic and O. Schuster, Chem. Phys., 2005, 311, 151 CrossRef CAS PubMed; (c) H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2008, 37, 1931 RSC; (d) A. Otero-de-la-Roza, J. D. Mallory and E. R. Johnson, J. Chem. Phys., 2014, 140, 18A504 CrossRef PubMed.
- P. Pyykkö, W. Schneider, A. Bauer, A. Bayler and H. Schmidbaur, Chem. Commun., 1997, 1111 RSC.
- (a) R. Usón, A. Laguna and M. Laguna, Inorg. Synth., 1989, 26, 85 CrossRef; (b) M.-C. Brandys, M. C. Jennings and R. J. Puddephatt, J. Chem. Soc., Dalton Trans., 2000, 4601 RSC.
- SMART version 5.628, Bruker AXS Inc., Madison, WI, 2001 Search PubMed.
- SAINT+ version 6.22a, Bruker AXS Inc., Madison, WI, 2001 Search PubMed.
- G. W. Sheldrick, SADABS version 2.10, University of Göttingen, 2001 Search PubMed.
- SHELXTL version 6.14, Bruker AXS Inc., Madison, WI, 2000 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed; (b) A. D. Becke, Phys. Rev. A, 1988, 38, 3098 CrossRef CAS; (c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- (a) K. A. Peterson, D. Figgen, E. Goll, H. Stoll and M. Dolg, J. Chem. Phys., 2003, 119, 11113 CrossRef CAS PubMed; (b) K. A. Peterson, J. Chem. Phys., 2003, 119, 11099 CrossRef CAS PubMed; (c) D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227 CrossRef CAS PubMed; (d) K. A. Peterson and C. Puzzarini, Theor. Chem. Acc., 2005, 114, 283 CrossRef CAS; (e) K. A. Peterson, B. C. Shepler, D. Figgen and H. Stoll, J. Phys. Chem. A, 2006, 110, 13877 CrossRef CAS PubMed.
- (a) T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef PubMed; (b) D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1993, 98, 1358 CrossRef CAS PubMed.
- (a) D. Feller, J. Comput. Chem., 1996, 17, 1571 CrossRef CAS; (b) K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected crystallographic data; Computational details. CCDC 1029440–1029445. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03201b |
| This journal is © The Royal Society of Chemistry 2015 |