
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold(I) thiotetrazolates as thioredoxin reductase inhibitors and antiproliferative agents†
Tatiyana V.
Serebryanskaya
abc,
Alexander S.
Lyakhov
b,
Ludmila S.
Ivashkevich
b,
Julia
Schur
a,
Corazon
Frias
d,
Aram
Prokop
d and
Ingo
Ott
*a
aInstitute of Medicinal and Pharmaceutical Chemistry, Technische Universität Braunschweig, Beethovenstrasse 55, 38106 Braunschweig, Germany. E-mail: ingo.ott@tu-bs.de
bResearch Institute for Physical Chemical Problems of the Belarusian State University, 14 Leningradskaya st., 220030 Minsk, Belarus
cInstitute of Chemistry, Saint Petersburg State University, 198504 Stary Petergof, Russian Federation
dDepartment of Paedriatric Oncology, Children's Hospital Cologne, Amsterdamer Strasse 59, 50735 Cologne, Germany
First published on 21st November 2014
Abstract
Gold(I) complexes with phosphane and thiotetrazolate ligands were prepared and investigated as a new type of bioactive gold metallodrugs. The complexes triggered very efficient inhibition of the enzyme thioredoxin reductase (TrxR), which is an important molecular target for gold species. Strong cytotoxic effects were observed in MDA-MB-231 breast adenocarcinoma and HT-29 colon carcinoma cells, and the complexes also caused strong effects in vincristine resistant Nalm-6 leukemia cells. Cellular uptake studies showed elevated cellular gold levels for complexes containing a triphenylphosphane ligand, whereas trifurylphosphane analogues accumulated at significantly lower cellular concentrations.
Introduction
The therapeutic use of gold and its complexes dates back to thousands of years ago and was also a hot topic in alchemy during medieval ages and the Renaissance.1 However, the elaboration of gold based antiproliferative agents is a relatively young and intensively developing area of medicinal and pharmaceutical chemistry.2–5 The precise mechanisms of antitumor activity of gold compounds have not been completely understood. As a result of a series of mechanistic studies it was suggested that binding of gold to the selenoenzyme thioredoxin reductase (TrxR) leads to its inhibition, which results in the alteration of mitochondrial functions, increased formation of reactive oxygen species and finally causes cell death via apoptosis.6,7 Both gold(I) and gold(III) derivatives were found to be efficient inhibitors of thioredoxin reductase,6 yet gold(III) based complexes are nowadays considered as prodrugs that are reduced before binding to give gold(I) species.8The choice of ligands appears to be one of the most challenging tasks in the design of novel gold based antitumor agents. Owing to the “soft acidic” nature of gold(I), its coordination compounds are highly prone to rapid metabolic transformations via interaction with numerous sulfur containing proteins and peptides in vivo.2 Therefore, stability is a crucial point in the design of gold(I) species, and usually, typical “soft bases” such as phosphorus- and sulphur-containing nucleophiles are utilized as ligands to enhance the stability of the resulting gold(I) complexes under physiological conditions. Gold(I) phosphane complexes are the most frequently investigated group of gold based compounds.3,9–15 Many of them, including auranofin (Fig. 1) and chlorido gold(I) phosphane (1, Fig. 1) complexes, were shown to be effective antiproliferative agents and potent TrxR inhibitors.6,7,16
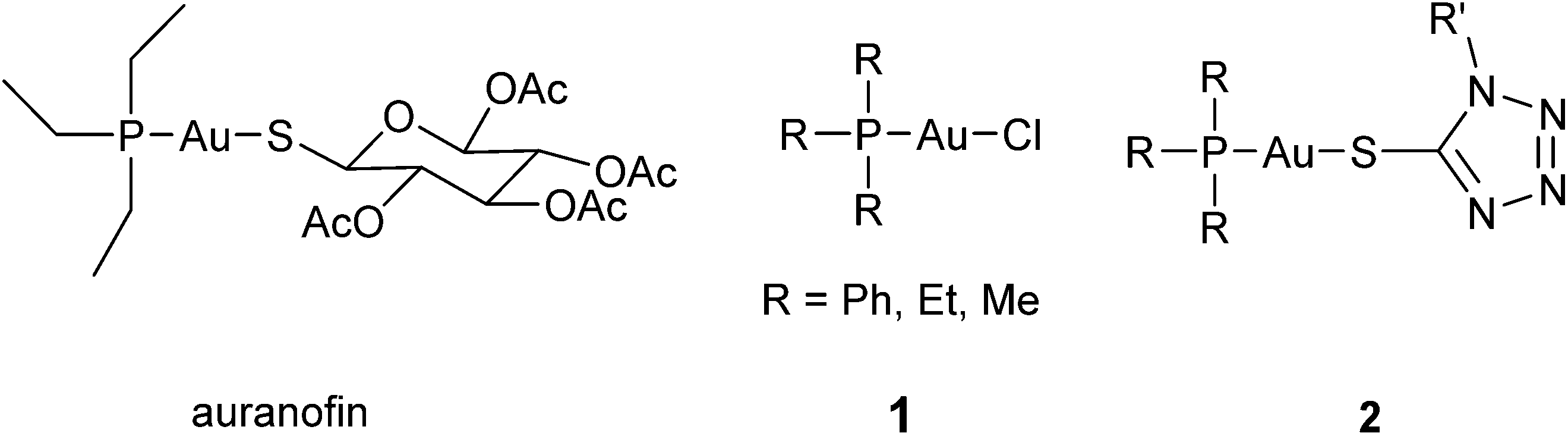 | ||
| Fig. 1 Examples of gold(I) phosphane complexes. | ||
Here we report on the synthesis and biological evaluation of a new series of tetrazole-containing gold(I) phosphane complexes (2, Fig. 1). Being rather new ligands in bioinorganic chemistry, tetrazole derivatives have been gaining increasing attention in recent years due to the development of more efficient synthesis methods.17 Moreover, the tetrazole ring is a well known bioisoster of the carboxylic group and several tetrazole-containing platinum complexes have earlier been reported as possessing a promising antiproliferative profile and low toxicity in vivo.18–20
Gold(I) complexes with polynitrogen-containing azoles have been relatively little studied so far. Among them, mixed-ligand phosphane gold(I) 5-thiotetrazolates 2 are among the most investigated groups.21 The obvious structural analogy between this group of gold(I) complexes and auranofin makes them interesting subjects for antitumor drug design. Despite this fact, there have been to date no data reported in the literature on their antiproliferative or other biological effects. In order to elucidate a possible structure–activity relationships within this group of gold(I) complexes, a panel of (phosphane)gold(I) 1-R-5-thiotetrazolates has been synthesized and characterized by means of spectroscopic and structural methods. Their antiproliferative activity against two human cancer cell lines as well as the inhibitory effects toward TrxR and glutathione reductase (GR) have been evaluated, whereupon cellular uptake studies were performed using atomic absorption spectroscopy (AAS). Additionally, the antiproliferative activity of the best candidates was investigated against resistant leukemia cell lines.
Results and discussion
Synthesis and characterization
Tetrazole-containing gold(I) complexes 2a–e bearing triphenylphosphane or trifurylphosphane ligands were synthesized, starting from the corresponding 1-substituted 5-thiotetrazoles 3a–c and chloridogold(I) phosphanes ClAuPR3 [R = Ph (1a), 2-furyl (1b)] in the presence of triethylamine according to Scheme 1. The synthesis of compounds 2a and 2b under similar conditions was reported elsewhere earlier.22,23 Complexes 2c–e were obtained for the first time.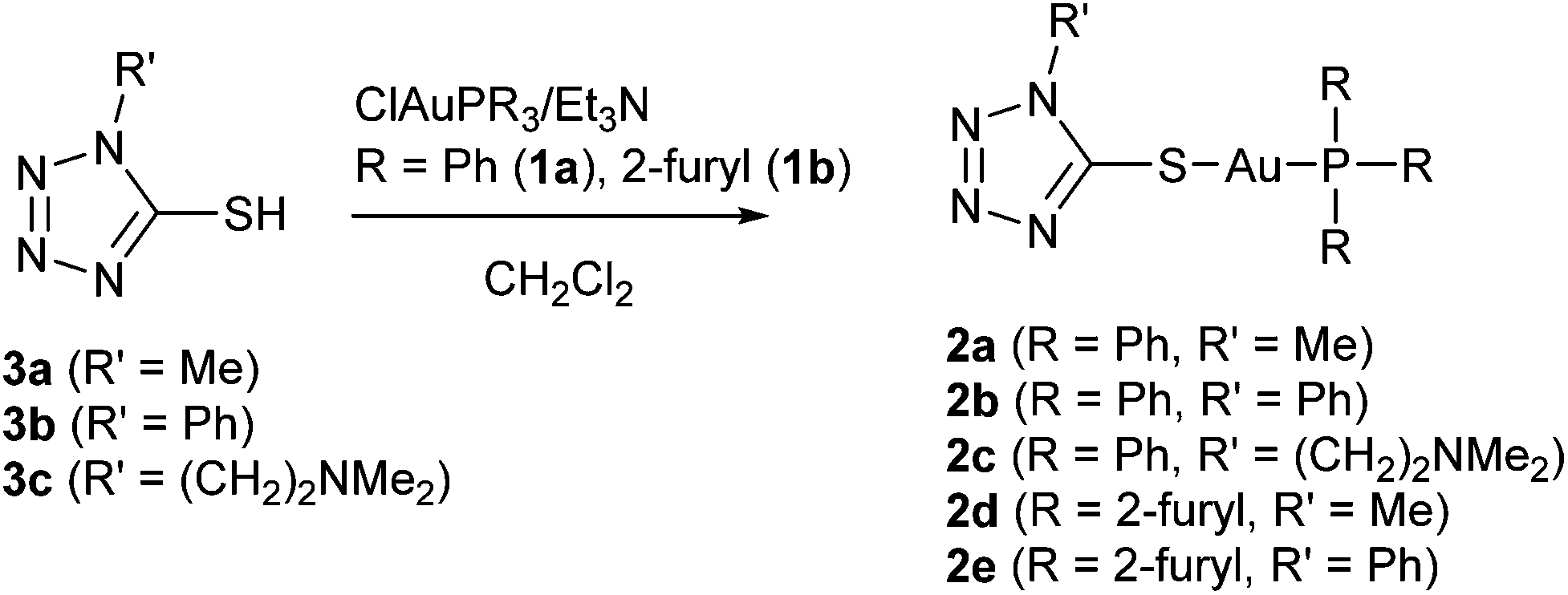 | ||
| Scheme 1 Synthesis of thiotetrazole-containing gold(I) phosphane complexes 2a–e. | ||
The composition and structure of the products were confirmed by elemental analysis, ESI mass spectrometry, and NMR spectroscopy (1H and 13C). Complexes were additionally characterized by means of IR spectroscopy and thermal analysis (TG/DSC). The molecular and crystal structure of 2e was additionally confirmed by single crystal X-ray diffraction. In general the spectral and structural characteristics of complexes 2a and 2b were found to be consistent with previously published results.23,24
ESI(+) mass spectra of 2a–e showed pseudomolecular ions [M + H]+ along with the respective [Au(PR3)2]+ species. Assignments of signals observed in 1H and 13C NMR spectra of all complexes were made based on previously published data23,25 and are in agreement with the suggested structures (see Scheme 1). In particular, 1H NMR spectra of 2a–e lack signals of NH protons that are observed in the spectra of uncoordinated tetrazolylthiones 3a–c at approx. 14 ppm. Coordination also leads to significant upfield shifts (approx. 5–6 ppm) of the C(5) signals, which are observed at approx. 158 ppm in the 13C NMR spectra of 2a–e (163–164 ppm in the spectra of 3a–c). Other resonances remain mainly unchanged when compared to the spectra of the starting materials. Due to 13C–31P coupling, phenyl and furyl carbons of the phosphane ligands appear as doublet resonances.
When compared with the spectra of the free ligands 3a–c, IR spectra of 2a–e show the same pattern of changes including disappearance of the intensive bands at 1510, 1350–1360, 1040–1050, and 790–800 cm−1 characteristic of the stretching and deformation vibrations of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S and N–CS fragments.22,23 New partially split bands at 690–710 cm−1 assigned to the stretching vibrations of the C–S bond23 were observed. Altogether, these changes support the formation of a covalent Au–S bond between the triarylphosphane gold(I) moiety and the thiolate function of the tetrazole-containing ligands.
S and N–CS fragments.22,23 New partially split bands at 690–710 cm−1 assigned to the stretching vibrations of the C–S bond23 were observed. Altogether, these changes support the formation of a covalent Au–S bond between the triarylphosphane gold(I) moiety and the thiolate function of the tetrazole-containing ligands.
According to the results of DSC/TG, all the complexes were found to be thermally stable up to 180–200 °C. Exothermal decomposition of 2a–c and 2e at higher temperatures is preceded by their melting at 180–195 °C, while 2d melts at a noticeably lower temperature (approx. 120 °C).
Crystal and molecular structures of complex 2e
Complex 2e crystallizes in the monoclinic space group C2/c, with eight formula units in the unit cell. Crystal data and structure refinement details for this compound are summarized in Table 1. Similarly to the previously reported complexes 2a and 2b,24 complex 2e presents a molecular complex showing a bidentate linear coordination environment of the gold atoms (Fig. 2a).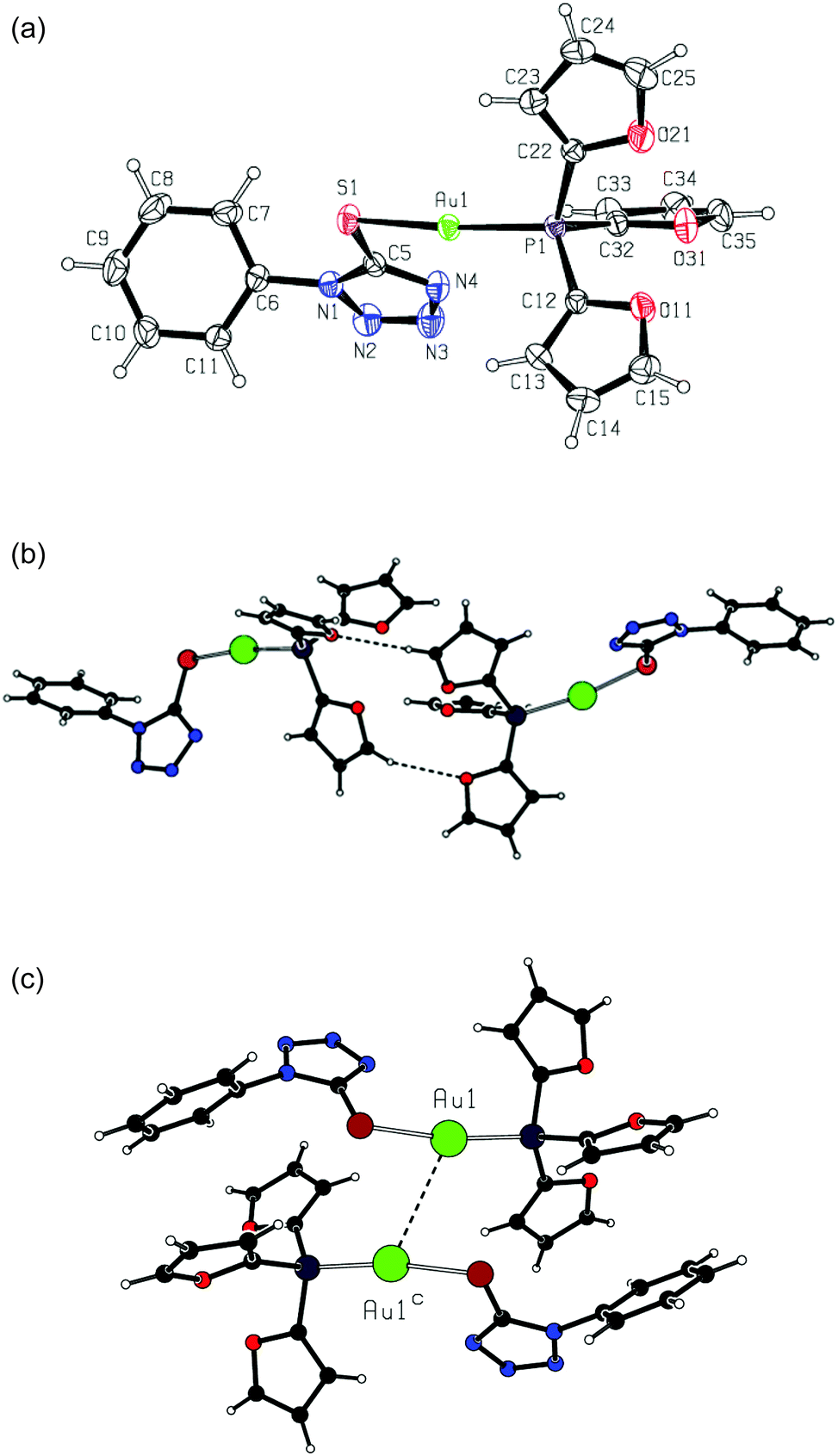 | ||
| Fig. 2 (a) Complex 2e, with the atom numbering scheme and displacement ellipsoids drawn at the 30% probability level; (b) hydrogen-bonded dimer in the crystal structure of 2e; (c) dimeric unit formed by aurophilic bonding in the crystal structure of 2e [symmetry transformation: (c) 1 − x, y, 1/2 − z]. | ||
| Formula | C19H14AuN4O3PS |
| Formula weight | 606.34 |
| T/K | 296(2) |
| Crystal system | Monoclinic |
| Space group | C2/c |
| Crystal size/mm | 0.35 × 0.34 × 0.12 |
| a/Å | 13.79810(10) |
| b/Å | 13.08130(10) |
| c/Å | 22.1996(3) |
| β/° | 91.0696(5) |
| V/Å3 | 4006.27(7) |
| Z | 8 |
| D c/g cm−3 | 2.011 |
| μ/mm−1 | 7.557 |
| Reflections | 5371 |
| Restraints | 0 |
| Parameters | 262 |
| R 1/wR2[I > 2σ(I)] | 0.0210/0.0484 |
| R 1/wR2 [all data] | 0.0283/0.0505 |
| Goodness-of-fit | 1.004 |
In complex 2e, the gold atom is in a standard linear coordination, being attached to the phosphorus atom with an Au1–P1 distance of 2.2475(6) Å and to the sulfur atom with an Au1–S1 distance of 2.3051(7) Å. The coordination angle P1–Au1–S1 takes on a value of 170.93(3)°. The geometries of the coordination units are very close in three compounds, 2a, 2b and 2e (see Table 2).
| Compound | 2a | 2b | 2e |
|---|---|---|---|
| a From ref. 24. | |||
| Au1–P1 | 2.261(1) | 2.265(2) | 2.2475(6) |
| Au1–S1 | 2.325(1) | 2.304(2) | 2.3051(7) |
| P1–Au1–S1 | 170.11(3) | 172.68(5) | 170.93(3) |
| C5–S1–Au1 | 97.5(1) | 105.3(2) | 99.28(9) |
| S1–C5 | 1.740(4) | 1.733(7) | 1.726(3) |
| N1–C5 | 1.342(5) | 1.346(9) | 1.350(3) |
| N1–N2 | 1.354(5) | 1.378(8) | 1.361(3) |
| N2–N3 | 1.289(5) | 1.295(9) | 1.285(4) |
| N3–N4 | 1.354(5) | 1.366(8) | 1.359(4) |
| N4–C5 | 1.331(5) | 1.346(8) | 1.326(3) |
In complex 2e, the oxygen atoms of the furane rings are oriented away from the gold atom. The angles Au1–P1–C12 [116.21(8)°] and Au1–P1–C22 [114.05(9)°] are larger than the tetrahedral standard, whereas the angle Au1–P1–C32 [107.76(9)°] is close to that but is somewhat smaller. The phosphane C–P–C valence angles, lying in a range of 103.83(12)–107.55(12)°, are smaller than the tetrahedral one. These geometric features are very similar to those in complexes 2a and 2b. The fragment AuPR3 (R = 2-furyl) displays a distorted non-propeller-like structure due to the large variation in dihedral angles Au–P–C–C [–5.8(3), 8.1(3) and 16.6(4)°].
As for the tetrazole ring geometry, it is typical of 1H-tetrazoles in the three complexes 2a, 2b and 2e (see Table 2), with the shortest bond N2–N3 and remaining bond distances lying in rather narrow ranges. In the crystal structure of complex 2e, there are non-classic intermolecular hydrogen bonds C35–H35⋯O11b between two neighboring complex molecules [symmetry transformation: (b) −x, y, −z + 1/2; hydrogen bond geometry: H35⋯O11b 2.55 Å, C35⋯O11b 3.424(3) Å, C35–H35⋯O11b 157°]. These bonds form dimeric entities comprising R22(14) hydrogen-bonded rings (Fig. 2b).
The complex molecules are bonded into dimeric units through very weak aurophilic bonding,26 with a separation of the gold atoms Au1⋯Au1c of 3.3892(5) Å (symmetry transformation as in Fig. 2c). These units are connected via the above mentioned hydrogen bonds to give polymeric chains running along the a axis.
TrxR and GR inhibition activity
As mentioned above, inhibition of the selenoenzyme thioredoxin reductase (TrxR) is considered to be an important mechanism of bioactivity of gold(I) species.3,6,7 Therefore, the potential of tetrazole-based gold(I) complexes to inhibit TrxR was studied on isolated rat liver TrxR using the DTNB (dithiobisnitrobenzoic acid) reduction assay. As far as selenocysteine is considered to be the relevant binding site for gold complexes, another structurally and functionally similar disulfide reductase, i.e. glutathione reductase (GR, from yeast), was included in the test as a reference. Instead of selenocysteine, GR contains a cysteine residue in its active site and thus can be used to check the specificity of TrxR inhibition by the tested compounds.28,29According to the results given in Table 3, tetrazole-containing gold(I) complexes turned out to be very effective inhibitors of TrxR with IC50 values in the low nanomolar range. With the exception of 2c, all complexes showed IC50 values in the range 30–55 nM and at least 10-fold selectivity for inhibition of TrxR over GR. In all cases 2a–e were more effective enzyme inhibitors than the respective chlorido complexes 1a and 1b.
| Compound | IC50 (μM) | Selectivity GR/TrxR (x-fold) | IC50 (μM) | ||
|---|---|---|---|---|---|
| TrxR | GR | MDA-MB-231 | HT-29 | ||
| 1a | 0.256 ± 0.00227 | 4.2 ± 0.727 | 16 | 9.5 ± 0.7 | 4.2 ± 0.910 |
| 1b | 0.207 ± 0.021 | n.d. | 9.9 ± 0.7 | 10.0 ± 1.8 | |
| 2a | 0.037 ± 0.010 | 0.97 ± 0.25 | 26 | 7.9 ± 1.9 | 12.0 ± 2.7 |
| 2b | 0.046 ± 0.012 | 0.55 ± 0.04 | 12 | 7.8 ± 2.0 | 10.2 ± 1.5 |
| 2c | 0.165 ± 0.065 | 0.66 ± 0.19 | 4 | 8.9 ± 1.1 | 8.6 ± 0.7 |
| 2d | 0.033 ± 0.005 | 0.43 ± 0.05 | 13 | 8.5 ± 1.5 | 13.3 ± 2.9 |
| 2e | 0.053 ± 0.001 | 0.51 ± 0.07 | 10 | 9.3 ± 0.2 | 11.5 ± 1.7 |
| 3a | n.d. | n.d. | — | >100 | >100 |
| 3b | n.d. | n.d. | — | >100 | >100 |
| 3c | n.d. | n.d. | — | >100 | >100 |
Antiproliferative activity
Antiproliferative activity of thiotetrazole-containing gold(I) complexes 2a–e against two human cancer cell lines (MDA-MB-231 and HT-29) was assessed using the crystal violet assay. Chlorido(triphenylphosphane)gold(I) (1a) and chlorido[tri(2-furyl)phosphane]gold(I) (1b) were included in the test as positive controls. According to the results (Table 3), the tetrazole-containing complexes 2a–e demonstrate considerable antiproliferative activity against both human cancer cell lines with IC50 values found in the micromolar or low micromolar range (approx. 8.0–13.0 μM). These values are comparable to those of the positive controls and the platinum anticancer drug cisplatin (IC50 values of 7.0 μM in HT-29 cells30 and 4.0 μM in MDA-MB-231 cells31). The metal-free thiotetrazole derivatives 3a–c were investigated as negative controls and showed no inhibition of tumor cell proliferation.Cellular uptake
In order to evaluate the extent of cellular uptake, we measured the gold content in HT-29 cells exposed to 10.0 μM (approx. equal to the IC50 calculated for this cell line) solutions of the tested compounds for a period of 1, 4 and 8 h by a method based on high resolution continuum source atomic absorption spectroscopy (HR-CS AAS).32,33 Results were corrected for the respective protein contents of the samples and are presented in Fig. 3 as nmol gold per milligram cellular protein. Besides, based on certain biophysical parameters of HT-29 cells,32 the nmol Au per mg protein values obtained were used to estimate the respective intracellular molar concentrations. | ||
| Fig. 3 Cellular gold content of HT-29 cells exposed to 10 μM of gold(I) complexes 2a–2e. | ||
As shown in Fig. 3, the highest values of intracellular accumulation were observed for triphenylphosphine gold(I) complexes 2a–c. The content of gold in HT-29 cells reached by these complexes after 1 h of exposure correlated to intracellular concentrations of 150 (2a), 265 (2b) and 210 (2c) μM, respectively. Taking into account the exposure concentration of 10.0 μM, one can conclude that for these complexes at least 15-fold accumulation can be achieved already within the first hour of the experiment. This is markedly higher than the 8-fold accumulation reported for the chlorido derivative 1a after 6 h of exposure.10 Drastically lower levels of cellular uptake were found for [tri(2-furyl)phosphane]gold(I) complexes 2d and 2e, which can be explained by their lower lipophilicity. Intracellular concentrations of 10 and 40 μM observed for 2d and 2e, respectively, after 1 h of exposure are 7–15 times lower in comparison with their triphenylphosphane-based analogs 2a and 2b.
Antiproliferative activity in a resistant cell line
Resistance to therapeutic agents is a major problem in current tumor chemotherapy. Hence, the development of drugs that can overcome drug resistance in tumor tissues is of major interest. Here we used wild type and vincristine resistant NALM-6 cells, which overexpress the drug efflux pump p-glycoprotein,33 to evaluate the possible overcoming of drug resistance by the gold complexes under study. Complexes 2a and 2d were selected for these experiments as examples with high (2a) and low (2d) cellular uptake but comparable toxicity and TrxR inhibitory potential (see above). Apoptosis induction was assessed as DNA fragmentation in the cells.In fact, both 2a and 2d triggered strong effects in wild type and vincristine resistant NALM-6 cells already in low micromolar concentrations (>50% DNA fragmentation at 1.0 μM with 2a and 2.5 μM with 2d), which clearly demonstrated that both complexes could overcome p-glycoprotein related multidrug resistance (see Fig. 4).
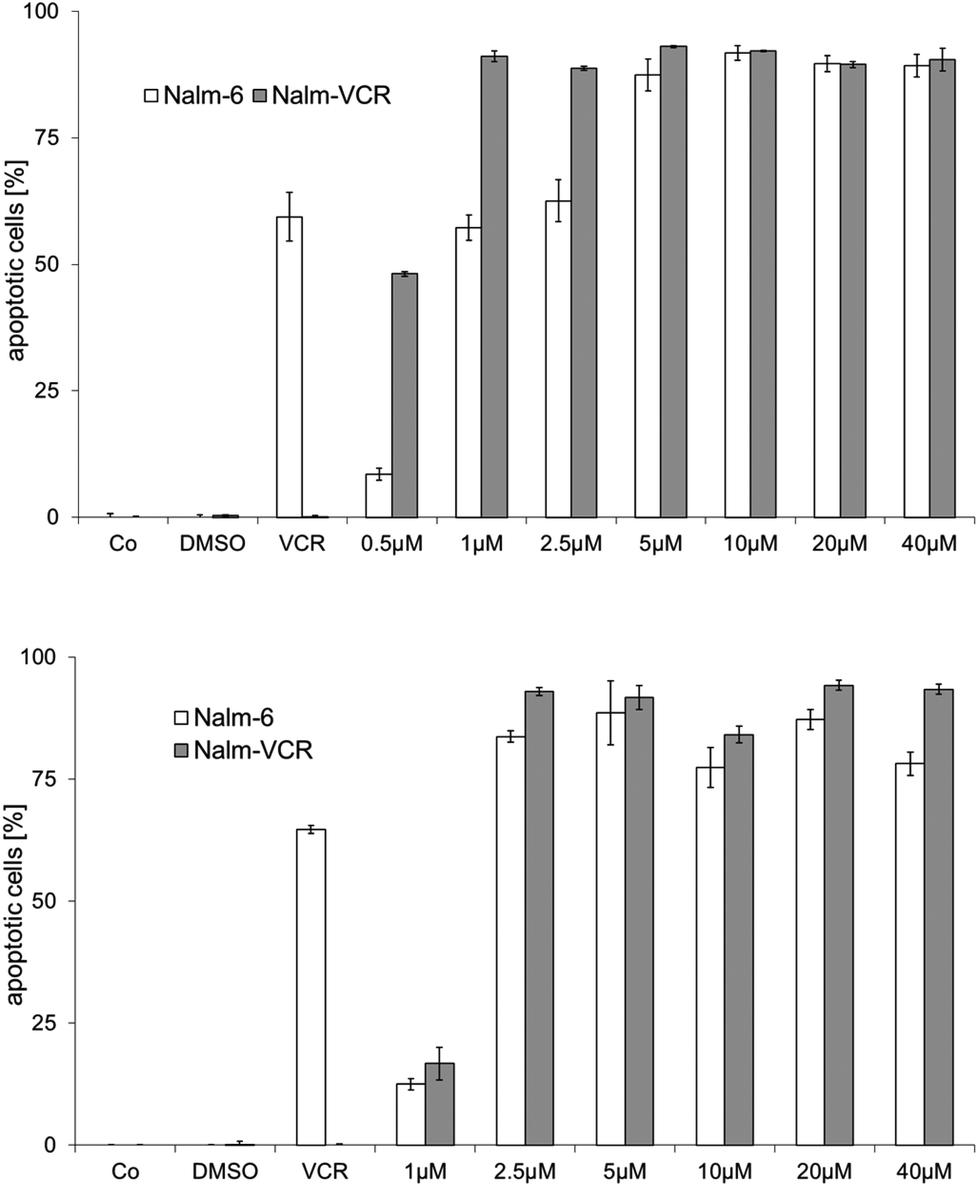 | ||
| Fig. 4 Effects of 2a (top) and 2d (bottom) on DNA fragmentation in wild type and vincristine resistant Nalm-6 cells after 72 h of exposure; Co: control; DMSO: cells treated with the same amount of DMSO; VCR: vincristine (20 nM). | ||
Conclusions
A series of different gold(I) phosphane complexes with thiotetrazolato ligands was prepared and biologically evaluated. The crystal structure of complex 2e showed the expected linear coordination geometry around the gold center. Interestingly, dimers were formed by intermolecular hydrogen bonding between the furane moieties.Complexes 2a–e turned out to be very effective inhibitors of the enzyme TrxR. Comparison with the respective chloride analogues 1a and 1b indicated that the thiotetrazolato ligand was important for this effect. The lowest activity in the series 2a–e was observed for 2c, which contains an aliphatic side chain with a tertiary nitrogen at the thiotetrazolato ligand. Notably, strong TrxR inhibition with thiophenolate containing gold(I) carbene complexes was also observed in a previous study.34 However, the enhanced activity against TrxR did not translate into elevated cytotoxicity since complexes 2a–e were of comparable activity with 1a and 1b in cell proliferation assays.
Cellular uptake studies showed a very high accumulation of the triphenylphosphane containing complexes 2a–c, whereas the triphenylfurane analogues 2d and 2e were found at significantly lower levels in the cells. It can be speculated that possible dimer formation by hydrogen bonding of the trifurylphosphane moiety might contribute to this effect besides differences in the lipophilicity. It should be mentioned that we observed higher cellular accumulation of triphenylphosphane gold complexes compared to trialkylphosphane derivatives in a number of recent studies10,11,35 and that lipophilicity is generally considered to be an important parameter for cellular uptake of gold phosphane species.36
Importantly, selected compounds 2a and 2d showed decent activity in p-glycoprotein overexpressing vincristine-resistant Nalm-6 cells, which indicates that this type of gold compounds might be useful for the design of resistance overcoming drugs.
In summary, gold complexes with thiotetrazolate represent a new promising class of gold species, which trigger important biological effects such as TrxR inhibition and cytotoxicity.
Experimental
Materials and methods
All reagents and solvents were used as received from Sigma, Aldrich, or Acros Organics. 1H and 13C NMR spectra were recorded on a Bruker DRX-400 AS NMR system. ESI(+) mass spectra were recorded on a Bruker micrOTOF instrument using dichloromethane as a solvent. The purity of the target compounds (>95%) was confirmed by elemental analysis. For all compounds undergoing biological evaluation, the experimental values differed less than 0.5% from the calculated ones.IR spectra of powder samples were registered on a Nicolet Thermo Avatar 330 spectrometer using a Smart Diffuse Reflection accessory in the range of 4000–400 cm−1. The TG and DSC curves were obtained with a NETZSCH STA 429 thermoanalyzer under a dynamic nitrogen atmosphere (heating rate of 10 K min−1, aluminum oxide, mass 5–6 mg and temperature range from room temperature to 600 °C).
Synthesis of gold(I) complexes 2a–e
Compounds 2a–c were obtained using the following general procedure. 0.5 mmol (248 mg) of chlorido(triphenylphosphane)gold(I) and 0.5 mmol of the corresponding 1-R′-5-thiotetrazole 3a–c were dissolved in 20 ml of CH2Cl2. Triethylamine (0.1 mL) was added dropwise to the prepared solution, and the mixture was stirred overnight at room temperature. Then the mixture was evaporated to dryness, and the residue was dissolved in hot THF. After cooling to room temperature, needle-like crystals of triethylammonium chloride formed and were separated by filtration. The filtrate was evaporated again, and the solid residue was recrystallized from CH2Cl2–hexane (2a and 2b) or acetonitrile (2c) to obtain the colorless product.Ntz) + ν(N–Ntz); 1101 (s), 1077 (m), 1026 (m), 996 (m), 969 (m), δ(CN4) + δ(C6H5); 749 (vs), ν(C–P); 693 (vs), ν(C–S).
Ntz) + ν(N–Ntz); 1103 (vs), 1079 (m), 1028 (m), 998 (m), 980 (w), δ(CN4) + δ(C6H5); 758 (vs), 748 (vs), ν(C–P); 713 (s), 696 (vs) ν(C–S).
Ntz) + ν(N–Ntz); 1100 (s), 1066 (m), 1023 (m), 997 (m), 982 (m), 920 (m), δ(CN4) + δ(C6H5); 777 (m), ν(C–N); 750 (vs), ν(C–P); 710 (s), 690 (vs) ν(C–S).
Complexes 2d and 2e were obtained using the same procedure starting from chlorido[tri(2-furyl)phosphane]gold(I) and the corresponding 1-R-5-thiotetrazole (R = Me (3a), Ph (3b)).
Ntz) + ν(N–Ntz); 1170 (m), ν(C–O); 1130 (m), 1110 (s), 910 (m), 881 (m), δ(CN4) + δ(furyl); 760 (s), ν(C–P); 705 (m) ν(C–S).
Crystallographic studies
Single crystal X-ray data of complex 2e were collected at room temperature on a SMART APEX II diffractometer using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). The reflection data were corrected on absorption. The structures were solved by direct methods with the program SIR200437 and refined on F2 by the full-matrix least squares technique with SHELXL-2013.38 All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were placed at calculated positions and refined using a “riding” model, with Uiso(H) = 1.2Ueq(C). Molecular graphics was performed with the program PLATON.39 Crystallographic data of complex 2e (excluding structure factors) have been deposited with the Cambridge Crystallographic Data Centre.TrxR/GR inhibition assay
To determine the inhibition of TrxR and GR an established microplate reader based assay was performed with minor modifications.12 For this purpose commercially available rat liver TrxR and baker yeast GR (both from Sigma-Aldrich) were used and diluted with distilled water to achieve concentrations of 0.4 U mL−1 (TrxR) and 20.0 U mL−1 (GR). The compounds were freshly dissolved as stock solutions in DMF. To each 25 μL aliquots of the enzyme solution, each 25 μL of potassium phosphate buffer pH 7.0 containing the compounds in graded concentrations or the vehicle (DMF) without compounds (control probe) were added and the resulting solutions (final concentration of DMF: max. 0.5% v/v) were incubated with moderate shaking for 75 min at 37 °C in a 96 well plate. To each well, 225 μL of the reaction mixture (1000 μL of the reaction mixture consisted of 500 μL potassium phosphate buffer pH 7.0, 80 μL of 100 mM EDTA solution pH 7.5, 20 μL bovine serum albumin solution 0.05%, 100 μL of 20 mM NADPH solution and 300 μL of distilled water) were added and the reaction was started by addition of 25 μL of an 20 mM ethanolic dithio-bis-2-nitrobenzoic acid (DTNB) solution. After proper mixing, the formation of 5-thio-2-nitrobenzoic acid (5-TNB) was monitored with a microplate reader (Perkin Elmer Victor X4) at 405 nm in 35 s intervals for 350 s. For each tested compound the non-interference with the assay components was confirmed by a negative control experiment using an enzyme free solution. The IC50 values were calculated as the concentration of the compound decreasing the enzymatic activity of the untreated control by 50% and are given as the means and error of 2–3 independent experiments.Cell culture and cytotoxicity assay in MDA-MB-231 and HT-29 cells
The antiproliferative effects in MDA-MB-231 and HT-29 cells after 72 h (HT-29) or 96 h (MDA-MB-231) exposure to the gold complexes were evaluated using the crystal violet assay.In short, HT-29 human colon carcinoma cells and MDA-MB-231 breast cancer cells were maintained in cell culture medium (minimum essential eagle medium supplemented with 2.2 g NaHCO3, 110 mg L−1 sodium pyruvate and 50 mg L−1 gentamicin sulfate adjusted to pH 7.4) containing 10% (v/v) fetal calf serum at 37 °C/5% CO2 and passaged once a week according to standard procedures. For the experiments, the compounds were prepared freshly as stock solutions in DMF and diluted with the cell culture medium to the final assay concentrations (0.1% v/v DMF). The assay procedure is described in more detail in our recent papers.10,40 The IC50 value was described as that concentration reducing the proliferation of untreated control cells by 50%.
Cellular uptake studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) for 15 min, pelleted, and resuspended in PBS containing 40 μg per mL of RNase A. After incubation for 30 min at 37 °C, cells were pelleted again and finally resuspended in PBS containing 50 μg per mL of propidium iodide. Nuclear DNA fragmentation was then quantified by flow cytometric determination of hypodiploid DNA. Data were collected and analyzed using an FACScan (Becton Dickinson, Heidelberg, Germany) equipped with the CELLQuest software. Data are given in % hypoploidy (subG1), which reflects the number of apoptotic cells.
:1, v/v) for 15 min, pelleted, and resuspended in PBS containing 40 μg per mL of RNase A. After incubation for 30 min at 37 °C, cells were pelleted again and finally resuspended in PBS containing 50 μg per mL of propidium iodide. Nuclear DNA fragmentation was then quantified by flow cytometric determination of hypodiploid DNA. Data were collected and analyzed using an FACScan (Becton Dickinson, Heidelberg, Germany) equipped with the CELLQuest software. Data are given in % hypoploidy (subG1), which reflects the number of apoptotic cells.
Acknowledgements
The postdoctoral fellowships for T.V.S. by German Academic Exchange Service (DAAD) and Saint Petersburg State University (12.50.1560.2013) as well as the financial support by Dr Kleist-Foundation (Berlin) are gratefully acknowledged. Mass-spectrometry was performed at the Center for Chemical Analysis and Material Research of St Petersburg State University.References
- R. Rubbiani, B. Wahrig and I. Ott, J. Biol. Inorg. Chem., 2014, 19, 961–965 CrossRef CAS PubMed.
- C. F. Shaw, Chem. Rev., 1999, 99, 2589–2600 CrossRef CAS PubMed.
- I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS PubMed.
- B. Bertrand and A. Casini, Dalton Trans., 2014, 43, 4209–4219 RSC.
- E. E. Langdon-Jones and S. J. Pope, Chem. Commun., 2014, 50, 10343–10354 RSC.
- A. Bindoli, M. P. Rigobello, G. Scutari, C. Gabbiani, A. Casini and L. Messori, Coord. Chem. Rev., 2009, 253, 1692–1707 CrossRef CAS PubMed.
- S. Gromer, L. D. Arscott, C. H. Williams, R. H. Schirmer and K. Becker, J. Biol. Chem., 1998, 273, 20096–20101 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, S. S.-Y. Chui and C.-M. Che, Angew. Chem., Int. Ed., 2013, 52, 1–5 CrossRef.
- O. Rackham, S. J. Nichols, P. J. Leedman, S. J. Berners-Price and A. Filipovska, Biochem. Pharmacol., 2007, 74, 992–1002 CrossRef CAS PubMed.
- H. Scheffler, Y. You and I. Ott, Polyhedron, 2010, 29, 66–69 CrossRef CAS PubMed.
- C. P. Bagowski, Y. You, H. Scheffler, D. H. Vlecken, D. J. Schmitz and I. Ott, Dalton Trans., 2009, 10799–10805 RSC.
- I. Ott, X. Qian, Y. Xu, D. H. W. Vlecken, I. J. Marques, D. Kubutat, J. Will, W. S. Sheldrick, P. Jesse, A. Prokop and C. P. Bagowski, J. Med. Chem., 2009, 52, 763–770 CrossRef CAS PubMed.
- Y. Wang, M. Liu, R. Cao, W. Zhang, M. Yin, X. Xiao, Q. Liu and N. Huang, J. Med. Chem., 2013, 56, 1455–1466 CrossRef CAS PubMed.
- T. Zou, C. T. Lum, C.-N. Lok, W.-P. To, K.-H. Low and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 5810–5814 CrossRef CAS PubMed.
- C. I. Yeo, K. K. Ooi, A. M. Akim, K. P. Ang, Z. A. Fairuz, S. N. Halim, S. W. Ng, H. L. Seng and E. R. Tiekink, J. Inorg. Biochem., 2013, 127, 24–38 CrossRef CAS PubMed.
- W. Fiskus, N. Saba, M. Shen, M. Ghias, J. Liu, S. D. Gupta, L. Chauhan, R. Rao, S. Gunewardena, K. Schorno, C. P. Austin, K. Maddocks, J. Byrd, A. Melnick, P. Huang, A. Wiestner and K. N. Bhalla, Cancer Res., 2014, 74, 2520–2532 CrossRef CAS PubMed.
- V. A. Ostrovskii, G. I. Koldobskii and R. E. Trifonov, Comprehensive Heterocyclic Chemistry III, Elsevier Ltd., 2008, vol. 15, pp. 257–423 Search PubMed.
- M. Uemura, T. Suzuki, K. Nishio, M. Chikuma and S. Komeda, Metallomics, 2012, 4, 686–692 RSC.
- S. Komeda, H. Takayama, T. Suzuki, A. Odani, T. Yamori and M. Chikuma, Metallomics, 2013, 5, 461–468 RSC.
- T. V. Serebryanskaya, T. Yung, A. A. Bogdanov, A. Shchebet, S. A. Johnsen, A. S. Lyakhov, L. S. Ivashkevich, Z. A. Ibrahimava, T. S. Garbuzenco, T. S. Kolesnikova, N. I. Melnova, P. N. Gaponik and O. A. Ivashkevich, J. Inorg. Biochem., 2013, 120, 44–53 CrossRef CAS PubMed.
- A. Ilie and K. Karaghiosoff, Phosphorus, Sulfur Silicon Relat. Elem., 2011, 186, 389–403 CrossRef CAS.
- W. Beck, K. Burger and M. Keubler, Z. Anorg. Allg. Chem., 1977, 428, 173–186 CrossRef CAS.
- A. Ilie, C. I. Rat, S. Scheutzow, C. Kiske, K. Lux, T. M. Klapotke, C. Silvestru and K. Karaghiosoff, Inorg. Chem., 2011, 50, 2675–2684 CrossRef CAS PubMed.
- H. Nöth, W. Beck and K. Burger, Eur. J. Inorg. Chem., 1998, 93–99 CrossRef.
- U. Monkowius, S. Nogai and H. Schmidbaur, Z. Naturforsch., B: Chem. Sci., 2003, 58, 751–758 CAS.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646–8657 CrossRef CAS PubMed.
- A.-B. Witte, K. Anestal, E. Jerremalm, H. Ehrsson and E. S. J. Arner, Free Radicals Biol. Med., 2005, 39, 696–703 CrossRef CAS PubMed.
- E. S. Arner and A. Holmgren, Eur. J. Biochem., 2000, 267, 6102–6109 CrossRef CAS.
- S. Schäfer, I. Ott, R. Gust and W. S. Sheldrick, Eur. J. Inorg. Chem., 2007, 3034–3046 CrossRef.
- I. Ott, K. Schmidt, B. Kircher, P. Schumacher, T. Wiglenda and R. Gust, J. Med. Chem., 2005, 48, 622–629 CrossRef CAS PubMed.
- I. Ott, H. Scheffler and R. Gust, ChemMedChem, 2007, 2, 702–707 CrossRef CAS PubMed.
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608–8618 CrossRef CAS PubMed.
- R. Rubbiani, E. Schuh, A. Meyer, J. Lemke, J. Wimberg, N. Metzler-Nolte, F. Meyer, F. Mohr and I. Ott, Med. Chem. Commun., 2013, 4, 942–948 RSC.
- R. Rubbiani, L. Salassa, A. de Almeida, A. Casini and I. Ott, ChemMedChem, 2014, 9, 1205–1210 CrossRef CAS PubMed.
- M. J. McKeage, S. J. Berners-Price, P. Galettis, R. J. Bowen, W. Brouwer, L. Ding, L. Zhuang and B. C. Baguley, Cancer Chemother. Pharmacol., 2000, 46, 343–350 CrossRef CAS PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. D. Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr. Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- K. Navakoski de Oliveira, V. Andermark, S. von Grafenstein, L. A. Onambele, G. Dahl, R. Rubbiani, G. Wolber, C. Gabbiani, L. Messori, A. Prokop and I. Ott, ChemMedChem, 2013, 8, 256–264 CrossRef CAS PubMed.
Footnote |
| † CCDC 1027451. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03105a |
| This journal is © The Royal Society of Chemistry 2015 |