
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coordination geometry of lead carboxylates – spectroscopic and crystallographic evidence†
Jaclyn
Catalano
ab,
Anna
Murphy
b,
Yao
Yao
b,
Glenn P. A.
Yap
b,
Nicholas
Zumbulyadis
c,
Silvia A.
Centeno
a and
Cecil
Dybowski
*b
aDepartment of Scientific Research, The Metropolitan Museum of Art, New York, NY 10028, USA
bDepartment of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA. E-mail: dybowski@udel.edu
cIndependent Researcher, Rochester, NY 14613, USA
First published on 15th December 2014
Abstract
Despite their versatility, only a few single-crystal X-ray structures of lead carboxylates exist, due to difficulties with solubility. In particular, the structures of long-chain metal carboxylates have not been reported. The lone electron pair in Pb(II) can be stereochemically active or inactive, leading to two types of coordination geometries commonly referred to as hemidirected and holodirected structures, respectively. We report 13C and 207Pb solid-state NMR and infrared spectra for a series of lead carboxylates, ranging from lead hexanoate (C6) to lead hexadecanoate (C18). The lead carboxylates based on consistent NMR parameters can be divided in two groups, shorter-chain (C6, C7, and C8) and longer-chain (C9, C10, C11, C12, C14, C16, and C18) carboxylates. This dichotomy suggests two modes of packing in these solids, one for the short-chain lead carboxylates and one for long-chain lead carboxylates. The consistency of the 13C and 207Pb NMR parameters, as well as the IR data, in each group suggests that each motif represents a structure characteristic of each subgroup. We also report the single-crystal X-ray diffraction structure of lead nonanoate (C9), the first single-crystal structure to have been reported for the longer-chain subgroup. Taken together the evidence suggests that the coordination geometry of C6–C8 lead carboxylates is hemidirected, and that of C9–C14, C16 and C18 lead carboxylates is holodirected.
Introduction
The long-chain carboxylates of divalent metal ions are of considerable technical and scientific interest. They have been used as secondary driers in linseed-oil- and alkyd-resin-based paints,1 as corrosion inhibitors for passivating lead metal surfaces,2 emulsifiers, grease thickeners, and dispersant agents.3,4 Lead carboxylates have also been used in metal organic frameworks based on aromatic5 and aliphatic6 lead(II) polycarboxylates because of their gas sorption properties, in the synthesis of monodispersed superconducting Pb(0) nanocrystals,7 and in octahedral clusters of PbSe nanocrystals for high-efficiency solar cells.8Lead carboxylates are a major concern in the conservation and preservation of oil paintings.9,10 Lead hexadecanoate, lead octadecanoate and, in some cases, lead azelate have been found in inclusions by a variety of techniques.11,12 Soaps (carboxylates of metal ions) form when saturated fatty acids, resulting from the hydrolysis of glycerides in the oil binder, react with metals to form inclusions that grow over time, compromising the condition of the artwork. Soaps may also remain evenly distributed in the paint layers, modifying their optical properties and leading to an increased transparency.10 Soaps formed in the paint layers may migrate to the surface of a painting where they react with gases in the environment, resulting in carbonates, sulfates, possibly oxalates, and other more stable carboxylates in the form of highly disruptive whitish crusts.13 Knowledge of the coordination environment and the crystal packing of these lead soaps may provide insights into the migration of these soaps and how to arrest these processes.
There has recently been a resurgence of interest in the coordination chemistry of divalent lead, motivated by the toxicity of lead and its widespread occurrence in the environment. A knowledge of the coordination properties leading to preferential binding of lead over other essential metal ions is crucial for understanding the toxicological properties of lead, the design of selective chelation-therapy agents,14 and the development of efficient chelating agents for the remediation of polluted water and soil.15,16 Considerable effort has been devoted to controlling the stereochemistry around Pb(II) synthetically.16–19 Surprisingly, there is a paucity of single-crystal X-ray diffraction data for Pb(II) carboxylates. Only a handful of structures have been reported, notably lead formate,20 lead acetate,20 lead crotonate,21 lead heptanoate,2,22 and lead azelate.23 The structures of aliphatic lead dicarboxylate metal–organic frameworks have also been reported.6
The ligand coordination number and geometry around Pb(II) are quite diverse, with at least five different coordination polyhedra reported in the literature where the coordination numbers vary anywhere between 2 and 10.24 The electron configuration of Pb(II) entails a 6s2 lone pair. The diversity of coordination geometries depends on whether the lone pair is stereochemically active or inactive. Shimoni-Livny et al. have identified two limiting cases for the diverse geometries.14 Using their terminology the two cases are named “holodirected, in which the bonds to ligand atoms are directed throughout the surface of an encompassing globe” and “hemidirected, in which the bonds to ligand atoms are directed throughout only part of the globe, that is, there is an identifiable void (or gap) in the distribution of bonds to the ligands”.14 Pb(II) compounds are hemidirected for low coordination numbers (2–5) and holodirected for high coordination numbers (9 and 10), but for intermediate coordination numbers (6–8), examples of both types of stereochemistry have been reported.6,20,25
Ab initio molecular orbital calculations and a survey of published structures have identified the primary factors determining coordination geometry.14,26 Hemidirected geometry is favored if the ligand coordination number is low, the ligands are hard, and there are attractive interactions between the ligands. In such complexes, the lone-pair orbital has p character and fewer electrons are transferred from the ligands to the bonding orbitals of Pb(II), resulting in bonds that are more strongly ionic. Holodirected geometry is favored when the coordination number is high and the ligands are soft and bulky, or when there are strong interligand repulsions. In holodirected structures, the lone pair has essentially no p character, more ligand electrons are transferred to Pb(II), and the bonds are more covalent. The energy cost of converting a hemidirected to a holodirected structure is on the order 8–12 kcal mol−1 in the absence of strong interligand interactions.
For most lead salts of saturated monocarboxylic acids, particularly those of longer chain length, information about coordination geometry is accessible only by ssNMR because of their insolubility and the difficulties encountered in attempting to grow sufficiently large single crystals for X-ray diffraction studies. Several solution- and solid-state NMR (ssNMR) studies have established correlations between the geometry around the lead coordination site and the observed values of 207Pb isotropic chemical shifts or principal components of 207Pb chemical-shift tensors in lead compounds and coordination polymers. Harrison et al. reported solution-state 207Pb chemical shifts for several Pb(II) complexes and their dissociation products in various solvents.27 They observed that species in which the lone pair is stereochemically inactive are characterized by strongly isotropically shielded 207Pb resonances. When the lone pair is stereochemically active, the chemical shifts are comparatively isotropically deshielded.27,28 Reger et al. observed a similar trend in a series of lead complexes with various poly(pyrazolyl) borate ligands.18
In this paper, we report the 207Pb and 13C ssNMR characterization of lead carboxylates of chain length C6, C7, C8, C9, C10, C11, C16 and C18. A clear division between two subgroups is observed from the NMR parameters based on carboxylate chain length and is supported by FTIR spectra. We have also determined the structure of lead nonanoate (C9) by X-ray diffraction, which demonstrates the relevance of the NMR properties to the local electronic structure.
Experimental section
Synthesis of materials
Lead carboxylates of hexanoic (caproic) (C6), heptanoic (enanthic) (C7), octanoic (caprylic) (C8), nonanoic (pelargonic) (C9), decanoic (capric) (C10), undecanoic (undecylic) (C11), hexadecanoic (palmitic) (C16), and octadecanoic (stearic) (C18) acids were synthesized by methods adapted from previously published protocols.29,30 Specifically, the lead carboxylates were synthesized from equimolar amounts (3.25 mmoles) of lead nitrate dissolved in 20 mL of water and the respective acid dissolved in 50 mL of ethanol, except that hexanoic acid was dissolved in 20 mL of ethanol. The two solutions were mixed with an equimolar amount of aqueous potassium hydroxide (5 M solution) and reacted for 20 minutes at 80 °C. The reaction mixture was cooled to room temperature, filtered, and the residue was washed with several portions of water, methanol, ethanol, and acetone. The resulting material was dried and recrystallized from ethanol.Analysis of materials
The purity of each compound was verified by infrared spectroscopy (FTIR) and powder X-ray diffraction (PXRD) analysis. The FTIR measurements were carried out with a Bruker Vertex70® spectrometer coupled to a Hyperion® microscope. The samples were mounted between the windows of a diamond anvil cell and observed in the transmission mode, with a spectral resolution of 4 cm−1. The PXRD measurements were performed using a Philips PW1835 diffractometer with Cu Kα (λ = 1.542 Å) radiation (45 kV acceleration voltage, and 35 mA current).NMR spectroscopy
13C and 207Pb ssNMR spectra of the lead carboxylates were recorded at 11.75 tesla (500.13 MHz proton frequency; 125.76 MHz carbon-13 frequency; 104.63 MHz lead-207 frequency) with a standard Bruker 4 mm probe. The protocols have been previously published.30 The spectra were obtained from approximately 100 mg of sample packed in a 4 mm rotor. Solid lead nitrate was used as a secondary external reference for the 207Pb spectra, the isotropic chemical shift being −3491 ppm relative to tetramethyllead (TML) at 298 K.31 A plot of spinning speed versus isotropic chemical shift of lead nitrate was obtained to compensate for the temperature increase due to spinning. Glycine was used as a secondary external reference for the 13C spectra, with an isotropic chemical shift of the carboxyl peak of 176 ppm relative to tetramethylsilane.13C NMR spectra were acquired using cross polarization and magic-angle spinning at 12 kHz (CPMAS). Acquisition parameters for these experiments included a contact time of 1 ms, 13C pulse width of 4 μs, and a recycle delay of 5 s. During acquisition, proton decoupling (SPINAL-64 at 100 kHz) was used to suppress dipolar couplings. The number of scans per spectrum was 1024.
207Pb spectra of the carboxylates were acquired by direct excitation using spin-temperature alteration with magic angle spinning (STA/MAS) (νrot = 11 or 12 kHz) to minimize the effects of ringdown of the probe circuits.32 General conditions for these experiments included a π pulse width of 8.5 μs, a delay of 1 ms, and a π/2 pulse width of 4.25 μs. High-power proton decoupling (SPINAL-64 at 100 kHz) was used to suppress dipolar couplings. The recycle delay in most experiments was 5 s and approximately 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans per spectrum were acquired.
000 scans per spectrum were acquired.
207Pb WURST-CPMG spectra of static samples were recorded using the parameters of MacGregor et al.33 WURST pulse widths were 50 μs, with pulse shapes created via the shape tool in Topspin 3.1.30 75 Meiboom–Gill loops were acquired for the WURST-CPMG experiments, with a 200-μs echo, and a sweep range of 0.5 MHz in all cases. The recycle delay was 7 s. High-power proton decoupling (CW at 100 kHz) was used to suppress dipolar couplings. For lead hexanoate, lead heptanoate and lead octanoate, multiple WURST-CPMG subspectra were collected at different carrier frequencies by shifting the carrier frequency a multiple of the spikelet separation (981.934 ppm) from spectrum to spectrum. Each spectrum contained the signal accumulated in 2048 scans. The collected subspectra were superimposed to form the final spectrum.
The analysis of the 207Pb chemical-shift tensors was performed by simultaneously fitting the envelope of the WURST-CPMG spectrum and the MAS sideband pattern. The isotropic chemical shift acquired in the MAS experiment was fixed in the fitting procedure, because it could be determined accurately. For compounds with smaller spans, an initial estimate of the principal components of the chemical-shift tensor was made with the Herzfeld–Berger method,34 as implemented in the program HBA 3.1 developed by Eichele.35 The final fit was refined by performing a simultaneous fit to the sideband patterns and the WURST-CPMG envelope. Fits to the WURST-CPMG envelopes were aided by simulation of the powder pattern with the program WSOLIDS.35
Single-crystal X-ray diffraction
Crystals, obtained by slow evaporation of a saturated solution prepared from 38 mg of lead nonanoate (C9) in 30 mL of toluene, were sectioned, mounted using viscous oil onto a plastic mesh, and cooled to the data collection temperature. Data were collected on a Bruker-AXS APEX CCD diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å and Cu-Kα (λ = 1.54178 Å) radiation. Unit cell parameters were obtained from 36 data frames, 0.3° ω, from three different sections of the Ewald sphere. The systematic absences in the diffraction data are consistent with P2/c (no. 13) and Pc (no. 7). Detailed information on data collection, fitting, and structure validation using the checkCIF report36 is included in the ESI.† Scattering factors are contained in the SHELXTL 6.12 program library.37 The CIF has been deposited under CCDC 1023395.Results and discussion
NMR spectroscopy
Carbon ssNMR provides information on the crystal packing of the lead carboxylates. Carbon ssNMR spectra (Fig. 1) show resonance doubling of the carbons closest to the lead site for all lead carboxylates studied, indicating two carboxylate conformations in the asymmetric unit. This result is in agreement with previous NMR reports for lead decanoate and octadecanoate,38 electron diffraction measurements of Langmuir-Blodgett films of lead octadecanoate,39 and the published crystal structure of lead heptanoate,22 as well as the crystal structure of lead nonanoate discussed below. There are two resonances in the carboxylate region of the spectrum with relative intensities of 1:1 for all the carboxylates. However, the series parses into two subseries based on the chemical-shift difference between these two resonances. For the long-chain carboxylates (C9, C10, C11, C16, and C18), the difference is in the range of 1.13–1.25 ppm. For the shorter-chain carboxylates (C6, C7, and C8), the chemical-shift difference of the two carboxylate forms is in the range of 0.50–0.69 ppm. A similar trend is seen for the α-carbon, with chemical-shift differences of 2.0–2.1 ppm for long-chain carboxylates, and 0.50–0.69 ppm for short-chain carboxylates.
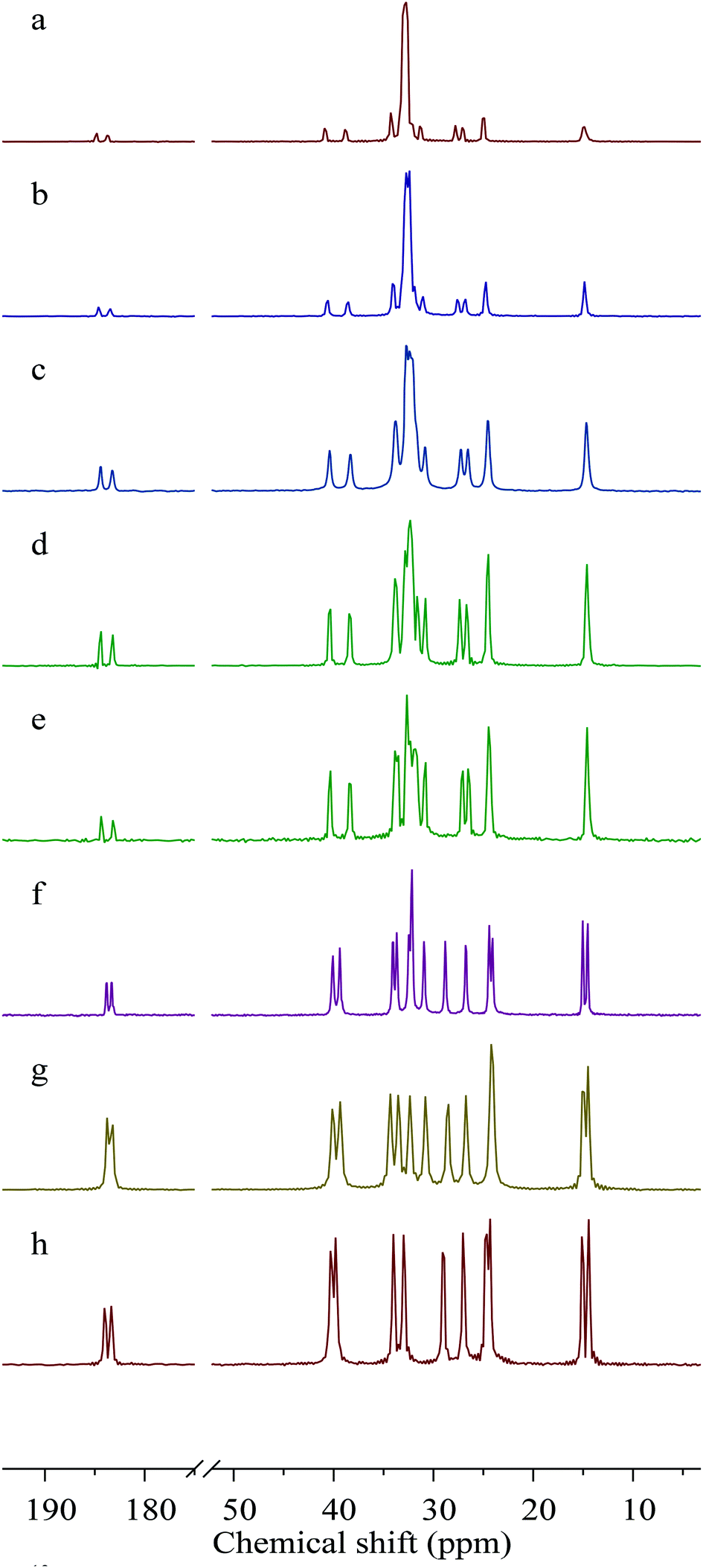 | ||
| Fig. 1 13C NMR spectra of lead carboxylates obtained with cross polarization and proton decoupling spinning at 12 kHz. (a) Lead octadecanoate, (b) lead hexadecanoate, (c) lead undecanoate, (d) lead decanoate, (e) lead nonanoate, (f) lead octanoate, (g) lead heptanoate, and (h) lead hexanoate. For carbons closest to the lead, the resonances are doubled, indicating two different carboxylate chain conformations in the unit cell. | ||
Carbon atoms further from the carboxylate center also show doubling of resonances but, as proposed by Burrows et al.,28 the doubling in these regions is not necessarily indicative of multiple carboxylate coordination geometries but rather it results from a distortion of the usual all-trans arrangement of such chains. Especially noteworthy is the observation that the methyl resonances of the long-chain carboxylates are singlets, whereas the short-chain carboxylates show doubling (1:1) of the methyl resonance, a fact demonstrating that there are two chain-end conformations for these short-chain members of the series.
Lead WURST-CPMG and STA/MAS (not shown) spectra demonstrate that the local lead environments in the long-chain lead carboxylates are different from those of the short-chain carboxylates. Fig. 2 shows the 207Pb WURDT-CPMG spectra of the C6–C11, C16 and C18 lead carboxylates, and the principal components of the chemical-shift tensors for the various carboxylates are reported in Table 1. The 207Pb chemical-shift tensors for lead hexadecanoate and lead octadecanoate in Table 1 have been previously reported and included for comparison.30 In every case, the 207Pb spectrum is fit as a single pattern implying the presence of a single lead center per unit cell. Due to the similarity between C11, C16, and C18, it is expected that similar spectra would be obtained for lead carboxylates of chain length C12–C15 and C17.
 | ||
| Fig. 2 207Pb NMR spectra of lead carboxylates obtained with WURST-CPMG. (a) Lead octadecanoate, (b) lead hexadecanoate, (c) lead undecanoate, (d) lead decanoate, (e) lead nonanoate, (f) lead octanoate, (g) lead heptanoate, and (h) lead hexanoate. The spectra show that the lead carboxylates can be divided into two subgroups, shorter chains (C6–C8) having a larger span than the long-chain carboxylates (C9–C11, C16, and C18). | ||
| Compound | δ 11 (ppm) | δ 22 (ppm) | δ 33 (ppm) | δ iso (ppm) | Ω (ppm) | κ |
|---|---|---|---|---|---|---|
| a The fitting procedure used the isotropic shift, as determined from a MAS spectrum of the material, with simulation of the values of δ11, δ22, and δ33 determined by the edges of the WURST-CPMG spectrum. Span (Ω) and skew (κ) were calculated. Ω = |δ33 − δ11| and κ = 3 × (δiso − δ22)/Ω. | ||||||
| Lead hexanoate | −100 ± 3 | −533 ± 8 | −2745 ± 5 | −1126 ± 3 | 2645 ± 6 | 0.67 ± 0.01 |
| Lead heptanoate | −40 ± 8 | −480 ± 10 | −2750 ± 5 | −1090 ± 3 | 2710 ± 9 | 0.68 ± 0.01 |
| Lead octanoate | 0 ± 4 | −469 ± 8 | −2740 ± 5 | −1070 ± 3 | 2740 ± 6 | 0.66 ± 0.01 |
| Lead nonanoate | −1819 ± 3 | −2035 ± 5 | −2565 ± 7 | −2140 ± 3 | 746 ± 8 | 0.42 ± 0.02 |
| Lead decanoate | −1836 ± 5 | −2047 ± 6 | −2575 ± 8 | −2153 ± 4 | 739 ± 9 | 0.43 ± 0.03 |
| Lead undecanoate | −1818 ± 3 | −2036 ± 5 | −2570 ± 7 | −2141 ± 3 | 752 ± 8 | 0.42 ± 0.02 |
| Lead hexadecanoate30 | −1820 ± 3 | −2013 ± 5 | −2560 ± 3 | −2131 ± 3 | 740 ± 4 | 0.48 ± 0.02 |
| Lead octadecanoate30 | −1810 ± 3 | −2007 ± 6 | −2555 ± 3 | −2124 ± 4 | 745 ± 4 | 0.47 ± 0.03 |
Based on the 207Pb parameters, the lead carboxylates fall into the same two subgroups as determined by the 13C NMR parameters. The C6, C7, and C8 carboxylates have spans of approximately 2700 ppm, which suggests that they are hemidirected. The longer-chain carboxylates have much smaller spans (on the order of 750 ppm), which suggests that these carboxylates are holodirected. The spans are correlated with the isotropic shifts, the hemidirected carboxylates having isotropic shifts around −1100 ppm and the holodirected carboxylates having isotropic shifts around −2125 ppm. In addition, the hemidirected carboxylates show skews in the range of 0.65–0.70, whereas the holodirected carboxylates have skews in the range of 0.42–0.48. Katz et al. found a similar dependence of the lead isotropic shifts, spans, and skews in two holodirected birefringent lead–gold cyanide complexes on the stereochemical activity.40,41 In another study, Briand et al. demonstrated that the very large spans and large skews of 207Pb chemical-shift tensors in 4-substituted pyridine adducts of lead thiolates are attributable to the presence of the stereochemically active lone pair.42 Earlier work has shown that the holodirected geometry around nine-coordinate Pb in PbCO343 results in a much narrower span for the 207Pb chemical-shift tensor.32 Similar studies of the thiourea and phenanthroline complexes of the lead halides show spans of the order of 600–700 ppm, suggesting a holodirected geometry in those cases.44,45
The difference between the 207Pb NMR properties of the two types of lead carboxylate structures (Table 1), whether in the span or the isotropic shift, is primarily the result of the values of differences in δ11 and δ22, the two least shielded components. The values of δ33, the most shielded component, are similar for all of the lead carboxylates. Both δ11 and δ22 are more shielded by approximately 1500 ppm in the longer-chain carboxylates than in the shorter-chain carboxylates. This difference in these two components is an indication of a difference in lead coordination geometry that is also reflected by the NMR properties of the carboxyl carbons. A similar change in coordination geometry around the metal ion when comparing the octanoate and nonanoate ligands is also encountered when comparing the Raman spectra of copper octanoate and copper decanoate.46,47
The precise point in the series at which a transition from short-chain to long-chain behavior is manifested depends on the manner in which one examines the material. The present ssNMR experiments are consistent with a change in conformational behavior or packing between C8 and C9. On the other hand, from infrared and X-ray studies at room temperature, Ellis et al. conclude that this transition occurs between C12 and C14.48 Based on the number of intermediate liquid crystalline phases detected by differential thermal analysis (DTA), Adeosun and Sime place the transition between C8 and C12, with C10 exhibiting an intermediate behavior.49 In these experiments,48,49 only the members of the series having even numbers of carbons were investigated. It may be that, as suggested by Arenas et al., the packing of the chains of lead carboxylates of C9–C12 is affected by the thermal history of the sample.50 This is not the case for lead carboxylates of chain length greater than 13 or less than 8, where no differences between the Raman spectra of the recrystallized and premelted samples are obtained.50
FTIR spectroscopy
Fig. 3 shows FTIR spectra of the lead carboxylates taken in our laboratory. The interpretation of the IR results is consistent with the NMR spectra showing that the lead carboxylates are divided into two subgroups. The absorption assigned to the antisymmetric carboxylate stretch consists of a distinct doublet for lead carboxylates C9, C10, C11, C16, and C18 with peaks at ∼1515 and 1540 cm−1 and for C6, C7, and C8 there is only a broad peak centered around 1525 cm−1. There are differences in the low-frequency region, as well, where bands are quite sensitive to the alkyl chain packing.48 In the C6, C7, and C8 carboxylates, a peak assigned to a CH2 rocking mode appears around 720 cm−1.51 In C9, C10, C11, C16 and C18 carboxylates, this region contains multiple features, consistent with the IR results of Mesubi et al. who found a singlet for lead carboxylates of C6 and C8 but a doublet for C10, C12, C14, and C16 carboxylates.51 In the range between 1150 and 1350 cm−1, C9, C10, C11, C16 and C18 carboxylates show the regular progression of bands associated with the wagging and twisting of the hydrocarbon chains.51 In this region, the smaller carboxylates do not show a particularly regular set of bands, demonstrating that the chain motions are different from those in the larger carboxylates.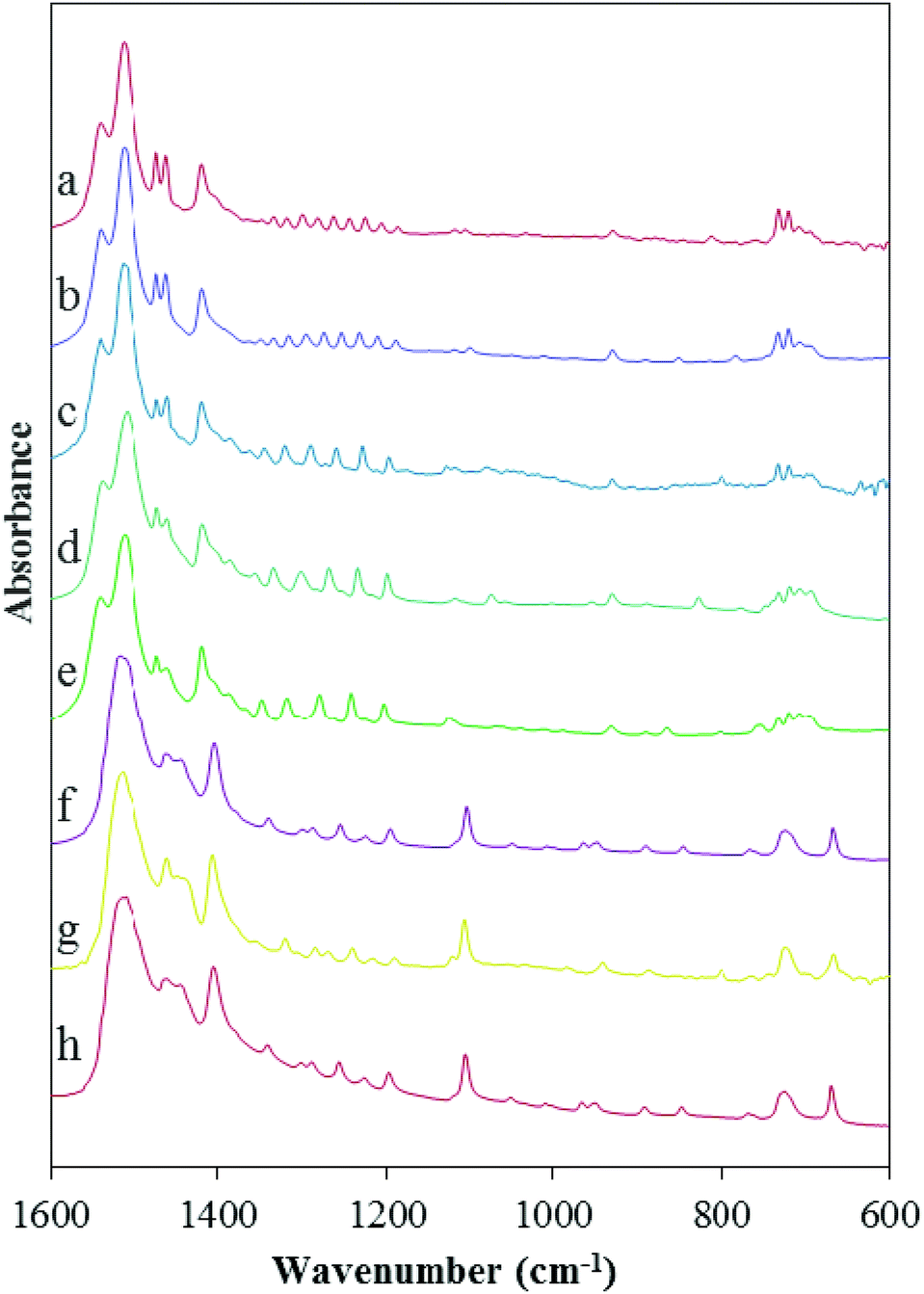 | ||
| Fig. 3 FTIR spectra of (a) lead octadecanoate, (b) lead hexadecanoate, (c) lead undecanoate, (d) lead decanoate, (e) lead nonanoate, (f) lead octanoate, (g) lead heptanoate, and (h) lead hexanoate. The FTIR spectra show differences in the two subgroups (short- and long-chain lead carboxylates) in the ranges 1515–1540 cm−1, 1150–1350 cm−1, and around 720 cm−1. | ||
Crystal structure of lead nonanoate
A consistent theme in studies of the spectroscopy of lead is the existence of two structural motifs around the lead center.14 The 207Pb NMR parameters and the FTIR data suggest that the carboxylates fall into two subgroups, one being C8 and shorter-chain Pb-carboxylates and the other being C9 and longer-chain salts. The structure of lead heptanoate by single-crystal X-ray diffraction has been reported.22 Of the chains greater than C7, only lead azelate has been previously reported,23 however it is a dicarboxylate, which may influence the packing. Thus, there is no representative single-crystal structure of a long-chain lead monocarboxylate.Lead nonanoate displays poor solubility in many common solvents and, as a result, is often deposited as powders. After several trials, thin plate-like crystals were afforded from a slow evaporation of a saturated toluene solution. Because of the large a-axis dimension, initial unit-cell determinations using Mo-kα (λ = 0.71073 Å) failed because of poor resolution of the virtually overlapping diffraction spots. Data collected using Cu-kα (λ = 1.54178 Å) allowed proper resolution of the diffraction pattern and thereby allowed successful unit cell measurement and subsequently successful integration of the two-dimensional frame data. The crystal data and refinement details of lead nonanoate are given in Table 2.
| Crystal system | Monoclinic |
| Space group | P2/c |
| Empirical formula | PbC18H34O4 |
| Temperature (K) | 200(2) |
| Wavelength (Å) | 0.71073 |
| Unit cell dimensions (Å, °) | a = 54.872(6) |
| b = 4.9603(5) | |
| c = 7.1628(7) | |
| β = 90.368(2) | |
| Cell volume (Å3) | 1949.54(3) |
| Density (g cm−3) | 1.777 |
| Z | 4 |
| Absorption coefficient (mm−1) | 8.670 |
| Min./Max. transmission | 0.4056/0.7456 |
| Reflections collected/unique | 25918/4459 |
| Completeness to θ = 24.242 (%) | 99.8 |
| Data/restraints/parameters | 4459/261/198 |
| R 1, wR2 (%) | 8.39, 16.48 |
| Goodness-of-fit on F2 | 1.311 |
The structure of lead nonanoate consists of lead sites coordinated with the oxygen of the carboxylate groups to form extended sheets, which are packed along the c direction to form the three-dimensional structure. Each lead center is apparently surrounded by six oxygen atoms in a distorted octahedral array (Fig. 4). A closer inspection of the octahedron and reported bond distances and angles in Table 3, however, reveals a remarkably large O1–Pb1–O2i angle, 114.0(3)° (ideally 90°) and O2 that is roughly in the same plane as Pb1, O1 and O2i. Furthermore the Pb1–O2 distance at 2.977(10) Å is comparable to Pb1–O2i while, in contrast, Pb1–O4 is much longer at 3.383(10) Å. This observation suggests that the lead coordination sphere could be properly viewed as seven coordinate.
 | ||
| Fig. 4 Solid-state molecular diagram and numbering scheme of lead nonanoate monomer, and coordination sphere of the lead atom, down the c axis showing the monodentate binding to each carboxylate group. The alkyl groups are aligned in an all-trans structure. Thermal ellipsoids are at 50% probability. H-atoms are depicted at an arbitrary radius. Symmetry operations: i = x, −y, z + 1/2, ii = x, y + 1, z. | ||
| Symmetry operations: i = x, −y, z + 1/2, ii = x, y + 1, z. | |||||
|---|---|---|---|---|---|
| Pb1–O1 | 2.660(10) | O1–Pb1–O3 | 90.7(3) | O3–Pb1–O4ii | 84.9(3) |
| Pb1–O3 | 2.403(10) | O1–Pb1–O1i | 95.3(2) | O4i–Pb1–O4ii | 99.9(3) |
| Pb1–O1i | 2.437(10) | O1–Pb1–O2i | 114.0(3) | O1i–Pb1–O4i | 78.1(3) |
| Pb1–O2i | 2.856(10) | O1–Pb1–O4ii | 86.0(3) | O1i–Pb1–O2i | 78.3(3) |
| Pb1–O4i | 2.583(10) | O3–Pb1–O1i | 91.1(4) | O4i–Pb1–O2i | 75.8(3) |
| Pb1–O4ii | 2.661(10) | O3–Pb1–O4i | 78.5(3) | O4ii–Pb1–O2i | 104.8 (3) |
In Fig. 5, we compare the local structures of lead heptanoate and lead nonanoate around the Pb site, including atoms within 3.1 Angstroms of the lead center. In the heptanoate (Fig. 5b), the structure is best described as lead coordinated to seven nearby oxygen atoms. The authors claim that the lead atom is coordinated to six oxygen atoms in a “very distorted polyhedron”.22 From Fig. 5b, one sees that the structure resembles a pentagonal pyramid with the apex of the pyramid being capped with two oxygen atoms, rather than one. The lead site in the nonanoate (Fig. 5a) is also 7-coordinate. It resembles a strained octahedral arrangement of oxygen atoms with an added oxygen atom or a distorted pentagonal bipyramid. Although neither structure has exact spherical or cubic point symmetry about the lead nucleus, from the comparison of the structures in Fig. 5, it is obvious that the local symmetry around the lead site in lead nonanoate is less asymmetric than that in lead heptanoate. A qualitative argument implies that the 207Pb NMR span in lead nonanoate should be smaller than the span for lead heptanoate, which is in good agreement with the spectroscopic results (Table 1). Thus, one sees that a large 207Pb span (∼2600–2700 ppm) is consistent with a hemidirected structure, whereas a “small” 207Pb span (∼730–750 ppm) is consistent with a holodirected structure. This suggests the C6, C7, and C8 lead carboxylates crystallize in a hemidirected structure, whereas C9, C10, C11, C16, and C18 crystallize in a holodirected structure. By analogy, the local structure of lead azelate, as demonstrated by the 207Pb NMR parameters,30 has a local coordination similar to that of the short-chain carboxylates, which is also supported by the crystal structure.23
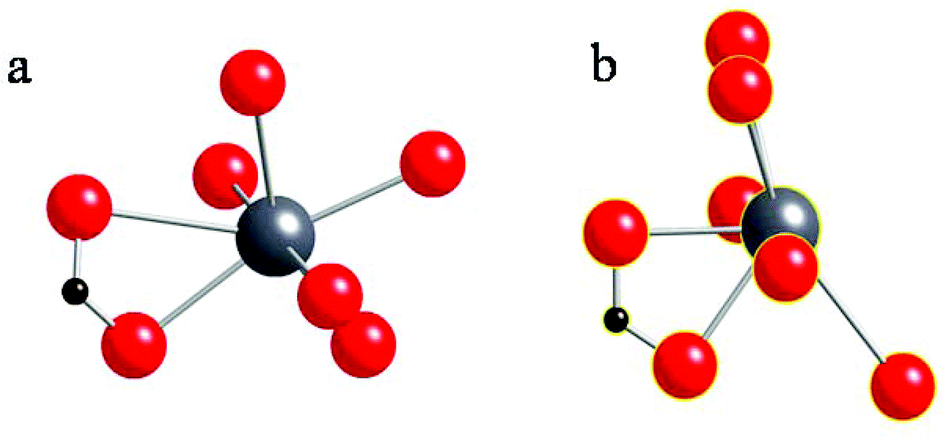 | ||
| Fig. 5 (a) The local structure around the lead atom in lead nonanoate obtained in the present study. (b) The local structure around the lead atom in lead heptanoate, as reported by Lacouture et al.22 | ||
Conclusions
Spectroscopic and X-ray diffraction analyses of a series of lead carboxylates have provided insight into the relationship between structure and spectroscopic parameters. The C6–C18 lead carboxylates can be classified into two subgroups based on the spectroscopic and diffraction parameters. In the short-chain group (i.e. C6–C8), the structure around the lead site is hemidirecting. The 207Pb NMR span is rather large (∼2600–2700 ppm) and the 13C NMR data show that there are two unique carboxylate and methyl groups per unit cell. The FTIR spectra show a broad peak for the asymmetric carboxylate stretch at 1525 cm−1, a sharp single CH2 rocking mode around 720 cm−1, and the twisting and bending modes of the chain (1150–1350 cm−1) indicate a complex motion of the alkyl chains.For the long-chain group (C9–C18), the 207Pb NMR span is relatively small (∼730–750 ppm), indicating a more nearly spherical local structure at the lead atom site, which would be termed holodirecting. The 13C NMR spectra show that there are two distinct carboxylates and the single resonance for the methyl groups at the ends of the aliphatic chains shows that the methyl groups are in similar environments, not affected by the structure at the carboxylate carbons/lead ions. The FTIR spectra show a doublet in the carboxylate asymmetric stretch region, and the bands due to the CH2 rocking around 720 cm−1 suggest that there are multiple slightly different modes. The positions of features assigned to twisting modes (1150–1350 cm−1) for these long-chain molecules show a regular progression, suggesting a systematic set of vibrational motions of the chains when the carboxylates are nine carbons or longer. The dicarboxylate, lead azelate, has NMR parameters like the short-chain carboxylates, indicating that the presence of two carboxylates on the chain is sufficient to move the local structure at the lead site into the hemidirected structure.
Due to the similarity in the NMR tensors of lead nonanoate and lead hexadecanoate and octadecanoate, we propose that the crystal structure of lead nonanoate serves as a model for the structure of lead hexadecanoate and lead octadecanoate. In addition, the NMR results directly correlate with features of the IR data. That connection to structure should allow the use of ATR (attenuated total reflectance)-FTIR as a non-invasive IR technique to study soap formation in paintings. This information may assist in future studies of the migration of lead soaps in paint films and in the development of treatments to arrest the soap deterioration process in oil paintings.
Acknowledgements
CD acknowledges the support of the National Science Foundation (NSF) under grants CHEM-0956006 and CHE-1139192. SC acknowledges the support of the NSF under grant CHEM-1139180. GPAY thanks the NSF for CRIF 1048367. We acknowledge helpful discussions with Shi Bai and Guangjin Hou about the development of wide-band inversion and the spectroscopy of 207Pb. We also acknowledge Joen Hermans (University of Amsterdam), Katrien Keune (University of Amsterdam and TU Delft), and Ronald Birke (City College of New York), for helpful initial discussions regarding this topic.References
- R. Vangorkum and E. Bouwman, Coord. Chem. Rev., 2005, 249, 1709–1728 CrossRef CAS.
- E. Rocca and J. Steinmetz, Corros. Sci., 2001, 43, 891–902 CrossRef CAS.
- R. G. Bossert, J. Chem. Educ., 1950, 27, 10 CrossRef CAS.
- M. S. Akanni, E. K. Okoh, H. D. Burrows and H. A. Ellis, Thermochim. Acta, 1992, 208, 1–41 CrossRef CAS.
- Y. Zhang, J. Wang, X. Yan, X. Liu, H. Zhou and A. Yuan, Microporous Mesoporous Mater., 2014, 184, 15–20 CrossRef CAS.
- A. Thirumurugan and C. N. R. Rao, J. Solid State Chem., 2008, 181, 1184–1194 CrossRef CAS.
- I. Resa, H. Moreira, B. Bresson, B. Mahler, B. Dubertret and H. Aubin, J. Phys. Chem. C, 2009, 113, 7120–7122 CAS.
- D. K. Britt, Y. Yoon, P. Ercius, T. D. Ewers and A. P. Alivisatos, Chem. Mater., 2013, 25, 2544–2548 CrossRef CAS.
- K. Keune, A. van Loon and J. J. Boon, Microsc. Microanal., 2011, 17, 696–701 CrossRef CAS PubMed.
- P. Noble, A. van Loon and J. J. Boon, ICOM Committee for Conservation 14th Triennnial Meeting, The Hague, 2005.
- C. Higgitt, M. Spring and D. Saunders, National Gallery Technical Bulletin, 2003, 24, 75–91 Search PubMed.
- K. Keune, PhD Thesis, University of Amsterdam, 2005.
- A. van Loon, PhD Thesis, University of Amsterdam, 2008.
- L. Shimoni-Livny, J. P. Glusker and C. W. Bock, Inorg. Chem., 1998, 37, 1853–1867 CrossRef CAS.
- D. Esteban-Gómez, C. Platas-Iglesias, T. Enríquez-Pérez, F. Avecilla, A. de Blas and T. Rodríguez-Blas, Inorg. Chem., 2006, 45, 5407–5416 CrossRef PubMed.
- A. Pellissier, Y. Bretonnière, N. Chatterton, J. Pécaut, P. Delangle and M. Mazzanti, Inorg. Chem., 2007, 46, 3714–3725 CrossRef CAS PubMed.
- D. L. Reger, J. E. Collins, A. L. Rheingold, L. M. Liable-Sands and G. P. A. Yap, Inorg. Chem., 1997, 36, 345–351 CrossRef CAS.
- D. L. Reger, M. F. Huff, A. L. Rheingold and B. S. Haggerty, J. Am. Chem. Soc., 1992, 114, 579–584 CrossRef CAS.
- D. L. Reger, T. D. Wright, C. A. Little, J. J. S. Lamba and M. D. Smith, Inorg. Chem., 2001, 40, 3810–3814 CrossRef CAS PubMed.
- P. G. Harrison and A. T. Steel, J. Organomet. Chem., 1982, 239, 105–113 CrossRef CAS.
- W. Clegg, I. R. Little and B. P. Straughan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1319–1322 CrossRef.
- F. Lacouture, M. François, C. Didierjean, J.-P. Rivera, E. Rocca and J. Steinmetz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2001, 57, 530–531 CAS.
- M. J. Plater, B. De Silva, T. Gelbrich, M. B. Hursthouse, C. L. Higgitt and D. R. Saunders, Polyhedron, 2003, 22, 3171–3179 CrossRef CAS.
- R. L. Davidovich, V. Stavila, D. V. Marinin, E. I. Voit and K. H. Whitmire, Coord. Chem. Rev., 2009, 253, 1316–1352 CrossRef CAS.
- M. L. Hu, A. Morsali and L. Aboutorabi, Coord. Chem. Rev., 2011, 255, 2821–2859 CrossRef CAS.
- A. Walsh and G. W. Watson, J. Solid State Chem., 2005, 178, 1422–1428 CrossRef CAS.
- P. G. Harrison, M. A. Healy and A. T. Steel, J. Chem. Soc., Dalton Trans., 1983, 1845–1848 RSC.
- H. D. Burrows, C. Geraldes, T. J. T. Pinheiro, R. K. Harris and A. Sebald, Liq. Cryst., 1988, 3, 853–860 CrossRef CAS.
- L. Robinet and M. C. Corbeil, Stud. Conserv., 2003, 48, 23–40 CAS.
- J. Catalano, Y. Yao, A. Murphy, N. Zumbulyadis, S. A. Centeno and C. Dybowski, Appl. Spectrosc., 2014, 68, 280–286 CrossRef CAS PubMed.
- G. Neue and C. Dybowski, Solid State Nucl. Magn. Reson., 1997, 7, 333–336 CrossRef CAS PubMed.
- G. Neue, C. Dybowski, M. L. Smith, M. A. Hepp and D. L. Perry, Solid State Nucl. Magn. Reson., 1996, 6, 241–250 CrossRef CAS PubMed.
- A. W. MacGregor, L. A. O'Dell and R. W. Schurko, J. Magn. Reson., 2011, 208, 103–113 CrossRef CAS PubMed.
- J. Herzfeld and A. E. Berger, J. Chem. Phys., 1980, 73, 6021–6030 CrossRef CAS.
- K. Eichele, HBA 3.1 and WSOLIDS, University of Tubingen, Tubingen, 2013 Search PubMed.
- Check.def File Version 5/2/14, http://checkcif.iucr.org.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. Feio, H. D. Burrows, C. F. G. C. Geraldes and T. J. T. Pinheiro, Liq. Cryst., 1991, 9, 417–432 CrossRef CAS.
- J. F. Stephens and C. Tuck-Lee, J. Appl. Crystallogr., 1969, 2, 1–10 CrossRef CAS.
- M. J. Katz, P. M. Aguiar, R. J. Batchelor, A. A. Bokov, Z. G. Ye, S. Kroeker and D. B. Leznoff, J. Am. Chem. Soc., 2006, 128, 3669–3676 CrossRef CAS PubMed.
- M. J. Katz, V. K. Michaelis, P. M. Aguiar, R. Yson, H. Lu, H. Kaluarachchi, R. J. Batchelor, G. Schreckenbach, S. Kroeker, H. H. Patterson and D. B. Leznoff, Inorg. Chem., 2008, 47, 6353–6363 CrossRef CAS PubMed.
- G. G. Briand, A. D. Smith, G. Schatte, A. J. Rossini and R. W. Schurko, Inorg. Chem., 2007, 46, 8625–8637 CrossRef CAS PubMed.
- G. Chevrier, G. Giester, G. Heger, D. Jarosch, M. Wildner and J. Zemann, Z. Kristallogr., 1992, 199, 67–74 CrossRef CAS.
- C. Dybowski, A. Glatfelter, S. Bai, D. Martinez, S. Segarra and D. L. Perry, Mater. Res. Soc. Symp. Proc., 2007, 984 Search PubMed.
- A. Glatfelter, S. Bai, O. Dmitrenko, D. L. Perry, S. E. Van Bramer and C. Dybowski, Can. J. Chem., 2011, 89, 863–869 CrossRef CAS.
- T. R. Lomer and K. Perera, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2912–2913 CrossRef.
- T. R. Lomer and K. Perera, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 2913–2915 CrossRef.
- H. A. Ellis, N. A. S. White, R. A. Taylor and P. T. Maragh, J. Mol. Struct., 2005, 738, 205–210 CrossRef CAS.
- S. O. Adeosun and S. Sime, Thermochim. Acta, 1976, 17, 351 CrossRef CAS.
- A. S. Arenas, M. V. Garcia, M. I. Redondo, J. A. R. Cheda, M. V. Roux and C. Turrion, Liq. Cryst., 1995, 18, 431–441 CrossRef CAS.
- M. A. Mesubi, J. Mol. Struct., 1982, 81, 61–71 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed X-ray crystallographic data collection and fitting. CCDC 1023395. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt03075c |
| This journal is © The Royal Society of Chemistry 2015 |