
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unprecedented silicon(II)→calcium complexes with N-heterocyclic silylenes†
Burgert
Blom
*a,
Günter
Klatt
b,
Daniel
Gallego
a,
Gengwen
Tan
a and
Matthias
Driess
*a
aTechnische Universität Berlin, Department of Chemistry: Metalorganics and Inorganic Materials, Sekr. C2, Strasse des 17. Juni 135, 10623 Berlin, Germany. E-mail: burgert.blom@tu-berlin.de; matthias.driess@tu-berlin.de; Fax: (+49) 30-314-29732
bPhysikalisch-Chemisches Institut der Universität Heidelberg, Im Neuenheimer Feld 229, D-69120 Heidelberg, Germany
First published on 4th November 2014
Abstract
The first N-heterocyclic silylene (NHSi) complexes of any s-block element to date are reported for calcium: [(η5-C5Me5)2Ca←:Si(O-C6H4-2-tBu){(NtBu)2CPh}] (6) and [(η5-C5Me5)2Ca←:Si(NtBuCH)2] (7). Complexes 6 and 7 are isolable in a facile way upon reaction of the corresponding free N-heterocyclic silylenes (NHSis) with [(η5-C5Me5)2Ca] (2). Complexes 6 and 7 were fully characterised by spectroscopic means and the single crystal X-ray diffraction analysis of 6 is also reported. Analysis of the bonding situation by DFT methods including a Bader Atoms in molecules (AIM) analysis is also reported. The bonding interaction between the Si and Ca centres in complexes 6 and 7 can best be viewed as σ-donor–acceptor interactions, with a considerable ionic contribution in the bond. The reactivity towards the oxygen containing substrates THF and benzophenone is also discussed.
Introduction
N-heterocyclic silylene (NHSi) complexes have been reported across the transition metals,1 and a plethora of studies have been carried out to date, even highlighting their potential applications in catalysis.2 These efforts have been heralded by the high abundance, low toxicity and cost of silicon. Remarkably, no reports of NHSi complexes for any s-block elements exist, particularly for the alkaline-earth metals, although two seminal reports for f-block NHSi complexes have been reported.3 This is somewhat surprising since employing environmentally benign alkaline earth metals, such as calcium, might enable access to inexpensive and environmentally friendly NHSi complexes for catalysis, stoichiometric transformations, or as precursors for silicate glasses. Herein we report the first group 2 (alkaline earth metal) calcium NHSi complexes readily accessible in a quantitative fashion by simple coordination reactions with the corresponding NHSis. Preliminary reactivity studies are also reported in addition to the theoretical calculations by DFT methods elucidating the bonding situation between the Ca and Si centres.Results and discussion
The starting point in our study was to investigate the reaction of the chlorosilylene of Roesky :Si(Cl)(PhC(NtBu)2)4 (1) with [(η5-C5Me5)2Ca] (2), since earlier investigations showed that 1 is robust in coordination to a range of transition metals.5 The reaction thereof with 2, however, affords an inseparable mixture of the novel dimer [(η5-C5Me5)CaCl(thf)2]2 (3) (Fig. 1) and :Si(Cp*)(PhC(NtBu)2) (4), when dissolved in thf, by salt metathesis as opposed to the desired coordination compound [(η5-C5Me5)2Ca←:Si(Cl){(NtBu)2CPh}]6 (see ESI†).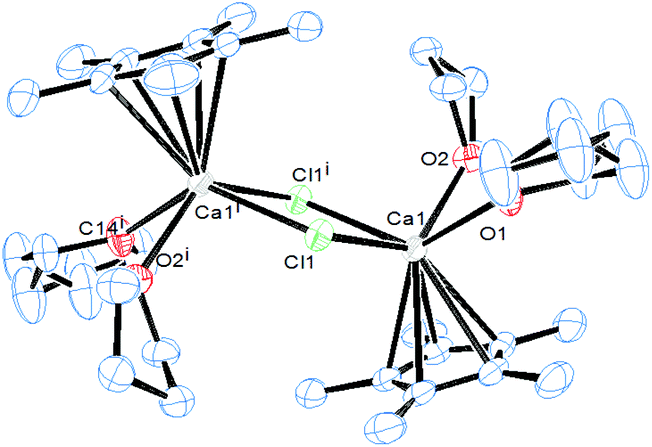 | ||
| Fig. 1 ORTEP representation of the molecular structure of dimer 3 in the solid state, resulting from the reaction of the chlorosilylene 1 with [(η5-C5Me5)2Ca] (2). Thermal ellipsoids are set at the 50% probability interval and H atoms are omitted for clarity. | ||
In order to suppress this metathetical reaction, the NH-silylene 1 was functionalised with an aryloxy substituent affording the novel NHSi :Si(O-C6H4-2-tBu){(NtBu)2CPh} (5), since cleavage of the Si–O bond in the latter is highly unlikely, due to the oxophilic nature of silicon. Indeed this strategy enables facile and near quantitative entry to the first s-block silylene complex: [(η5-C5Me5)2Ca←:Si(O-C6H4-2-tBu){(NtBu)2CPh}] (6) as a colourless solid, upon reaction of 5 with 2 in toluene at room temperature (Scheme 1). Strikingly, preferential coordination to Ca occurs via the Si atom and not the O atom in compound 6. This is likely due to the high steric demand of the aryloxy substituent precluding O coordination.
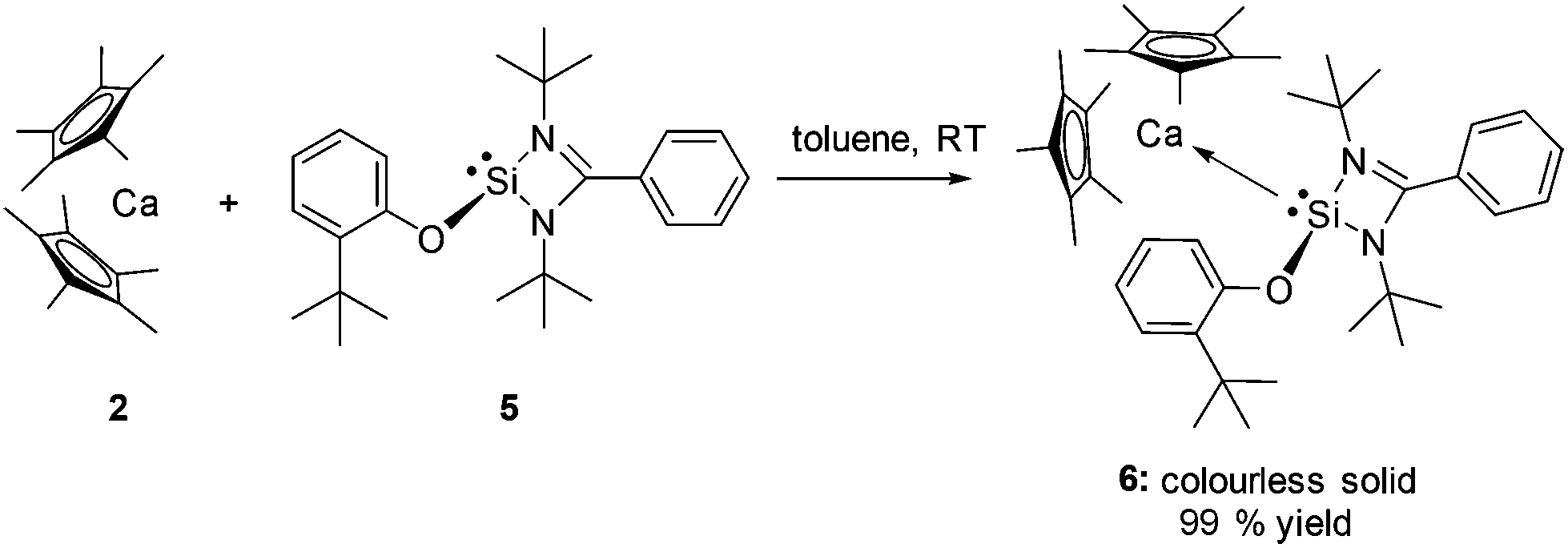 | ||
| Scheme 1 Synthetic entry to the first NHSi calcium complex by coordination of 5 to calcium precursor 2. | ||
The same synthetic methodology can also be employed to prepare the three coordinate NHSi complex [(η5-C5Me5)2Ca←:Si(NtBuCH)2] (7) by reaction of the West silylene :Si(NtBuCH)2 (8)7 with the calcium precursor 2 in an analogous way. This affords compound 7 quantitatively as a colourless solid (Scheme 2).
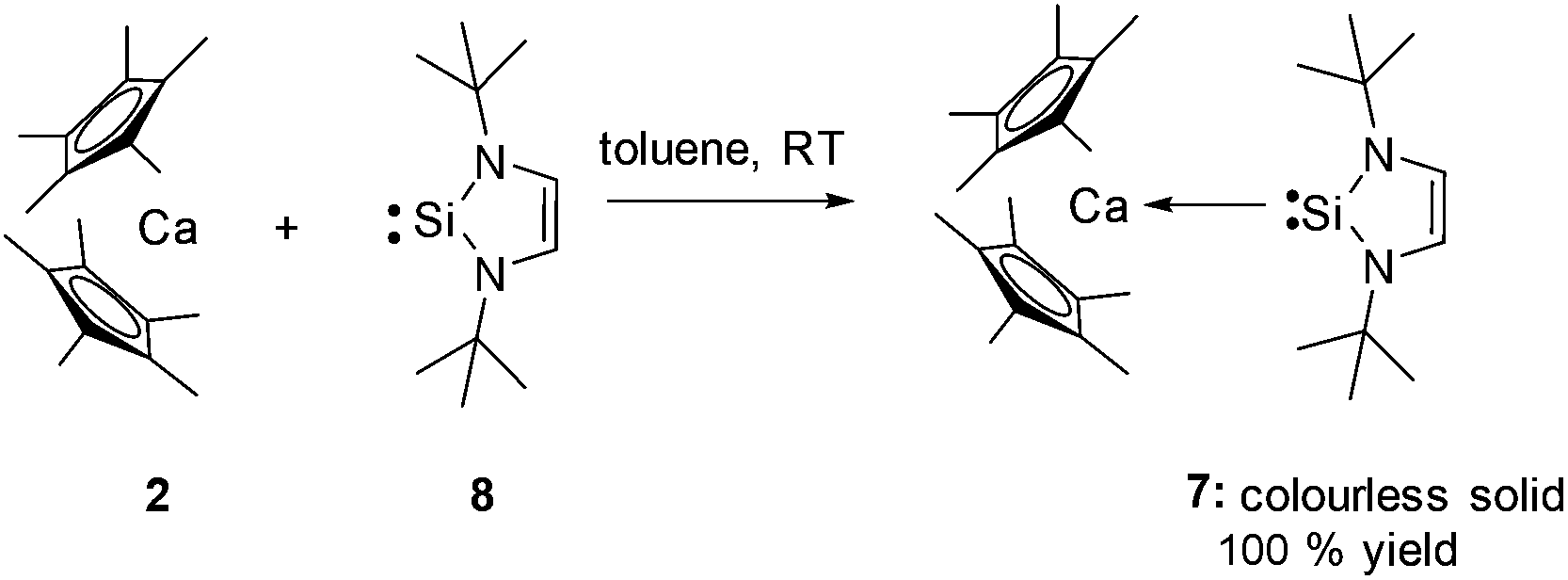 | ||
| Scheme 2 Synthetic entry to the three coordinate NHSi calcium complex 7. | ||
Complexes 6 and 7 are remarkable examples of molecular compounds featuring Ca–Si bonds, which are exceptionally rare.8 The most diagnostic spectroscopic feature for compounds 6 and 7 is the 29Si NMR shift in solution (6: δ = −13.7 ppm; 7: δ = 81 ppm, both in C6D6). Strikingly, these chemical shifts are only marginally deshielded from the uncoordinated NHSis (δ = −22.8 and δ = 78.3 ppm7 respectively) in both cases. This observation is in contrast to d-block NHSi complexes where a dramatic deshielding effect is observed on coordination and suggests only a very weak interaction between the Si and Ca centres in both complexes.
Dissociation in solution can be ruled out since 29Si MAS-solid state NMR spectroscopy revealed an identical chemical shift as to the solution spectrum for compound 7. Crystals of compound 6 suitable for single crystal X-ray diffraction analysis were obtained from a concentrated toluene solution with slow cooling to −30 °C. In the case of compound 7 a similar procedure also consistently afforded crystals visually of high quality, but the quality of the diffraction data was rather low (R1 = 0.1855 for I > 2σ(I)), despite several measurements and many other crystallisation procedures. Despite this, the structural motif was unambiguously located in the structure solution (see ESI†), and the metrical parameters are in good agreement with the calculated structure (BP86-D3 functional plus DZP basis set). Fig. 2 depicts the solid state structure of compound 6 and the geometry optimised structure of 7.
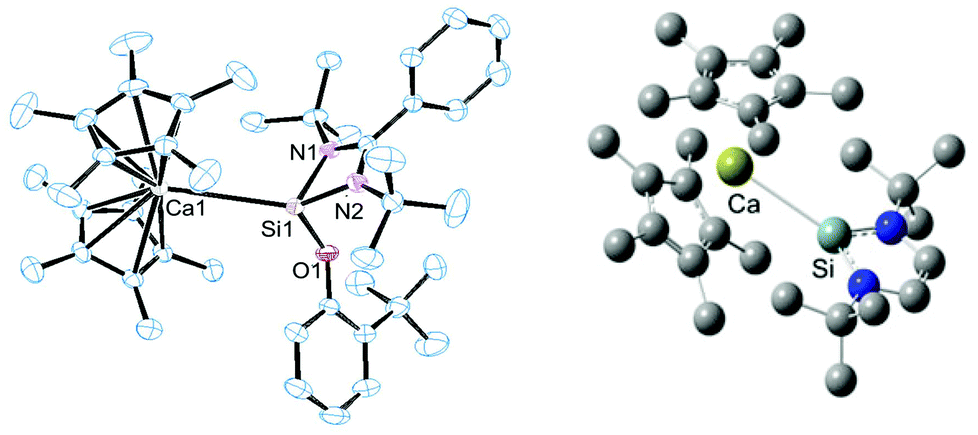 | ||
| Fig. 2 ORTEP representation of the molecular structure of compound 6 in the solid state (left). Thermal ellipsoids set at 50% probability level and H atoms omitted for clarity. The geometry optimised structure of 7 (right) (BP86-D3 functional plus DZP basis set). For 6: d(Ca–Si) = 3.2732(5) Å. 7: d(Cal–Si) 3.090 Å (calcd). | ||
Both compounds feature rather long Ca–Si bond lengths: in compound 7, bearing a three-coordinate Si atom, this distance is shorter by 18 pm compared to compound 6, indicating stronger coordination in the former compound. The observed difference in bond length might be due to sterics as compound 6 bears a substantially more sterically congested NHSi than compound 7 which would impede coordination thereby resulting in an elongated bond length. The bond lengths are somewhat longer than those reported in the calcium silyl complex Ca(thf)3(Si(SiMe3)3)2 at 3.042(9) and 3.086(9) Å respectively.8a Moreover, in both compounds, this bond distance is significantly larger than the sum of the single bond covalent radii proposed by Pyykkö and Atsumi9 for Ca (1.71 Å) and Si (1.16 Å) = 2.87 Å which led us to question whether it is merely an electrostatic interaction, with no covalent character. In order to address this point we performed DFT calculations to elucidate the nature of the bonding interaction between Si and Ca in both compounds (BP86-D3 and the DZP basis).
In compound 6, the LUMO, E = −0.085 eV, is located mostly on the phenyl residue while the HOMO, E = −0.146 eV, mostly involves the Cp* ligands with some involvement of the Ca centre (Fig. 3). Similarly, in compound 7 the LUMO, E = −0.063 eV, is mostly Si(p) + 2 × N(p) + 2 × C(p) while the HOMO, E = −0.151 eV, is also mostly located on the Cp* ligands with some involvement of the Ca centre and (Fig. 4).
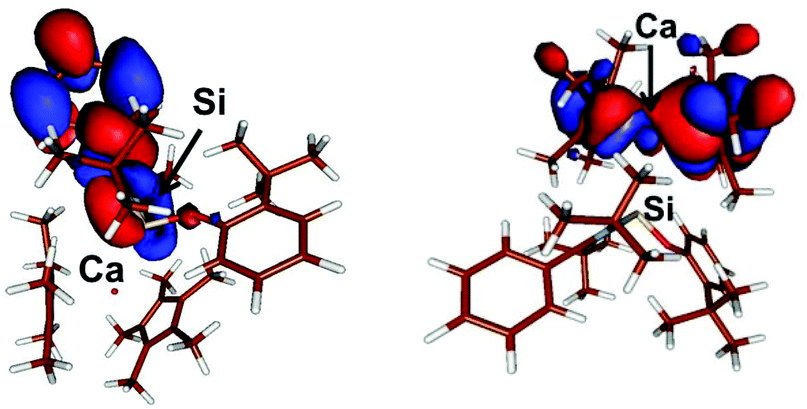 | ||
| Fig. 3 Plot of the boundary surfaces of the LUMO (left) (E = −0.085 eV) and HOMO (right) (E = −0.146 eV) in compound 6. | ||
 | ||
| Fig. 4 Plot of the boundary surfaces of the LUMO (left) (E = −0.063 eV) and HOMO (right) (E = −0.151 eV) in compound 7. | ||
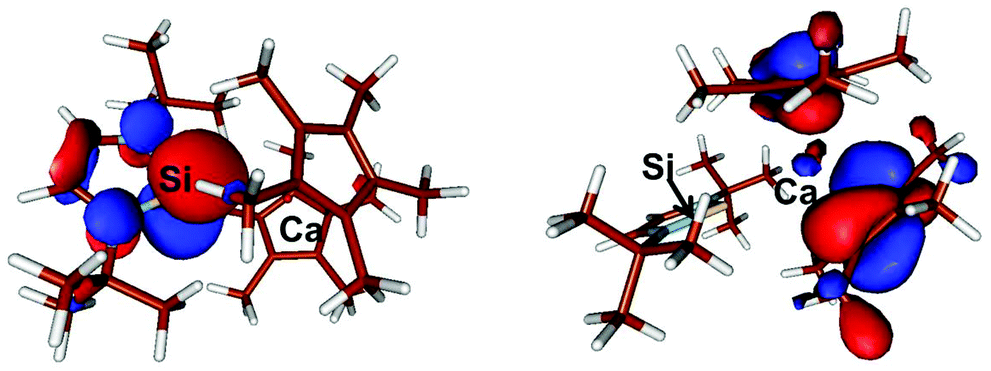
The most interesting MO in both compounds signifying the Si–Ca interaction is in both cases the HOMO−5 6: E = −0.207 eV; 7: E = −0.230 eV, which clearly shows a donor–acceptor interaction between Si and Ca in both compounds (Fig. 5). Notably, in both cases some contribution arises from the Ca centre in the MO. Moreover, the calculated Wiberg-bond index (WBI) of compound 6 = 0.47 while in compound 7, in accord with the decreased Ca–Si bond length observed experimentally, is slightly higher at 0.53. These indices are indicative of a covalent bonding interaction between the Ca and Si centres in both compounds.
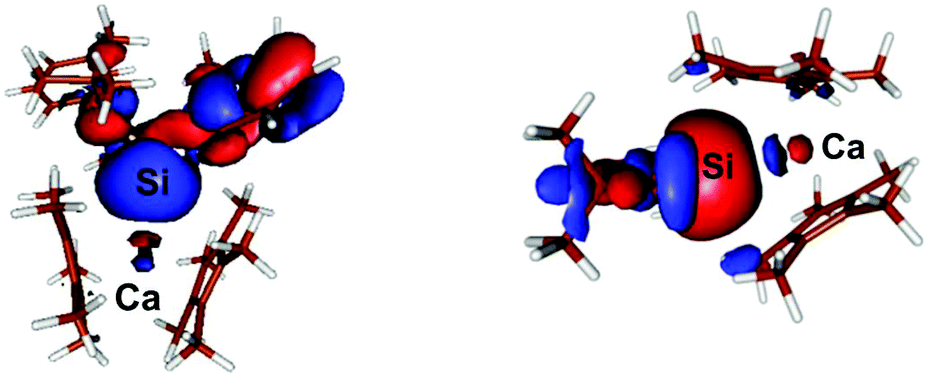 | ||
| Fig. 5 Plot of the boundary surfaces of the HOMO−5 in 6 (left) occupancy is 1.91, with 86% Si sp- and 14% Ca spd-contribution. In 7 (right) occupancy is 1.95, with 85% Si sp- and 15% Ca spd-contribution. | ||
In contrast, a Bader atoms in molecules (AIM) analysis10 of compound 7 (see ESI† and Fig. 6) revealed a (3, −1) bond critical point (BCP) between the Ca and Si centres with ρ(r) = 0.0175 a.u. and ∇2ρ(r) = 0.0657 a.u. at the bond critical point. These values indicate a closed-shell (ionic) interaction between the two centres. Moreover the ratio of the eigenvalues |λ1|/λ3 = 0.127 (λ1 = −0.0115, λ2 = −0.0130, λ3 = 0.0902) is also indicative of a close-shell interaction. Hence the AIM analysis points to a substantial ionic character in the Si–Ca interaction; at odds with the positive WBI clearly indicating covalency in the bond. These results collectively show that the Ca–Si bonding interaction is perhaps therefore best described as a σ-donor acceptor interaction between Si and Ca which bears considerable ionic character.
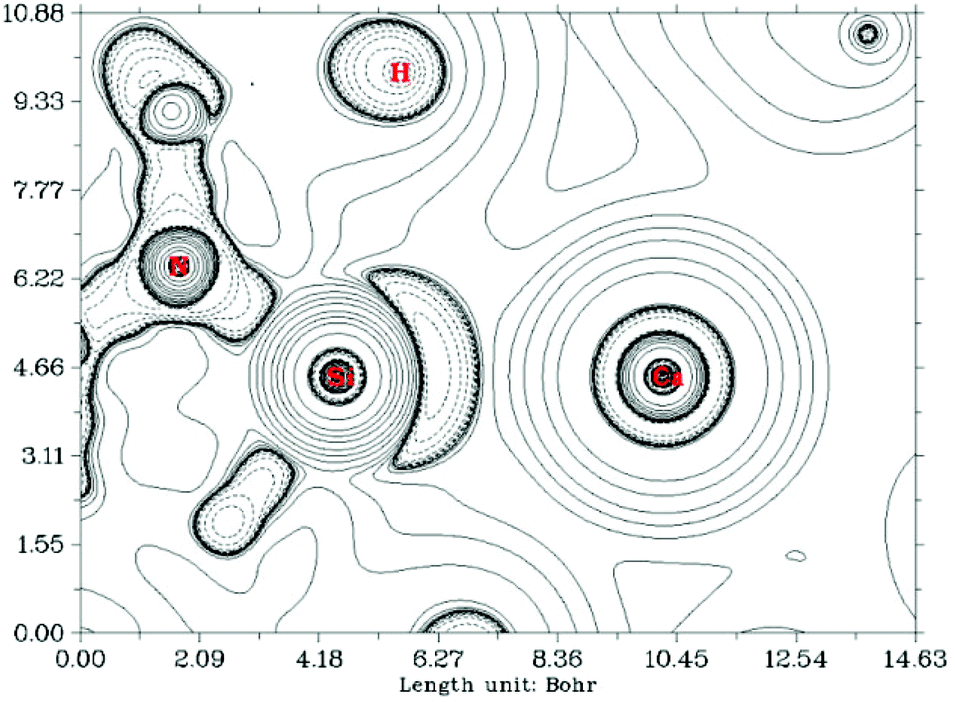 | ||
| Fig. 6 Contour map of the Laplacian in compound 7. Negative values are indicated by a dotted line (where there is charge concentration), and solid lines where the Laplacian is positive (charge depletion). | ||
The labile coordination of the NHSi in compound 7 was shown by the reaction with THF where immediate NHSi elimination results with concomitant formation of [(η5-C5Me5)2Ca(thf)2] (2·2thf), observed in the 1H NMR spectra. This result is certainly due to the highly oxophilic nature of the Ca centre. We next attempted a cycloaddition reaction of benzophenone with 7 with the goal of forming the cycloaddition product 9 (see Scheme 3). Even in this instance, the NHSi 8 is eliminated with concomitant formation of a dark purple ketone adduct [(η5-C5Me5)2Ca←:O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CPh2] (10) (Fig. 7).11 The isolation and formation of 10 in this reaction dramatically highlights the weak coordination of the NHSi to Ca, which is even eliminated by a ketone coordination.12 Again, the oxophilic nature of the Ca centre likely drives this process.
CPh2] (10) (Fig. 7).11 The isolation and formation of 10 in this reaction dramatically highlights the weak coordination of the NHSi to Ca, which is even eliminated by a ketone coordination.12 Again, the oxophilic nature of the Ca centre likely drives this process.
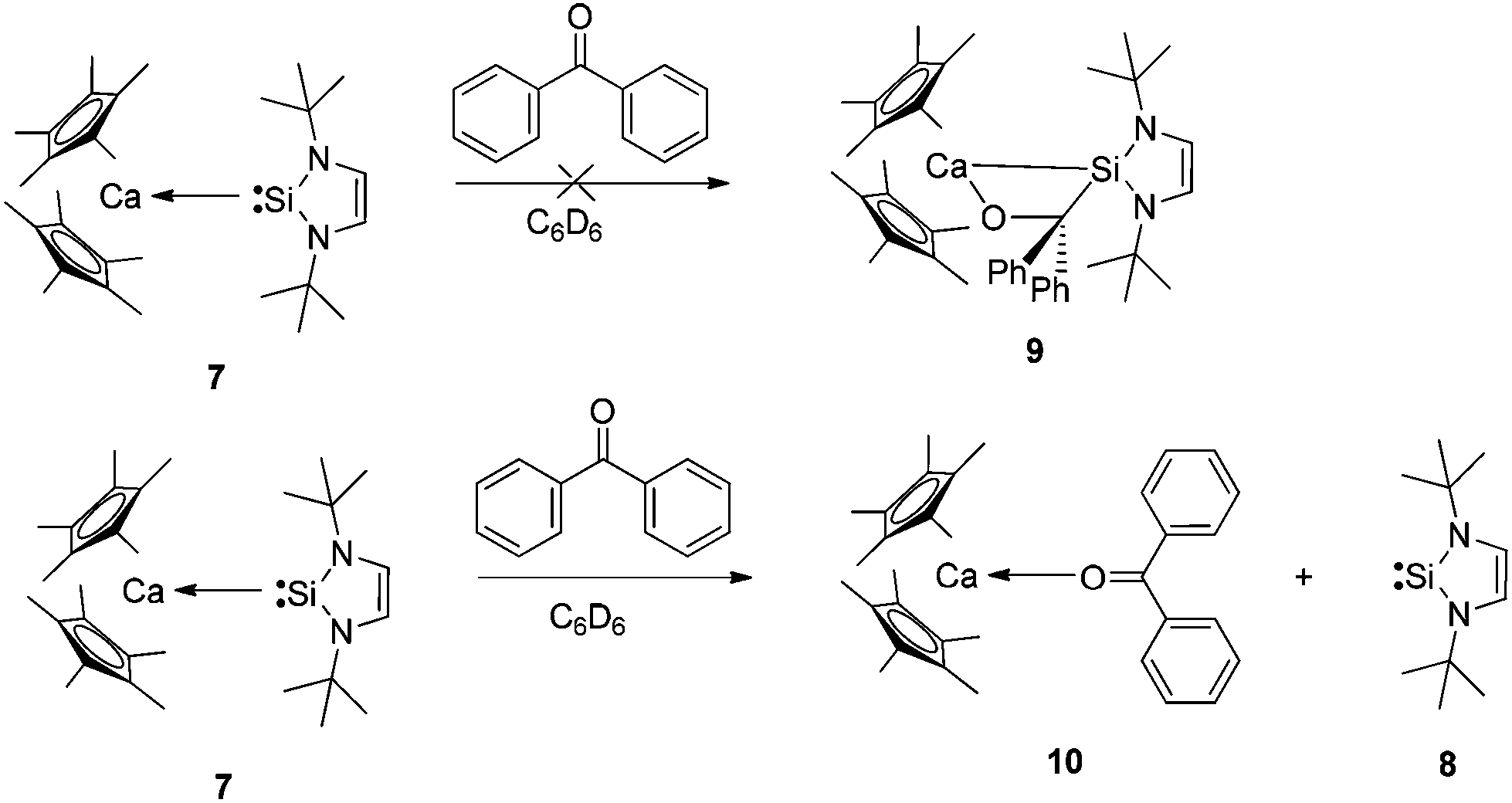 | ||
| Scheme 3 Reaction of compound 7 with benzophenone does not afford the expected cycloaddition product (9), rather the formation of the ketone adduct 10 with concomitant elimination of NHSi 8 is observed. | ||
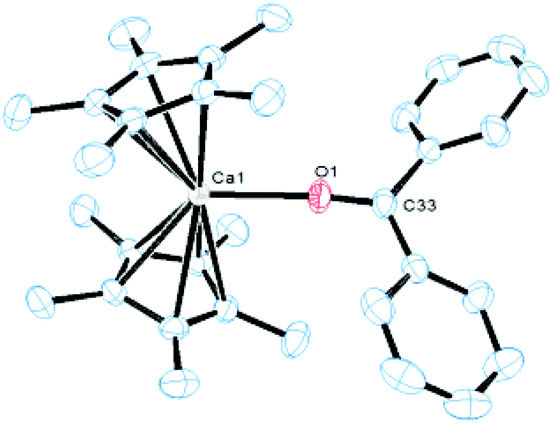 | ||
| Fig. 7 ORTEP representation of the molecular structure of ketone adduct 10 in the solid state. Thermal ellipsoids set at the 50% probability level, H atoms omitted for clarity. Selected bond lengths (Å): Ca1–O1 2.293(2), O1–C33 1.238(3). | ||
Conclusion
The first examples of s-block N-heterocyclic silylene complexes (NHSi) have been reported for the group 2 alkaline-earth metal element calcium. These complexes are also the only existing examples of any s-block NHSi coordination compound to date. The four or three coordinate NHSi complexes can be prepared in a quantitative fashion upon reaction of the free NHSis with the readily available precursor [(η5-C5Me5)2Ca] affording [(η5-C5Me5)2Ca←:Si(O-C6H4-2-tBu){(NtBu)2CPh}] (6) and [(η5-C5Me5)2Ca←:Si(NtBuCH)2] (7) respectively. Both compounds exhibit remarkably long Ca–Si bond lengths, but on the basis of DFT investigations these can be considered simple donor–acceptor interactions from Si to Ca, with considerable ionic character. The NHSi in compound 7 is very labile to substitution, and is readily eliminated by THF and even benzophenone, affording in the latter case the ketone adduct: [(η5-C5Me5)2Ca←:OCPh2] (10). This showcases the rather low bond energy of the Ca–Si bond. We are currently exploring NHSis that bind more strongly to the Ca centre which might enable novel Ca, Si mediated processes, without NHSi elimination. We are also currently extending this synthetic approach to other group 2 elements (Ba and Sr) and will report these finding in due course.
Experimental section
All experiments were carried out under dry oxygen-free nitrogen using standard Schlenk techniques. Solvents were dried by standard methods and freshly distilled and degassed prior to use. The NMR spectra were recorded on Bruker spectrometers (AV400 or AV200) referenced to residual solvent signals as internal standards (1H NMR: C6D6, 7.15; THF-d8 3.58 (left signal) ppm and 13C{H} NMR: C6D6, 128.0; THF-d8 67.6 (left signal) ppm) or with an external standard (SiMe4 for 29Si NMR). Concentrated solutions of samples were sealed off in a Young-type NMR tube for measurements. Signals were unambiguously assigned by a combination of H,H COSY, HSQC and HMBC correlation 2D experiments. Melting points were recorded on a “Melting point tester” device from BSGT company and are uncorrected. All the samples are sealed off in capillary under vacuum and each sample was measured in duplicate. High resolution ESI mass spectra were recorded on an Orbitrap LTQ XL of Thermo Scientific mass spectrometer and the raw data evaluated using the Xcalibur computer program. For the single crystal X-ray structure analyses the crystals were each mounted on a glass capillary in perfluorinated oil and measured in a cold N2 flow. The data were collected on an Agilent Technologies SuperNova (single source) at 150 K (CuKα radiation, λ = 1.5418 Å) and refined on F2 with the SHELX-971 software package. The positions of the H atoms were calculated and considered isotropically according to a riding model. Solid state 29Si{1H} static and MAS cross polarization (CP) measurements were carried out on a Bruker Avance II spectrometer at an external magnetic field of 9.4 T (i.e. a 29Si resonance frequency of 79.46 MHz) using a standard Bruker 4 mm double-resonance H-X MAS probe. The CP spectra were recorded with a cross polarization time of 2 ms and composite pulse 1H decoupling was applied during the acquisition. The spectra were referenced externally to TMS (tetramethylsilane) using TKS (tetrakis(trimethylsilyl)silane) as a secondary reference. Benzophenone was purchased from Aldrich and used as received. [(η5-C5Me5)2Ca] (2) was prepared from Ca{N(SiMe3)2}2 upon reaction with Cp*H and purified by sublimation as a white crystalline solid as previously reported by Tanner et al.13 The NHSi 14 and 87a was prepared by literature procedures.CH). 13C{1H} NMR (100.2 MHz, C6D6, 298 K, ppm): δ = 11.8 (s, 2 × C5(CH3)5), 32.1 (s, 2 × C(CH3)3), 54.4 (s, 2 × C(CH3)3), 113.6 (s, 2 × C5(CH3)5), 120.8 (s, 1 × CHCH). 29Si{1H} NMR (79.5 MHz, C6D6, 298 K, ppm): δ = 81.0 ppm. 29Si{1H}-MAS-NMR: δ = 81.4 ppm ESI-MS (THF): 507.3449 (expt.) 507.3397 (calcd) [M + H]+.
CPh2] (10). The identity of 10 was confirmed by the synthesis of 10 directly from the reaction of [(η5-C5Me5)2Ca] (2) with Benzophenone as follows: A Schlenk tube was charged with [(η5-C5Me5)2Ca] (2) (0.412 g, 1.326 mmol) and benzophenone (0.240 g, 1.326 mmol). The two solids were mixed by stirring at room temperature and a colour change (in the solid state) was immediately observed to bright purple. Toluene (10 mL) was added to the purple solid and the formed purple solution stirred at room temperature for 1 h. The solvent was removed in vacuo affording the product as a purple solid. Yield: 0.650 g, 100%). 1H NMR (400 MHz, C6D6, 298 K, ppm): δ = 2.07 (s, 30H, 2 × 2 × C5(CH3)5), 7.00 (t, 4H, 3J(H,H) = 7.8 Hz, 2 × C2,6–H, Ph), 7.11 (t, 2H, 3J(H,H) = 7.8 Hz, 2 × C4–H, Ph), 7.46 (d, 4H, 3J(H,H) = 7.8 Hz, 2 × C3,5–H, Ph). 13C{1H} NMR (100.2 MHz, C6D6, 298 K, ppm): δ = 11.0 (s, 2 × C5(CH3)5), 112.5, (s, 2 × C5(CH3)5), 129.0 (s, 2 × C2,6, Ph), 130.6 (s, 2 × C3,5, Ph), 134.4 (s, 2 × C4, Ph), 201.6 (s, 1 × CO⋯Ca). ESI-MS(THF): 491.2613 (expt.) 491.2621 (calcd) [M − H−]+, 183.0800 (expt.) 183.0804 (calcd) [C13H10O + H]+/[free benzophenone + H]+.
Acknowledgements
The authors wish to acknowledge the Unicat cluster of excellence for funding and Dr J.D. Epping for the Solid-state NMR measurement. Dr S. Kohl is thanked for a generous gift of Cp*2Ca. Dr J. Li (TU Berlin) is thanked for performing the AIM analysis of 7.Notes and references
- (a) B. Blom, M. Stoelzel and M. Driess, Chem. – Eur. J, 2013, 19, 40 CrossRef CAS PubMed; (b) B. Blom and M. Driess, Struct. Bonding, 2014, 156, 85 CrossRef CAS.
- B. Blom, D. Gallego and M. Driess, Inorg. Chem. Front., 2014, 1, 134 RSC.
- (a) X. Cai, B. Gehrhus, P. B. Hitchcock and M. F. Lappert, Can. J. Chem., 2000, 78, 1484 CrossRef CAS; (b) W. J. Evans, J. M. Perotti, J. W. Ziller, D. F. Moser and R. West, Organometallics, 2003, 22, 1160 CrossRef CAS.
- (a) C.-W. So, H. W. Roesky, J. Magull and R. B. Oswald, Angew. Chem., 2006, 118, 4052 ( Angew. Chem., Int. Ed. , 2006 , 45 , 3948 ) CrossRef; (b) S. S. Sen, H. W. Roesky, D. Stern, J. Henn and D. Stalke, J. Am. Chem. Soc., 2010, 132, 1123 CrossRef CAS PubMed.
- See as selected examples: (a) R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, J. Am. Chem. Soc., 2012, 134, 2423 CrossRef CAS PubMed; (b) B. Blom, S. Inoue, D. Gallego and M. Driess, Chem. – Eur. J., 2012, 18, 13355 CrossRef CAS PubMed; (c) R. Azhakar, R. S. Ghadwal, H. W. Roesky, J. Hey and D. Stalke, Chem. – Asian. J., 2012, 7, 528 CrossRef CAS PubMed; (d) B. Blom, S. Enthaler, S. Inoue, E. Irran and M. Driess, J. Am. Chem. Soc., 2013, 135, 6703 CrossRef CAS PubMed; (e) G. Tan, B. Blom, D. Gallego and M. Driess, Organometallics, 2014, 33, 363 CrossRef CAS.
- Recently similar reactivity has been reported with Zinc. See: S. Schäfer, R. Köppe, M. T. Gamer and P. W. Roesky, Chem. Commun., 2014, 50, 11401 RSC.
- (a) M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belaykov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691 CrossRef CAS; (b) M. Haaf, A. Schmiedl, T. A. Schmedake, D. R. Powell, A. J. Millevolte, M. Denk and R. West, J. Am. Chem. Soc., 1998, 120, 12714 CrossRef CAS.
- Only very few reports exist of well-defined molecular compounds featuring Ca–Si bonds. See as examples: (a) W. Teng and K. Ruhlandt-Senge, Organometallics, 2004, 23, 2694 CrossRef CAS; (b) K. Yan, G. Schoendorff, B. M. Upton, A. Ellern, T. L. Windus and A. D. Sadow, Organometallics, 2013, 32, 1300 CrossRef CAS; (c) K. Takanashi, V. Y. Lee, T. Yokoyama and A. Sekiguchi, J. Am. Chem. Soc., 2009, 131, 916 CrossRef CAS PubMed; (d) V. Leich, T. P. Spaniol, L. Maron and J. Okuda, Chem. Commun., 2014, 50, 2311 RSC.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 186 CrossRef PubMed.
- See as examples: (a) R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS; (b) R. F. W. Bader and H. Essen, J. Chem. Phys., 1984, 80, 1943 CrossRef CAS.
- Compound 10 is surprisingly the first ketone adduct of Cp*2Ca. Ketone adducts or ketyl complexes of calcium have been reported but are somewhat rare. See: (a) D. Cook, Can. J. Chem., 1963, 41, 515 CrossRef CAS; (b) D. Cook, Can. J. Chem., 1963, 41, 522 CrossRef CAS; (c) Z. Hou and Y. Wakatsuki, Chem. – Eur. J., 1997, 3, 1005 CrossRef CAS; (d) L. Orzechowski and S. Harder, Organometallics, 2007, 26, 2144 CrossRef CAS; (e) V. Vyas, P. Raizada and U. Sharma, Int. J. Electrochem., 2011, 798321 Search PubMed.
- The synthesis of ketone adduct 10 can also be achieved by the reaction of benzophenone with 2, affording 10 quantitatively. This reaction can remarkably even be carried out in the solid state without any solvent.
- P. S. Tanner, D. J. Burkey and T. P. Hanusa, Polyhedron, 1995, 14, 331 CrossRef CAS.
- R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Organometallics, 2012, 31, 4588 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and ESI-MS spectra and the crystal structure of dimer 3 and compound 7, along with crystallographic details of compounds 3, 6 and 7 and 10 are available. Details concerning the DFT calculations of 6 and 7 and tables of Z-matrices are also included. CCDC 1021899 (3) CCDC 1021900 (6) and CCDC 1021901 (10). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03148b |
| This journal is © The Royal Society of Chemistry 2015 |