
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic oxygenation of sp3 “C–H” bonds with Ir(III) complexes of chelating triazoles and mesoionic carbenes†
Stephan
Hohloch
a,
Selina
Kaiser
a,
Fenja Leena
Duecker
a,
Aljoša
Bolje
b,
Ramananda
Maity
a,
Janez
Košmrlj
b and
Biprajit
Sarkar
*a
aInstitut für Chemie und Biochemie, Anorganische Chemie, Freie Universität Berlin, Fabeckstraße 34-36, D-14195, Berlin, Germany. E-mail: biprajit.sarkar@fu-berlin.de
bFaculty of Chemistry and Chemical Technology, University of Ljubljana, Aškerčeva 5, SI-1000, Ljubljana, Slovenia
First published on 30th October 2014
Abstract
Cp*-Ir(III) complexes with additional chelating ligands are known active pre-catalysts for the oxygenation of C–H bonds. We present here eight examples of such complexes where the denticity of the chelating ligands has been varied from the well-known 2,2′-bpy through pyridyl-triazole, bi-triazole to ligands containing pyridyl-triazolylidene, triazolyl-triazolylidene and bi-triazolylidenes. Additionally, we also compare the catalytic results to complexes containing chelating cyclometallated ligands with additional triazole or triazolylidene donors. Single crystal X-ray structural data are presented for all the new complexes that contain one or more triazolylidene donors of the mesoionic carbene type. We present the first example of a metal complex containing a chelating triazole-triazolylidene ligand. The results of the catalytic screening show that complexes containing unsymmetrical donors of the pyridyl-triazole or pyridyl-triazolylidene types are the most potent pre-catalysts for the C–H oxygenation of cyclooctane in the presence of either m-CPBA or NaIO4 as a sacrificial oxidant. These pre-catalysts can also be used to oxygenate C–H bonds in other substrates such as fluorene and ethyl benzene. The most potent pre-catalysts presented here work with a lower catalyst loading and under milder conditions while delivering better product yields in comparison with related literature known Ir(III) pre-catalysts. These results thus point to the potential of ligands with unsymmetrical donors obtained through the click reaction in oxidation catalysis.
Introduction
1,2,3-Triazoles, the product of the best “click” reaction known so far, have found extensive use as ligands in coordination and organometallic chemistry.1 Furthermore, 1,2,3-triazoles can be easily methylated to generate 1,2,3-triazolium salts, which are direct precursors of 1,2,3-triazol-5-ylidenes, compounds that have been variously called abnormal or mesoionic carbenes.2 Due to their mesoionic character, such 1,2,3-triazolylidenes have been shown to have excellent σ-donor properties.2 Metal complexes of chelating pyridyl-triazoles have been widely studied.3 On the triazolylidene front, most efforts have focused on monodentate ligand systems,2 with the use of chelating ligand motifs being still rather limited.3b,4Within the last 5 years, iridium based catalysts have gained special interest for their potential in water oxidation catalysis.5,6 The direct oxygenation of inactivated C–H bonds to obtain alcohols or ketones can also be catalysed using similar iridium catalysts.7 In 2010, Crabtree and co-workers were able to catalytically oxidize cis-decaline to the corresponding alcohol with retention of the stereocenter using Ir(III) pre-catalysts.7a Three years later the same group reported on sodium periodate promoted oxidation of cyclooctane to cyclooctanone using these pre-catalysts.7b They found complexes C1 and C2 to be the most active complexes for such transformations (Scheme 1).
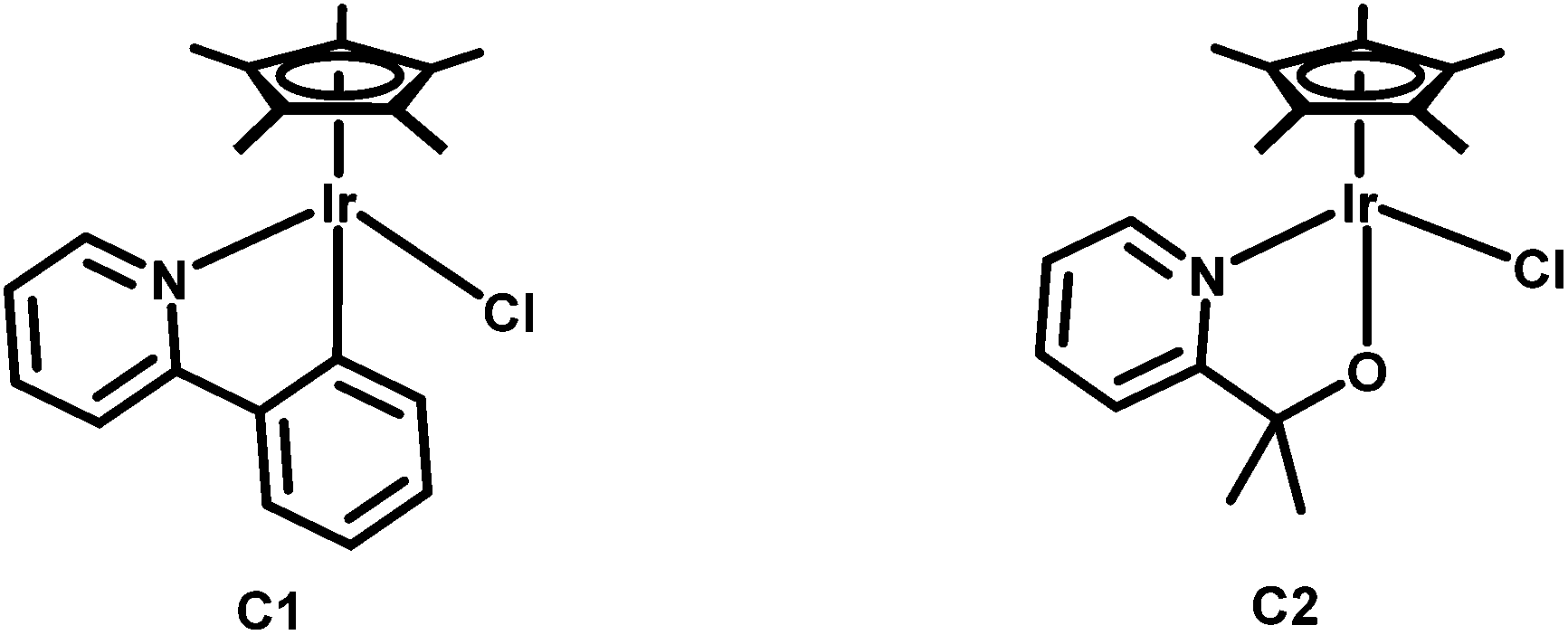 | ||
| Scheme 1 IrCp*-(pre)catalysts reported by Crabtree et al. for the catalytic oxidation of inactivated alkanes.7b | ||
Interestingly, although N-heterocyclic carbenes and mesoionic carbenes, especially 1,2,3-triazolylidenes, have already proven to generate very active complexes for the water oxidation reaction,8 in direct oxidations of alkanes, they have not found any use so far. Some of us have recently reported on the selective synthesis of pyridyl-triazolium salts, which are useful precursors for the generation of metal complexes containing chelating pyridyl-triazolylidene ligands.4c In the following, we present the results for the catalytic direct oxygenation of sp3 C–H bonds in the presence of a sacrificial oxidant using eight different Cp*-Ir(III) (Cp* = pentamethylcyclopentadienyl) pre-catalysts with the ligands shown in Scheme 2. The ligands have been systematically varied in terms of their donor properties as well as their coordination symmetry (the same or different donor atoms within the same ligand).
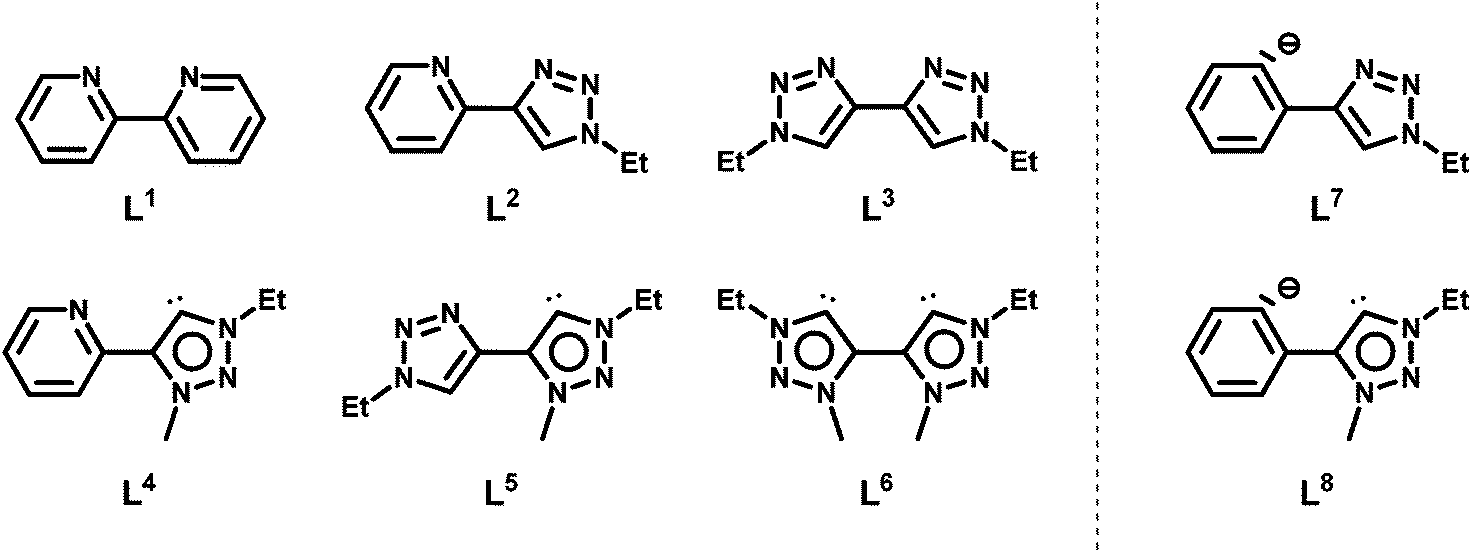 | ||
| Scheme 2 Ligand systems used in this work. | ||
Results and discussion
Synthesis and crystal structures
The synthesis of ligands L1, L2,4gL7 and L8,4d has been reported previously. The synthesis of all other ligands or ligand precursors followed literature known procedures as shown in the Experimental section.Complexes 1–3 with ligands L1–L3 containing only N-donors were obtained by the direct reaction of [Cp*ClIr(μ-Cl)2IrClCp*] with the ligands, followed by salt metathesis.9,3b Complexes 4–6 that contain one or more mesoionic carbene donors were synthesized by a transmetallation route using silver oxide. Complexes 7 and 8 containing cyclometallated ligands have been reported by us previously.4d The identity and purity of all complexes were established by 1H-, 13C-NMR spectroscopy and mass spectrometry (see ESI†). The existence of the carbene complexes 4, 5 and 6 was unambiguously proven by 13C-NMR spectroscopy, displaying resonances at δ 156.5, 154.5 and 155.4 ppm respectively for the carbene C atom.
For complexes 4–6 that display one or more mesoionic carbene donors, we were able to obtain single crystals suitable for an X-ray diffraction analysis (Fig. 1). An analysis of the bond lengths in 4, 5·CHCl3·0.25H2O and 6·CH2Cl2 shows that the double bonds within the triazolylidene ligands are delocalized as is expected for complexes of this type.8 On the other hand, the triazole moiety in 5·CHCl3·0.25H2O shows the expected azo character. The Ir–C and Ir–N distances are all in the expected range (Table 1).3b,8 Complex 5 is the first example of a metal complex containing a triazole-triazolylidene type chelating ligand.
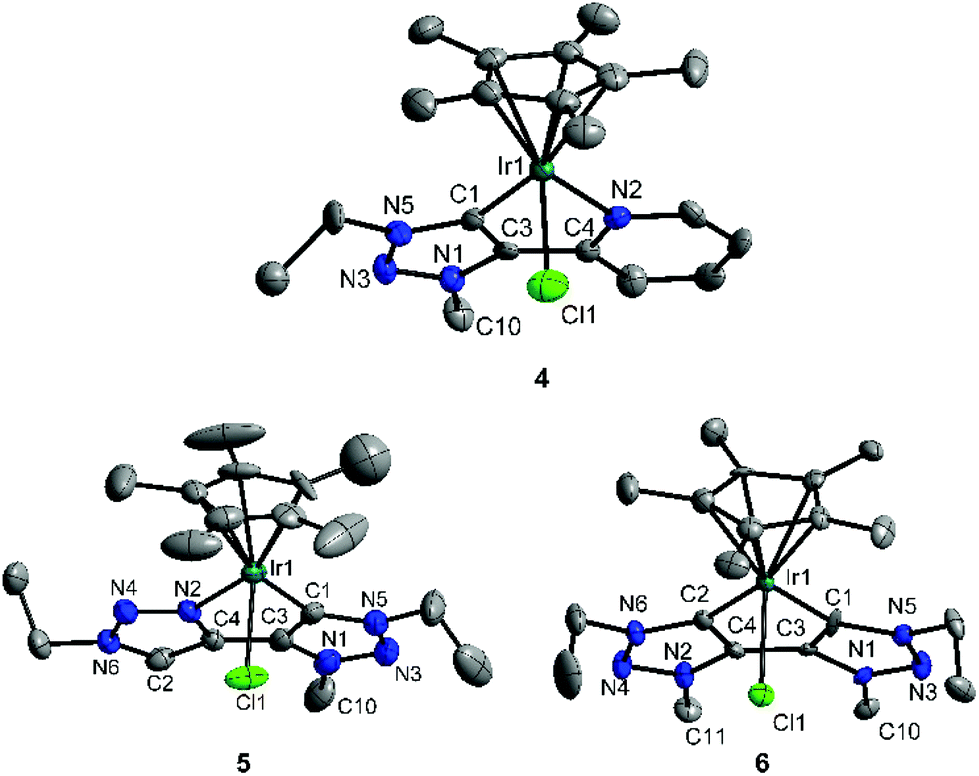 | ||
| Fig. 1 ORTEP plot of 4, 5·CHCl3·0.25H2O and 6·CH2Cl2. Hydrogen atoms, counter ions and solvent molecules are omitted for clarity. Thermal ellipsoids are drawn at 50% probability. | ||
| Atoms | 4 | 5·CHCl3·0.25H2O | 6·CH2Cl2 |
|---|---|---|---|
| N2–Ir1 | 2.14(1) | 2.10(1) | — |
| C1–Ir1 | 2.03(1) | 2.04(1) | 2.03(1) |
| C2–Ir1 | — | — | 2.05(1) |
| C3–C4 | 1.45(1) | 1.42(1) | 1.42(1) |
| Cl1–Ir1 | 2.39(1) | 2.42(1) | 2.40(1) |
| C1–C3 | 1.37(1) | 1.38(1) | 1.41(1) |
| C2–C4 | — | 1.37(1) | 1.41(1) |
| C3–N1 | 1.35(1) | 1.36(1) | 1.38(1) |
| C4–N2 | 1.36(1) | 1.36(1) | 1.35(1) |
| N1–N3 | 1.32(1) | 1.33(1) | 1.32(1) |
| N2–N4 | — | 1.31(1) | 1.33(1) |
| N3–N5 | 1.34(1) | 1.33(1) | 1.33(1) |
| N4–N6 | — | 1.35(1) | 1.34(1) |
| N5–C1 | 1.36(1) | 1.36(1) | 1.37(1) |
| N6–C2 | — | 1.33(1) | 1.35(1) |
| Ir–Cp*(cent) | 1.817(1) | 1.808(1) | 1.849(1) |
| C3–C1–N5 | 102.8(7) | 102.6(7) | 102.9(8) |
| C4–C2–N6 | — | 105.4(7) | 101.7(8) |
| N2–Ir1–C1 | 76.2(3) | 75.7(3) | — |
| C1–Ir1–C2 | — | — | 76.5(4) |
Catalysis
We next turned to the catalytic performances of these complexes in the direct oxygenation of inactivated alkanes. Two different sacrificial oxidants, m-CPBA (3-chloroperoxybenzoic acid) in non-aqueous solutions (Method A)10 and NaIO4 in aqueous solutions (Method B, Scheme 3),7b were chosen for this purpose.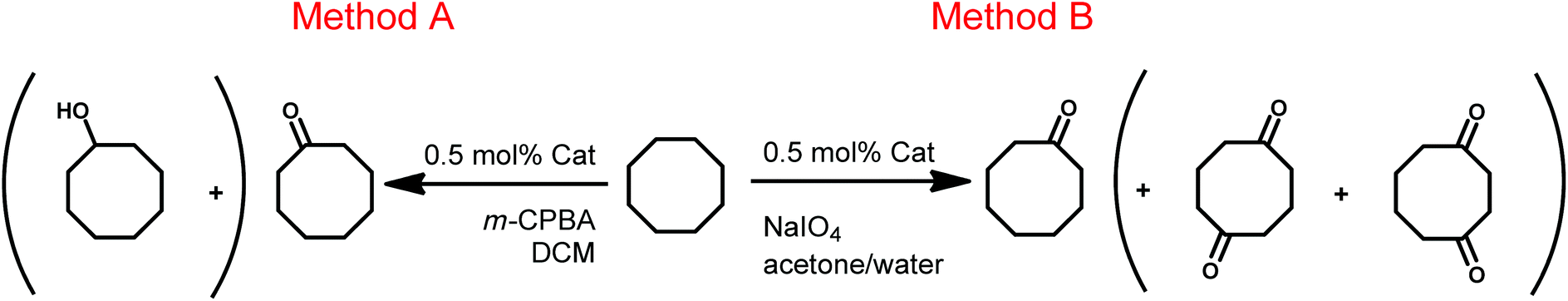 | ||
| Scheme 3 General conditions for Method A and Method B. | ||
Table 2 summarizes the obtained conversion for the m-CPBA supported oxidation reaction. All complexes show reasonably high conversions, ranging from 22% to 59% conversion of the starting materials. Remarkably, although we have used an excess of the oxidant, a lot of “under-oxidized” cyclooctanol was detected after workup and in selected cases was also found to be the main product of the reaction. For example, if the cyclometallated complex 7 was applied, cyclooctanol was obtained in a conversion of 35% while only 16% cyclooctanone was observed. Complexes with two different donor atoms (with unsymmetrical ligands) showed the highest overall conversion of cyclooctane (except for 5).
| Cat | Overalla [%] | Cyclooctanonea [%] | Cyclooctanola [%] |
|---|---|---|---|
| a All reactions were carried out using 1 mmol of cyclooctanone and 4 equivalents of the oxidant at 45 °C overnight with 0.5 mol% catalyst. Conversions were determined by proton NMR spectroscopy averaged from 3 independent runs using 0.4 eq. of 1,2-dichlorobenzene as an internal standard. | |||
| 1 | 22 | 10 | 12 |
| 2 | 45 | 27 | 18 |
| 3 | 38 | 14 | 24 |
| 4 | 59 | 40 | 19 |
| 5 | 24 | 13 | 11 |
| 6 | 32 | 23 | 9 |
| 7 | 51 | 16 | 35 |
| 8 | 47 | 27 | 20 |
The most active complex with m-CPBA as an oxidant was found to be the pyridyl-triazolylidene complex 4, displaying overall conversions of 59% with cyclooctanone as the desired main product in 40% conversion. The second most active complex was the pyridyl-triazole complex 2, displaying an overall conversion of 45% with 27% cyclooctanone. However, for complex 2 the selectivity is low, since the amount of “under-oxidized” cyclooctanol is relatively high for that catalyst (Table 2).
After establishing the unsymmetrical pyridyl containing complexes 2 and 4 as the most active complexes for the m-CPBA promoted oxidation reaction, we next tested the activity of complexes 1–8 with sodium periodate as a sacrificial oxidant. The results are summarized in Table 3.
| Cat | Overalla [%] | Cyclooctanonea [%] | Cyclooctanedioneb [%] |
|---|---|---|---|
| a All reactions were carried out using 1 mmol of cyclooctanone and 10 equivalents of the oxidant. Conversions were determined by proton NMR spectroscopy averaged from 3 independent runs using 0.2 eq. of 1,4-dibromobenzene as an internal standard. b Conversions to cyclooctanedione are given as the sum of the conversions to 1,4- and 1,5-cyclooctanedione. | |||
| 1 | 24 | 21 | 3 |
| 2 | 62 | 56 | 6 |
| 3 | 28 | 27 | 1 |
| 4 | 62 | 55 | 7 |
| 5 | 21 | 20 | 1 |
| 6 | 32 | 30 | 2 |
| 7 | 37 | 35 | 2 |
| 8 | 32 | 31 | 1 |
Again all complexes displayed reasonable activity for this transformation. In agreement with Crabtree's report from 20137b we also found the over-oxidation to the two-fold oxidized 1,4- and 1,5-cyclooctanediones, but only upto a maximum of 7% yield. Complexes 1, 3 and 6 with symmetrical ligands showed a lower activity than the complexes having a non-symmetrical chelator, 2 and 4. Excluded from this trend is the triazol-triazolylidene complex 5 showing only a minor activity. Using sodium periodate in an aqueous solution the cyclometallated complexes 7 and 8 also show less overall conversion compared to the m-CPBA promoted oxidation (Tables 2 and 3). This is most likely related to the fact that under aqueous conditions, water oxidation occurs as a side reaction. Cyclometallated Ir(III) complexes have already been proven to give very active catalysts for the water oxidation reaction.8 With an overall conversion of 62% and formation of 56 and 55% cyclooctanone respectively, the pyridine containing complexes 2 and 4 have the highest catalytic potential under these conditions as well. Except for complexes 7 and 8 all other complexes showed higher conversions to cyclooctanone if method B is applied. Furthermore, using method B the reaction is more selective than when using method A. Method B delivers large conversions to cyclooctanone. Reactions without any iridium complexes resulted in no conversions at all, no matter which conditions were applied. The use of the [IrCp*Cl2]2 precursor or IrCl3 hydrate as a catalyst for this transformation has already been explored by Crabtree et al. and resulted in only small conversions of the starting materials (6–17%).7b Both these experiments indicate that both the metal and the additional chelating ligands are crucial for the catalytic oxidations of C–H bonds.
We further focused on optimizing the reaction towards higher conversions and a better atom efficiency with the most efficient catalysts 2 and 4. Since applying method B results in higher conversions with better selectivities, we focused our studies exclusively on those conditions.
At first we investigated the reaction time to see whether overnight reaction times are really needed and also studied the influence of the amount of oxidant used in catalysis. Hence, we recorded a time-conversion diagram with 0.5 mol% of complex 4 as a catalyst, with 10 equivalents of sodium periodate at 45 °C. As can be seen from Fig. 2, after 10 hours, the reaction does not proceed any further. At that point we added another 5 equivalents of sodium periodate. After the addition of more oxidants (Fig. 2, dotted line), the reaction starts to proceed again. This shows that indeed the conversion determining step is the amount of oxidant used, while the catalyst stays active over time. It could be seen that the reaction should be run for at least 10 hours to achieve good conversions and hence we continued to carry out the subsequent reactions with overnight reaction times.
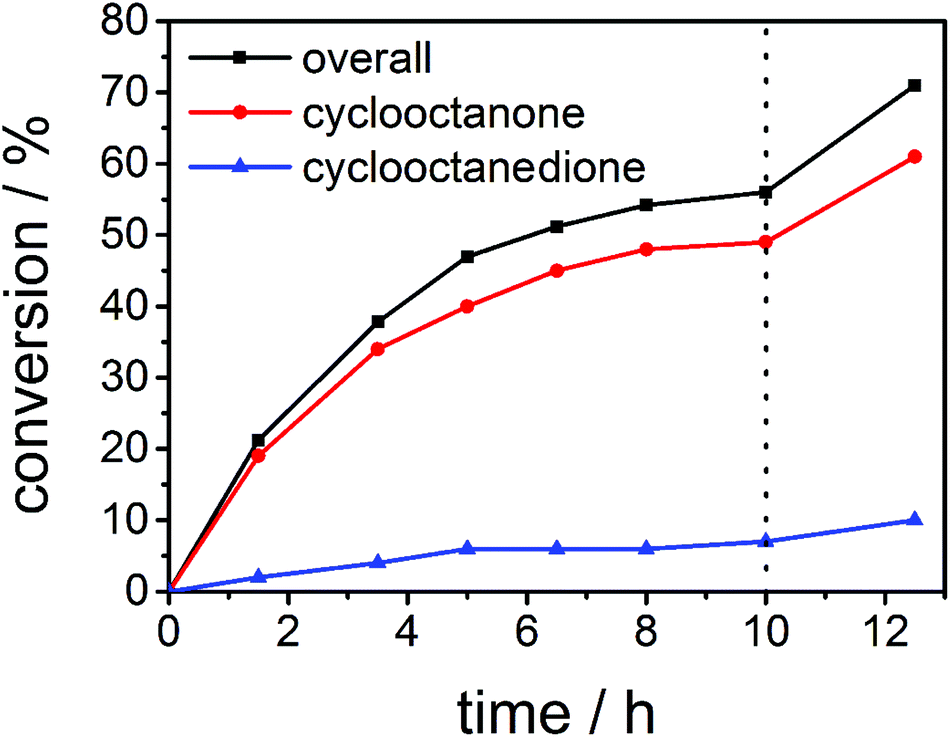 | ||
| Fig. 2 Time conversion for the catalytic oxidation of cyclooctane using 0.5 mol% 4 with 10 equivalents of sodium periodate at 45 °C. The dotted line indicates the addition of another 5 equivalents of sodium periodate. | ||
We found that lowering by 50% the amount of oxidant used has strong effects on the catalytic performances of 2 and 4 at 45 °C. The overall conversions achieved decreased by approximately 20% (Table 4). Also, when the temperature is decreased from 45 °C to room temperature a strong difference in reactivity is observed between complexes 2 and 4. For 2, the conversion decreases from 56% of cyclooctanone to only 12% if 10 eq. of the oxidant is used. In contrast, if the pyridyl-triazolylidene complex 4 is used as a pre-catalyst, lowering the temperature has only minor effects. The conversion to cyclooctanone decreases only from 55% to 46% using 10 eq. of the oxidant. Furthermore, reducing the amount of oxidant used to only 5 equivalents still afforded 38% conversion to the desired product (Table 4). The above data thus show that the pyridyl-triazolylidene complex 4 is the best pre-catalyst.
| Cat | Temp. [°C] | NaIO4 [eq.] | Overall [%] | Cyclooctanone [%] | Cyclooctanedione [%] |
|---|---|---|---|---|---|
| 2 | 45 | 10 | 62 | 56 | 6 |
| 45 | 5 | 40 | 35 | 5 | |
| RT | 10 | 13 | 12 | 1 | |
| RT | 5 | 8.5 | 8 | 0.5 | |
| 4 | 45 | 10 | 62 | 55 | 7 |
| 45 | 5 | 42 | 35 | 7 | |
| RT | 10 | 53 | 46 | 7 | |
| RT | 5 | 42 | 38 | 4 | |
In the next step, we investigated the effect of catalyst loading on the conversion. The results are summarized in Table 5.
| Loading [mol%] | Overalla [%] | Cyclooctanonea [%] | Cyclooctadionea [%] |
|---|---|---|---|
a All reactions were carried out using 1 mmol of the cyclooctane at room temperature in an acetone–water mixture 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Conversions were determined by proton NMR spectroscopy averaged from 3 independent experiments using 0.2 eq. of 1,4-dibromobenzene as an internal standard. :1. Conversions were determined by proton NMR spectroscopy averaged from 3 independent experiments using 0.2 eq. of 1,4-dibromobenzene as an internal standard.
|
|||
| 2 | 60 | 49 | 11 |
| 1 | 67 | 57 | 10 |
| 0.5 | 42 | 38 | 4 |
| 0.1 | 9.4 | 9 | 0.4 |
From Table 5 it can be seen that using 1 mol% of complex 4 with only 5 equivalents of the oxidant at room temperature we were now able to achieve the highest conversion of all our test reactions. Furthermore we were able to show that with increasing the catalyst loading to 2 mol%, the catalytic activity seems to decrease again, which might be attributed to the fact that at higher catalyst loadings the water oxidation side reaction might become the favored reaction.7b,8 Decreasing the catalyst loading to only 0.1 mol% results in only minor conversions of the starting material.
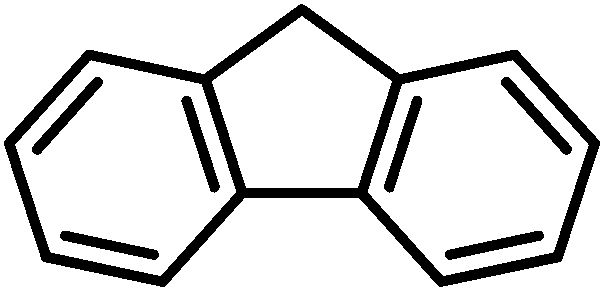
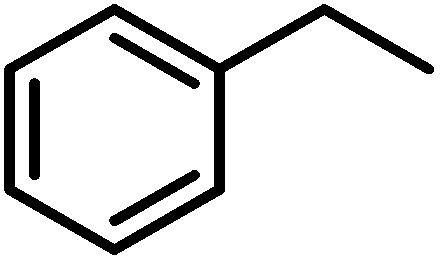
Finally, we turned our attention to the substrate scope of the reaction. Fluorene and ethyl benzene were tested with special interest in the latter, since the desired product would be acetophenone, which is a major starting material in organic synthesis. For fluorene, performing this reaction at room temperature with only 0.5 mol% of the catalyst we were not able to obtain good conversions. Raising the temperature to 45 °C using 0.5 mol% of 4 we were already able to achieve a moderate conversion of 52%. Increasing the catalyst loading to 1 mol% we were able to achieve excellent conversions of 87% in an average of 2 runs (Table 6). Switching to ethyl benzene as the substrate we were able to obtain the same trends. While performing the reaction at room temperature with low catalyst loadings we could only find moderate conversions of 78% of the starting material and 17% of acetophenone. Increasing the temperature results in higher conversions to acetophenone (Table 6). The best results were again obtained using 1 mol% of the catalyst at 45 °C. Hence, these conditions afforded an average conversion of 90% of ethyl benzene, and 69% of acetophenone was found in the solution after the reaction. Despite all our efforts, we were not able to determine the fate of the starting material that is not converted to acetophenone.
| Substrate | Loading [mol%] | Temp. [°C] | Overalla [%] | Acetophenonea [%] |
|---|---|---|---|---|
| a All reactions were carried out using 1 mmol of the substrate. Conversions were determined by NMR spectroscopy using 0.2 eq. of 4-bromobenzaldehyde as an internal standard. | ||||
|
0.5 | RT | 11 | — |
| 0.5 | 45 | 52 | — | |
| 1 | 45 | 87 | — | |
|
0.5 | RT | 78 | 17 |
| 0.5 | 45 | 95 | 47 | |
| 1 | 45 | 90 | 69 | |
Conclusions
We have reported here on five new Ir(III) complexes (2–6) with chelating “click”-derived 1,2,3-triazoles and 1,2,3-triazolylidenes. This is the first report of a complex where a mono-methylated triazol-triazolylidene N/C-donor (L5) is bound to only one metal center in a chelating fashion (complex 5). A total of eight complexes were tested towards their catalytic activity in the direct oxidation of cyclooctane under two different conditions with 0.5 mol% of the catalyst at elevated temperatures. The complexes (2 and 4) with unsymmetrical ligands form much better catalysts compared to complexes 1, 3 and 6 with symmetrical ligands. Optimizing the catalytic conditions, we were able to produce cyclooctanone in high yields, using mild conditions (room temperature) and low catalyst loadings (1 mol%). Additionally, we were able to show that other substrates such as fluorene and ethylbenzene can be converted to their corresponding ketones. Compared to Crabtree's system,7b under optimized conditions we achieved about the same conversion (57%) as them (54%) but by using only a fifth of the amount of catalyst and half the equivalents of the oxidant.7b In comparison with general literature reports, this result is one of the best as well. There are a few reports on using ruthenium,10,11a,b iron,11c–e manganese11f,g or copper11h based catalyst systems to reach higher conversion but the method also requires the use of higher catalyst loadings of up to 10 mol% and usually uses relatively complicated ligand systems. In view of the importance of direct C–H oxidations, we are trying to further optimize the reaction conditions with the unsymmetrical, pyridine containing ligands, especially for the use of lighter alkanes and to extend the methodology presented here to the oxidation of sp2 carbons.Experimental section
Materials and physical methods
[IrCp*Cl2]2 and bipyridine (L1) were purchased from Merck. L2,4gL7 and L8,4d were synthesized according to known procedures. The complex [IrCp*Cl(L1)] PF6 (1) was synthesised as reported before.9 All the reagents were used as supplied. The solvents used for metal complex synthesis were dried and distilled under argon and degassed by common techniques prior to use. 1H and 13C NMR spectra were recorded on a Jeol ECS 400 spectrometer or a Bruker Avance 700 spectrometer. Mass spectrometry was performed on an Agilent 6210 ESI-TOF.Synthesis of ligands
:1 as eluents. The desired product was obtained as a light yellow oil. Yield: 83% (397 mg, 2.29 mmol). 1H NMR (400 MHz, CDCl3; 25 °C, TMS): δ (ppm) = 1.60 (t, J = 7.4 Hz, 3H, CH3); 4.48 (q, J = 7.4 Hz, 2H, CH2); 7.17–7.24 (m, 1H, py-H); 7.77 (m, 1H, py-H); 8.08–8.22 (m, 2H, py-H + triazole-5H); 8.57 (m, 1H, py-H) 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 15.5 (CH3), 45.5 (CH2), 120.3 (py-C), 121.4 (py-C), 122.9 (triazole-4C), 137.0 (triazole-5C), 149.4 (py-C), 150.4 (py-C) MS (ESI): m/z cacld 175.0984 found 175.0988 [C9H10N4 + H+].
:1 as an eluent. The desired product was obtained as a white powder. Yield: 72% (414 mg, 2.16 mmol). 1H NMR (400 MHz, CDCl3; 25 °C, TMS): δ (ppm) = 1.55 (t, J = 7.4 Hz, 6H, CH3); 4.43 (q, J = 7.4 Hz, 4H, CH2); 8.03 (s, 2H, triazole-5H) 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 15.6 (CH3), 45.5 (CH2), 112.0 (triazole-4C), 140.3 (triazole-5C), MS (ESI): m/z calcd 193.1202, found 193.1202 [C8H12N6 + H+].
:1. The product was obtained as a light brown solid. Yield: 90% (248 mg, 0.9 mmol). 1H NMR (400 MHz, CDCl3; 25 °C, TMS): δ (ppm) = 1.64 (t, J = 7.2 Hz, 3H, CH3); 4.56 (s, 3H, N-CH3); 4.65 (q, J = 7.2 Hz, 2H, CH2); 7.37–7.45 (m, 1H, aryl-H); 7.85–7.91 (m, 1H, aryl-H); 7.96–8.00 (m 1H, aryl-H); 8.65–8.69 (m, 1H, Py-H); 9.04 (s, 1H, triazol-5H) 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ (ppm) = 14.5 (CH3), 41.2 (N-CH3), 50.1 (N-CH2), 125.5 (py-C), 125.9 (py-C), 129.7 (triazole-4C), 138.6 (triazole-5C), 140.9(py-C), 143.0 (py-C), 149.8 (py-C), MS (ESI): m/z calcd 189.1140, found 189.1140 [C10H13N4+].
Synthesis of the iridium complexes
Complexes with C^N or C^C coordination motifs
The complexes were synthesized according to literature known procedures.3b The corresponding ligands (2 eq., 0.2 mmol) were mixed with basic silver(I) oxide (7 eq., 163 mg, 0.7 mmol) and a chloride source, potassium chloride (20 eq., 155 mg, 2 mmol), and were dissolved, under nitrogen, in acetonitrile (10 ml). The mixtures were stirred under the exclusion of light for 2 days. Afterwards the black mixture was filtered and all solvents were removed under high vacuum. The remaining white-yellowish solids were then dissolved in dichloromethane (15 ml) and [Ir(Cp*)Cl2]2 (1 eq., 80 mg, 0.1 mmol) was added, and the mixture was again stirred for 2 days under the exclusion of light. The precipitated silver(I) chloride was filtered off through a pad of Celite and all volatiles were removed. The crude product was then dissolved in methanol (5 ml) and KPF6 (8 eq., 147 mg, 0.8 mmol) was added, and the solution was stirred for another 20 minutes before water (80 ml) was slowly added to cause precipitation of the desired complexes. The yellow solids were filtered and dried under air. When precipitation from water was not successful, the aqueous phase was extracted with dichloromethane (3 times × 20 ml). The organic layer was collected, dried over NaSO4 and the solvents were evaporated unitl only 5 ml of dichloromethane was left. Addition of hexane (50 ml) to that solution caused precipitation of the desired complexes and the yellow solids were collected by filtration. The complexes were obtained as yellow powders in moderate yields of 40% and higher.X-ray crystallography
X-ray quality crystals of 4 and 6 were obtained by layering concentrated solutions in dichloromethane with hexane at 8 °C. Single crystals of 5 were obtained by layering a chloroform solution with hexane at room temperature. X-ray data were collected on a Bruker Smart AXS. Data were collected at 140(2) K using graphite-monochromated Mo Kα radiation (λα = 0.71073 Å). The strategy for the data collection was evaluated using the Smart software. The data were collected by the standard ‘omega scan techniques’, and were scaled and reduced using Saint+ software. The structures were solved by direct methods using SHELXS-97 and refined by full matrix least-squares with SHELXL-97, refining on F2.13 CCDC 1002759, 1010867 and 1002758 contain the CIF files for 4, 5·CHCl3·0.25H2O and 6·CH2Cl2 respectively.Acknowledgements
We are grateful to the Fonds der Chemischen Industrie (FCI) and the Freie Universität Berlin for the financial support of this project. A. B. and J. K. acknowledge the financial support from the Ministry of Education, Science and Sport, Republic of Slovenia, the Slovenian Research Agency (grant P1-0230), and the Slovene Human Resources Development and Scholarship Fund, Public Call for Scholarships or Grants for the Research Cooperation of Doctoral Students Abroad in 2012, no. 11012-16/2013.References
- For selected reviews see: (a) L. Liang and D. Astruc, Chem. Rev., 2011, 255, 2933 CAS; (b) H. Struthers, T. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675 RSC; (c) D. Schweinfurth, N. Deibel, F. Weisser and B. Sarkar, Nach. Chem., 2011, 59, 937 CrossRef CAS; (d) J. D. Crowley and D. McMorran in Topics in Heterocyclic Chemistry, ed. J. Košmrlj, Springer, Berlin/Heidelberg, 2012, vol. 22, p. 31 Search PubMed; (e) Y. H. Lau, P. J. Rutledge, M. Watkinson and M. H. Todd, Chem. Soc. Rev., 2011, 40, 2848 RSC.
- For selected examples see: (a) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534 CrossRef CAS PubMed; (b) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145 RSC; (c) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755 CrossRef CAS PubMed; (d) S. Hohloch, C.-Y. Su and B. Sarkar, Eur. J. Inorg. Chem., 2011, 3067 CrossRef CAS; (e) S. Hohloch, L. Nauton, F. Cisnetti and A. Gautier, Tetrahedron Lett., 2013, 54, 1808 CrossRef CAS PubMed; (f) S. Hohloch, D. Scheiffele and B. Sarkar, Eur. J. Inorg. Chem., 2013, 3956 CrossRef CAS; (g) S. Hohloch, W. Frey, C.-Y. Su and B. Sarkar, Dalton Trans., 2013, 42, 11355 RSC; (h) G. G. Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759 CrossRef PubMed; (i) K. J. Kilpin, U. S. D. Paul, A.-L. Lee and J. D. Crowley, Chem. Commun., 2011, 47, 328 RSC.
- For selected examples see: (a) D. Schweinfurth, N. Büttner, S. Hohloch, N. Deibel, J. Klein and B. Sarkar, Organometallics, 2013, 32, 5834 CrossRef CAS; (b) S. Hohloch, L. Suntrup and B. Sarkar, Organometallics, 2013, 32, 7376 CrossRef CAS; (c) S. Hohloch, D. Schweinfurth, M. G. Sommer, F. Weisser, N. Deibel, F. Ehret and B. Sarkar, Dalton Trans., 2014, 43, 4437 RSC; (d) U. Monkowius, S. Ritter, B. König, M. Zabel and H. Yersin, Eur. J. Inorg. Chem., 2007, 4597 CrossRef CAS; (e) J. M. Fletcher, B. J. Bumgarner, N. D. Engels and D. A. Skoglund, Organometallics, 2008, 27, 5430 CrossRef CAS; (f) C. E. Welby, S. Grkinic, A. Zahid, B. S. Uppal, E. A. Gibson, C. R. Rice and P. I. P. Elliot, Dalton Trans., 2012, 41, 7637 RSC; (g) C. E. Welby, L. Gilmartin, R. R. Marriott, A. Zahid, C. R. Rice, E. A. Gibson and P. I. P. Elliott, Dalton Trans., 2013, 42, 13527 RSC.
- For selected examples see: (a) A. Bolje, S. Hohloch, D. Urankar, A. Pevec, M. Gazvoda, B. Sarkar and J. Košmrlj, Organometallics, 2014, 33, 2588 CrossRef CAS; (b) S. Hohloch, L. Hettmanczyk and B. Sarkar, Eur. J. Inorg. Chem., 2014, 3164 CrossRef CAS; (c) A. Bolje and J. Košmrlj, Org. Lett., 2013, 15, 5084 CrossRef CAS PubMed; (d) R. Maity, S. Hohloch, C.-Y. Su, M. van der Meer and B. Sarkar, Chem. – Eur. J., 2014, 20, 9952 CrossRef CAS PubMed; (e) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Organometallics, 2011, 30, 6017 CrossRef CAS PubMed; (f) J. M. Aizpurua, M. Sgartzazu-Aizourua, Z. Monasterio, I. Azcune, C. Mendicute, J. I. Miranda, E. Garcia-Lecina, A. Altube and R. M. Fratila, Org. Lett., 2012, 14, 1866 CrossRef CAS PubMed; (g) L. Bernet, R. Lalrempuia, W. Ghattas, H. Müller-Bunz, L. Vigara, A. Llobet and M. Albrecht, Chem. Commun., 2011, 47, 8058 RSC; (h) M. Delganod-Rebollo, D. Canseco-Gonzales, M. Hollering, H. Müller-Bunz and M. Albrecht, Dalton Trans., 2014, 43, 4462 RSC.
- For selected examples see: (a) N. D. McDaniel, F. J. Coughlin, L. L. Tinker and S. Bernhard, J. Am. Chem. Soc., 2008, 130, 210 CrossRef CAS PubMed; (b) J. F. Hull, D. Balcells, J. D. Blakemore, C. D. Incarvito, O. Eisenstein, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2009, 131, 8730 CrossRef CAS PubMed; (c) T. P. Brewster, J. D. Blakemore, N. D. Schley, C. D. Incarvito, N. Hazari, G. W. Brudvig and R. H. Crabtree, Organometallics, 2011, 30, 965 CrossRef CAS; (d) J. D. Blakemore, N. D. Schley, M. N. Kushner-Lenhoff, A. M. Winter, F. D'Souza, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2012, 51, 7749 CrossRef CAS PubMed.
- For selected examples see: (a) B. B. Grothjahn, D. B. Brown, J. K. Martin, D. C. Marelius, M.-C. Abadjian, H. N. Tran, G. Kalyuzhny, K. S. Vecchio, Z. G. Specht, S. A. Cortes-Llama, V. Miranda-Soto, C. van Niekerk, C. E. Moore and A. L. Rheingold, J. Am. Chem. Soc., 2011, 133, 19024 CrossRef PubMed; (b) A. J. Ingram, A. B. Wolk, C. Flender, J. Zhang, C. J. Johnson, U. Hintermair, R. H. Crabtree, M. A. Johnson and R. N. Zare, Inorg. Chem., 2014, 53, 423 CrossRef CAS PubMed; (c) U. Hintermair, S. W. Sheehan, A. R. Parent, D. H. Ess, D. T. Richens, P. H. Vaccaro, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 2013, 135, 10837 CrossRef CAS PubMed; (d) A. Savini, A. Bucci, G. Bellachioma, L. Rocchigiani, C. Zuccaccia, A. Llobet and A. Macchioni, Eur. J. Inorg. Chem., 2014, 690 CrossRef CAS; (e) R. H. Crabtree, J. Organomet. Chem., 2014, 751, 174 CrossRef CAS PubMed.
- For selected examples see: (a) M. Zhou, N. D. Schley and R. H. Crabtree, J. Am. Chem. Soc., 2010, 132, 12550 CrossRef CAS PubMed; (b) M. Zhou, U. Hintermair, B. G. Hashiguchi, A. R. Parent, S. M. Hashmi, M. Elimelech, R. A. Periana, G. W. Brudvig and R. H. Crabtree, Organometallics, 2013, 32, 957 CrossRef CAS; (c) M. Zhou, D. Balcells, A.-R. Parent, R. H. Crabtree and O. Eisenstein, ACS Catal., 2012, 2, 208 CrossRef CAS; (d) B. G. Hashiguchi, S. M. Bischoff, M. M. Konnick and R. A. Periana, Acc. Chem. Res., 2012, 45, 885 CrossRef CAS PubMed.
- For selected examples see: (a) R. Lalrempuia, N. D. McDaniel, H. Müller-Bunz, S. Bernhard and M. Albrecht, Angew. Chem., Int. Ed., 2010, 49, 9765 CrossRef CAS PubMed; (b) A. Petronilho, J. A. Woods, S. Bernhard and M. Albrecht, Eur. J. Inorg. Chem., 2014, 708 CrossRef CAS; (c) J. A. Woods, R. Lalremupia, N. D. McDaniel, H. Müller-Bunz, M. Albrecht and S. Bernhard, Energy Environ. Sci., 2014, 7, 2316 RSC.
- (a) W. Kaim, R. Reinhardt and M. Sieger, Inorg. Chem., 1994, 33, 4453 CrossRef CAS; (b) M.-T. Youinou and R. Ziessel, J. Organomet. Chem., 1989, 363, 197 CrossRef CAS.
- M. Yamaguchi, H. Kouska, S. Izawa, Y. Ichii, T. Kumano, D. Masui and T. Yamaghishi, Inorg. Chem., 2006, 45, 8342 CrossRef CAS PubMed.
- For selected examples see: (a) S. Murahashi, Y. Oda, N. Komiya and T. Naota, Tetrahedron Lett., 1994, 35, 7953 CrossRef CAS; (b) C. S. Yi, K.-H. Kwon and D. W. Lee, Org. Lett., 2009, 11, 1567 CrossRef CAS PubMed; (c) K. E. Djernes, M. Padilla, M. Mettry, M. C. Young and R. J. Hooley, Chem. Commun., 2012, 48, 11576 RSC; (d) V. Mirkhani, M. Moghadam, S. Tangestaninejad, I. Mohammadpoor-Baltork and N. Rasouli, Catal. Commun., 2008, 9, 2171 CrossRef CAS PubMed; (e) I. Prat, L. Gomez, M. Canta, X. Ribas and M. Costas, Chem. – Eur. J., 2013, 19, 1908 CrossRef CAS PubMed; (f) Z. Li, C.-G. Xia and C.-Z. Xu, Tetrahedron Lett., 2003, 44, 9229 CrossRef CAS PubMed; (g) D. Shen, C. Miao, S. Wang, C. Xia and W. Sun, Org. Lett., 2014, 16, 1108 CrossRef CAS PubMed; (h) G. Pozzi, M. Cavazzini, S. Quici and S. Fontana, Tetrahedron Lett., 1997, 38, 7605 CrossRef CAS.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornoe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef CAS PubMed.
- G. M. Sheldrick, SHELX-97, Program for Crystal Structure Refinement, Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallography table and NMR spectra. CCDC 1002759, 1010867 and 1002758. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02879a |
| This journal is © The Royal Society of Chemistry 2015 |